
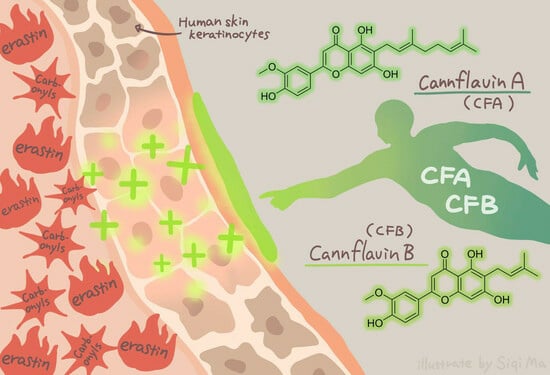
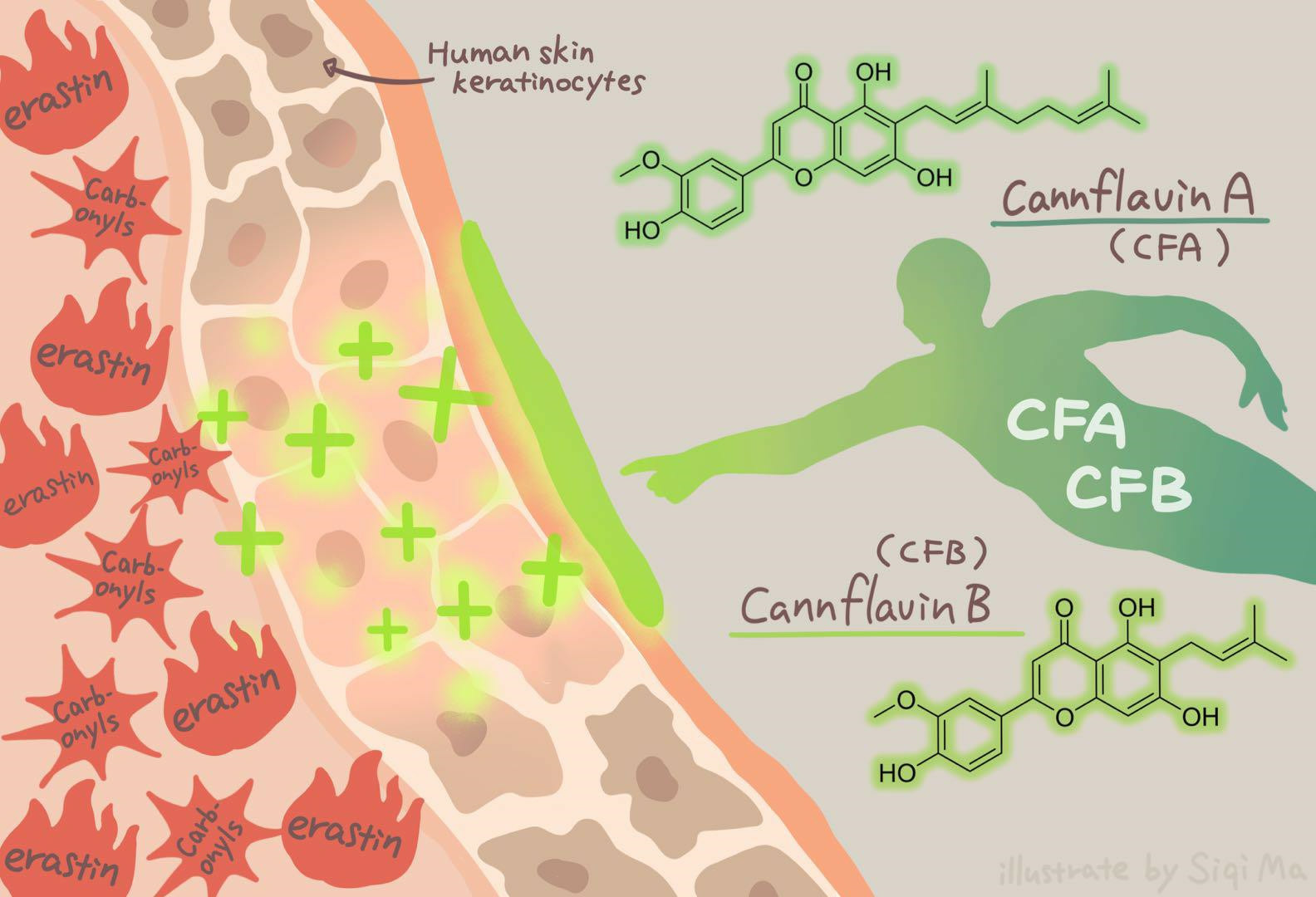
Cannflavins A and B with Anti-Ferroptosis, Anti-Glycation, and Antioxidant Activities Protect Human Keratinocytes in a Cell Death Model with Erastin and Reactive Carbonyl Species
 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
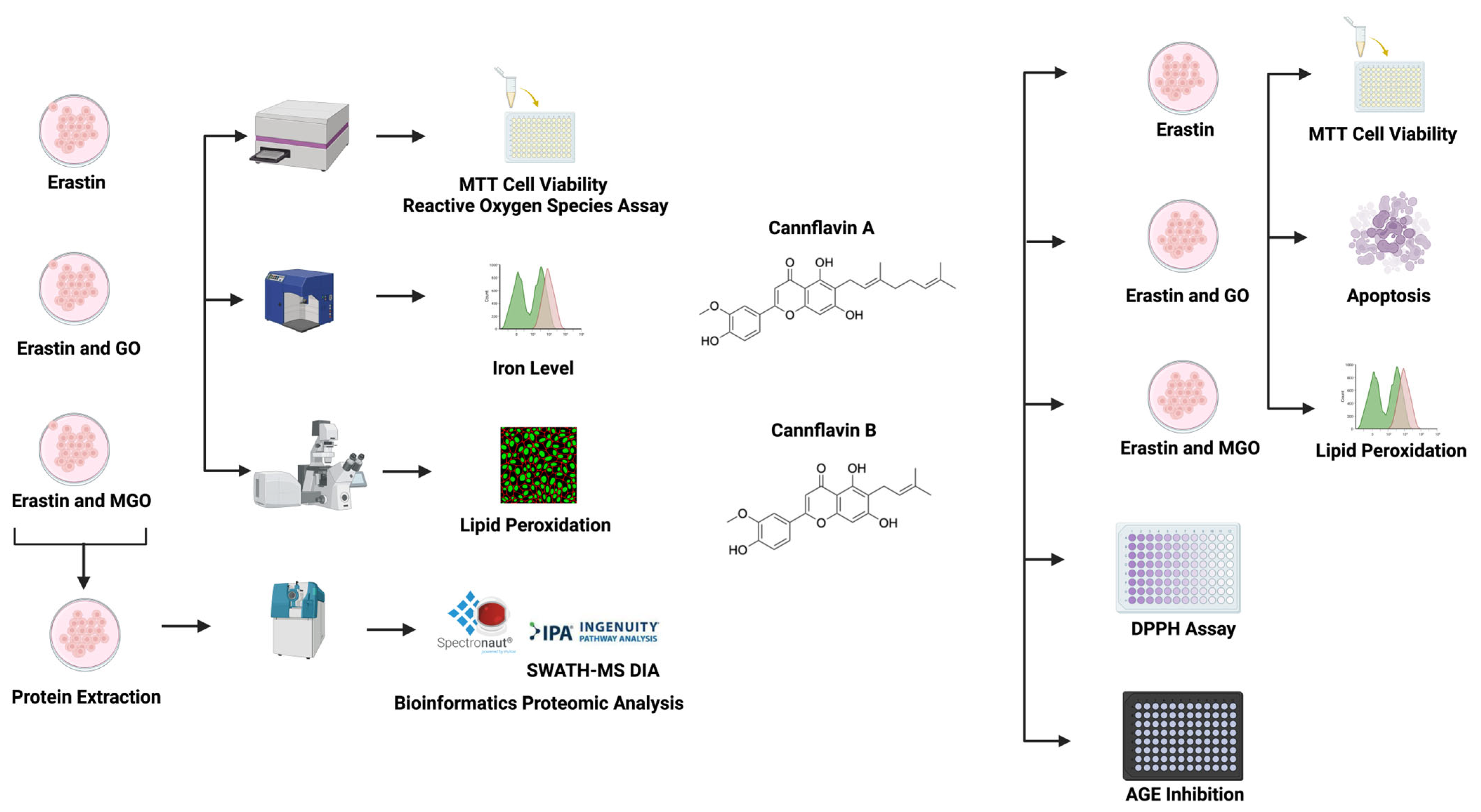
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture and Viability
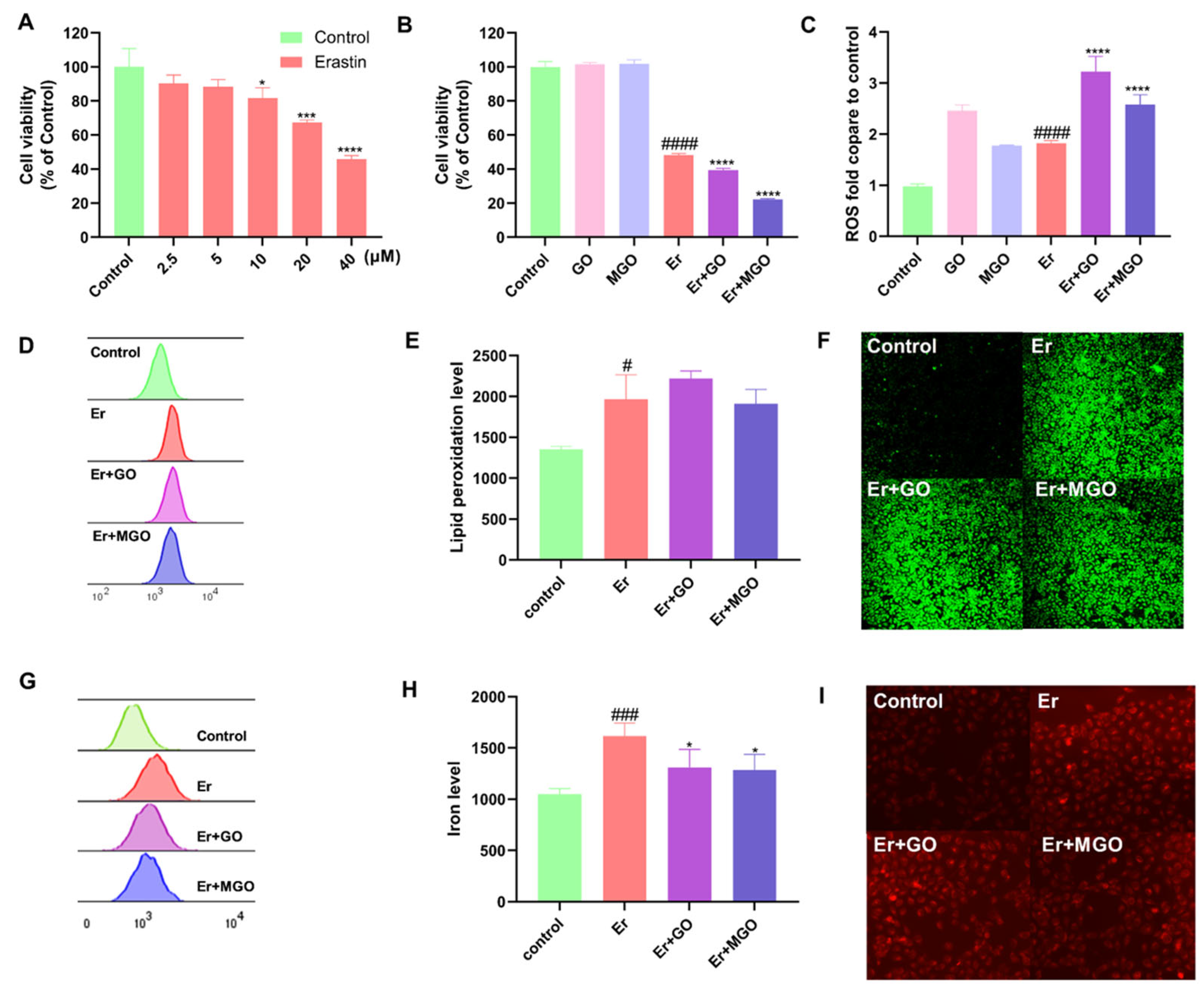
2.3. Measurement of ROS Level
2.4. Cellular Lipid Peroxide Assessment
2.5. Detection of the Cellular Iron Level
2.6. Homogenate Preparation
2.7. Trypsin Digestion
2.8. LC-MS/MS with SWATH Acquisition
2.9. Data Processing and Data-Independent Acquisition Analysis
2.10. Bioinformatic Analysis
2.11. Free Radical (DPPH) Scavenging Assay
2.12. €-MGO Assay
2.13. Apoptosis and Necrosis Assays
2.14. Statistical Analysis
3. Results
3.1. RCSs Exacerbated Overall Cell Death but Did Not Affect Ferroptosis in Human Keratinocytes
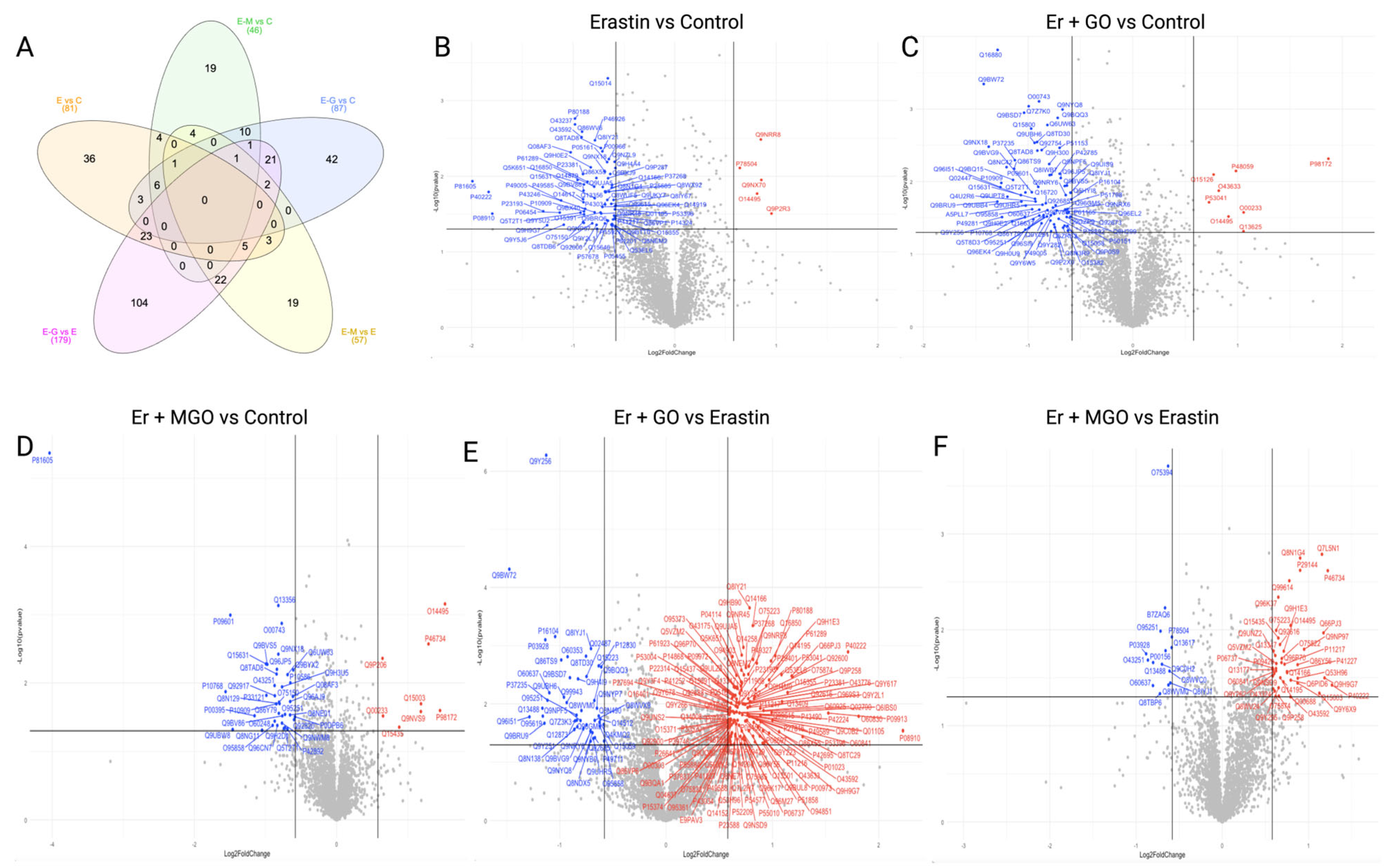
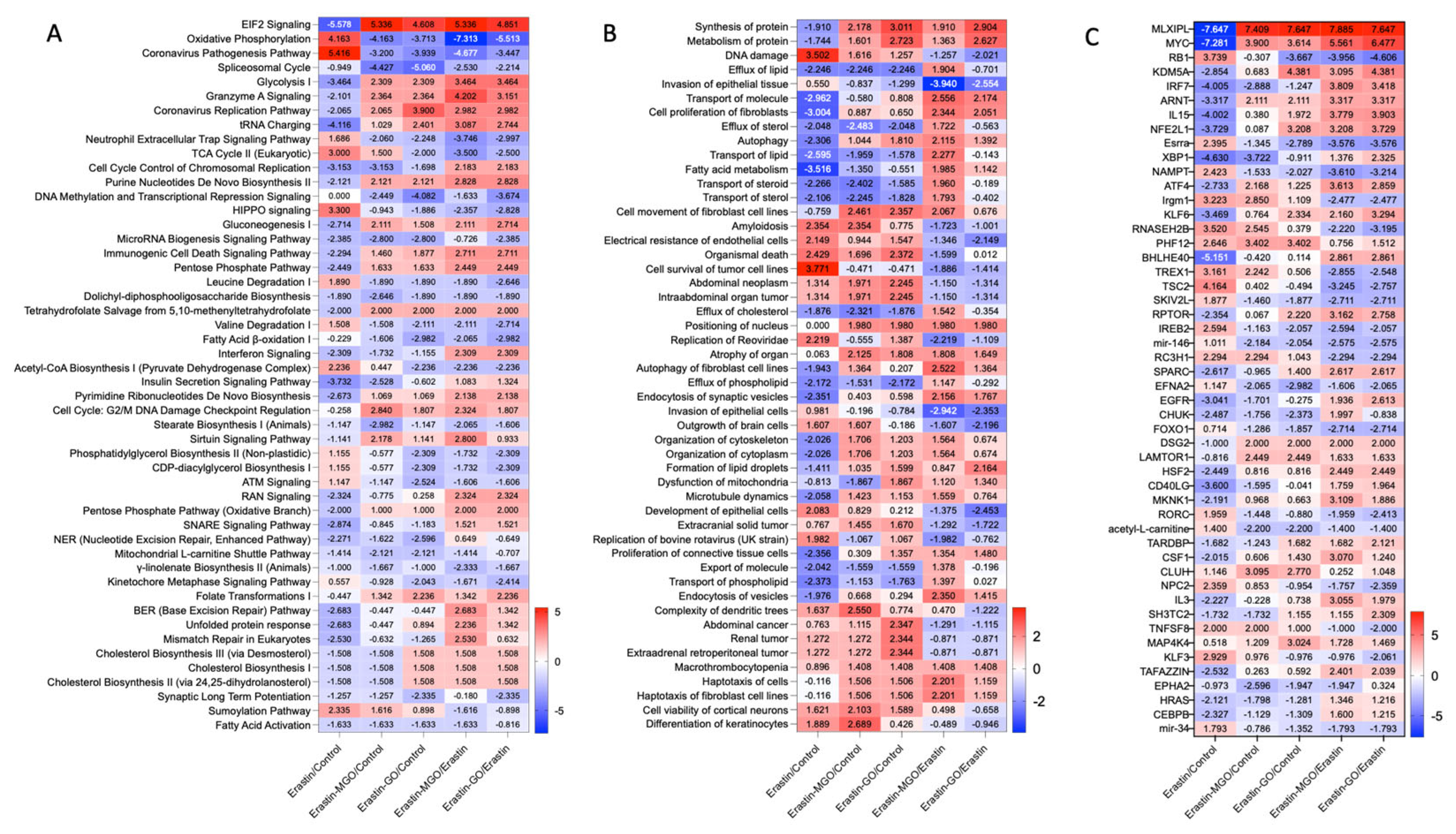
3.2. Erastin and RCSs Altered Protein Expression in Human Keratinocytes
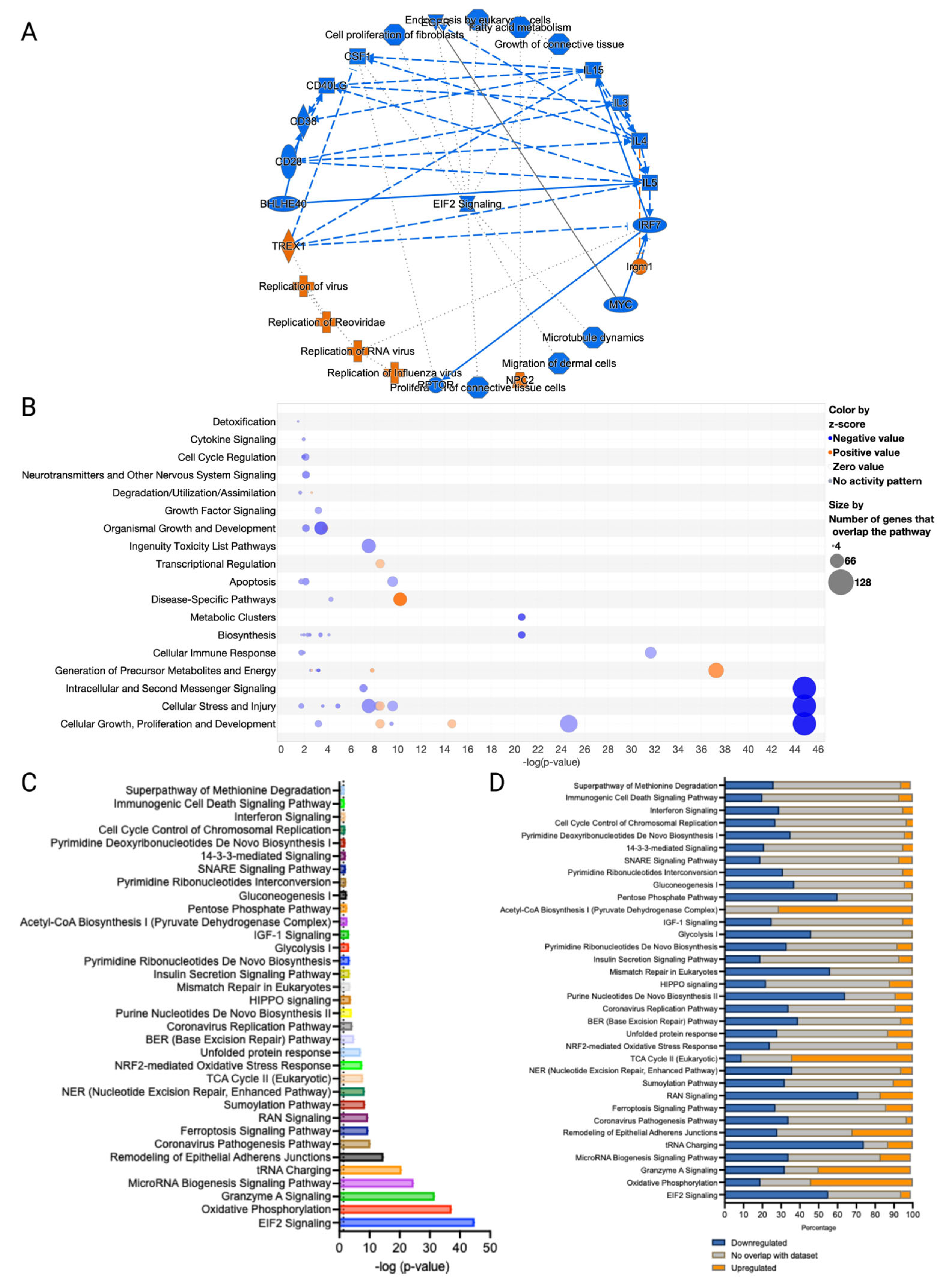
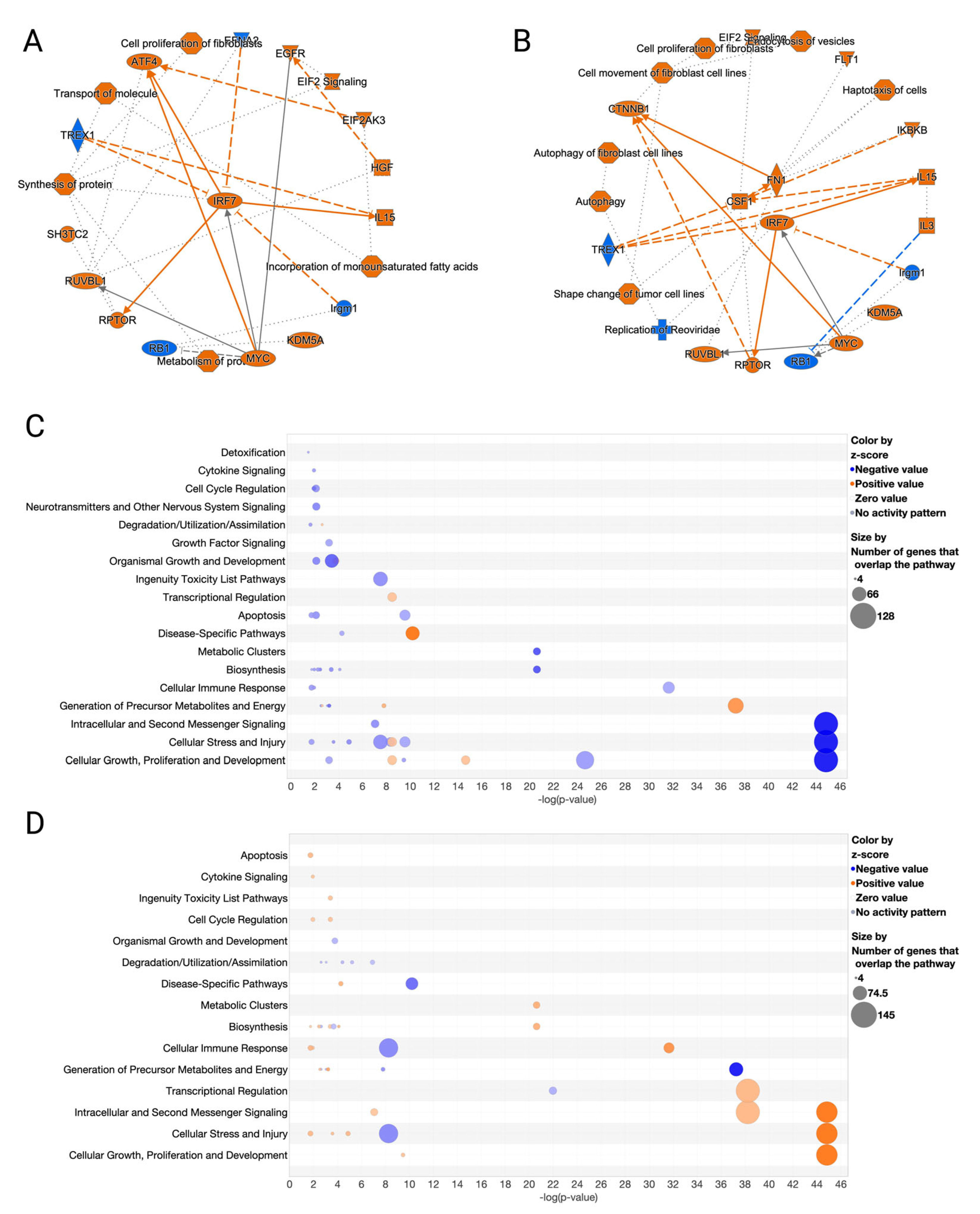
3.3. Canonical Pathways and Upstream Regulators in Ferroptotic HaCaT Cells
3.4. GO and MGO Had Distinct Influences on Pathways Involved in Erastin-Induced Cell Death
3.5. Erastin and RCSs Had Distinct Changes in Canonical Pathways and Upstream Regulators
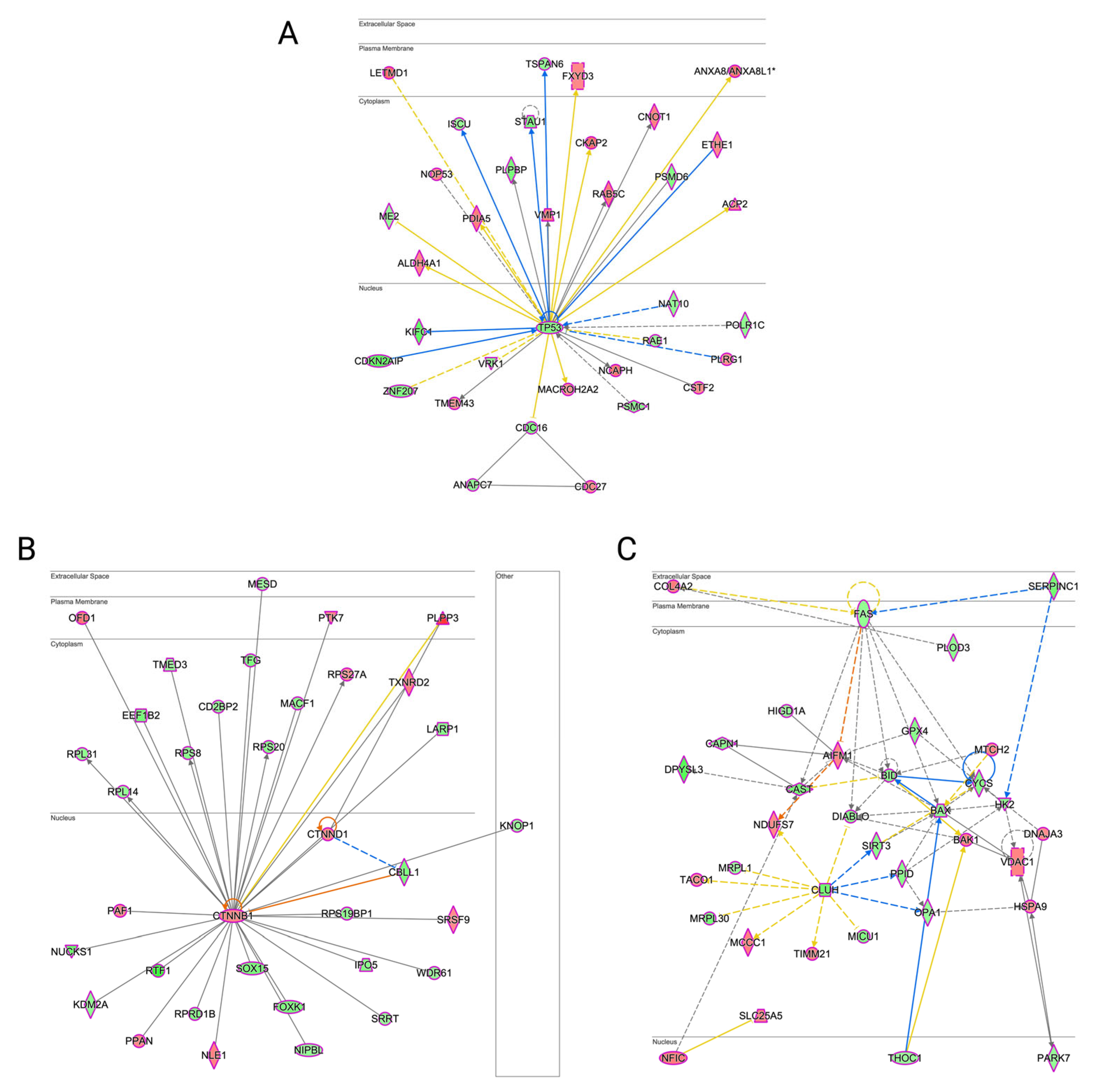
3.6. Erastin-Induced Ferroptosis and RCSs-Induced Cell Death Had a Different Impact on Molecular Networks Related to Cell Death and Survival
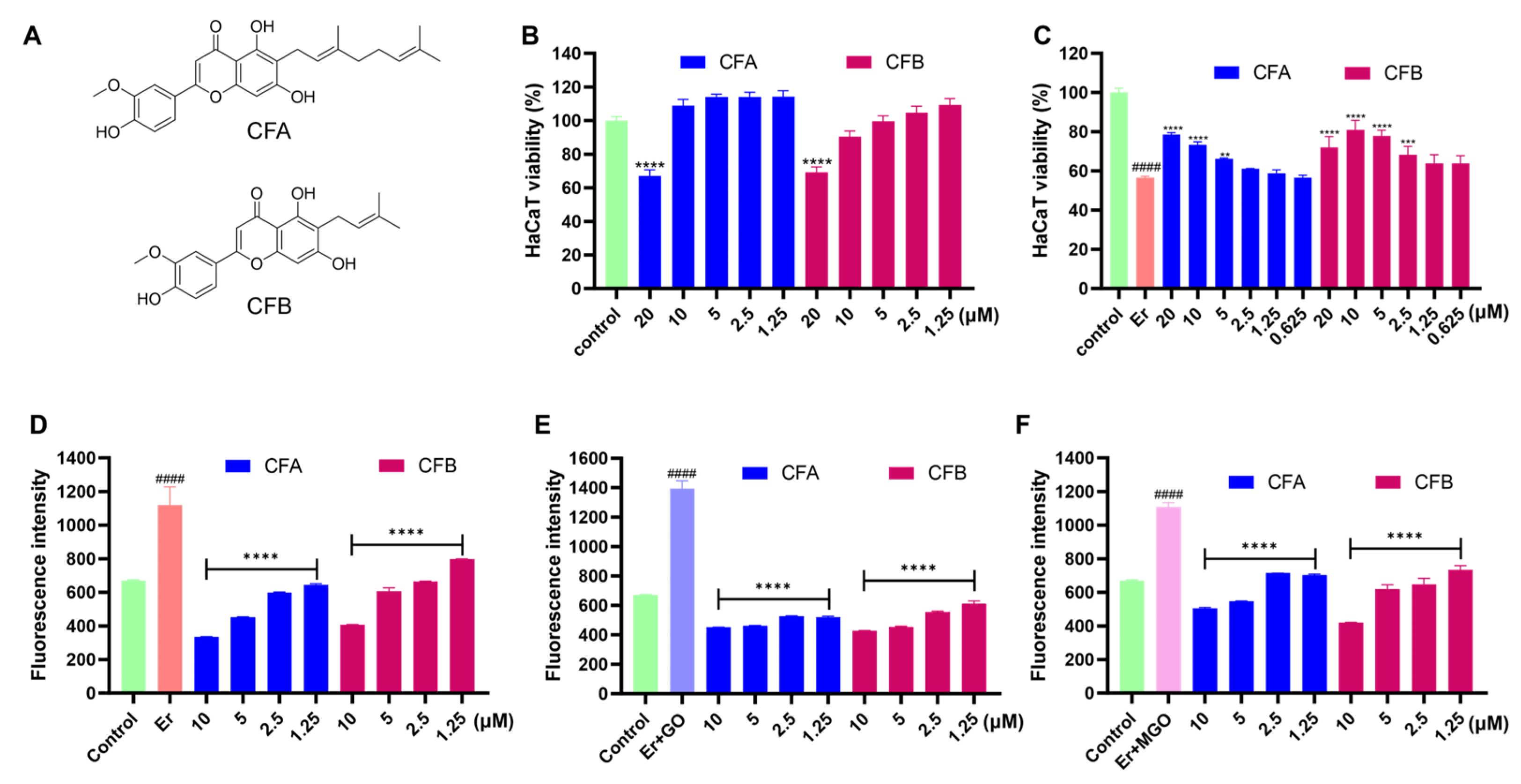
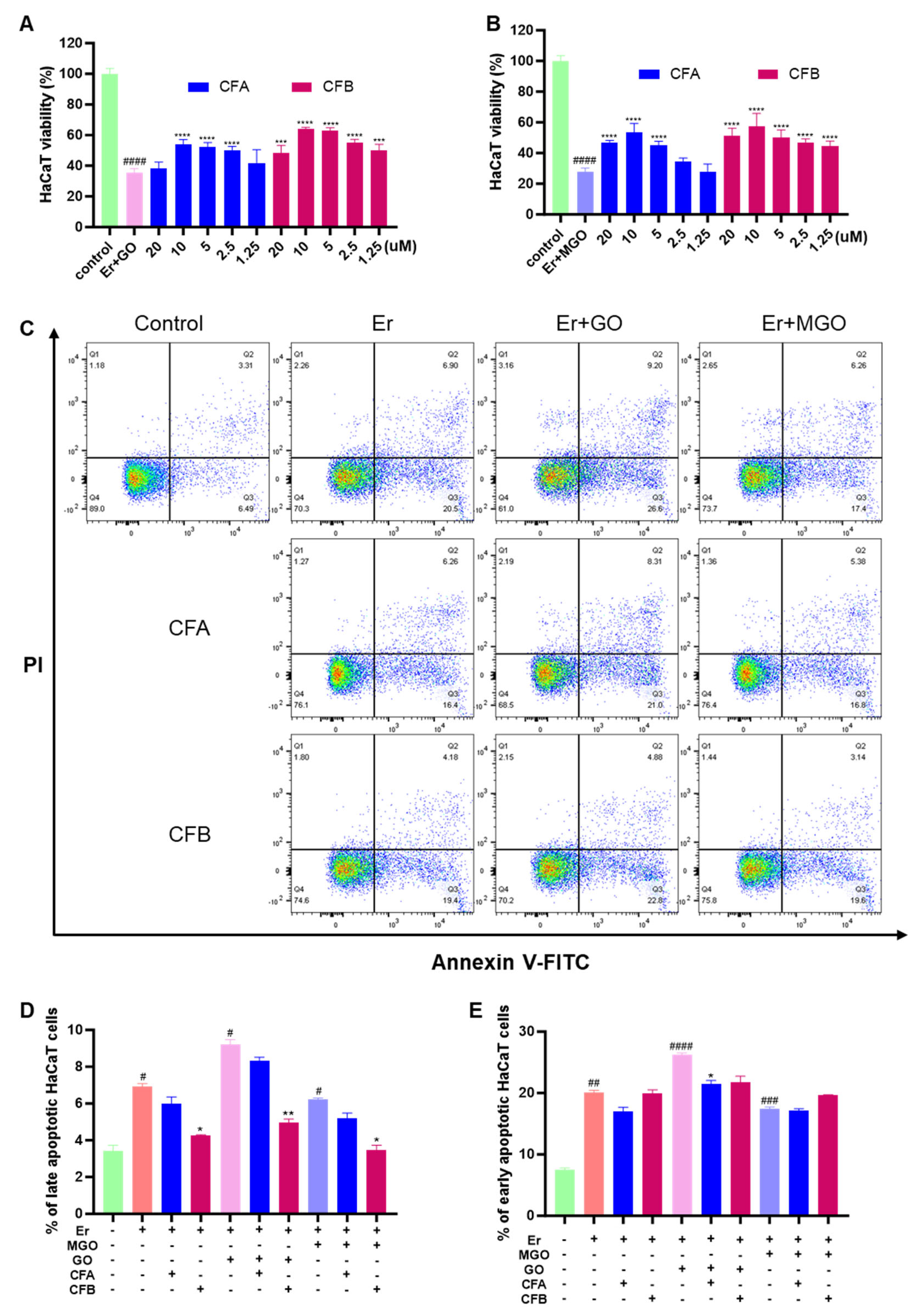
3.7. CFA and CFB Alleviated Erastin-Induced Cytotoxicity in HaCaT Cells by Reducing Cellular Lipid Peroxidation
3.8. CFA and CFB Ameliorated Cytotoxicity Induced by Er + GO and Er + MGO in HaCaT Cells
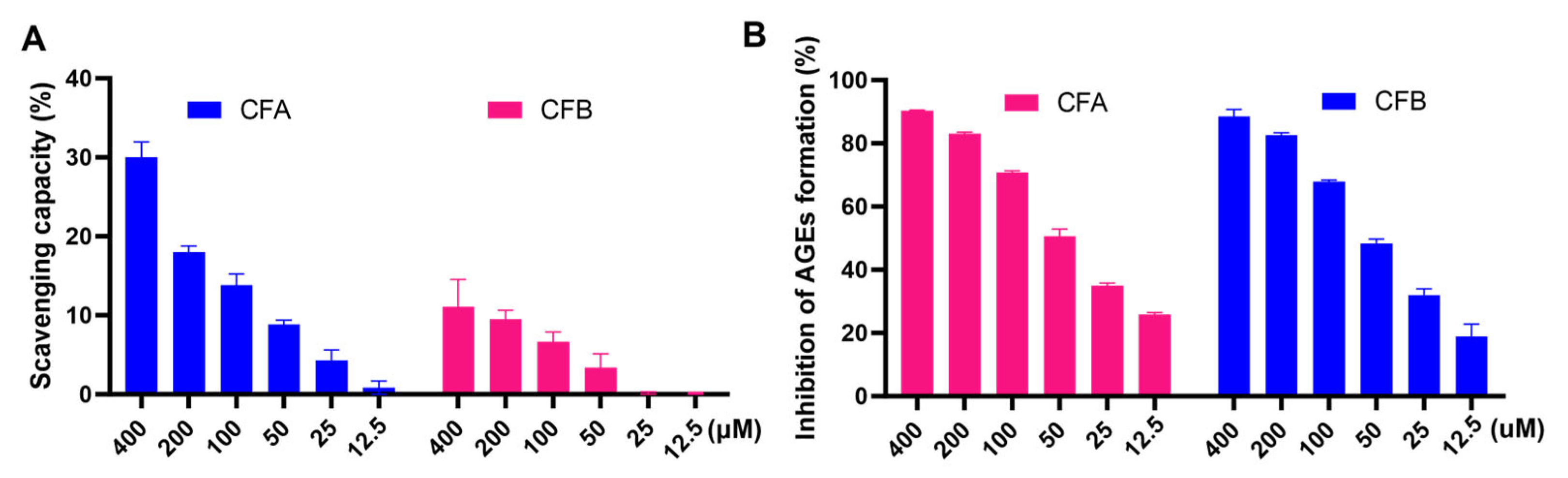
3.9. CFA and CFB Scavenged Free Radicals and Inhibited the Formation of AGEs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AGEs | advanced glycation endproducts |
| ARNT | aryl hydrocarbon receptor nuclear translocator |
| ATF4 | activating transcription factor 4 |
| BHLHE40 | basic helix–loop–helix family member e40 |
| CD | cluster of differentiation |
| CD40LG | cd40 ligand |
| CDKN2AIP | CDKN2A interacting protein |
| ChREBP | carbohydrate-response element-binding protein |
| CSF1 | colony stimulating factor 1 |
| CTNNB1 | catenin beta 1 |
| DIA | data-independent acquisition |
| DOC | sodium deoxycholate |
| DPPH | 2,2-diphenyl-1-picrylhydrazyl |
| EFNA2 | ephrin A2 |
| EGFR | epidermal growth factor receptor |
| EIF2 | eukaryotic initiation factor 2 |
| EIF2AK3 | eukaryotic translation initiation factor 2 alpha kinase 3 |
| FDR | false discovery rate |
| FN1 | fibronectin 1 |
| GADD34 | growth arrest and DNA damage-inducible protein |
| GO | glyoxal |
| GOLD | glyoxal-lysine dimer |
| GPX4 | glutathione peroxidase 4 |
| GSH | glutathione |
| HGF | hepatocyte growth factor |
| HSA | human serum albumin |
| IAA | Iodoacetamide |
| IKBKB | inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase beta |
| IL | interleukin |
| IRF7 | interferon regulatory factor 7 |
| Irgm1 | immunity-related GTPase M 1 |
| ISCU | iron-sulfur cluster assembly enzyme |
| KDM5A | lysine demethylase 5A |
| KLF6 | Kruppel-like factor 6 |
| MGO | methylglyoxal |
| MLXIPL | Max-like protein X interacting protein-like |
| MOLD | methylglyoxal-lysine dimer |
| MS | mass spectra |
| NEF2L1 | nuclear factor erythroid 2-related factor 1 |
| PSM | peptide-spectrum match |
| RB1 | retinoblastoma 1 |
| RCSs | reactive carbonyl species |
| RPTOR | regulatory-associated protein of mTOR |
| RUVBL1 | RuvB-like 1 |
| SLC7A11 | cystine/glutamate transporter |
| SWATH | sequential window acquisition of all theoretical |
| TP53 | tumor protein p53 |
| TREX1 | three prime repair exonuclease 1 |
References
- Janzowski, C.; Glaab, V.; Mueller, C.; Straesser, U.; Kamp, H.G.; Eisenbrand, G. α,β-Unsaturated Carbonyl Compounds: Induction of Oxidative DNA Damage in Mammalian Cells. Mutagenesis 2003, 18, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Shangari, N.; O’Brien, P.J. The Cytotoxic Mechanism of Glyoxal Involves Oxidative Stress. Biochem. Pharmacol. 2004, 68, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Protein and Nucleotide Damage by Glyoxal and Methylglyoxal in Physiological Systems—Role in Ageing and Disease. Drug Metab. Drug Interact. 2008, 23, 125–150. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Guo, H.; Dain, J.A.; Wan, Y.; Gao, X.-H.; Chen, H.-D.; Seeram, N.P.; Ma, H. Cytoprotective Effects of a Proprietary Red Maple Leaf Extract and Its Major Polyphenol, Ginnalin A, against Hydrogen Peroxide and Methylglyoxal Induced Oxidative Stress in Human Keratinocytes. Food Funct. 2020, 11, 5105–5114. [Google Scholar] [CrossRef]
- de Faria Lopes, L.; Jandova, J.; Justiniano, R.; Perer, J.; Baptista, M.S.; Wondrak, G.T. The Glycolysis-Derived α-Dicarbonyl Metabolite Methylglyoxal Is a UVA-Photosensitizer Causing the Photooxidative Elimination of HaCaT Keratinocytes with Induction of Oxidative and Proteotoxic Stress Response Gene Expression. Photochem. Photobiol. 2023, 99, 826–834. [Google Scholar] [CrossRef]
- Radjei, S.; Friguet, B.; Nizard, C.; Petropoulos, I. Prevention of Dicarbonyl-Mediated Advanced Glycation by Glyoxalases: Implication in Skin Aging. Biochem. Soc. Trans. 2014, 42, 518–522. [Google Scholar] [CrossRef]
- Pageon, H.; Zucchi, H.; Dai, Z.; Sell, D.R.; Strauch, C.M.; Monnier, V.M.; Asselineau, D. Biological Effects Induced by Specific Advanced Glycation End Products in the Reconstructed Skin Model of Aging. BioResearch Open Access 2015, 4, 54–64. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Tang, D.; Kroemer, G. Ferroptosis. Curr. Biol. 2020, 30, R1292–R1297. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. OncoTargets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Jiang, L.; Chen, X.; Wu, Q.; Zhu, H.; Du, C.; Ying, M.; He, Q.; Zhu, H.; Yang, B.; Cao, J. The C Terminus of DJ-1 Determines Its Homodimerization, MGO Detoxification Activity and Suppression of Ferroptosis. Acta Pharmacol. Sin. 2021, 42, 1150–1159. [Google Scholar] [CrossRef]
- Csekes, E.; Račková, L. Skin Aging, Cellular Senescence and Natural Polyphenols. Int. J. Mol. Sci. 2021, 22, 12641. [Google Scholar] [CrossRef]
- Menaa, F.; Menaa, A.; Tréton, J. Chapter 63—Polyphenols against Skin Aging. In Polyphenols in Human Health and Disease; Watson, R.R., Preedy, V.R., Zibadi, S., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 819–830. ISBN 978-0-12-398456-2. [Google Scholar]
- Cao, C.; Xiao, Z.; Wu, Y.; Ge, C. Diet and Skin Aging—From the Perspective of Food Nutrition. Nutrients 2020, 12, 870. [Google Scholar] [CrossRef] [PubMed]
- Masaki, H. Role of Antioxidants in the Skin: Anti-Aging Effects. J. Dermatol. Sci. 2010, 58, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Gkogkolou, P.; Böhm, M. Advanced Glycation End Products. Derm. -Endocrinol. 2012, 4, 259–270. [Google Scholar] [CrossRef]
- Roberts, M.J.; Wondrak, G.T.; Laurean, D.C.; Jacobson, M.K.; Jacobson, E.L. DNA Damage by Carbonyl Stress in Human Skin Cells. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2003, 522, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Hadshiew, I.M.; Eller, M.S.; Gilchrest, B.A. Skin Aging and Photoaging: The Role of DNA Damage and Repair. Am. J. Contact Dermat. 2000, 11, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Li, H.; Go, Y.; Chan, X.H.; Huang, Q.; Wu, J. Research Advances on the Damage Mechanism of Skin Glycation and Related Inhibitors. Nutrients 2022, 14, 4588. [Google Scholar] [CrossRef]
- Ma, H.; Liu, W.; Frost, L.; Kirschenbaum, L.J.; Dain, J.A.; Seeram, N.P. Glucitol-Core Containing Gallotannins Inhibit the Formation of Advanced Glycation End-Products Mediated by Their Antioxidant Potential. Food Funct. 2016, 7, 2213–2222. [Google Scholar] [CrossRef]
- Liu, W.; Ma, H.; Frost, L.; Yuan, T.; Dain, J.A.; Seeram, N.P. Pomegranate Phenolics Inhibit Formation of Advanced Glycation Endproducts by Scavenging Reactive Carbonyl Species. Food Funct. 2014, 5, 2996–3004. [Google Scholar] [CrossRef]
- Liu, C.; Guo, H.; DaSilva, N.A.; Li, D.; Zhang, K.; Wan, Y.; Gao, X.-H.; Chen, H.-D.; Seeram, N.P.; Ma, H. Pomegranate (Punica granatum) Phenolics Ameliorate Hydrogen Peroxide-Induced Oxidative Stress and Cytotoxicity in Human Keratinocytes. J. Funct. Foods 2019, 54, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xu, Y.; Kirk, R.D.; Li, H.; Li, D.; DaSilva, N.A.; Bertin, M.J.; Seeram, N.P.; Ma, H. Inhibitory Effects of Skin Permeable Glucitol-Core Containing Gallotannins from Red Maple Leaves on Elastase and Their Protective Effects on Human Keratinocytes. J. Funct. Foods 2020, 75, 104208. [Google Scholar] [CrossRef]
- Guo, H.; Liu, C.; Tang, Q.; Li, D.; Wan, Y.; Li, J.-H.; Gao, X.-H.; Seeram, N.P.; Ma, H.; Chen, H.-D. Pomegranate (Punica granatum) Extract and Its Polyphenols Reduce the Formation of Methylglyoxal-DNA Adducts and Protect Human Keratinocytes against Methylglyoxal-Induced Oxidative Stress. J. Funct. Foods 2021, 83, 104564. [Google Scholar] [CrossRef]
- Li, H.; Roy, T.; Boateng, S.T.; He, H.; Liu, C.; Liu, W.; Li, D.; Wu, P.; Seeram, N.P.; Chamcheu, J.C.; et al. Standardized Pomegranate (Pomella®) and Red Maple (Maplifa®) Extracts and Their Phenolics Protect Type I Collagen by the Inhibition of Matrix Metalloproteinases, Collagenase, and Collagen Cross-Linking. Molecules 2022, 27, 7919. [Google Scholar] [CrossRef] [PubMed]
- Anjo, S.I.; Santa, C.; Manadas, B. SWATH-MS as a Tool for Biomarker Discovery: From Basic Research to Clinical Applications. Proteomics 2017, 17, 1600278. [Google Scholar] [CrossRef]
- Puopolo, T.; Chang, T.; Liu, C.; Li, H.; Liu, X.; Wu, X.; Ma, H.; Seeram, N.P. Gram-Scale Preparation of Cannflavin A from Hemp (Cannabis sativa L.) and Its Inhibitory Effect on Tryptophan Catabolism Enzyme Kynurenine-3-Monooxygenase. Biology 2022, 11, 1416. [Google Scholar] [CrossRef]
- Bollinger, A.; Brandt, O.N.; Stettler, L.D.; Ream, A.; Kopysciansky, V.T.; Nelson, C.A.; Slitt, A.; Ma, J.; Ann Tom, L.; Ma, H.; et al. Sulfide-Linked 3,4,5-Trimethoxyphenyl-Thiosemicarbazide/Triazole Hybrids: Synthesis, Antioxidant, Antiglycation, DNA Cleavage and DNA Molecular Docking Studies. Results Chem. 2023, 5, 100806. [Google Scholar] [CrossRef]
- Liu, C.; Li, H.; Xu, F.; Jiang, X.; Ma, H.; Seeram, N.P. Cannabidiol Protects Human Skin Keratinocytes from Hydrogen-Peroxide-Induced Oxidative Stress via Modulation of the Caspase-1–IL-1β Axis. J. Nat. Prod. 2021, 84, 1563–1572. [Google Scholar] [CrossRef]
- Liang, Z.; Wu, Q.; Wang, H.; Tan, J.; Wang, H.; Gou, Y.; Cao, Y.; Li, Z.; Zhang, Z. Silencing of lncRNA MALAT1 Facilitates Erastin-Induced Ferroptosis in Endometriosis through miR-145-5p/MUC1 Signaling. Cell Death Discov. 2022, 8, 190. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, H.; Wang, F.; Xu, L.; Yan, Y.; Tong, X.; Yan, H. GPX7 Is Targeted by miR-29b and GPX7 Knockdown Enhances Ferroptosis Induced by Erastin in Glioma. Front. Oncol. 2022, 11, 802124. [Google Scholar] [CrossRef]
- Collins, B.C.; Hunter, C.L.; Liu, Y.; Schilling, B.; Rosenberger, G.; Bader, S.L.; Chan, D.W.; Gibson, B.W.; Gingras, A.-C.; Held, J.M.; et al. Multi-Laboratory Assessment of Reproducibility, Qualitative and Quantitative Performance of SWATH-Mass Spectrometry. Nat. Commun. 2017, 8, 291. [Google Scholar] [CrossRef] [PubMed]
- van der Spek, S.J.F.; Gonzalez-Lozano, M.A.; Koopmans, F.; Miedema, S.S.M.; Paliukhovich, I.; Smit, A.B.; Li, K.W. Age-Dependent Hippocampal Proteomics in the APP/PS1 Alzheimer Mouse Model: A Comparative Analysis with Classical SWATH/DIA and directDIA Approaches. Cells 2021, 10, 1588. [Google Scholar] [CrossRef]
- Navarro, P.; Kuharev, J.; Gillet, L.C.; Bernhardt, O.M.; MacLean, B.; Röst, H.L.; Tate, S.A.; Tsou, C.-C.; Reiter, L.; Distler, U.; et al. A Multicenter Study Benchmarks Software Tools for Label-Free Proteome Quantification. Nat. Biotechnol. 2016, 34, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Martín-Saiz, L.; Guerrero-Mauvecin, J.; Martín-Sanchez, D.; Fresnedo, O.; Gómez, M.J.; Carrasco, S.; Cannata-Ortiz, P.; Ortiz, A.; Fernandez, J.A.; Sanz, A.B. Ferrostatin-1 Modulates Dysregulated Kidney Lipids in Acute Kidney Injury. J. Pathol. 2022, 257, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.; Wan, C.; Thomas, A.; Seeram, N.P.; Ma, H. Phenolic Compounds Isolated and Identified from Amla (Phyllanthus emblica) Juice Powder and Their Antioxidant and Neuroprotective Activities. Nat. Prod. Commun. 2018, 13, 1934578X1801301. [Google Scholar] [CrossRef]
- Liu, W.; Cai, A.; Carley, R.; Rocchio, R.; Petrovas, Z.M.; Chartier, C.A.; Meng, X.; Su, J.; Cho, B.P.; Dain, J.A.; et al. Bioactive Anthraquinones Found in Plant Foods Interact with Human Serum Albumin and Inhibit the Formation of Advanced Glycation Endproducts. J. Food Bioact. 2018, 4, 130–138. [Google Scholar] [CrossRef]
- Quan, K.K.; Kusewitt, D.F.; Hudson, L.G. Glyoxal Leads to Defective Keratinocyte Migration and Down-Regulation of Snai2. J. Dermatol. Sci. 2014, 73, 166–169. [Google Scholar] [CrossRef]
- Halkoum, R.; Salnot, V.; Capallere, C.; Plaza, C.; L’honoré, A.; Pays, K.; Friguet, B.; Nizard, C.; Petropoulos, I. Glyoxal Induces Senescence in Human Keratinocytes through Oxidative Stress and Activation of the Protein Kinase B/FOXO3a/p27KIP1 Pathway. J. Investig. Dermatol. 2022, 142, 2068–2078.e7. [Google Scholar] [CrossRef]
- Proud, C.G. eIF2 and the Control of Cell Physiology. Semin. Cell Dev. Biol. 2005, 16, 3–12. [Google Scholar] [CrossRef]
- Singh, C.R.; Lee, B.; Udagawa, T.; Mohammad-Qureshi, S.S.; Yamamoto, Y.; Pavitt, G.D.; Asano, K. An eIF5/eIF2 Complex Antagonizes Guanine Nucleotide Exchange by eIF2B during Translation Initiation. EMBO J. 2006, 25, 4537–4546. [Google Scholar] [CrossRef]
- Moon, S.L.; Parker, R. EIF2B2 Mutations in Vanishing White Matter Disease Hypersuppress Translation and Delay Recovery during the Integrated Stress Response. RNA 2018, 24, 841–852. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, J.; Liu, J.; Li, S.; Zhou, S.; Zhang, C.; Wang, Y.; Shi, J.; Liu, J.; Wu, Q. RNA-Seq Analysis of the Protection by Dendrobium Nobile Alkaloids against Carbon Tetrachloride Hepatotoxicity in Mice. Biomed. Pharmacother. 2021, 137, 111307. [Google Scholar] [CrossRef]
- Morris, E.J.; Dyson, N.J. Retinoblastoma Protein Partners. In Advances in Cancer Research; Academic Press: Cambridge, MA, USA, 2001; Volume 82, pp. 1–54. [Google Scholar]
- Chang, X.; Tian, C.; Jia, Y.; Cai, Y.; Yan, P. MLXIPL Promotes the Migration, Invasion, and Glycolysis of Hepatocellular Carcinoma Cells by Phosphorylation of mTOR. BMC Cancer 2023, 23, 176. [Google Scholar] [CrossRef]
- Herman, M.A.; Peroni, O.D.; Villoria, J.; Schön, M.R.; Abumrad, N.A.; Blüher, M.; Klein, S.; Kahn, B.B. A Novel ChREBP Isoform in Adipose Tissue Regulates Systemic Glucose Metabolism. Nature 2012, 484, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Cao, Y.; Shu, L.; Zhu, Y.; Peng, Q.; Ran, L.; Wu, J.; Luo, Y.; Zuo, G.; Luo, J.; et al. Long Non-Coding RNA (lncRNA) H19 Induces Hepatic Steatosis through Activating MLXIPL and mTORC1 Networks in Hepatocytes. J. Cell. Mol. Med. 2020, 24, 1399–1412. [Google Scholar] [CrossRef]
- Agius, L. Dietary Carbohydrate and Control of Hepatic Gene Expression: Mechanistic Links from ATP and Phosphate Ester Homeostasis to the Carbohydrate-Response Element-Binding Protein. Proc. Nutr. Soc. 2016, 75, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Alborzinia, H.; Flórez, A.F.; Kreth, S.; Brückner, L.M.; Yildiz, U.; Gartlgruber, M.; Odoni, D.I.; Poschet, G.; Garbowicz, K.; Shao, C.; et al. MYCN Mediates Cysteine Addiction and Sensitizes Neuroblastoma to Ferroptosis. Nat. Cancer 2022, 3, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Qiu, J.; Lu, X.; Li, G. C-MYC Inhibited Ferroptosis and Promoted Immune Evasion in Ovarian Cancer Cells through NCOA4 Mediated Ferritin Autophagy. Cells 2022, 11, 4127. [Google Scholar] [CrossRef]
- Yumnam, S.; Subedi, L.; Kim, S.Y. Glyoxalase System in the Progression of Skin Aging and Skin Malignancies. Int. J. Mol. Sci. 2021, 22, 310. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Achatz, M.I.W.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 Mutations in Human Cancers: Functional Selection and Impact on Cancer Prognosis and Outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef]
- Tong, W.-H.; Rouault, T.A. Functions of Mitochondrial ISCU and Cytosolic ISCU in Mammalian Iron-Sulfur Cluster Biogenesis and Iron Homeostasis. Cell Metab. 2006, 3, 199–210. [Google Scholar] [CrossRef]
- Ledinek, Ž.; Sobočan, M.; Knez, J. The Role of CTNNB1 in Endometrial Cancer. Dis. Markers 2022, 2022, e1442441. [Google Scholar] [CrossRef]
- Lalier, L.; Cartron, P.-F.; Juin, P.; Nedelkina, S.; Manon, S.; Bechinger, B.; Vallette, F.M. Bax Activation and Mitochondrial Insertion during Apoptosis. Apoptosis 2007, 12, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a P53-Mediated Activity during Tumour Suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Wang, G.; Sun, L.; Wang, S.; Guo, J.; Xiao, R.; Li, W.; Qi, W.; Qiu, W. Ferroptosis-related Long Non-coding RNAs and the Roles of LASTR in Stomach Adenocarcinoma. Mol. Med. Rep. 2022, 25, 118. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox Signaling and Unfolded Protein Response Coordinate Cell Fate Decisions under ER Stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Chen, H.; Mai, Z.; Sun, H.; Xu, L.; Wu, G.; Tu, Z.; Cheng, X.; Wang, X.; Chen, T. Bim- and Bax-Mediated Mitochondrial Pathway Dominates Abivertinib-Induced Apoptosis and Ferroptosis. Free Radic. Biol. Med. 2022, 180, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Kalimuthu, K.; Park, Y.S.; Luo, X.; Choudry, M.H.A.; Bartlett, D.L.; Lee, Y.J. BAX-Dependent Mitochondrial Pathway Mediates the Crosstalk between Ferroptosis and Apoptosis. Apoptosis 2020, 25, 625–631. [Google Scholar] [CrossRef]
- Huang, S.-M.; Chuang, H.-C.; Wu, C.-H.; Yen, G.-C. Cytoprotective Effects of Phenolic Acids on Methylglyoxal-Induced Apoptosis in Neuro-2A Cells. Mol. Nutr. Food Res. 2008, 52, 940–949. [Google Scholar] [CrossRef]
- Ellis, E.M. Reactive Carbonyls and Oxidative Stress: Potential for Therapeutic Intervention. Pharmacol. Ther. 2007, 115, 13–24. [Google Scholar] [CrossRef]
- Vander Jagt, D.L. Glyoxalase II: Molecular Characteristics, Kinetics and Mechanism. Biochem. Soc. Trans. 1993, 21, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Domaszewska-Szostek, A.; Puzianowska-Kuźnicka, M.; Kuryłowicz, A. Flavonoids in Skin Senescence Prevention and Treatment. Int. J. Mol. Sci. 2021, 22, 6814. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Fan, Z.; Yang, Z.; Huang, T.; Tong, Y.; Yang, D.; Mao, X.; Yang, M. Flavonoids—Natural Gifts to Promote Health and Longevity. Int. J. Mol. Sci. 2022, 23, 2176. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.; Jakus, J.; Portillo, M.; Gvirtz, R.; Ogen-Shtern, N.; Silberstein, E.; Ayzenberg, T.; Rozenblat, S. In Vitro, Ex Vivo, and Clinical Evaluation of Anti-Aging Gel Containing EPA and CBD. J. Cosmet. Dermatol. 2023. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 586578. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Cidlowski, J.A. Apoptosis and Glutathione: Beyond an Antioxidant. Cell Death Differ. 2009, 16, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zeng, J.; Liu, Y.; Liang, M.; Liu, Q.; Li, Z.; Zhao, X.; Chen, D. Inhibitory Effect and Mechanism of Action of Quercetin and Quercetin Diels-Alder Anti-Dimer on Erastin-Induced Ferroptosis in Bone Marrow-Derived Mesenchymal Stem Cells. Antioxidants 2020, 9, 205. [Google Scholar] [CrossRef]
- Liu, J.; Li, X.; Cai, R.; Ren, Z.; Zhang, A.; Deng, F.; Chen, D. Simultaneous Study of Anti-Ferroptosis and Antioxidant Mechanisms of Butein and (S)-Butin. Molecules 2020, 25, 674. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Deng, N.; Puopolo, T.; Jiang, X.; Seeram, N.P.; Liu, C.; Ma, H. Cannflavins A and B with Anti-Ferroptosis, Anti-Glycation, and Antioxidant Activities Protect Human Keratinocytes in a Cell Death Model with Erastin and Reactive Carbonyl Species. Nutrients 2023, 15, 4565. https://doi.org/10.3390/nu15214565
Li H, Deng N, Puopolo T, Jiang X, Seeram NP, Liu C, Ma H. Cannflavins A and B with Anti-Ferroptosis, Anti-Glycation, and Antioxidant Activities Protect Human Keratinocytes in a Cell Death Model with Erastin and Reactive Carbonyl Species. Nutrients. 2023; 15(21):4565. https://doi.org/10.3390/nu15214565
Chicago/Turabian StyleLi, Huifang, Ni Deng, Tess Puopolo, Xian Jiang, Navindra P. Seeram, Chang Liu, and Hang Ma. 2023. "Cannflavins A and B with Anti-Ferroptosis, Anti-Glycation, and Antioxidant Activities Protect Human Keratinocytes in a Cell Death Model with Erastin and Reactive Carbonyl Species" Nutrients 15, no. 21: 4565. https://doi.org/10.3390/nu15214565