
A High-Performance Liquid Chromatography with Photodiode Array Detection Method for Simultaneous Determination of Three Compounds Isolated from Wikstroemia ganpi: Assessment of the Effects on Cytochrome P450-Mediated Metabolism In Vitro and In Vivo
 and
and
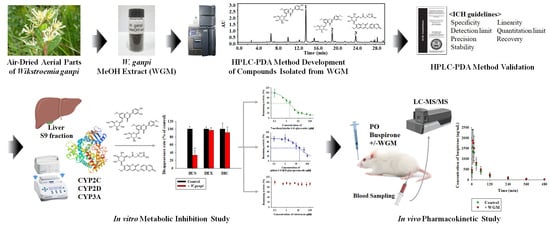
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Animal
2.2. Plant Materials
2.3. Extraction and Isolation
2.4. Preparation of Sample and Reference Solutions
2.5. Determination of HPLC-PDA Conditions
2.6. HPLC-PDA Method Validation
2.7. In Vitro Metabolic Inhibition Study in Rat Liver S9 Fraction
2.8. In Vivo Pharmacokinetic Study in Rats
2.9. Pharmacokinetic Analysis and Statistic
3. Results and Discussion
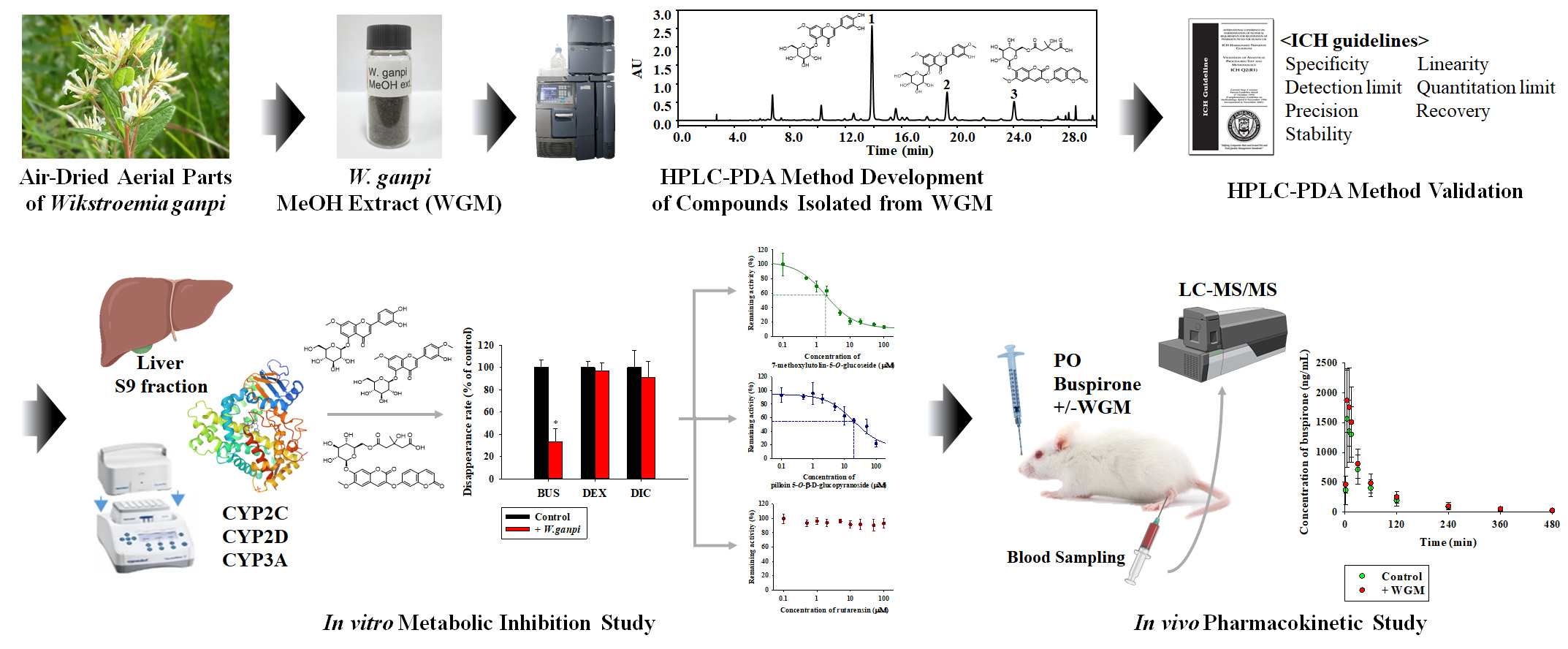
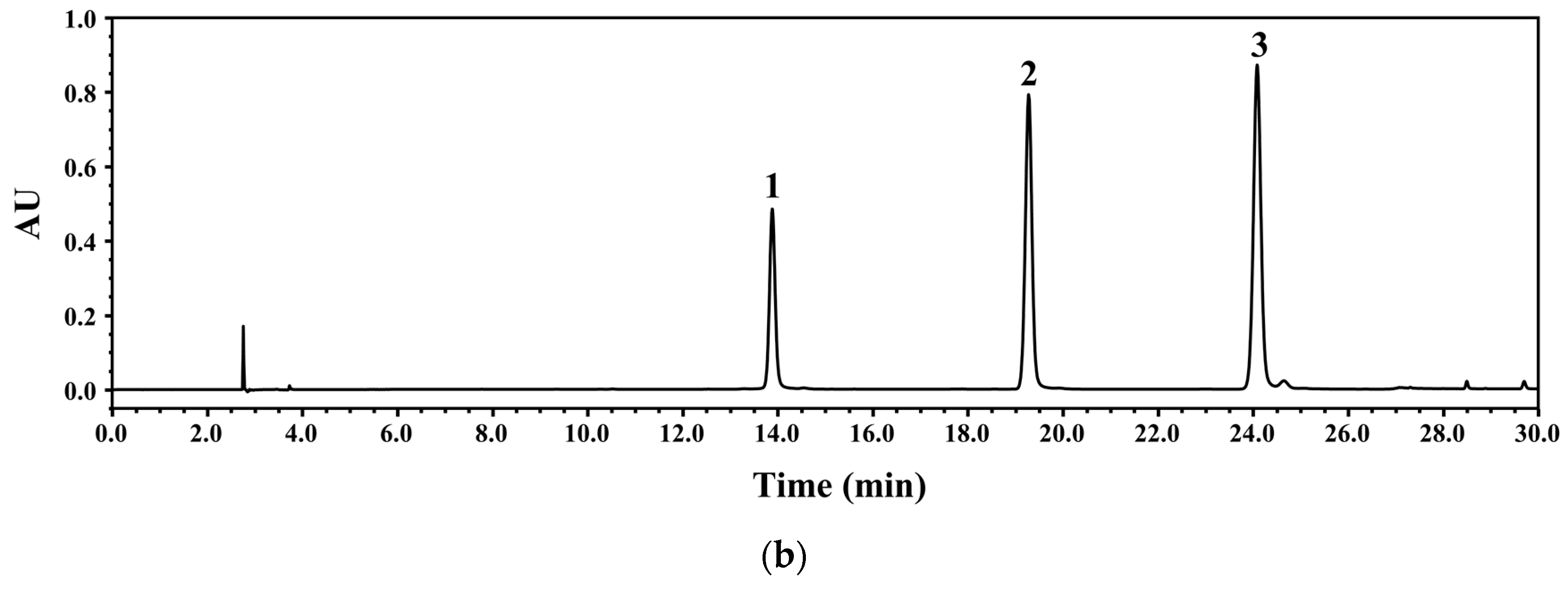
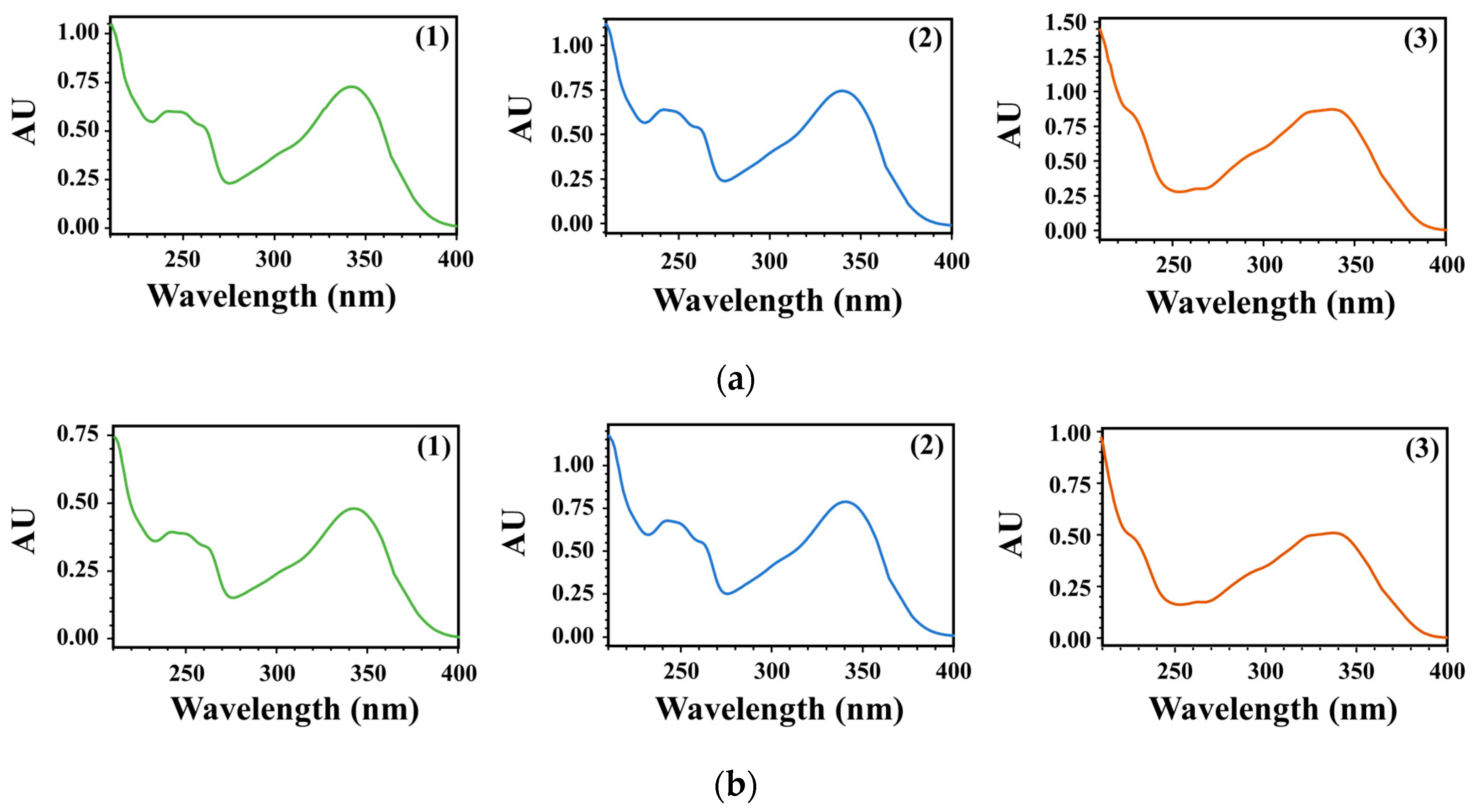
3.1. Identification and Purification of Compounds
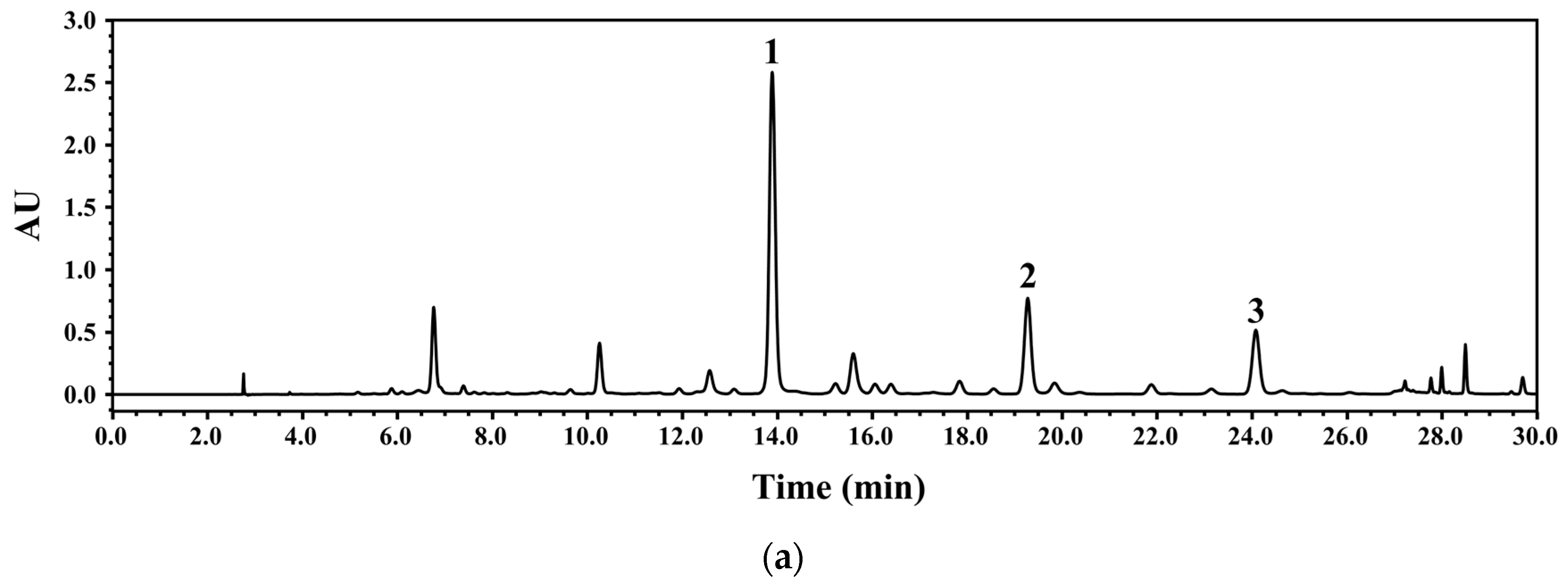
3.2. Development of HPLC-PDA Conditions
3.3. Validation of Methods
3.4. Chemical Profiling of WGM
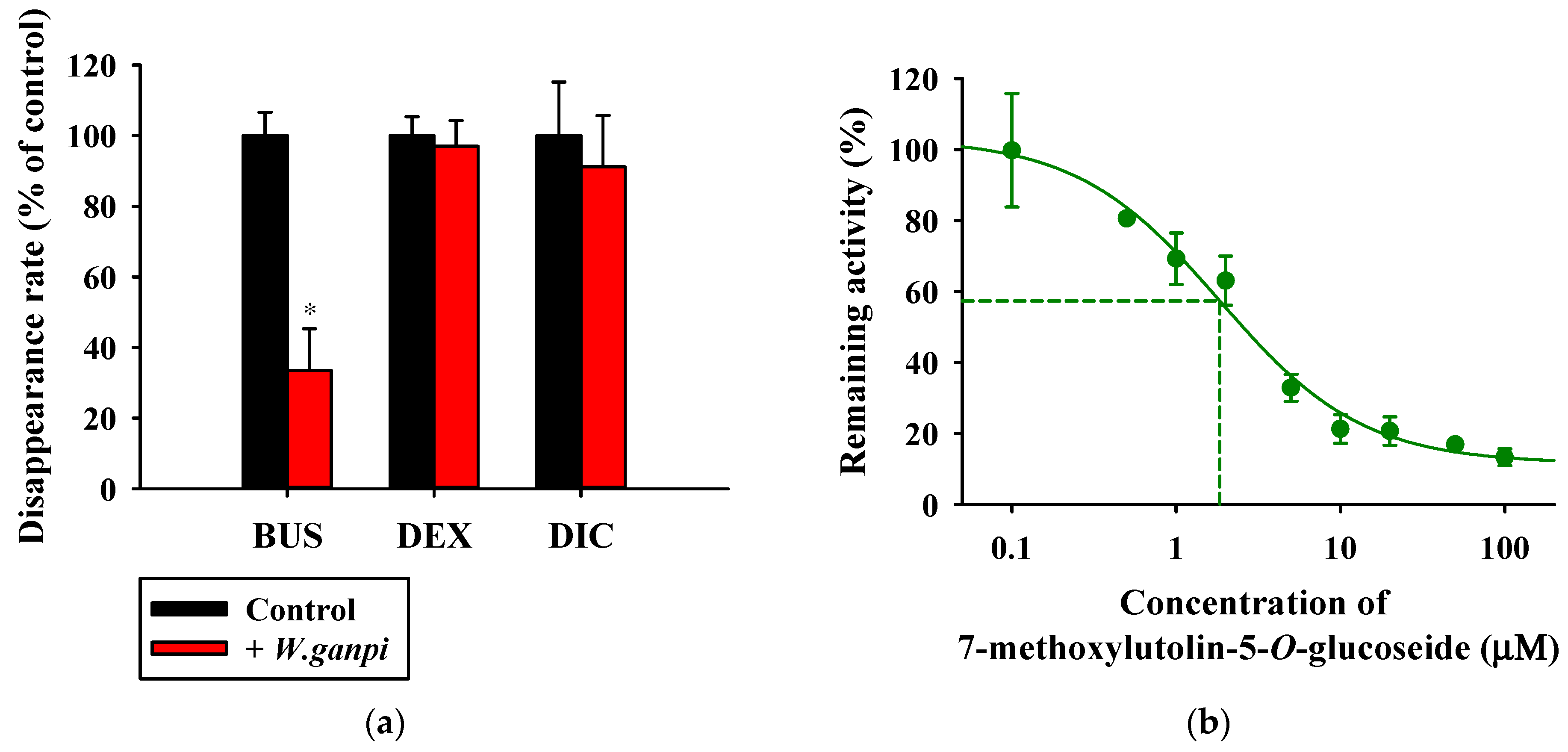
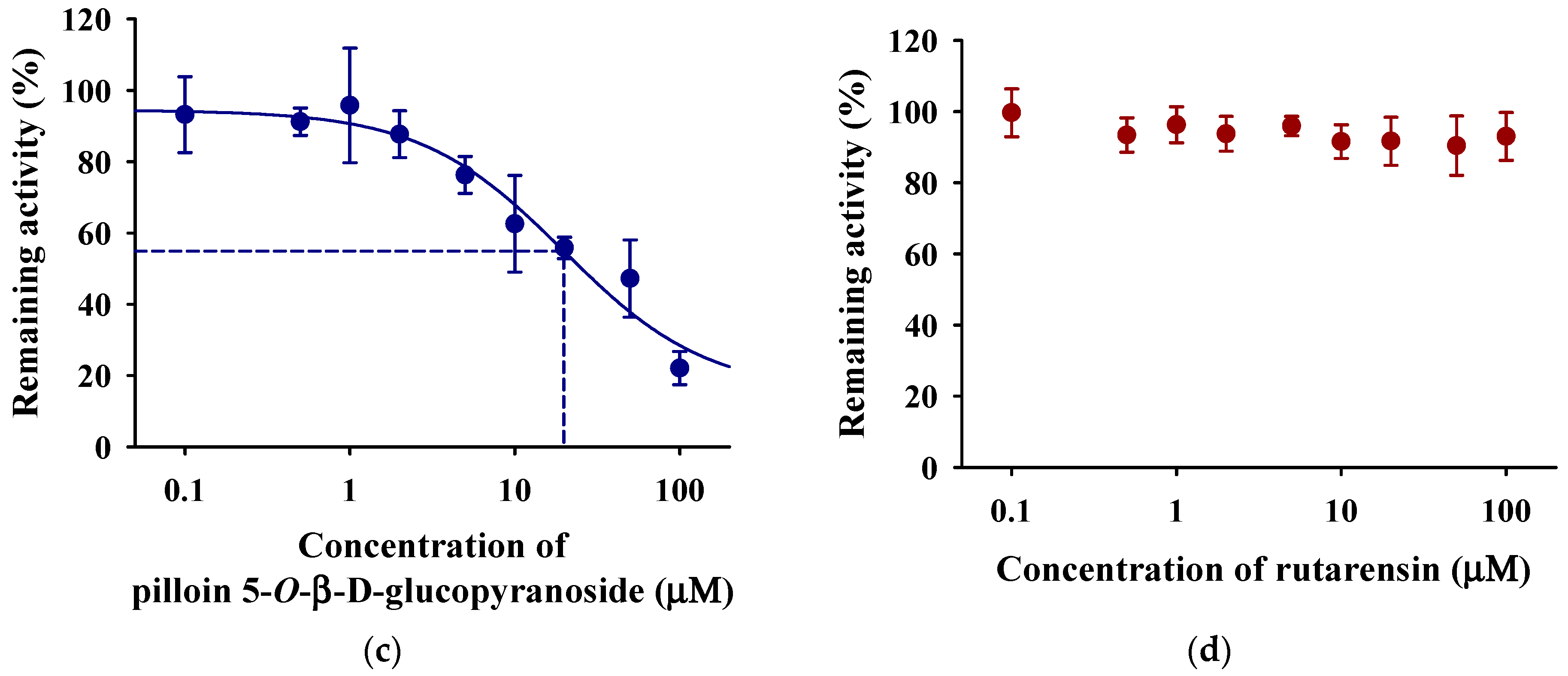
3.5. Inhibitory Effects of WGM and Marker Compounds on Cytochrome P450 Activity in the Rat Liver S9 Fractions
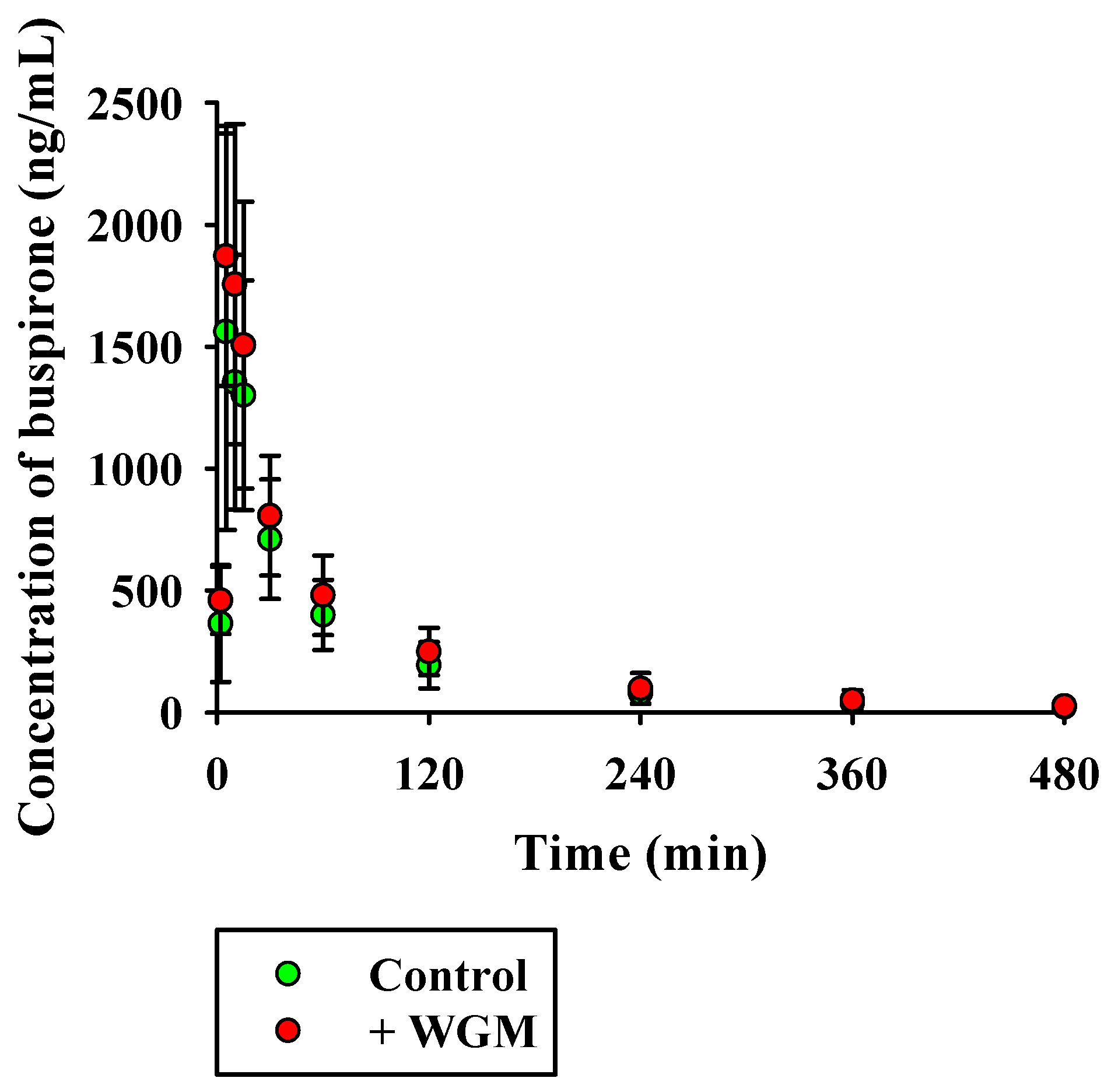
3.6. In Vivo Pharmacokinetic Studies in Rats
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Devkota, H.P.; Yoshizaki, K.; Yahara, S. Pilloin 5-O-Beta-D-Glucopyranoside from the Stems of Diplomorpha ganpi. Biosci. Biotechnol. Biochem. 2012, 76, 1555–1557. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhang, Y.; Lee, S.Y. Complete Plastomes of Six Species of Wikstroemia (Thymelaeaceae) Reveal Paraphyly with the Monotypic Genus Stellera. Sci. Rep. 2021, 11, 13608. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, L.Y.; Zeng, L.H.; Zhang, C.; Hu, J.L.; Li, X.R. Sikokianin D, a New C-3/C-3″-Biflavanone from the Roots of Wikstroemia indica. Molecules 2012, 17, 7792–7797. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Chen, I.S.; Peng, C.F. Structural Elucidation and Bioactivity of Biflavonoids from the Stems of Wikstroemia taiwanensis. Int. J. Mol. Sci. 2012, 13, 1029–1038. [Google Scholar] [CrossRef]
- Chen, Y.; Fu, W.W.; Sun, L.X.; Wang, Q.; Qi, W.; Yu, H. A New Coumarin from Wikstroemia indica (L.) C. A. Mey. Chin. Chem. Lett. 2009, 20, 592–594. [Google Scholar] [CrossRef]
- Liao, S.G.; Wu, Y.; Yue, J.M. Lignans from Wikstroemia hainanensis. Helv. Chim. Acta 2006, 89, 73–80. [Google Scholar] [CrossRef]
- Kim, T.Y.; Park, N.J.; Jegal, J.; Choi, S.; Lee, S.W.; Hang, J.; Kim, S.N.; Yang, M.H. Chamaejasmine Isolated from Wikstroemia dolichantha Diels Suppresses 2,4-Dinitrofluoro-Benzene-Induced Atopic Dermatitis in SKH-1 Hairless Mice. Biomolecules 2019, 9, 697. [Google Scholar] [CrossRef]
- Wang, L.Y.; Unehara, T.; Kitanaka, S. Anti-Inflammatory Activity of New Guaiane Type Sesquiterpene from Wikstroemia indica. Chem. Pharm. Bull. 2005, 53, 137–139. [Google Scholar] [CrossRef]
- Lee, K.H.; Tagahara, K.; Suzuki, H.; Wu, R.Y.; Haruna, M.; Hall, I.H.; Huang, H.C.; Ito, K.; Iida, T.; Lai, J.S. Antitumor Agents. 49 Tricin, Kaempferol-3-O-Beta-D-Glucopyranoside and (+)-Nortrachelogenin, Antileukemic Principles from Wikstroemia indica. J. Nat. Prod. 1981, 44, 530–535. [Google Scholar] [CrossRef]
- Duh, C.Y.; Phoebe, C.H., Jr.; Pezzuto, J.M.; Kinghorn, A.D.; Farnsworth, N.R. Plant Anticancer Agents, XLII. Cytotoxic Constituents from Wikstroemia elliptica. J. Nat. Prod. 1986, 49, 706–709. [Google Scholar] [CrossRef]
- Hu, K.; Kobayashi, H.; Dong, A.; Iwasaki, S.; Yao, X. Antifungal, Antimitotic and Anti-HIV-1 Agents from the Roots of Wikstroemia indica. Planta Med. 2000, 66, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Jegal, J.; Park, N.J.; Jo, B.G.; Kim, T.Y.; Bong, S.K.; Choi, S.; Paik, J.H.; Kim, J.W.; Kim, S.N.; Yang, M.H. Wikstroemiaganpi Extract Improved Atopic Dermatitis-like Skin Lesions via Suppression of Interleukin-4 in 2,4-Dinitrochlorobenzene-Induced SKH-1 Hairless Mice. Molecules 2021, 26, 2016. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, E.; Arshad, M.; Khan, M.; Amjad, M.S.; Sadaf, H.; Riaz, I.; Sabir, S.; Ahmed, N.; Sabaoon. Secondary Metabolites and Their Multidimensional Prospective in Plant Life. J. Pharmacogn. Phytochem. 2017, 6, 205–214. [Google Scholar]
- Jayachandran Nair, C.V.; Ahamad, S.; Khan, W.; Anjum, V.; Mathur, R. Development and Validation of High-Performance Thin-Layer Chromatography Method for Simultaneous Determination of Polyphenolic Compounds in Medicinal Plants. Pharmacogn. Res. 2017, 9, S67–S73. [Google Scholar]
- Papuc, C.; Goran, G.V.; Predescu, C.N.; Nicorescu, V.; Stefan, G. Plant Polyphenols as Antioxidant and Antibacterial Agents for Shelf-Life Extension of Meat And Meat Products: Classification, Structures, Sources, and Action Mechanisms. Compr. Rev. Food Sci. Food. Saf. 2017, 16, 1243–1268. [Google Scholar] [CrossRef]
- Yi, L.Z.; Ma, S.S.; Ren, D.B. Phytochemistry and Bioactivity of Citrus Flavonoids: A Focus on Antioxidant, Anti-Inflammatory, Anticancer and Cardiovascular Protection Activities. Phytochem. Rev. 2017, 16, 479–511. [Google Scholar] [CrossRef]
- Garg, S.S.; Gupta, J.; Sharma, S.; Sahu, D. An Insight into the Therapeutic Applications of Coumarin Compounds and Their Mechanisms of Action. Eur. J. Pharm. Sci. 2020, 152, 105424. [Google Scholar] [CrossRef]
- Pochet, L.; Doucet, C.; Dive, G.; Wouters, J.; Masereel, B.; Reboud-Ravaux, M.; Pirotte, B. Coumarinic Derivatives as Mechanism-Based Inhibitors of Alpha-Chymotrypsin and Human Leukocyte Elastase. Bioorg. Med. Chem. 2000, 8, 1489–1501. [Google Scholar] [CrossRef]
- Joshi, D.D. Herbal Drugs and Fingerprints: Evidence Based Herbal Drugs; Springer: New Delhi, India, 2012. [Google Scholar]
- Krishnan, P.; Kruger, N.J.; Ratcliffe, R.G. Metabolite Fingerprinting and Profiling in Plants Using NMR. J. Exp. Bot. 2005, 56, 255–265. [Google Scholar] [CrossRef]
- Jo, B.-G.; Kang, K.-H.; Yang, M.H. Development and Validation of a HPLC–PDA Method for the Simultaneous Determination of Berberine, Palmatine, Geniposide, and Paeoniflorin in Haedoksamul-Tang. Appl. Sci. 2020, 10, 5482. [Google Scholar] [CrossRef]
- Wu, L.Y.; Gao, H.Z.; Wang, X.L.; Ye, J.H.; Lu, J.L.; Liang, Y.R. Analysis of Chemical Composition of Chrysanthemum indicum Flowers by GC/MS and HPLC. J. Med. Plants Res. 2010, 4, 421–426. [Google Scholar]
- Patel, R.P.; Narkowicz, C.; Hutchinson, J.P.; Hilder, E.F.; Jacobson, G.A. A Simple Capillary Electrophoresis Method for the Rapid Separation and Determination of Intact Low Molecular Weight and Unfractionated Heparins. J. Pharm. Biomed. Anal. 2008, 46, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Gad, H.A.; El-Ahmady, S.H.; Abou-Shoer, M.I.; Al-Azizi, M.M. Application of Chemometrics in Authentication of Herbal Medicines: A Review. Phytochem. Anal. 2013, 24, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Pons, M.N.; Le Bonte, S.; Potier, O. Spectral Analysis and Fingerprinting for Biomedia Characterisation. J. Biotechnol. 2004, 113, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Francisco, M.L.D.; Resurreccion, A.V.A. Development of a Reversed-Phase High Performance Liquid Chromatography (RP-HPLC) Procedure for the Simultaneous Determination of Phenolic Compounds in Peanut Skin Extracts. Food Chem. 2009, 117, 356–363. [Google Scholar] [CrossRef]
- Vorauer-Uhl, K.; Wagner, A.; Katinger, H. Long Term Stability of Rh-Cu/Zn-Superoxide Dismutase (SOD)-Liposomes Prepared by the Cross-Flow Injection Technique Following International Conference on Harmonisation (ICH)-Guidelines. Eur. J. Pharm. Biopharm. 2002, 54, 83–87. [Google Scholar] [CrossRef]
- Rozet, E.; Ceccato, A.; Hubert, C.; Ziemons, E.; Oprean, R.; Rudaz, S.; Boulanger, B.; Hubert, P. Analysis of Recent Pharmaceutical Regulatory Documents on Analytical Method Validation. J. Chromatogr. A 2007, 1158, 111–125. [Google Scholar] [CrossRef]
- Liu, J.; Liu, H.; Zeng, Q. The Effect of Naringenin on the Pharmacokinetics of Ibrutinib in Rat: A Drug-Drug Interaction Study. Biomed. Chromatogr. 2019, 33, e4507. [Google Scholar] [CrossRef]
- Bibi, Z. Role of Cytochrome P450 in Drug Interactions. Nutr. Metab. 2008, 5, 27. [Google Scholar] [CrossRef]
- Dores, A.R.; Peixoto, M.; Castro, M.; Sa, C.; Carvalho, I.P.; Martins, A.; Maia, E.; Praca, I.; Marques, A. Knowledge and Beliefs about Herb/Supplement Consumption and Herb/Supplement-Drug Interactions among the General Population, Including Healthcare Professionals and Pharmacists: A Systematic Review and Guidelines for a Smart Decision System. Nutrients 2023, 15, 2298. [Google Scholar] [CrossRef]
- Wang, X.; Fa, J.; Zhang, Y.; Huang, S.; Liu, J.; Gao, J.; Xing, L.; Liu, Z.; Wang, X. Evaluation of Herb-Drug Interaction Between Danshen and Rivaroxaban in Rat and Human Liver Microsomes. Front. Pharmacol. 2022, 13, 950525. [Google Scholar] [CrossRef] [PubMed]
- Amaeze, O.; Eng, H.; Horlbogen, L.; Varma, M.V.S.; Slitt, A. Cytochrome P450 Enzyme Inhibition and Herb-Drug Interaction Potential of Medicinal Plant Extracts Used for Management of Diabetes in Nigeria. Eur. J. Drug Metab. Pharmacokinet. 2021, 46, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yang, X.; Zhou, J.; Tang, L.; Xia, B.; Hu, M.; Zhou, F.; Liu, Z. The Exposure of Highly Toxic Aconitine does not Significantly Impact the Activity and Expression of Cytochrome P450 3A in Rats Determined by a Novel Ultra Performance Liquid Chromatography-Tandem Mass Spectrometric Method of a Specific Probe Buspirone. Food Chem. Toxicol. 2013, 51, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Cho, S.S.; Cho, H.J.; Yoon, I.S. Modulation of Hepatic Cytochrome P450 Enzymes by Curcumin and Its Pharmacokinetic Consequences in Sprague-Dawley Rats. Pharmacogn. Mag. 2015, 11, S580–S584. [Google Scholar] [PubMed]
- ICH Harmonisation for Better Health. Validation of Analytical Procedures: Text and Methodology Q2(R1). 2005. Available online: https://www.ich.org/page/quality-guidelines (accessed on 10 March 2023).
- Yasuda, K.; Ueno, S.; Ueda, E.; Nishikawa, M.; Takeda, K.; Kamakura, M.; Ikushiro, S.; Sakaki, T. Influence of Sesamin on CYP2C-Mediated Diclofenac Metabolism: In Vitro and In Vivo Analysis. Pharmacol. Res. Perspect. 2015, 3, e00174. [Google Scholar] [CrossRef]
- Seo, S.W.; Han, D.G.; Baek, Y.M.; Park, M.C.; Yoo, J.W.; Jung, Y.; Maeng, H.J.; Myung, H.; Yoon, I.S. Investigation of the Factors Responsible for the Low Oral Bioavailability of Alizarin Using a Sensitive LC-MS/MS Method: In Vitro, In Situ, and In Vivo Evaluations. Drug Dev. Res. 2023, 84, 579–591. [Google Scholar] [CrossRef]
- Knights, K.M.; Stresser, D.M.; Miners, J.O.; Crespi, C.L. In Vitro Drug Metabolism Using Liver Microsomes. Curr. Protoc. Pharmacol. 2016, 74, 7–8. [Google Scholar] [CrossRef]
- Seo, S.W.; Han, D.G.; Choi, E.; Park, T.; Byun, J.H.; Cho, H.J.; Jung, I.H.; Yoon, I.S. Development and Application of a Physiologically Based Pharmacokinetic Model for Entrectinib in Rats and Scale-Up to Humans: Route-Dependent Gut Wall Metabolism. Biomed. Pharmacother. 2022, 146, 112520. [Google Scholar] [CrossRef]
- Seo, S.W.; Han, D.G.; Choi, E.; Seo, M.J.; Song, I.S.; Yoon, I.S. Factors Determining the Oral Absorption and Systemic Disposition of Zeaxanthin in Rats: In Vitro, In Situ, and In Vivo Evaluations. Pharm. Biol. 2022, 60, 2266–2275. [Google Scholar] [CrossRef]
- Jeon, S.W.; Park, J.H.; Kim, J.E.; Park, Y.J. Design of Experiment (DoE)-Based Formulation Design of Bepotastine Sustained-Release Tablet and In Vitro-In Vivo Pharmacokinetic Correlation. J. Pharm. Investig. 2023, 53, 407–416. [Google Scholar] [CrossRef]
- Lee, T.J.; Kim, D.; Kim, J.C.; Ro, S.W.; Na, D.H. Formulation Development and Pharmacokinetic Evaluation of Enteric-Coated Dexrabeprazole Tablets. J. Pharm. Investig. 2023, 53, 323–331. [Google Scholar] [CrossRef]
- Xu, W.Z.; Jin, H.Z.; Zhang, W.D.; Hu, X.I.; Zhang, W.; Fu, J.J.; Su, J.; Yan, S.K.; Shen, Y.H. Chemical Constituents from Daphne pedunculata. Chem. Nat. Compd. 2009, 45, 417–419. [Google Scholar] [CrossRef]
- Yang, Q.Y.; Tian, X.Y.; Fang, W.S. Bioactive Coumarins from Boenninghausenia sessilicarpa. J. Asian. Nat. Prod. Res. 2007, 9, 59–65. [Google Scholar] [CrossRef] [PubMed]
- DuBois, B.N.; Mehvar, R. UPLC-MS/MS Analysis of Dextromethorphan-O-Demethylation Kinetics in Rat Brain Microsomes. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1096, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.W.; Pang, N.H.; Xu, R.A.; Qu, G.E.; Tang, C.R. Inhibition of Axitinib on Buspirone Metabolism In Vitro and In Vivo. Drug. Des. Devel. Ther. 2022, 16, 2031–2042. [Google Scholar] [CrossRef]
- Wu, J.; Cheng, Z.; Chen, H.; Zhang, G.; He, S.; Liu, S.; Lin, Z.; Zhu, L.; Liu, Z.; Lin, N.; et al. The Significant Inhibition on CYP3A caused by Radix Aconiti Single Herb is not Observed in the Wutou Decoction: The Necessity of Combination Therapy of Radix Aconiti. J. Ethnopharmacol. 2015, 170, 251–254. [Google Scholar] [CrossRef]
- Choi, M.K.; Lee, J.; Song, I.S. Pharmacokinetic Modulation of Substrate Drugs via the Inhibition of Drug-Metabolizing Enzymes and Transporters Using Pharmaceutical Excipients. J. Pharm. Investig. 2023, 53, 1–18. [Google Scholar] [CrossRef]
- Xia, F.; Chen, X.Y. Effect of Intestinal Cytochrome P450 3A on Phytochemical Presystemic Metabolism. Chin. J. Integr. Med. 2005, 11, 232–236. [Google Scholar]
- Xie, Y.; Zhang, D.; Zhang, J.; Yuan, J. Metabolism, Transport and Drug-Drug Interactions of Silymarin. Molecules 2019, 24, 3693. [Google Scholar] [CrossRef]
- Choi, Y.H. Interpretation of Drug Interaction Using Systemic and Local Tissue Exposure Changes. Pharmaceutics 2020, 12, 417. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound a | Linear Range (μg/mL) | Regression Equation b | r2 | DL (μg/mL) | QL (μg/mL) |
|---|---|---|---|---|---|
| 1 | 5.00–600.00 | y = 20,102.01x + 91,314.67 | 0.9995 | 1.19 | 3.59 |
| 2 | 5.00–600.00 | y = 18,769.28x + 9052.25 | 0.9995 | 0.72 | 2.19 |
| 3 | 5.00–600.00 | y = 14,803.15x + 46,931.92 | 0.9995 | 3.34 | 10.12 |
| Compound a | Concentration (μg/mL) | Intra-Day (n = 5) | Inter-Day (n = 5) | ||
|---|---|---|---|---|---|
| Mean ± SD (μg/mL) | RSD (%) | Mean ± SD (μg/mL) | RSD (%) | ||
| 1 | 9.70 | 10.6 ± 0.1 | 1.24 | 10.6 ± 0.1 | 0.94 |
| 97.0 | 95.7 ± 0.5 | 0.47 | 96.2 ± 0.8 | 0.87 | |
| 291 | 291 ± 1 | 0.26 | 293± 4 | 1.43 | |
| 2 | 9.70 | 9.40 ± 0.15 | 1.55 | 9.40 ± 0.10 | 1.11 |
| 97.0 | 97.4 ± 0.4 | 0.45 | 97.5 ± 0.6 | 0.60 | |
| 291 | 291 ± 2 | 0.54 | 296 ± 5 | 1.68 | |
| 3 | 9.80 | 11.2 ± 0.1 | 1.22 | 11.2 ± 0.1 | 0.78 |
| 98.0 | 96.0 ± 0.5 | 0.51 | 97.1 ± 1.0 | 1.04 | |
| 294 | 295 ± 2 | 0.75 | 294 ± 2 | 0.80 | |
| Compound a | Original Conc. (μg/mL) | Spiked Conc. (μg/mL) | Found Conc. ± SD (μg/mL) | Recovery ± SD (%) | RSD (%) |
|---|---|---|---|---|---|
| 1 | 804 | 11.6 | 817 ± 1 | 113 ± 2 | 1.86 |
| 58.2 | 873 ± 1 | 118 ± 1 | 0.70 | ||
| 116 | 915 ± 1 | 95.3 ± 1.3 | 1.39 | ||
| 2 | 196 | 11.6 | 208 ± 0 | 105 ± 1 | 0.98 |
| 58.2 | 255 ± 0 | 100 ± 1 | 1.16 | ||
| 116 | 311 ± 0 | 98.8 ± 0.5 | 0.48 | ||
| 3 | 173 | 11.8 | 183 ± 0 | 95.5 ± 1.6 | 1.68 |
| 58.8 | 229 ± 0 | 93.4 ± 1.2 | 1.33 | ||
| 118 | 285 ± 0 | 96.1 ± 0.3 | 0.36 |
| Solutions | Compound a | Temp. | Peak Area (%) | RSD (%) | |||
|---|---|---|---|---|---|---|---|
| 6 h | 24 h | 48 h | 72 h | ||||
| WGM | 1 | R.T. | 100.80 | 100.10 | 97.43 | 100.99 | 1.33 |
| 4 °C | 100.10 | 98.87 | 98.63 | 101.60 | 1.28 | ||
| 2 | R.T. | 101.41 | 101.08 | 98.97 | 103.35 | 1.52 | |
| 4 °C | 100.32 | 99.18 | 98.86 | 102.02 | 1.34 | ||
| 3 | R.T. | 101.30 | 101.14 | 98.98 | 103.56 | 1.58 | |
| 4 °C | 100.19 | 99.30 | 98.91 | 102.15 | 1.38 | ||
| Reference | 1 | R.T. | 99.46 | 98.56 | 100.34 | 99.03 | 0.76 |
| 4 °C | 99.15 | 96.60 | 96.58 | 97.39 | 1.46 | ||
| 2 | R.T. | 101.71 | 103.18 | 107.58 | 103.48 | 2.57 | |
| 4 °C | 99.29 | 102.15 | 104.42 | 101.79 | 1.93 | ||
| 3 | R.T. | 96.77 | 98.82 | 98.54 | 98.72 | 1.19 | |
| 4 °C | 98.02 | 99.40 | 98.78 | 101.22 | 1.21 | ||
| Compound a | Amount | ||
|---|---|---|---|
| Mean (mg/g) | SD | %RSD | |
| 1 | 51.81 | 0.05 | 0.09 |
| 2 | 19.36 | 0.02 | 0.10 |
| 3 | 17.10 | 0.02 | 0.11 |
| Parameter | BUS Alone | BUS with WGM |
|---|---|---|
| AUCinf (μg·min/mL) | 98.2 ± 39.7 | 119 ± 50 |
| t1/2 (min) | 111 ± 23 | 103 ± 27 |
| Cmax (ng/mL) | 1629 ± 762 | 2016 ± 619 |
| Tmax (min) | 10 (5–15) | 5 (5–10) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keem, M.-J.; Seo, S.-W.; Kim, T.; Jo, B.-G.; Kim, S.-N.; Yoon, I.-S.; Yang, M.H. A High-Performance Liquid Chromatography with Photodiode Array Detection Method for Simultaneous Determination of Three Compounds Isolated from Wikstroemia ganpi: Assessment of the Effects on Cytochrome P450-Mediated Metabolism In Vitro and In Vivo. Nutrients 2023, 15, 4061. https://doi.org/10.3390/nu15184061
Keem M-J, Seo S-W, Kim T, Jo B-G, Kim S-N, Yoon I-S, Yang MH. A High-Performance Liquid Chromatography with Photodiode Array Detection Method for Simultaneous Determination of Three Compounds Isolated from Wikstroemia ganpi: Assessment of the Effects on Cytochrome P450-Mediated Metabolism In Vitro and In Vivo. Nutrients. 2023; 15(18):4061. https://doi.org/10.3390/nu15184061
Chicago/Turabian StyleKeem, Min-Ji, Seong-Wook Seo, Taeyoung Kim, Beom-Geun Jo, Su-Nam Kim, In-Soo Yoon, and Min Hye Yang. 2023. "A High-Performance Liquid Chromatography with Photodiode Array Detection Method for Simultaneous Determination of Three Compounds Isolated from Wikstroemia ganpi: Assessment of the Effects on Cytochrome P450-Mediated Metabolism In Vitro and In Vivo" Nutrients 15, no. 18: 4061. https://doi.org/10.3390/nu15184061