
Hyperhomocysteinemia in Adult Patients: A Treatable Metabolic Condition
 , , and
, , and
Abstract
:1. Introduction
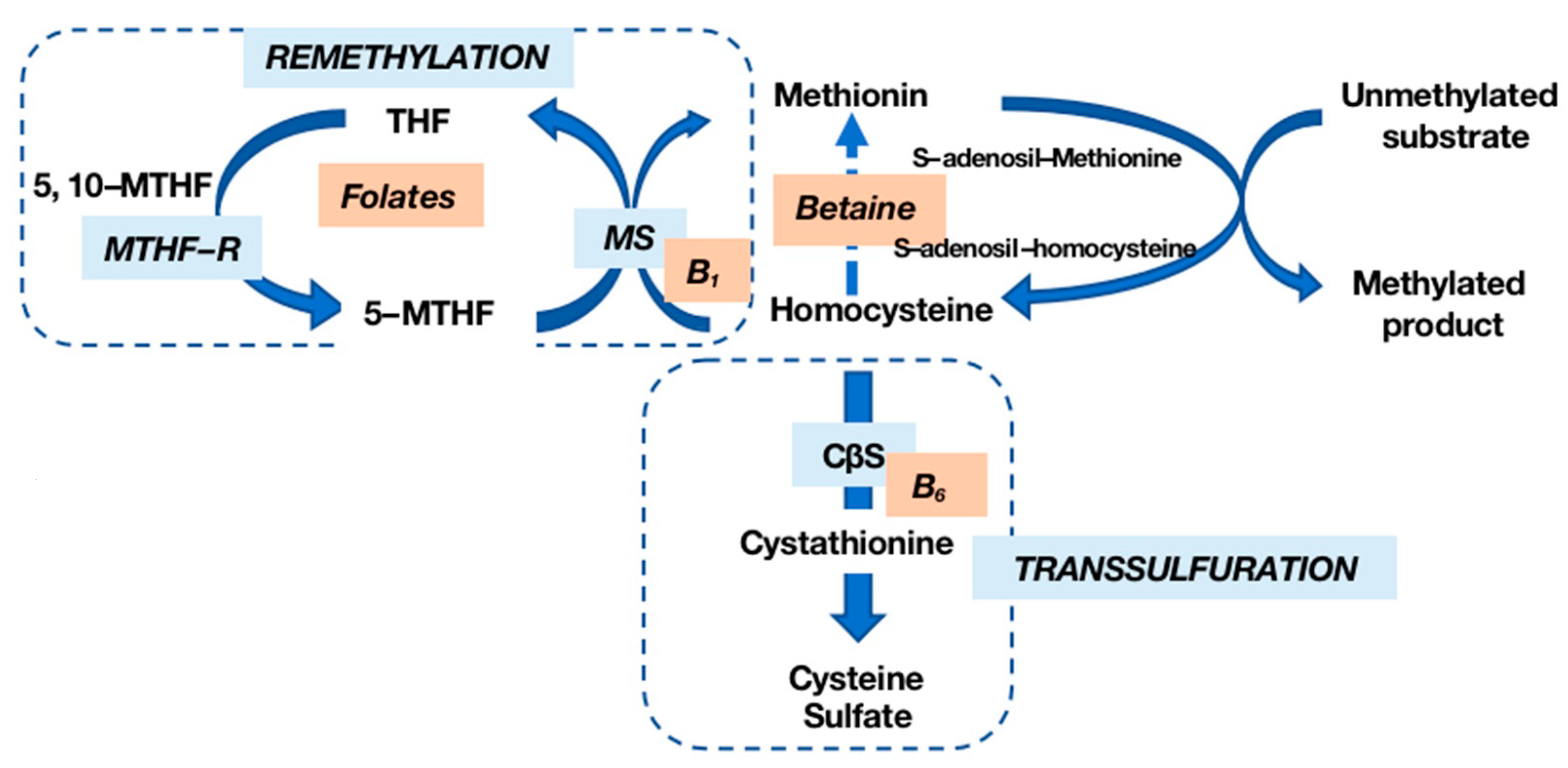
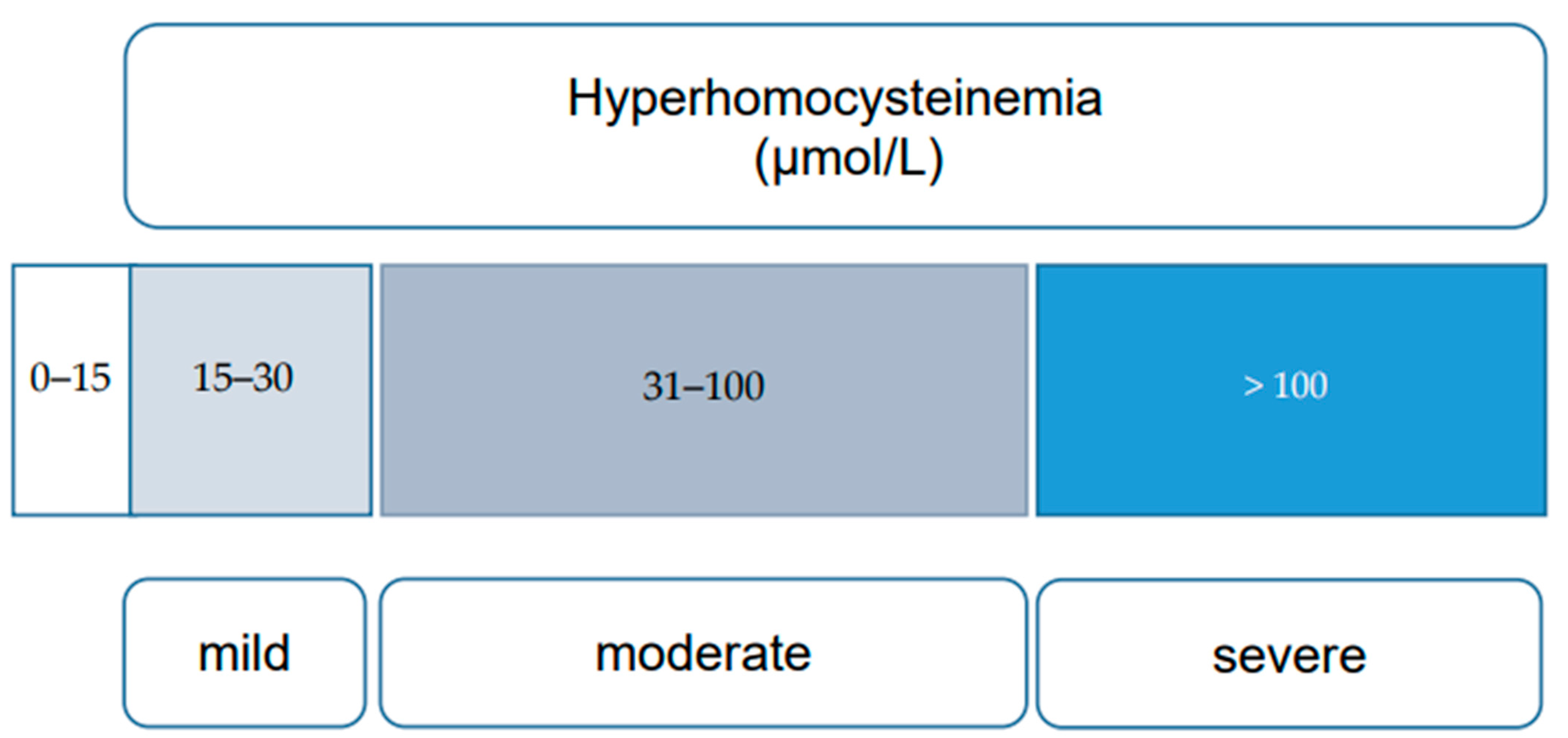
2. Types of Hyperhomocysteinemias (HHcy)
3. Role of Homocysteine in Disease Processes (Toxicity)
4. Clinical Manifestations of HHcy
5. Specific Organ or System Manifestations (Table 2)
5.1. Vascular

{kind=link}
{kind=link}
{kind=link}
| HCU and Hematological/Vascular Pathology | HCU and Ophthalmological Pathology |
Classical HCU
| Classical HCU
|
| HCU and Neuropsychiatric Pathology | HCU and Fertility |
Classical HCU
| HCU due to Remethylation Defects
|
| HCU and Renal Disease | Others (Skeletal, Hearing Loss, …) |
Hyperhomocysteinemia
| Skeletal
|
5.2. Hematological
5.3. Marfanoid Habit and Osteoporosis
5.4. Neuropsychiatric Abnormalities
5.5. Kidney Disease
5.6. Ocular Abnormalities
5.7. Hearing Loss
5.8. Reproductive Medicine and Pregnancy
6. Management of Patients with Suspected HHcy
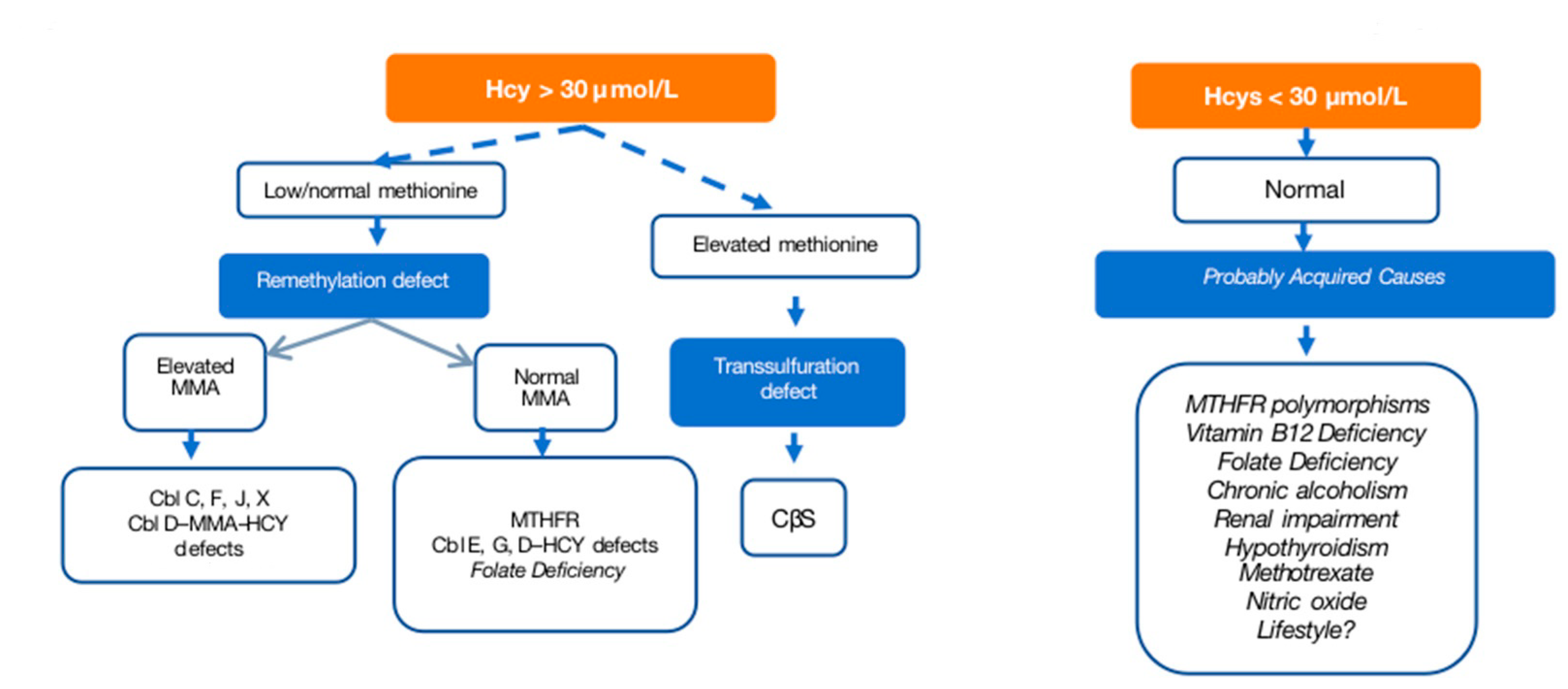
6.1. Evaluation
6.2. Treatment
7. Hypoprotein Diet
8. Conclusions
Funding
Conflicts of Interest
References
- Lee, M.; Hong, K.-S.; Chang, S.-C.; Saver, J.L. Efficacy of homocysteine-lowering therapy with folic acid in stroke prevention. Stroke 2010, 41, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, T.; Zheng, Y.; Muka, T.; Troup, J.; Hu, F.B. Folic Acid Supplementation and the Risk of Cardiovascular Diseases: A Meta-Analysis Of Randomized Controlled Trials. J. Am. Heart Assoc. 2016, 5, e003768. [Google Scholar] [CrossRef] [PubMed]
- Martí-Carvajal, A.J.; Solà, I.; Lathyris, D.; Dayer, M. Homocysteine-lowering interventions for preventing cardiovascular events. Cochrane Database Syst. Rev. 2017, 8, CD006612. [Google Scholar] [CrossRef] [PubMed]
- Huemer, M.; Diodato, D.; Schwahn, B.; Schiff, M.; Bandeira, A.; Benoist, J.; Burlina, A.; Cerone, R.; Couce, M.L.; Garcia-Cazorla, A.; et al. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J. Inherit. Metab. Dis. 2017, 40, 21–48. [Google Scholar] [CrossRef]
- Veeranki, S.; Gandhapudi, S.K.; Tyagi, S.C.; Majumder, A.; Singh, M.; George, A.K. Interactions of hyperhomocysteinemia and T cell immunity in causation of hypertension. Can. J. Physiol. Pharmacol. 2017, 95, 239–246. [Google Scholar] [CrossRef]
- Bostom, A.G.; Carpenter, M.A.; Kusek, J.W.; Levey, A.S.; Hunsicker, L.; Pfeffer, M.A.; Selhub, J.; Jacques, P.F.; Cole, E.; Gravens-Mueller, L.; et al. Homocysteine-lowering and cardiovascular disease outcomes in kidney transplant recipients: Primary results from the Folic Acid for Vascular Outcome Reduction in Transplantation trial. Circulation 2011, 123, 1763–1770. [Google Scholar] [CrossRef]
- Park, W.-C.; Chang, J.-H. Clinical Implications of Methylenetetrahydrofolate Reductase Mutations and Plasma Homocysteine Levels in Patients with Thromboembolic Occlusion. Vasc. Spec. Int. 2014, 30, 113–119. [Google Scholar] [CrossRef]
- Malinow, M.R.; Bostom, A.G.; Krauss, R.M. Homocyst(e)ine, diet, and cardiovascular diseases: A statement for healthcare professionals from the Nutrition Committee, American Heart Association. Circulation 1999, 99, 178–182. [Google Scholar] [CrossRef]
- Wilcken, D.E.L.; Wilcken, B. The natural history of vascular disease in homocystinuria and the effects of treatment. J. Inherit. Metab. Dis. 1997, 20, 295–300. [Google Scholar] [CrossRef]
- Smith, A.D.; Smith, S.M.; de Jager, C.A.; Whitbread, P.; Johnston, C.; Agacinski, G.; Oulhaj, A.; Bradley, K.M.; Jacoby, R.; Refsum, H. Homocysteine-lowering by b vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: A randomized controlled trial. PLoS ONE 2010, 5, e12244. [Google Scholar] [CrossRef]
- Cylwik, B.; Chrostek, L. Disturbances of folic acid and homocysteine metabolism in the context of alcohol abuse. Pol. Merkur. Lekarski. 2011, 30, 295–299. (In Polish) [Google Scholar] [PubMed]
- Hirschowitz, B.I.; Worthington, J.; Mohnen, J. Vitamin B12 deficiency in hypersecretors during long-term acid suppression with proton pump inhibitors. Aliment. Pharmacol. Ther. 2008, 27, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Cianciolo, G.; De Pascalis, A.; Di Lullo, L.; Ronco, C.; Zannini, C.; La Manna, G. Folic Acid and Homocysteine in Chronic Kidney Disease and Cardiovascular Disease Progression: Which Comes First? Cardiorenal Med. 2017, 7, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Smach, M.A.; Jacob, N.; Golmard, J.-L.; Charfeddine, B.; Lammouchi, T.; Ben Othman, L.; Dridi, H.; Bennamou, S.; Limem, K. Folate and homocysteine in the cerebrospinal fluid of patients with Alzheimer’s disease or dementia: A case control study. Eur. Neurol. 2011, 65, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Dietrich-Muszalska, A.; Malinowska, J.; Olas, B.; Głowacki, R.; Bald, E.; Wachowicz, B.; Rabe-Jabłońska, J. Oxidative stress may be induced by elevated homocysteine in schizophrenic patients. Neurochem. Res. 2012, 37, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- LeBoff, M.S.; Narweker, R.; LaCroix, A.; Wu, L.; Jackson, R.; Lee, J.; Bauer, D.C.; Cauley, J.; Kooperberg, C.; Lewis, C.; et al. Homocysteine levels and the risk of hip fracture in postmenopausal women. J. Clin. Endocrinol. Metab. 2009, 94, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.A.M.; Kožich, V.; Santra, S.; Andria, G.; Ben-Omran, T.I.M.; Chakrapani, A.B.; Crushell, E.; Henderson, M.J.; Hochuli, M.; Huemer, M.; et al. Guidelines for the diagnosis and management of cystathionine beta-synthase deficiency. J. Inherit. Metab. Dis. 2017, 40, 49–74. [Google Scholar] [CrossRef] [PubMed]
- Suárez García, I.; Gómez Cerezo, J.F.; Ríos Blanco, J.J.; Barbado Hernández, F.J.; Vázquez Rodríguez, J.J. Homocysteine: The cardiovascular risk factor of the next millennium? An. Med. Interna. 2001, 18, 211–217. (In Spanish) [Google Scholar]
- Nuño-Ayala, M.; Carnicera, R.; Guzmán, M.A.; Guillén, N.; Navarro, M.A.; Arnal, A.; Osada, J. Homocysteine: Current panorama and the mouse’s contribution to its study. Clín. Investig. Arterioscler. 2010, 22, 200–219. [Google Scholar] [CrossRef]
- Couce, M.L.; Balcells, S.; Sánchez-Pintos, P.; Aldámiz Echevarría, L.; del Toro, M. Protocol for homocystinuria. In Protocols for the Diagnosis and Treatment of Congenital Metabolic Errors (AECOM), 2nd ed.; Ortega, D., Ed.; Ergon: Madrid, Spain, 2018; pp. 167–179. [Google Scholar]
- Mudd, H.; Levy, H.L.; Skovby, F. Disorders of transulfuration. In The Metabolic and Molecular Basis of Inherited Diseases, 7th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 1995; pp. 1279–1327. [Google Scholar]
- Heydrick, S.J.; Weiss, N.; Thomas, S.R.; Cap, A.P.; Pimentel, D.R.; Loscalzo, J.; Keaney, J.F. L-Homocysteine and L-homocystine stereospecifically induce endothelial nitric oxide synthase-dependent lipid peroxidation in endothelial cells. Free. Radic. Biol. Med. 2004, 36, 632–640. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine is a protein amino acid in humans. Implications for homocysteine-linked disease. J. Biol. Chem. 2002, 277, 30425–30428. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, D.W. Cellular mechanisms of homocysteine pathogenesis in atherosclerosis. In Homocysteine in Health and Disease; Carmel, R., Jacobsen, D., Eds.; Cambridge University Press: Cambridge, UK, 2001; pp. 425–440. [Google Scholar]
- Wijerathne, C.U.B.; Hewage, S.M.; Siow, Y.L.; O, K. Kidney Ischemia-Reperfusion Decreases Hydrogen Sulfide and Increases Oxidative Stress in the Heart. Biomolecules. Biomolecules 2020, 10, 1565. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, A.; Samra, Y.A.; Elsherbiny, N.M.; Al-Shabrawey, M. Implication of Hyperhomocysteinemia in Blood Retinal Barrier (BRB) Dysfunction. Biomolecules 2020, 10, 1119. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.H. The reaction of homocysteine with aldehyde: An explanation of the collagen defects in homocystinuria. Clin. Chim. Acta 1973, 45, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Lubec, B.; Fang-Kircher, S.; Lubec, T.; Blom, H.; Boers, G. Evidence for McKusick’s hypothesis of deficient collagen cross-linking in patients with homocystinuria. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1996, 1315, 159–162. [Google Scholar] [CrossRef]
- Mudd, S.H.; Skovby, F.; Levy, H.L.; Pettigrew, K.D.; Wilcken, B.; Pyeritz, R.E.; Andria, G.; Boers, G.H.; Bromberg, I.L.; Cerone, R. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am. J. Hum. Genet. 1985, 37, 1–31. [Google Scholar]
- Ovechkin, A.V.; Tyagi, N.; Sen, U.; Lominadze, D.; Steed, M.M.; Moshal, K.S.; Tyagi, S.C.; Beard, R.S.; Bearden, S.E.; Kumar, M.; et al. 3-Deazaadenosine mitigates arterial remodeling and hypertension in hyperhomocysteinemic mice. Am. J. Physiol. Cell Mol. Physiol. 2006, 291, L905–L911. [Google Scholar] [CrossRef]
- Giusti, B.; Marcucci, R.; Lapini, I.; Sestini, I.; Lenti, M.; Yacoub, M.; Pepe, G. Role of hyperhomocysteinemia in aortic disease. Cell. Mol. Biol. 2004, 50, 945–952. [Google Scholar]
- Takagi, H.; Umemoto, T. Homocysteinemia is a risk factor for aortic dissection. Med. Hypotheses 2005, 64, 1007–1010. [Google Scholar] [CrossRef]
- Tribouilloy, C.M.; Peltier, M.; Peltier, M.C.I.; Trojette, F.; Andrejak, M.; Lesbre, J.-P.M. Plasma homocysteine and severity of thoracic aortic atherosclerosis. Chest 2000, 118, 1685–1689. [Google Scholar] [CrossRef]
- Novaro, G.M.; Aronow, H.D.; Mayer-Sabik, E.; Griffin, B.P. Plasma homocysteine and calcific aortic valve disease. Heart 2004, 90, 802–803. [Google Scholar] [CrossRef] [PubMed]
- Chicoine, E.; Estève, P.-O.; Robledo, O.; Van Themsche, C.; Potworowski, E.F.; St-Pierre, Y. Evidence for the role of promoter methylation in the regulation of MMP-9 gene expression. Biochem. Biophys. Res. Commun. 2002, 297, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Zaina, S.; Lindholm, M.W.; Lund, G. Nutrition and aberrant DNA methylation patterns in atherosclerosis: More than just hyperhomocysteinemia? J. Nutr. 2005, 135, 5–8. [Google Scholar] [CrossRef]
- Krishna, S.M.; Dear, A.E.; Norman, P.E.; Golledge, J. Genetic and epigenetic mechanisms and their possible role in abdominal aortic aneurysm. Atherosclerosis 2010, 212, 16–29. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, Y.; Zhan, P.; Sun, W.; Dong, C.; Liu, X.; Yang, Y.; Wang, X.; Xie, Y.; Gao, C.; et al. Histone deacetylase 9 exacerbates podocyte injury in hyperhomocysteinemia through epigenetic repression of Klotho. Pharmacol. Res. 2023, 198, 107009. [Google Scholar] [CrossRef]
- Sánchez, O.; Fabregate, R.; Sabán Ruiz, J. Metabolic Factors I: Homocysteine, lipoproteins. In Global Control of Cardiometabolic Risk. Endothelial Dysfunction as the Preferential Target. Vol. I. Physiopathological, Clinical, and Diagnostic Bases of Cardiovascular Risk Factors. Pathogenesis; Sabán Ruiz, J., Ed.; Ediciones Díaz de Santos: Madrid, Spain, 2009; pp. 609–622. [Google Scholar]
- Urreizti, R.; Asteggiano, C.; Bermudez, M.; Córdoba, A.; Szlago, M.; Grosso, C.; de Kremer, R.D.; Vilarinho, L.; D’almeida, V.; Martínez-Pardo, M.; et al. The p.T191M mutation of the CBS gene is highly prevalent among homocystinuric patients from Spain, Portugal and South America. J. Hum. Genet. 2006, 51, 305–313. [Google Scholar] [CrossRef]
- Magner, M.; Krupková, L.; Honzík, T.; Zeman, J.; Hyánek, J.; Kožich, V. Vascular presentation of cystathionine beta-synthase deficiency in adulthood. J. Inherit. Metab. Dis. 2010, 34, 33–37. [Google Scholar] [CrossRef]
- Linnebank, M.; Junker, R.; Nabavi, D.G.; Linnebank, A.; Koch, H.G. Isolated thrombosis due to the cystathionine β-synthase mutation c.833T>C (I278T). J. Inherit. Metab. Dis. 2003, 26, 509–511. [Google Scholar] [CrossRef]
- Mudd, S.H. Vascular disease and homocysteine metabolism. N. Engl. J. Med. 1985, 313, 751–753. [Google Scholar] [CrossRef]
- Yap, S.; Boers, G.H.; Wilcken, B.; Wilcken, D.E.; Brenton, D.P.; Lee, P.J.; Walter, J.H.; Howard, P.M.; Naughten, E.R. Vascular outcome in patients with homocystinuria due to cystathionine β-synthase deficiency treated chronically. Arter. Thromb. Vasc. Biol. 2001, 21, 2080–2085. [Google Scholar] [CrossRef]
- Andria, G.; Fowler, B.; Sebastio, G. Disorders of sulfur amino acid metabolism. In Inborn Metabolic Diseases, 4th ed.; Fernandes, J., Saudubray, J.M., Van den Berghe, G., Walter, J.H., Eds.; Springer: Berlin, Germany, 2006; pp. 273–282. [Google Scholar]
- Abbott, M.H.; Folstein, S.E.; Abbey, H.; Pyeritz, R.E.; Opitz, J.M. Psychiatric manifestations of homocystinuria due to cystathionine β-synthase deficiency: Prevalence, natural history, and relationship to neurologic impairment and vitamin B6-responsiveness. Am. J. Med. Genet. 1987, 26, 959–969. [Google Scholar] [CrossRef]
- Colafrancesco, G.; Di Marzio, G.M.; Abbracciavento, G.; Stoppioni, V.; Leuzzi, V.; Ferrara, M. Acute psychosis in an adolescent with undiagnosed homocystinuria. Eur. J. Pediatr. 2015, 174, 1263–1266. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Stewart, P.M. Homocystinuria and psychiatric disorder: A case report. Pathology 1999, 31, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Sedel, F.; Baumann, N.; Turpin, J.; Lyon-Caen, O.; Saudubray, J.; Cohen, D. Psychiatric manifestations revealing inborn errors of metabolism in adolescents and adults. J. Inherit. Metab. Dis. 2007, 30, 631–641. [Google Scholar] [CrossRef]
- Shankar, A.; Wang, J.J.; Chua, B.; Rochtchina, E.; Flood, V.; Mitchell, P. Positive association between plasma homocysteine level and chronic kidney disease. Kidney Blood Press. Res. 2008, 31, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.; Nie, J. Homocysteine in renal injury. Kidney Dis. 2016, 2, 80–87. [Google Scholar] [CrossRef]
- Kalantari, S.; Brezzi, B.; Bracciamà, V.; Barreca, A.; Nozza, P.; Vaisitti, T.; Amoroso, A.; Deaglio, S.; Manganaro, M.; Porta, F.; et al. Adult-onset CblC deficiency: A challenging diagnosis involving different adult clinical specialists. Orphanet J. Rare Dis. 2022, 17, 1–18. [Google Scholar] [CrossRef]
- Castelli, E.; Terrone, C.; Faraone, N.; Tizzani, A. Renal infarction in a hyperhomocysteinemic patient. Nephron 2002, 92, 749–750. [Google Scholar] [CrossRef]
- Guevara-Márquez, Y.C.; Vela-Amieva, M.; Juárez Echenique, J.C.; Ordaz Favila, J.C.; Belmont-Martínez, L. Ophthalmic manifestations of inborn errors of metabolism. Acta Pediátrica De México 2013, 34, 212–224. [Google Scholar]
- Huang, X.; Yang, Y.; Duan, Y.; Kuang, Y.-Q.; Lin, D. Homocysteine in retinal artery occlusive disease: A meta-analysis of cohort studies. Sci. Rep. 2017, 7, 15708. [Google Scholar] [CrossRef]
- Gómez-Ulla de Irazazábal, F. Management of Retinal Vein Occlusions. In Clinical Practice Guidelines of the SERV; Spanish Society of Retina and Vitreous: Santiago de Complostela, Spain, 2015; ISBN 978-84-606-5721-7. Available online: www.serv.es (accessed on 1 November 2023).
- Baumgartner, M.R.; Hörster, F.; Dionisi-Vici, C.; Haliloglu, G.; Karall, D.; Chapman, K.A.; Huemer, M.; Hochuli, M.; Assoun, M.; Ballhausen, D.; et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis. 2014, 9, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Weisfeld-Adams, J.D.; McCourt, E.A.; Diaz, G.A.; Oliver, S.C. Ocular disease in the cobalamin C defect: A review of the literature and a suggested framework for clinical surveillance. Mol. Genet. Metab. 2015, 114, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.P.; Thompson, A.H.; Sloan, J.L.; Manoli, I.; Carrillo-Carrasco, N.; Zein, W.M.; Venditti, C.P. Ophthalmic Manifestations and Long-Term Visual Outcomes in Patients with Cobalamin C Deficiency. Ophthalmology 2016, 123, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.O.; Pillers, D.-A.M.; Steiner, R.D.; Bottiglieri, T.; Rosenblatt, D.S.; Debley, J.; Gibson, K.M. Potential for misdiagnosis due to lack of metabolic derangement in combined methylmalonic aciduria/hyperhomocysteinemia (cblC) in the neonate. J. Perinatol. 2003, 23, 384–386. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.C.; Morel, C.F.; Scharer, G.; Yang, M.; Lerner-Ellis, J.P.; Rosenblatt, D.S.; Thomas, J.A. Late-onset combined homocystinuria and methylmalonic aciduria (cblC) and neuropsychiatric disturbance. Am. J. Med. Genet. Part A 2007, 143, 2430–2434. [Google Scholar] [CrossRef]
- Carrillo-Carrasco, N.; Chandler, R.J.; Venditti, C.P. Combined methylmalonic acidemia and homocystinuria, cblC type. I. Clinical presentations, diagnosis and management. J. Inherit. Metab. Dis. 2011, 35, 91–102. [Google Scholar] [CrossRef]
- Partearroyo, T.; Vallecillo, N.; Mandruzzato, G.; Celaya, A.M.; Zeisel, S.H.; Pajares, M.A.; Murillo-Cuesta, S.; Bermúdez-Muñoz, J.M.; LaRosa, L.R.-D.; Varela-Moreiras, G.; et al. Betaine-homocysteine S -methyltransferase deficiency causes increased susceptibility to noise-induced hearing loss associated with plasma hyperhomocysteinemia. FASEB J. 2019, 33, 5942–5956. [Google Scholar] [CrossRef]
- Canto-Cetina, T.; Polanco-Reyes, L.; Ballote-Zapata, M.; Ordóñez-Luna, M. Homocisteína y perfil de lípidos en embarazo normal y embarazo complicado con preeclampsia. Rev. Esp. Med. Quir. 2014, 19, 423–430. [Google Scholar]
- van Gool, J.D.; Hirche, H.; Lax, H.; De Schaepdrijver, L. Folic acid and primary prevention of neural tube defects: A review. Reprod. Toxicol. 2018, 80, 73–84. [Google Scholar] [CrossRef]
- Kožich, V.; Sokolová, J.; Morris, A.A.M.; Pavlíková, M.; Gleich, F.; Kölker, S.; Krijt, J.; Dionisi-Vici, C.; Baumgartner, M.R.; Blom, H.J.; et al. Cystathionine β-synthase deficiency in the E-HOD registry-part I: Pyridoxine responsiveness as a determinant of biochemical and clinical phenotype at diagnosis. J. Inherit. Metab. Dis. 2020, 44, 677–692. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Schiff, M.; Guffon, N.; Nadjar, Y.; García-Cazorla, A.; Casanova, M.M.-P.; Cano, A.; Couce, M.L.; Dalmau, J.; Peña-Quintana, L.; et al. Betaine anhydrous in homocystinuria: Results from the RoCH registry. Orphanet J. Rare Dis. 2019, 14, 66. [Google Scholar] [CrossRef] [PubMed]



| Disorders of Hcy Transsulfuration (Decreased Cystathionine-β-Synthase (CβS) Activity) | Disorders of Hcy Remethylation (Altered Methionine Synthase (MS) or Methyltetrahydrofolate Reductase (MTHFR) Activities) |
|---|---|
|
|
| Disorder | Hcy | Met | Cy | MMA | Vit.B12 | Folates | Macrocytic Anemia |
|---|---|---|---|---|---|---|---|
| CβS deficiency | ↑ | N/↑ | ↓ | N | N | N | − |
| Cbl-C | ↑ | ↓ | N/↑ | ↑ | N | N | +/− |
| Cbl-D/Cbl-F | ↑ | ↓ | N/↑ | ↑ | N | N | + |
| Cbl-E/Cbl-G | ↑ | ↓/N | N/↑ | N | N | N | + |
| MTHFR deficiency | ↑ | ↓ | N/↑ | N | N | N | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Lamuño, D.; Arrieta-Blanco, F.J.; Fuentes, E.D.; Forga-Visa, M.T.; Morales-Conejo, M.; Peña-Quintana, L.; Vitoria-Miñana, I. Hyperhomocysteinemia in Adult Patients: A Treatable Metabolic Condition. Nutrients 2024, 16, 135. https://doi.org/10.3390/nu16010135
González-Lamuño D, Arrieta-Blanco FJ, Fuentes ED, Forga-Visa MT, Morales-Conejo M, Peña-Quintana L, Vitoria-Miñana I. Hyperhomocysteinemia in Adult Patients: A Treatable Metabolic Condition. Nutrients. 2024; 16(1):135. https://doi.org/10.3390/nu16010135
Chicago/Turabian StyleGonzález-Lamuño, Domingo, Francisco Jesús Arrieta-Blanco, Elena Dios Fuentes, María Teresa Forga-Visa, Monstserrat Morales-Conejo, Luis Peña-Quintana, and Isidro Vitoria-Miñana. 2024. "Hyperhomocysteinemia in Adult Patients: A Treatable Metabolic Condition" Nutrients 16, no. 1: 135. https://doi.org/10.3390/nu16010135