
Beta Cell Dysfunction in Youth- and Adult-Onset Type 2 Diabetes: An Extensive Narrative Review with a Special Focus on the Role of Nutrients
 , , and
, , and
Abstract
:1. Introduction
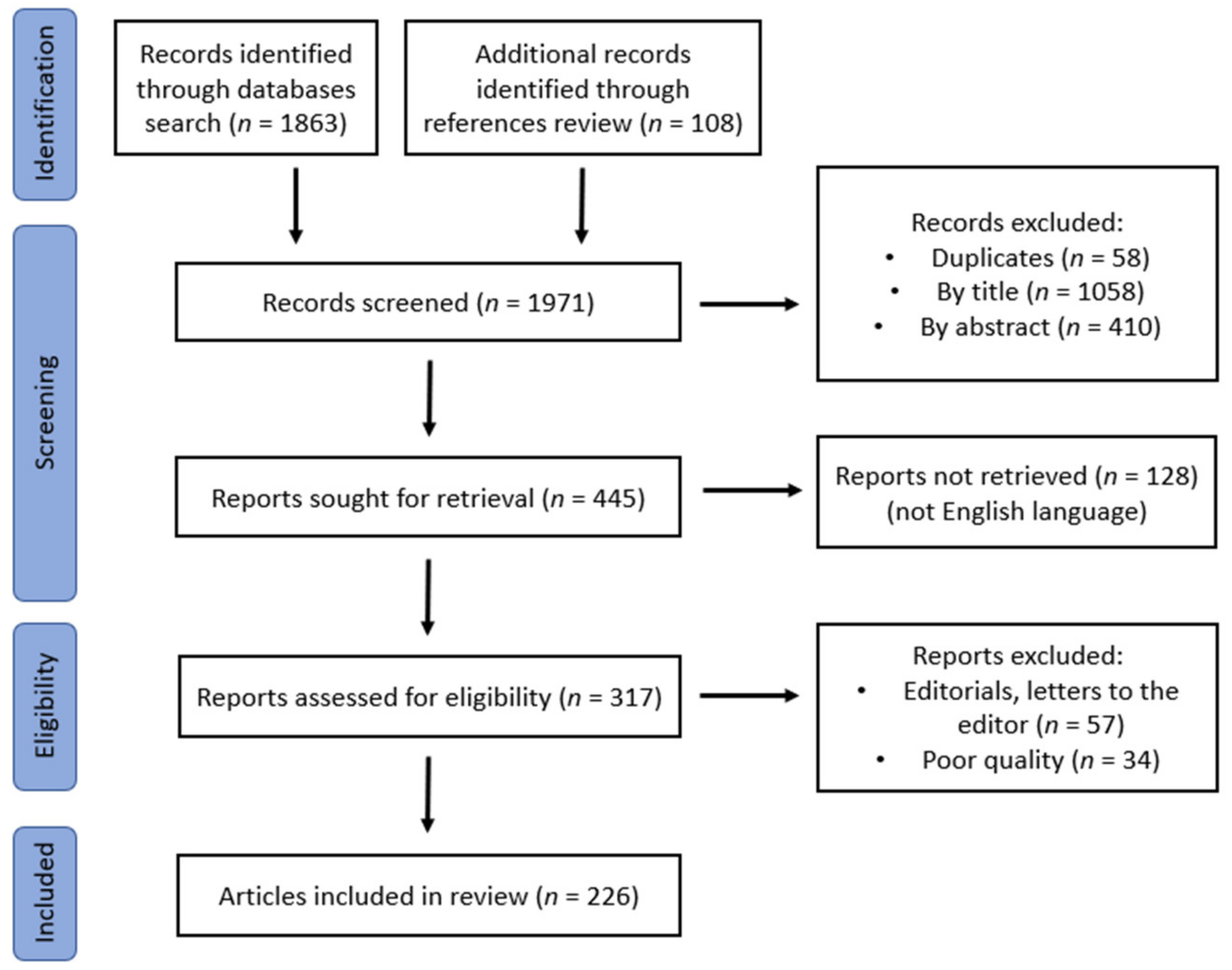
2. Materials and Methods
3. Results
4. Discussion
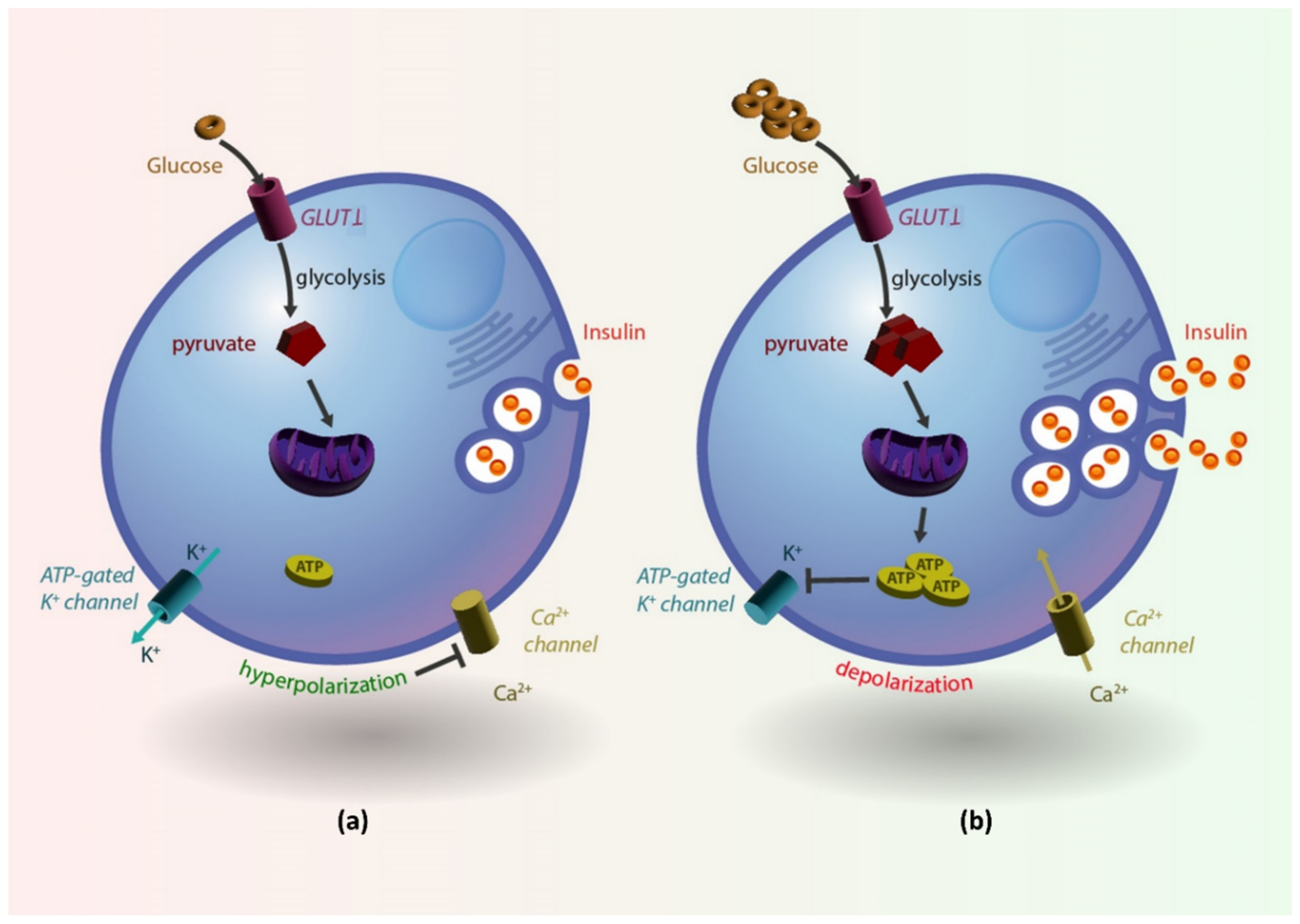
4.1. Normal Insulin Production by Pancreatic β-Cells
4.2. Clinical Evidence of the β-Cell Dysfunction Role in T2D
4.3. Studies Connecting Nutrient Levels with T2D Risk
4.4. Genetics, and Epigenetics Role in β-Cell Dysfunction
4.5. Defective Insulin Secretion from β-Cells before, at the Time of, and after T2D Diagnosis
4.6. Defective Insulin Production, Processing, and Secretion at the Cellular Level
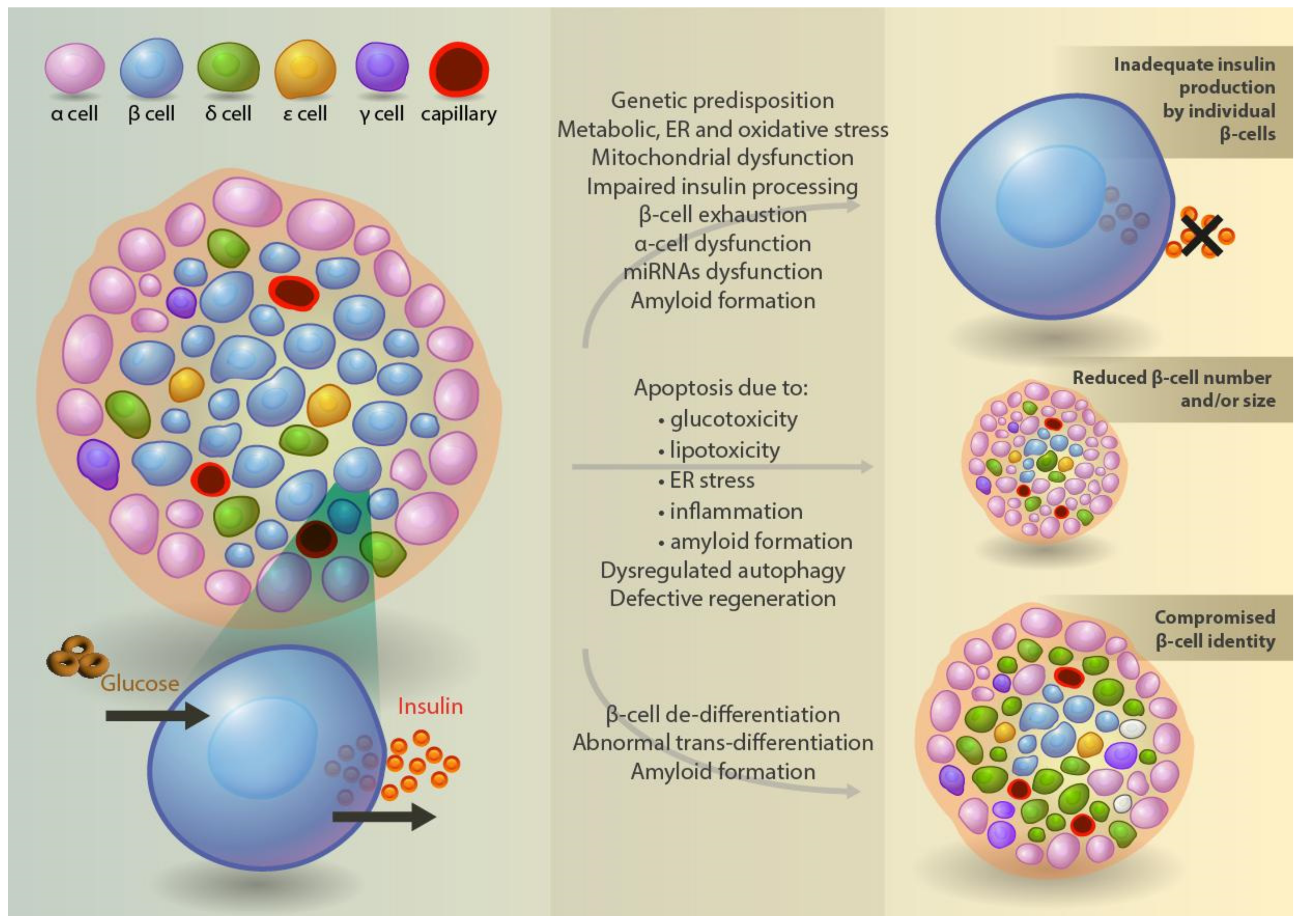
4.7. β-Cell Mass in Type 2 Diabetes
4.8. Mechanisms of β-Cell Death and Regeneration
4.9. Trans- and De-Differentiation of β-Cells
4.10. Role of Islet Inflammation
4.11. Role of α- and Other Islet Cell Dysfunction in Diabetes Pathogenesis
4.12. Role of Islet Amyloid Polypeptide
4.13. Role of miRNAs Dysfunction
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galicia-Garcia, U.; Benito-Vicente, A.; Jebari, S.; Larrea-Sebal, A.; Siddiqi, H.; Uribe, K.B.; Ostolaza, H.; Martín, C. Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2020, 21, 6275. [Google Scholar] [CrossRef]
- Mayer-Davis, E.J.; Lawrence, J.M.; Dabelea, D.; Divers, J.; Isom, S.; Dolan, L.; Imperatore, G.; Linder, B.; Marcovina, S.; Pettitt, D.J.; et al. Incidence Trends of Type 1 and Type 2 Diabetes among Youths, 2002–2012. N. Engl. J. Med. 2017, 376, 1419–1429. [Google Scholar] [CrossRef] [PubMed]
- Wagenknecht, L.E.; Lawrence, J.M.; Isom, S.; Jensen, E.T.; Dabelea, D.; Liese, A.D.; Dolan, L.M.; Shah, A.S.; Bellatorre, A.; Sauder, K.; et al. Trends in incidence of youth-onset type 1 and type 2 diabetes in the USA, 2002–2018: Results from the population-based SEARCH for Diabetes in Youth study. Lancet Diabetes Endocrinol. 2023, 11, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, M.; Long, Z.; Ning, H.; Li, J.; Cao, Y.; Liao, Y.; Liu, G.; Wang, F.; Pan, A. Global burden of type 2 diabetes in adolescents and young adults, 1990–2019: Systematic analysis of the Global Burden of Disease Study 2019. BMJ 2022, 379, e072385. [Google Scholar] [CrossRef]
- Daniels, N.F.; Burrin, C.; Chan, T.; Fusco, F. A Systematic Review of the Impact of the First Year of COVID-19 on Obesity Risk Factors: A Pandemic Fueling a Pandemic? Curr. Dev. Nutr. 2022, 6, nzac011. [Google Scholar] [CrossRef] [PubMed]
- Woolford, S.J.; Sidell, M.; Li, X.; Else, V.; Young, D.R.; Resnicow, K.; Koebnick, C. Changes in Body Mass Index Among Children and Adolescents During the COVID-19 Pandemic. JAMA 2021, 326, 1434. [Google Scholar] [CrossRef]
- Perng, W.; Conway, R.; Mayer-Davis, E.; Dabelea, D. Youth-Onset Type 2 Diabetes: The Epidemiology of an Awakening Epidemic. Diabetes Care 2023, 46, 490–499. [Google Scholar] [CrossRef]
- Al-Saeed, A.H.; Constantino, M.I.; Molyneaux, L.; D’souza, M.; Limacher-Gisler, F.; Luo, C.; Wu, T.; Twigg, S.M.; Yue, D.K.; Wong, J. An Inverse Relationship Between Age of Type 2 Diabetes Onset and Complication Risk and Mortality: The Impact of Youth-Onset Type 2 Diabetes. Diabetes Care 2016, 39, 823–829. [Google Scholar] [CrossRef]
- The RISE Consortium; Ehrmann, D.A.; Temple, K.A.; Rue, A.; Barengolts, E.; Mokhlesi, B.; Van Cauter, E.; Sam, S.; Miller, M.A.; Kahn, S.E.; et al. Metabolic Contrasts Between Youth and Adults With Impaired Glucose Tolerance or Recently Diagnosed Type 2 Diabetes: II. Observations Using the Oral Glucose Tolerance Test. Diabetes Care 2018, 41, 1707–1716. [Google Scholar] [CrossRef]
- Edelstein, S.L.; Kahn, S.E.; Arslanian, S.A.; Buchanan, T.A.; Ehrmann, D.A.; Nadeau, K.J.; Palmer, J.P.; Utzschneider, K.M. Metabolic Contrasts Between Youth Adults with Impaired Glucose Tolerance or Recently Diagnosed Type 2 Diabetes: I. Observations Using the Hyperglycemic Clamp. Diabetes Care 2018, 41, 1696–1706. [Google Scholar]
- Bacha, F.; Gungor, N.; Lee, S.; Arslanian, S.A. Progressive deterioration of β-cell function in obese youth with type 2 diabetes. Pediatr. Diabetes 2012, 14, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Arslanian, S.; Pyle, L.; Payan, M.; Bacha, F.; Caprio, S.; Haymond, M.W.; TODAY Study Group. Effects of metformin, metformin plus rosiglitazone, and metformin plus lifestyle on insulin sensitivity and β-cell function in TODAY. Diabetes Care 2013, 36, 1749–1757. [Google Scholar]
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. N. Am. 2004, 88, 787–835. [Google Scholar] [CrossRef] [PubMed]
- Dubey, P.; Thakur, V.; Chattopadhyay, M. Role of Minerals and Trace Elements in Diabetes and Insulin Resistance. Nutrients 2020, 12, 1864. [Google Scholar] [CrossRef]
- Black, M.H.; Watanabe, R.M.; Trigo, E.; Takayanagi, M.; Lawrence, J.M.; Buchanan, T.A.; Xiang, A.H. High-Fat Diet Is Associated with Obesi-ty-Mediated Insulin Resistance and β-Cell Dysfunction in Mexican Americans. J. Nutr. 2013, 143, 479–485. [Google Scholar] [CrossRef]
- Moore, W.T.; Bowser, S.M.; Fausnacht, D.W.; Staley, L.L.; Suh, K.-S.; Liu, D. Beta Cell Function and the Nutritional State: Dietary Factors that Influence Insulin Secretion. Curr. Diabetes Rep. 2015, 15, 76. [Google Scholar] [CrossRef]
- Pfeifer, M.A.; Halter, J.B.; Porte, D., Jr. Insulin secretion in diabetes mellitus. Am. J. Med. 1981, 70, 579–588. [Google Scholar] [CrossRef]
- Weir, G.C.; Gaglia, J.; Bonner-Weir, S. Inadequate β-cell mass is essential for the pathogenesis of type 2 diabetes. Lancet Diabetes Endocrinol. 2020, 8, 249–256. [Google Scholar] [CrossRef]
- Holman, R.R.; Clark, A.; Rorsman, P. β-cell secretory dysfunction: A key cause of type 2 diabetes. Lancet Diabetes Endocrinol. 2020, 8, 370. [Google Scholar] [CrossRef]
- Ramchandani, N.; Ellis, M.K.; Jain, S.; Bhandari, S.; Anhalt, H.; Maclaren, N.K.; Ten, S. Basal Insulin Requirements on Continuous Subcutaneous Insulin Infusion During the First 12 Months After Diagnosis of Type 1 Diabetes Mellitus. J. Diabetes Sci. Technol. 2010, 4, 610–614. [Google Scholar] [CrossRef]
- Lang, D.A.; Matthews, D.R.; Peto, J.; Turner, R.C. Cyclic Oscillations of Basal Plasma Glucose and Insulin Concentrations in Human Beings. N. Engl. J. Med. 1979, 301, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Gilon, P.; Ravier, M.A.; Jonas, J.-C.; Henquin, J.-C. Control Mechanisms of the Oscillations of Insulin Secretion In Vitro and In Vivo. Diabetes 2002, 51, S144–S151. [Google Scholar] [CrossRef] [PubMed]
- Henquin, J.-C.; Ishiyama, N.; Nenquin, M.; Ravier, M.A.; Jonas, J.-C. Signals and Pools Underlying Biphasic Insulin Secretion. Diabetes 2002, 51, S60–S67. [Google Scholar] [CrossRef] [PubMed]
- Lyon, J.; Fox, J.E.M.; Spigelman, A.F.; Kim, R.; Smith, N.; O’Gorman, D.; Kin, T.; Shapiro, A.M.J.; Rajotte, R.V.; MacDonald, P. Research-Focused Isolation of Human Islets From Donors With and Without Diabetes at the Alberta Diabetes Institute IsletCore. Endocrinology 2015, 157, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Perley, M.J.; Kipnis, D.M. Plasma insulin responses to oral and intravenous glucose: Studies in normal and diabetic sujbjects. J. Clin. Invest. 1967, 46, 1954–1962. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 2018, 20 (Suppl. S1), 5–21. [Google Scholar] [CrossRef]
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010, 1, 8–23. [Google Scholar] [CrossRef]
- Pingitore, A.; Gonzalez-Abuin, N.; Ruz-Maldonado, I.; Huang, G.C.; Frost, G.; Persaud, S.J. Short chain fatty acids stimulate insulin secretion and reduce apoptosis in mouse and human islets in vitro: Role of free fatty acid receptor 2. Diabetes Obes. Metab. 2018, 21, 330–339. [Google Scholar] [CrossRef]
- Henquin, J.-C.; Dufrane, D.; Nenquin, M. Nutrient Control of Insulin Secretion in Isolated Normal Human Islets. Diabetes 2006, 55, 3470–3477. [Google Scholar] [CrossRef]
- Singh, A.; Gibert, Y.; Dwyer, K.M. The adenosine, adrenergic and opioid pathways in the regulation of insulin secretion, beta cell proliferation and regeneration. Pancreatology 2018, 18, 615–623. [Google Scholar] [CrossRef]
- Di Cairano, E.S.; Moretti, S.; Marciani, P.; Sacchi, V.F.; Castagna, M.; Davalli, A.; Folli, F.; Perego, C. Neurotransmitters and Neuropeptides: New Players in the Control of Islet of Langerhans’ Cell Mass and Function. J. Cell. Physiol. 2016, 231, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Thurmond, D.C. Mechanisms of biphasic insulin-granule exocytosis—Roles of the cytoskeleton, small GTPases and SNARE proteins. J. Cell Sci. 2009, 122, 893. [Google Scholar] [CrossRef] [PubMed]
- Haffner, S.M.; Miettinen, H.; Gaskill, S.P.; Stern, M.P. Decreased insulin secretion and increased insulin resistance are independently related to the 7-year risk of NIDDM in Mexican-Americans. Diabetes 1995, 44, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Weyer, C.; Bogardus, C.; Mott, D.M.; Pratley, R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Investig. 1999, 104, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Tabák, A.G.; Jokela, M.; Akbaraly, T.N.; Brunner, E.J.; Kivimäki, M.; Witte, D.R. Trajectories of Glycemia, Insulin Sensitivity and Insulin Secretion Preceding the Diagnosis of Type 2 Diabetes: The Whitehall II Study. Lancet 2009, 373, 2215–2221. [Google Scholar] [CrossRef]
- Kitabchi, A.E.; Temprosa, M.; Knowler, W.C.; Kahn, S.E.; Fowler, S.E.; Haffner, S.M.; Andres, R.; Saudek, C.; Edelstein, S.L.; Arakaki, R.; et al. Role of insulin secretion and sensitivity in the evolution of type 2 diabetes in the diabetes prevention program: Effects of lifestyle intervention and metformin. Diabetes 2005, 54, 2404–2414. [Google Scholar]
- Giannini, C.; Weiss, R.; Cali, A.; Bonadonna, R.; Santoro, N.; Pierpont, B.; Shaw, M.; Caprio, S. Evidence for early defects in insulin sensitivity and secretion before the onset of glucose dysregulation in obese youths: A longitudinal study. Diabetes 2012, 61, 606–614. [Google Scholar] [CrossRef]
- Goran, M.I.; Bergman, R.N.; Avila, Q.; Watkins, M.; Ball, G.D.; Shaibi, G.Q.; Weigensberg, M.J.; Cruz, M.L. Impaired glucose tolerance and reduced beta-cell function in overweight Latino children with a positive family history for type 2 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 207–212. [Google Scholar] [CrossRef]
- Saad, R.; Gungor, N.; Arslanian, S. Progression from normal glucose tolerance to type 2 diabetes in a young girl: Longitudinal changes in insulin sensitivity and secretion assessed by the clamp technique and surrogate estimates. Pediatr. Diabetes 2005, 6, 95–99. [Google Scholar] [CrossRef]
- Burns, S.F.; Bacha, F.; Lee, S.J.; Tfayli, H.; Gungor, N.; Arslanian, S.A. Declining β-Cell Function Relative to Insulin Sensitivity With Escalating OGTT 2-h Glucose Concentrations in the Nondiabetic Through the Diabetic Range in Overweight Youth. Diabetes Care 2011, 34, 2033–2040. [Google Scholar] [CrossRef]
- Sjaarda, L.A.; Michaliszyn, S.F.; Lee, S.; Tfayli, H.; Bacha, F.; Farchoukh, L.; Arslanian, S.A. HbA1c Diagnostic Categories and β-Cell Function Relative to Insulin Sensitivity in Overweight/Obese Adolescents. Diabetes Care 2012, 35, 2559–2563. [Google Scholar] [CrossRef] [PubMed]
- Bacha, F.; Gungor, N.; Lee, S.; Arslanian, S.A. In vivo insulin sensitivity and secretion in obese youth: What are the differences between normal glucose tolerance, impaired glucose tolerance, and type 2 diabetes? Diabetes Care 2009, 32, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Lachin, J.M.; Zinman, B.; Haffner, S.M.; Aftring, R.P.; Paul, G.; Kravitz, B.G.; Herman, W.H.; Viberti, G.; Holman, R.R.; et al. Effects of rosiglitazone, glyburide, and metformin on β-cell function and insulin sensitivity in ADOPT. Diabetes 2011, 60, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- The RISE Consortium; Nadeau, K.J.; Hannon, T.S.; Edelstein, S.L.; Arslanian, S.A.; Caprio, S.; Leschek, E.W.; Zeitler, P.S.; Buchanan, T.A.; Ehrmann, D.A.; et al. Impact of Insulin and Metformin Versus Metformin Alone on β-Cell Function in Youth With Impaired Glucose Tolerance or Recently Diagnosed Type 2 Diabetes. Diabetes Care 2018, 41, 1717–1725. [Google Scholar] [CrossRef]
- Hannon, T.S.; Edelstein, S.L.; Arslanian, S.A.; Caprio, S.; Zeitler, P.S.; Buchanan, T.A.; Ehrmann, D.A.; Mather, K.J.; Tripputi, M.; Kahn, S.E.; et al. Withdrawal of medications leads to worsening of OGTT parameters in youth with impaired glucose tolerance or recently-diagnosed type 2 diabetes. Pediatr. Diabetes 2020, 21, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Badaru, A.; Klingensmith, G.J.; Dabelea, D.; Mayer-Davis, E.J.; Dolan, L.; Lawrence, J.M.; Marcovina, S.; Beavers, D.; Rodriguez, B.L.; Imperatore, G.; et al. Correlates of Treatment Patterns Among Youth With Type 2 Diabetes. Diabetes Care 2013, 37, 64–72. [Google Scholar] [CrossRef]
- Pittas, A.G.; Lau, J.; Hu, F.B.; Dawson-Hughes, B. The Role of Vitamin D and Calcium in Type 2 Diabetes. A Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 2007, 92, 2017–2029. [Google Scholar] [CrossRef]
- Hajhashemy, Z.; Rouhani, P.; Saneei, P. Dietary calcium intake in relation to type-2 diabetes and hyperglycemia in adults: A sys-tematic review and dose–response meta-analysis of epidemiologic studies. Sci. Rep. 2022, 12, 1050. [Google Scholar] [CrossRef]
- Gijsbers, L.; Ding, E.L.; Malik, V.S.; De Goede, J.; Geleijnse, J.M.; Soedamah-Muthu, S.S. Consumption of dairy foods and diabetes incidence: A dose-response meta-analysis of observational studies. Am. J. Clin. Nutr. 2016, 103, 1111–1124. [Google Scholar] [CrossRef]
- Aune, D.; Norat, T.; Romundstad, P.; Vatten, L.J. Dairy products and the risk of type 2 diabetes: A systematic review and dose-response meta-analysis of cohort studies. Am. J. Clin. Nutr. 2013, 98, 1066–1083. [Google Scholar] [CrossRef]
- Becerra-Tomás, N.; Estruch, R.; Bulló, M.; Casas, R.; Díaz-López, A.; Basora, J.; Fitó, M.; Serra-Majem, L.; Salas-Salvadó, J. Increased Serum Calcium Levels and Risk of Type 2 Diabetes in Individuals at High Cardiovascular Risk. Diabetes Care 2014, 37, 3084–3091. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Xun, P.; Bae, J.C.; Kim, J.H.; Kim, D.J.; Yang, K.; He, K. Circulating calcium levels and the risk of type 2 diabetes: A systematic review and meta-analysis. Br. J. Nutr. 2019, 122, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Javed, A.; Vella, A.; Balagopal, P.B.; Fischer, P.R.; Weaver, A.L.; Piccinini, F.; Man, C.D.; Cobelli, C.; Giesler, P.D.; Laugen, J.M.; et al. Cholecalciferol Supplementation Does Not Influence β-Cell Function and Insulin Action in Obese Adolescents: A Prospective Double-Blind Randomized Trial. J. Nutr. 2015, 145, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Kayaniyil, S.; Vieth, R.; Retnakaran, R.; Knight, J.A.; Qi, Y.; Gerstein, H.C.; Perkins, B.A.; Harris, S.B.; Zinman, B.; Hanley, A.J. Association of vitamin D with insulin resistance and beta-cell dysfunction in subjects at risk for type 2 diabetes. Diabetes Care 2010, 33, 1379–1381. [Google Scholar] [CrossRef] [PubMed]
- Kayaniyil, S.; Retnakaran, R.; Harris, S.B.; Vieth, R.; Knight, J.A.; Gerstein, H.C.; Perkins, B.A.; Zinman, B.; Hanley, A.J. Prospective associations of vitamin D with β-cell function and glycemia: The PROspective Metabolism and ISlet cell Evaluation (PROMISE) cohort study. Diabetes 2011, 60, 2947–2953. [Google Scholar] [CrossRef]
- Mitri, J.; Dawson-Hughes, B.; Hu, F.B.; Pittas, A.G. Effects of vitamin D and calcium supplementation on pancreatic β cell function, insulin sensitivity, and glycemia in adults at high risk of diabetes: The Calcium and Vitamin D for Diabetes Mellitus (CaDDM) randomized controlled trial. Am. J. Clin. Nutr. 2011, 94, 486–494. [Google Scholar] [CrossRef]
- Jiang, R.; Manson, J.E.; Meigs, J.B.; Ma, J.; Rifai, N.; Hu, F.B. Body Iron Stores in Relation to Risk of Type 2 Diabetes in Apparently Healthy Women. JAMA 2004, 291, 711–717. [Google Scholar] [CrossRef]
- Noetzli, L.J.; Mittelman, S.D.; Watanabe, R.M.; Coates, T.D.; Wood, J.C. Pancreatic iron and glucose dysregulation in thalassemia major. Am. J. Hematol. 2011, 87, 155–160. [Google Scholar] [CrossRef]
- Huth, C.; Beuerle, S.; Zierer, A.; Heier, M.; Herder, C.; Kaiser, T.; Koenig, W.; Kronenberg, F.; Oexle, K.; Rathmann, W.; et al. Biomarkers of iron metabolism are independently associated with impaired glucose metabolism and type 2 diabetes: The KORA F4 study. Eur. J. Endocrinol. 2015, 173, 643–653. [Google Scholar] [CrossRef]
- Lopez-Ridaura, R.; Willett, W.C.; Rimm, E.B.; Liu, S.; Stampfer, M.J.; Manson, J.E.; Hu, F.B. Magnesium Intake and Risk of Type 2 Diabetes in Men and Women. Diabetes Care 2004, 27, 134–140. [Google Scholar] [CrossRef]
- Hata, A.; Doi, Y.; Ninomiya, T.; Mukai, N.; Hirakawa, Y.; Hata, J.; Ozawa, M.; Uchida, K.; Shirota, T.; Kitazono, T.; et al. Magnesium intake decreases Type 2 diabetes risk through the improvement of insulin resistance and inflammation: The Hisayama Study. Diabet. Med. 2013, 30, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Bleys, J.; Navas-Acien, A.; Guallar, E. Serum Selenium and Diabetes in U.S. Adults. Diabetes Care 2007, 30, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zeng, C.; Gong, Q.-Y.; Yang, H.-B.; Li, X.-X.; Lei, G.-H.; Yang, T.-B. The association between dietary selenium intake and diabetes: A cross-sectional study among middle-aged and older adults. Nutr. J. 2015, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Stranges, S.; Sieri, S.; Vinceti, M.; Grioni, S.; Guallar, E.; Laclaustra, M.; Muti, P.; Berrino, F.; Krogh, V. A prospective study of dietary selenium intake and risk of type 2 diabetes. BMC Public Health 2010, 10, 564. [Google Scholar] [CrossRef]
- Stranges, S.; Marshall, J.R.; Natarajan, R.; Donahue, R.P.; Trevisan, M.; Combs, G.F.; Cappuccio, F.P.; Ceriello, A.; Reid, M.E. Effects of long-term selenium supplementation on the incidence of type 2 diabetes: A randomized trial. Ann. Intern. Med. 2007, 147, 217–223. [Google Scholar] [CrossRef]
- Lu, C.-W.; Chang, H.-H.; Yang, K.-C.; Kuo, C.-S.; Lee, L.-T.; Huang, K.-C. High serum selenium levels are associated with increased risk for diabetes mellitus independent of central obesity and insulin resistance. BMJ Open Diabetes Res. Care 2016, 4, e000253. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Duntas, L.H.; Rayman, M.P. The role of selenium in type-2 diabetes mellitus and its metabolic comorbidities. Redox Biol. 2022, 50, 102236. [Google Scholar] [CrossRef]
- Park, K.; Rimm, E.B.; Siscovick, D.S.; Spiegelman, D.; Manson, J.E.; Morris, J.S.; Hu, F.B.; Mozaffarian, D. Toenail Selenium and Incidence of Type 2 Diabetes in U.S. Men and Women. Diabetes Care 2012, 35, 1544–1551. [Google Scholar] [CrossRef]
- Sun, Q.; van Dam, R.M.; Willett, W.C.; Hu, F.B. Prospective Study of Zinc Intake and Risk of Type 2 Diabetes in Women. Diabetes Care 2009, 32, 629–634. [Google Scholar] [CrossRef]
- Drake, I.; Hindy, G.; Ericson, U.; Orho-Melander, M. A prospective study of dietary and supplemental zinc intake and risk of type 2 diabetes depending on genetic variation in SLC30A8. Genes Nutr. 2017, 12, 30. [Google Scholar] [CrossRef]
- Klein, B.E.K.; Klein, R.; Moss, S.E.; Cruickshanks, K.J. Parental History of Diabetes in a Population-Based Study. Diabetes Care 1996, 19, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Barnett, A.H.; Eff, C.; Leslie, R.D.G.; Pyke, D.A. Diabetes in identical twins. Diabetologia 1981, 20, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Forouhi, N.G.; Wareham, N.J. Epidemiology of diabetes. Medicine 2014, 42, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E. Is Reduced First-Phase Insulin Release the Earliest Detectable Abnormality in Individuals Destined to Develop Type 2 Diabetes? Diabetes 2002, 51 (Suppl. S1), S117–S121. [Google Scholar] [CrossRef]
- Lyssenko, V.; Jonsson, A.; Almgren, P.; Pulizzi, N.; Isomaa, B.; Tuomi, T.; Berglund, G.; Altshuler, D.; Nilsson, P.; Groop, L. Clinical risk factors, DNA variants, and the development of type 2 diabetes. N. Engl. J. Med. 2008, 359, 2220–2232. [Google Scholar] [CrossRef]
- Altshuler, D.; Hirschhorn, J.N.; Klannemark, M.; Lindgren, C.M.; Vohl, M.C.; Nemesh, J. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat. Genet. 2000, 26, 76–80. [Google Scholar] [CrossRef]
- Gloyn, A.L.; Weedon, M.N.; Owen, K.R.; Turner, M.J.; Knight, B.A.; Hitman, G.; Walker, M.; Levy, J.C.; Sampson, M.; Halford, S.; et al. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 2003, 52, 568–572. [Google Scholar] [CrossRef]
- Grant, S.F.A.; Thorleifsson, G.; Reynisdottir, I.; Benediktsson, R.; Manolescu, A.; Sainz, J.; Helgason, A.; Stefansson, H.; Emilsson, V.; Helgadottir, A.; et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat. Genet. 2006, 38, 320–323. [Google Scholar] [CrossRef]
- Radha, V.; Vimaleswaran, K.S.; Babu, H.N.S.; Abate, N.; Chandalia, M.; Satija, P.; Grundy, S.M.; Ghosh, S.; Majumder, P.P.; Deepa, R.; et al. Role of genetic polymorphism peroxisome proliferator-activated receptor-gamma2 Pro12Ala on ethnic susceptibility to diabetes in South-Asian and Caucasian subjects: Evidence for heterogeneity. Diabetes Care 2006, 29, 1046–1051. [Google Scholar] [CrossRef]
- Mahajan, A.; Wessel, J.; Willems, S.M.; Zhao, W.; Robertson, N.R.; Chu, A.Y.; Gan, W.; Kitajima, H.; Taliun, D.; Rayner, N.W.; et al. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat. Genet. 2018, 50, 559–571. [Google Scholar] [CrossRef]
- Wheeler, E.; Marenne, G.; Barroso, I. Genetic aetiology of glycaemic traits: Approaches and insights. Hum. Mol. Genet. 2017, 26, R172–R184. [Google Scholar] [CrossRef] [PubMed]
- Vujkovic, M.; Keaton, J.M.; Lynch, J.A.; Miller, D.R.; Zhou, J.; Tcheandjieu, C.; Huffman, J.E.; Assimes, T.L.; Lorenz, K.; Zhu, X.; et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 2020, 52, 680–691. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Todd, J. The Genetics of Type 2 Diabetes in Youth: Where We Are and the Road Ahead. J. Pediatr. 2022, 247, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Cropano, C.; Santoro, N.; Groop, L.; Man, C.D.; Cobelli, C.; Galderisi, A.; Kursawe, R.; Pierpont, B.; Goffredo, M.; Caprio, S. The rs7903146 Variant in the TCF7L2 Gene Increases the Risk of Prediabetes/Type 2 Diabetes in Obese Adolescents by Impairing β-Cell Function and Hepatic Insulin Sensitivity. Diabetes Care 2017, 40, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Chen, L.; Todd, J.; Divers, J.; Gidding, S.; Chernausek, S.; Gubitosi-Klug, R.A.; Kelsey, M.M.; Shah, R.; Black, M.H.; et al. The First Genome-Wide Association Study for Type 2 Diabetes in Youth: The Progress in Diabetes Genetics in Youth (ProDiGY) Consortium. Diabetes 2021, 70, 996–1005. [Google Scholar] [CrossRef]
- Schuch, N.J.; Garcia, V.C.; Vívolo, S.R.G.F.; Martini, L.A. Relationship between Vitamin D Receptor gene polymorphisms and the components of metabolic syndrome. Nutr. J. 2013, 12, 96. [Google Scholar] [CrossRef]
- Sentinelli, F.; Bertoccini, L.; Barchetta, I.; Capoccia, D.; Incani, M.; Pani, M.; Loche, S.; Angelico, F.; Arca, M.; Morini, S.; et al. The vitamin D receptor (VDR) gene rs11568820 variant is associated with type 2 diabetes and impaired insulin secretion in Italian adult subjects, and associates with increased cardio-metabolic risk in children. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 407–413. [Google Scholar] [CrossRef]
- Zeitz, U.; Weber, K.; Soegiarto, D.W.; Wolf, E.; Balling, R.; Erben, R.G. Impaired insulin secretory capacity in mice lacking a functional vitamin D receptor. FASEB J. 2003, 17, 509–511. [Google Scholar] [CrossRef]
- Aravindhan, S.; Almasoody, M.F.M.; Selman, N.A.; Andreevna, A.N.; Ravali, S.; Mohammadi, P.; Eslami, M.M.; Razi, B.; Aslani, S.; Imani, D. Vitamin D Receptor gene polymorphisms and susceptibility to type 2 diabetes: Evidence from a meta-regression and meta-analysis based on 47 studies. J. Diabetes Metab. Disord. 2021, 20, 845–867. [Google Scholar] [CrossRef]
- Chen, B.; Cheng, L.; Zhang, D.; Zhou, L.; Zhao, J. Association between SLC30A8 rs13266634 Polymorphism and Type 2 Diabetes Risk: A Meta-Analysis. Experiment 2015, 21, 2178–2189. [Google Scholar] [CrossRef]
- Dong, F.; Zhang, B.H.; Zheng, S.L.; Huang, X.X.; Du, X.B.; Zhu, K.H.; Chen, X.J.; Wu, J.; Liu, D.D.; Wen, Z.H.; et al. Association between SLC30A8 rs13266634 polymorphism and risk of T2DM and IGR in Chinese population: A systematic review and meta-analysis. Front. Endocrinol. 2018, 9, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Mashal, S.; Khanfar, M.; Al-Khalayfa, S.; Srour, L.; Mustafa, L.; Hakooz, N.M.; Zayed, A.A.; Khader, Y.S.; Azab, B. SLC30A8 gene polymorphism rs13266634 associated with increased risk for developing type 2 diabetes mellitus in Jordanian population. Gene 2020, 768, 145279. [Google Scholar] [CrossRef] [PubMed]
- Wijesekara, N.; Dai, F.F.; Hardy, A.B.; Giglou, P.R.; Bhattacharjee, A.; Koshkin, V.; Chimienti, F.; Gaisano, H.Y.; Rutter, G.A.; Wheeler, M.B. Beta cell-specific Znt8 deletion in mice causes marked defects in insulin processing, crystallisation and secretion. Diabetologia 2010, 53, 1656–1668. [Google Scholar] [CrossRef] [PubMed]
- Simmons, R.A.; Simmons, R. Developmental Origins of β-Cell Failure in Type 2 Diabetes: The Role of Epigenetic Mechanisms. Pediatr. Res. 2007, 61, 64–67. [Google Scholar] [CrossRef]
- Dabelea, D.; Hanson, R.L.; Lindsay, R.S.; Pettitt, D.J.; Imperatore, G.; Gabir, M.M.; Roumain, J.; Bennett, P.H.; Knowler, W.C. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: A study of discordant sibships. Diabetes 2000, 49, 2208–2211. [Google Scholar] [CrossRef]
- Sobngwi, E.; Boudou, P.; Mauvais-Jarvis, F.; Leblanc, H.; Velho, G.; Vexiau, P.; Porcher, R.; Hadjadj, S.; Pratley, R.; Tataranni, P.A.; et al. Effect of a diabetic environment in utero on pre-disposition to type 2 diabetes. Lancet 2003, 361, 1861–1865. [Google Scholar] [CrossRef]
- Shah, R.D.; Chernausek, S.D.; El Ghormli, L.; Geffner, M.E.; Keady, J.; Kelsey, M.M.; Farrell, R.; Tesfaldet, B.; Tryggestad, J.B.; Van Name, M.; et al. Maternal Diabetes in Youth-Onset Type 2 Diabetes Is Associated With Progressive Dysglycemia and Risk of Complications. J. Clin. Endocrinol. Metab. 2022, 108, 1120–1131. [Google Scholar] [CrossRef]
- Gautier, J.-F.; Wilson, C.; Weyer, C.; Mott, D.; Knowler, W.C.; Cavaghan, M.; Polonsky, K.S.; Bogardus, C.; Pratley, R.E. Low Acute Insulin Secretory Responses in Adult Offspring of People With Early Onset Type 2 Diabetes. Diabetes 2001, 50, 1828–1833. [Google Scholar] [CrossRef]
- Singh, R.; Pearson, E.; Avery, P.J.; McCarthy, M.I.; Levy, J.C.; Hitman, G.A.; Sampson, M.; Walker, M.; Hattersley, A.T. Reduced beta cell function in offspring of mothers with young-onset type 2 diabetes. Diabetologia 2006, 49, 1876–1880. [Google Scholar] [CrossRef]
- Whincup, P.H.; Kaye, S.J.; Owen, C.G.; Huxley, R.; Cook, D.G.; Anazawa, S.; Barrett-Connor, E.; Bhargava, S.K.; Birgisdottir, B.E.; Carlsson, S.; et al. Birth weight and risk of type 2 diabetes: A systematic review. JAMA 2008, 300, 2886–2897. [Google Scholar]
- Mohan, R.; Baumann, D.; Alejandro, E.U. Fetal undernutrition, placental insufficiency, and pancreatic β-cell development programming in utero. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R867. [Google Scholar] [CrossRef]
- Giapros, V.; Vavva, E.; Siomou, E.; Kolios, G.; Tsabouri, S.; Cholevas, V.; Bairaktari, E.; Tzoufi, M.; Challa, A. Low-birth-weight, but not catch-up growth, correlates with insulin resistance and resistin level in SGA infants at 12 months. J. Matern. Neonatal Med. 2016, 30, 1771–1776. [Google Scholar] [CrossRef]
- Brufani, C.; Grossi, A.; Fintini, D.; Tozzi, A.; Nocerino, V.; Patera, P.I.; Ubertini, G.; Porzio, O.; Barbetti, F.; Cappa, M. Obese children with low birth weight demonstrate impaired beta-cell function during oral glucose tolerance test. J. Clin. Endocrinol. Metab. 2009, 94, 4448–4452. [Google Scholar] [CrossRef] [PubMed]
- Van Assche, F.A.; Prins FDe Aerts, L.; Verjans, M. The endocrine pancreas in small-for-dates infants. Br. J. Obstet. Gynaecol. 1977, 84, 751–753. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Alvarez, J.; Conget, I.; Rasschaert, J.; Sener, A.; Gomis, R.; Malaisse, W.J. Enzymatic, metabolic and secretory patterns in human islets of Type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1994, 37, 177–181. [Google Scholar] [CrossRef]
- Deng, S.; Vatamaniuk, M.; Huang, X.; Doliba, N.; Lian, M.-M.; Frank, A.; Velidedeoglu, E.; Desai, N.M.; Koeberlein, B.; Wolf, B.; et al. Structural and Functional Abnormalities in the Islets Isolated From Type 2 Diabetic Subjects. Diabetes 2004, 53, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Del Guerra, S.; Lupi, R.; Marselli, L.; Masini, M.; Bugliani, M.; Sbrana, S.; Torri, S.; Pollera, M.; Boggi, U.; Mosca, F.; et al. Functional and Molecular Defects of Pancreatic Islets in Human Type 2 Diabetes. Diabetes 2005, 54, 727–735. [Google Scholar] [CrossRef]
- Gungor, N.; Arslanian, S. Progressive beta cell failure in type 2 diabetes mellitus of youth. J. Pediatr. 2004, 144, 656–659. [Google Scholar] [CrossRef]
- Mohan, V.; Amutha, A.; Ranjani, H.; Unnikrishnan, R.; Datta, M.; Anjana, R.M.; Staimez, L.; Ali, M.K.; Narayan, K.V. Associations of β-Cell Function and Insulin Resistance with Youth-Onset Type 2 Diabetes and Prediabetes Among Asian Indians. Diabetes Technol. Ther. 2013, 15, 315–322. [Google Scholar] [CrossRef]
- Cali’, A.M.G.; Bonadonna, R.C.; Trombetta, M.; Weiss, R.; Caprio, S. Metabolic Abnormalities Underlying the Different Prediabetic Phenotypes in Obese Adolescents. J. Clin. Endocrinol. Metab. 2008, 93, 1767–1773. [Google Scholar] [CrossRef]
- Elder, D.A.; Herbers, P.M.; Weis, T.; Standiford, D.; Woo, J.G.; D’alessio, D.A. β-Cell Dysfunction in Adolescents and Adults with Newly Diagnosed Type 2 Diabetes Mellitus. J. Pediatr. 2012, 160, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Cali, A.M.; Man, C.D.; Cobelli, C.; Dziura, J.; Seyal, A.; Shaw, M.; Allen, K.; Chen, S.; Caprio, S. Primary defects in beta-cell function further exacerbated by worsening of insulin resistance mark the development of impaired glucose tolerance in obese adolescents. Diabetes Care 2009, 32, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Hannon, T.S.; Janosky, J.; Arslanian, S.A. Longitudinal Study of Physiologic Insulin Resistance and Metabolic Changes of Puberty. Pediatr. Res. 2006, 60, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Caprio, S.; Trombetta, M.; Taksali, S.E.; Tamborlane, W.V.; Bonadonna, R. Beta-cell function across the spectrum of glucose tolerance in obese youth. Diabetes 2005, 54, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, K.M.; Tripputi, M.T.; Kozedub, A.; Mather, K.J.; Nadeau, K.J.; Edelstein, S.L.; Hannon, T.S.; Arslanian, S.A.; Cree-Green, M.; Buchanan, T.A.; et al. β-cells in youth with impaired glucose tolerance or early type 2 diabetes secrete more insulin and are more responsive than in adults. Pediatr. Diabetes 2020, 21, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.; Andrikopoulos, S.; Gunton, J. First phase insulin secretion and type 2 diabetes. Curr. Mol. Med. 2013, 13, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Bonner-Weir, S. Reduced glucose-induced first-phase insulin release is a danger signal that predicts diabetes. J. Clin. Investig. 2021, 131, e150022. [Google Scholar] [CrossRef] [PubMed]
- Matveyenko, A.V.; Liuwantara, D.; Gurlo, T.; Kirakossian, D.; Dalla Man, C.; Cobelli, C.; White, M.F.; Copps, K.D.; Volpi, E.; Fujita, S.; et al. Pulsatile portal vein insulin delivery enhances hepatic insulin action and signaling. Diabetes 2012, 61, 2269–2279. [Google Scholar] [CrossRef]
- Wahren, J.; Kallas, Å. Loss of Pulsatile Insulin Secretion: A Factor in the Pathogenesis of Type 2 Diabetes? Diabetes 2012, 61, 2228–2229. [Google Scholar] [CrossRef]
- Marchetti, P.; Del Guerra, S.; Marselli, L.; Lupi, R.; Masini, M.; Pollera, M.; Bugliani, M.; Boggi, U.; Vistoli, F.; Mosca, F.; et al. Pancreatic Islets from Type 2 Diabetic Patients Have Functional Defects and Increased Apoptosis That Are Ameliorated by Metformin. J. Clin. Endocrinol. Metab. 2004, 89, 5535–5541. [Google Scholar] [CrossRef]
- Prentki, M.; Peyot, M.L.; Masiello, P.; Murthy Madiraju, S.R. Nutrient-Induced Metabolic Stress, Adaptation, Detoxification, and Toxicity in the Pancreatic β-Cell. Diabetes 2020, 69, 279–290. [Google Scholar] [CrossRef]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel Excess and β-Cell Dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Lytrivi, M.; Castell, A.-L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2019, 432, 1514–1534. [Google Scholar] [CrossRef] [PubMed]
- Roma, L.P.; Jonas, J.C. Nutrient Metabolism, Subcellular Redox State, and Oxidative Stress in Pancreatic Islets and β-Cells. J. Mol. Biol. 2020, 432, 1461–1493. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, D.A.; Shyng, S.-L. Ion Channels of the Islets in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1326–1346. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J.; Knop, F.K.; Vilsbøll, T.; Krarup, T.; Madsbad, S. Loss of Incretin Effect Is a Specific, Important, and Early Characteristic of Type 2 Diabetes. Diabetes Care 2011, 34, S251–S257. [Google Scholar] [CrossRef]
- Grespan, E.; Guolo, A.; Muscelli, E.; Ferrannini, E.; Mari, A. Loss of the Incretin Effect in Type 2 Diabetes: A Systematic Review and Meta-analysis. J. Clin. Endocrinol. Metab. 2022, 107, 2092–2100. [Google Scholar] [CrossRef]
- Michaliszyn, S.F.; Mari, A.; Lee, S.; Bacha, F.; Tfayli, H.; Farchoukh, L.; Ferrannini, E.; Arslanian, S. β-Cell Function, Incretin Effect, and Incretin Hormones in Obese Youth Along the Span of Glucose Tolerance From Normal to Prediabetes to Type 2 Diabetes. Diabetes 2014, 63, 3846–3855. [Google Scholar] [CrossRef]
- Kahleova, H.; Tura, A.; Klementova, M.; Thieme, L.; Haluzik, M.; Pavlovicova, R.; Hill, M.; Pelikanova, T. A Plant-Based Meal Stimulates Incretin and Insulin Secretion More Than an Energy- and Macronutrient-Matched Standard Meal in Type 2 Diabetes: A Randomized Crossover Study. Nutrients 2019, 11, 486. [Google Scholar] [CrossRef]
- Nicolson, T.J.; Bellomo, E.A.; Wijesekara, N.; Loder, M.K.; Baldwin, J.M.; Gyulkhandanyan, A.V.; Koshkin, V.; Tarasov, A.I.; Carzaniga, R.; Kronenberger, K.; et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes 2009, 58, 2070–2083. [Google Scholar] [CrossRef]
- Li, Y.V. Zinc and insulin in pancreatic beta-cells. Endocrine 2014, 45, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, S.B.; Larsen, A.; Knuhtsen, A.; Rungby, J.; Smidt, K. Effects of zinc supplementation and zinc chelation on in vitro β-cell function in INS-1E cells. BMC Res. Notes 2014, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Slepchenko, K.G.; James, C.B.L.; Li, Y.V. Inhibitory effect of zinc on glucose-stimulated zinc/insulin secretion in an insulin-secreting β-cell line. Exp. Physiol. 2013, 98, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Priel, T.; Aricha-Tamir, B.; Sekler, I. Clioquinol attenuates zinc-dependent beta-cell death and the onset of insulitis and hyperglycemia associated with experimental type I diabetes in mice. Eur. J. Pharmacol. 2007, 565, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Al-Maroof, R.A.; Al-Sharbatti, S.S. Serum zinc levels in diabetic patients and effect of zinc supplementation on glycemic control of type 2 diabetics. Saudi Med. J. 2006, 27, 344–350. [Google Scholar]
- Simon, S.F.; Taylor, C.G. Dietary zinc supplementation attenuates hyperglycemia in db/db mice. Exp. Biol. Med. 2001, 226, 43–51. [Google Scholar] [CrossRef]
- Tang, X.-H.; Shay, N.F. Zinc Has an Insulin-Like Effect on Glucose Transport Mediated by Phosphoinositol-3-Kinase and Akt in 3T3-L1 Fibroblasts and Adipocytes. J. Nutr. 2001, 131, 1414–1420. [Google Scholar] [CrossRef]
- AAnderson, R. Nutritional factors influencing the glucose/insulin system: Chromium. J. Am. Coll. Nutr. 1997, 16, 404–410. [Google Scholar] [CrossRef]
- McCarty, M.F. The therapeutic potential of Glucose Tolerance Factor. Med. Hypotheses 1980, 6, 1177–1189. [Google Scholar] [CrossRef]
- Balk, E.M.; Tatsioni, A.; Lichtenstein, A.H.; Lau, J.; Pittas, A.G. Effect of Chromium Supplementation on Glucose Metabolism and LipidsA systematic review of randomized controlled trials. Diabetes Care 2007, 30, 2154–2163. [Google Scholar] [CrossRef]
- Sundaram, B.; Aggarwal, A.; Sandhir, R. Chromium picolinate attenuates hyperglycemia-induced oxidative stress in streptozotocin-induced diabetic rats. J. Trace Elem. Med. Biol. 2013, 27, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, S.; Norman, A.W. Dietary vitamin D is essential for normal insulin secretion from the perfused rat pancreas. J. Clin. Investig. 1984, 73, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Vangoitsenhoven, R.; Wolden-Kirk, H.; Lemaire, K.; Verstuyf, A.; Verlinden, L.; Yamamoto, Y.; Kato, S.; Van Lommel, L.; Schuit, F.; Van der Schueren, B.; et al. Effect of a transcriptional inactive or absent vitamin D receptor on beta-cell function and glucose homeostasis in mice. J. Steroid Biochem. Mol. Biol. 2016, 164, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Bornstedt, M.E.; Gjerlaugsen, N.; Pepaj, M.; Bredahl, M.K.L.; Thorsby, P.M. Vitamin D Increases Glucose Stimulated Insulin Secretion from Insulin Producing Beta Cells (INS1E). Int. J. Endocrinol. Metab. 2019, 17, e74255. [Google Scholar] [CrossRef]
- Marku, A.; Galli, A.; Marciani, P.; Dule, N.; Perego, C.; Castagna, M. Iron Metabolism in Pancreatic Beta-Cell Function and Dysfunction. Cells 2021, 10, 2841. [Google Scholar] [CrossRef]
- Banerji, M.A. Impaired beta-cell and alpha-cell function in African-American children with type 2 diabetes mellitus—”Flatbush diabetes”. J. Pediatr. Endocrinol. Metab. 2002, 15, S493–S501. [Google Scholar]
- Dmochowski, K.; Finegood, D.T.; Francombe, W.; Tyler, B.; Zinman, B. Factors determining glucose tolerance in patients with thalassemia major. J. Clin. Endocrinol. Metab. 1993, 77, 478–483. [Google Scholar]
- Buysschaert, M.; Paris, I.; Selvais, P.; Hermans, M.P. Clinical aspects of diabetes secondary to idiopathic haemochromatosis in French-speaking Belgium. Diabetes Metab. 1997, 23, 308–313. [Google Scholar]
- Platis, O.; Anagnostopoulos, G.; Farmaki, K.; Posantzis, M.; Gotsis, E.; Tolis, G. Glucose metabolism disorders improvement in patients with thalassaemia major after 24–36 months of intensive chelation therapy. Pediatr. Endocrinol. Rev. 2004, 2, 279–281. [Google Scholar]
- Abraham, D.; Rogers, J.; Gault, P.; Kushner, J.P.; McClain, D.A. Increased insulin secretory capacity but decreased insulin sensitivity after correction of iron overload by phlebotomy in hereditary haemochromatosis. Diabetologia 2006, 49, 2546–2551. [Google Scholar] [CrossRef]
- Qin, Y.; Huang, Y.; Li, Y.; Qin, L.; Wei, Q.; Chen, X.; Yang, C.; Zhang, M. Association between systemic iron status and β-cell function and insulin sensitivity in patients with newly diagnosed type 2 diabetes. Front. Endocrinol. 2023, 14, 1143919. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, P.; Ramasamy, J.; Vanitha, S.; Jacob, M.; Varghese, J. Impaired pancreatic beta-cell function after a single dose of oral iron: A before-and-after (pre–post) study. J. Hum. Nutr. Diet. 2022. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Tarim, O.; Küçükerdoğan, A.; Günay, U.; Eralp, O.; Ercan, I. Effects of iron deficiency anemia on hemoglobin A1c in type 1 diabetes mellitus. Pediatr. Int. 1999, 41, 357–362. [Google Scholar] [CrossRef]
- Santos, M.C.F.D.; Anderson, C.P.; Neschen, S.; Zumbrennen-Bullough, K.B.; Romney, S.J.; Kahle-Stephan, M.; Rathkolb, B.; Gailus-Durner, V.; Fuchs, H.; Wolf, E.; et al. Irp2 regulates insulin production through iron-mediated Cdkal1-catalyzed tRNA modification. Nat. Commun. 2020, 11, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Soliman, A.T.; De Sanctis, V.; Yassin, M.; Soliman, N. Iron deficiency anemia and glucose metabolism. Acta bio-medica : Atenei Parm. 2017, 88, 112–118. [Google Scholar] [CrossRef]
- Kahn, S.E.; Halban, P.A. Release of incompletely processed proinsulin is the cause of the disproportionate proinsulinemia of NIDDM. Diabetes 1997, 46, 1725–1732. [Google Scholar] [CrossRef]
- Røder, M.E.; Dinesen, B.; Hartling, S.G.; Houssa, P.; Vestergaard, H.; Sodoyez-Goffaux, F.; Binder, C. Intact proinsulin and beta-cell function in lean and obese subjects with and without type 2 diabetes. Diabetes Care 1999, 22, 609–614. [Google Scholar] [CrossRef]
- von Berghes, C.; Brabant, G.; Biebermann, H.; Krude, H.; Wiegand, S. Proinsulin and the proinsulin/insulin ratio in overweight and obese children and adolescents: Relation to clinical parameters, insulin resistance, and impaired glucose regulation. Pediatr. Diabetes 2011, 12, 242–249. [Google Scholar] [CrossRef]
- Zeitler, P.; El Ghormli, L.; Arslanian, S.; Caprio, S.; Isganaitis, E.; Kelsey, M.K.; Weinstock, R.S.; White, N.H.; Drews, K. Deterioration of Glycemic Control in Youth-Onset Type 2 Diabetes: What Are the Early and Late Predictors? J. Clin. Endocrinol. Metab. 2022, 107, e3384–e3394. [Google Scholar] [CrossRef]
- Clark, A.; Wells, C.A.; Buley, I.D.; Cruickshank, J.K.; Vanhegan, R.I.; Matthews, D.R.; Cooper, G.J.; Holman, R.R.; Turner, R.C. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: Quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988, 9, 151–159. [Google Scholar]
- Sakuraba, H.; Mizukami, H.; Yagihashi, N.; Wada, R.; Hanyu, C. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 2002, 45, 85–96. [Google Scholar] [CrossRef]
- Yoon, K.H.; Ko, S.H.; Cho, J.H.; Lee, J.M.; Ahn, Y.B.; Song, K.H.; Yoo, S.J.; Kang, M.I.; Cha, B.Y.; Lee, K.W.; et al. Selective β-Cell Loss and α-Cell Expansion in Patients with Type 2 Diabetes Mellitus in Korea. J. Clin. Endocrinol. Metab. 2003, 88, 2300–2308. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-Cell Deficit and Increased beta-Cell Apoptosis in Humans With Type 2 Diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.-C. Pancreatic β-cell mass in European subjects with type 2 diabetes. Diabetes, Obes. Metab. 2008, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Inaishi, J.; Saisho, Y. Beta-Cell Mass in Obesity and Type 2 Diabetes, and Its Relation to Pancreas Fat: A Mini-Review. Nutrients 2020, 12, 3846. [Google Scholar] [CrossRef]
- Matthews, K.A.; Rhoten, W.B.; Driscoll, H.K.; Chertow, B.S. Vitamin A Deficiency Impairs Fetal Islet Development and Causes Subsequent Glucose Intolerance in Adult Rats. J. Nutr. 2004, 134, 1958–1963. [Google Scholar] [CrossRef]
- Trasino, S.E.; Benoit, Y.D.; Gudas, L.J. Vitamin A Deficiency Causes Hyperglycemia and Loss of Pancreatic β-Cell Mass. J. Biol. Chem. 2015, 290, 1456–1473. [Google Scholar] [CrossRef]
- Hanley, S.C.; Austin, E.; Assouline-Thomas, B.; Kapeluto, J.; Blaichman, J.; Moosavi, M.; Petropavlovskaia, M.; Rosenberg, L. β-Cell Mass Dynamics and Islet Cell Plasticity in Human Type 2 Diabetes. Endocrinology 2010, 151, 1462–1472. [Google Scholar] [CrossRef]
- Marselli, L.; Suleiman, M.; Masini, M.; Campani, D.; Bugliani, M.; Syed, F.; Martino, L.; Focosi, D.; Scatena, F.; Olimpico, F.; et al. Are we overestimating the loss of beta cells in type 2 diabetes? Diabetologia 2014, 57, 362–365. [Google Scholar] [CrossRef]
- Cho, J.H.; Kim, J.W.; Shin, J.A.; Shin, J.; Yoon, K.H. β-cell mass in people with type 2 diabetes. J. Diabetes Investig. 2011, 2, 6–17. [Google Scholar] [CrossRef]
- Donath, M.Y.; Ehses, J.A.; Maedler, K.; Schumann, D.M.; Ellingsgaard, H.; Eppler, E.; Reinecke, M. Mechanisms of β-Cell Death in Type 2 Diabetes. Diabetes 2005, 54, S108–S113. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Eldor, R.; Abdul-Ghani, M. Pathophysiologic Approach to Therapy in Patients With Newly Diagnosed Type 2 Diabetes. Diabetes Care 2013, 36, S127–S138. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Welsh, N.; Jonas, J.C.; Jörns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of Pancreatic β-Cell Death in Type 1 and Type 2 Dia-betesMany Differences, Few Similarities. Diabetes 2005, 54, S97–S107. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H.E.; McKenzie, M.D.; Angstetra, E.; Campbell, P.D.; Kay, T.W. Beta cell apoptosis in diabetes. Apoptosis 2009, 14, 1389–1404. [Google Scholar] [CrossRef]
- Jung, T.W.; Lee, M.W.; Lee, Y.J.; Kim, S.M. Metformin prevents endoplasmic reticulum stress-induced apoptosis through AMPK-PI3K-c-Jun NH2 pathway. Biochem. Biophys. Res. Commun. 2012, 417, 147–152. [Google Scholar] [CrossRef]
- Wang, Y.; He, D.; Ni, C.; Zhou, H.; Wu, S.; Xue, Z.; Zhou, Z. Vitamin D induces autophagy of pancreatic β-cells and enhances insulin secretion. Mol. Med. Rep. 2016, 14, 2644–2650. [Google Scholar] [CrossRef]
- Ly, L.D.; Xu, S.; Choi, S.-K.; Ha, C.-M.; Thoudam, T.; Cha, S.-K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.-K.; Park, K.-S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef]
- Marrif, H.I.; Al-Sunousi, S.I. Pancreatic β cell mass death. Front. Pharmacol. 2016, 7, 83–99. [Google Scholar] [CrossRef]
- Sun, T.; Han, X. Death versus dedifferentiation: The molecular bases of beta cell mass reduction in type 2 diabetes. Semin. Cell Dev. Biol. 2019, 103, 76–82. [Google Scholar] [CrossRef]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Autophagy in health and disease. 1. Regulation and significance of autophagy: An overview. Am. J. Physiol. Physiol. 2010, 298, C776–C785. [Google Scholar] [CrossRef]
- Marasco, M.R.; Linnemann, A.K. β-Cell Autophagy in Diabetes Pathogenesis. Endocrinology 2018, 159, 2127–2141. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, J.; Park, K.; Lee, M.S. β-cell autophagy: Mechanism and role in β-cell dysfunction. Mol. Metab. 2019, 27, S92–S103. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; GangYi, Y.; QiNan, W. Autophagic dysfunction of β cell dysfunction in type 2 diabetes, a double-edged sword. Genes Dis. 2021, 8, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Masini, M.; Bugliani, M.; Lupi, R.; del Guerra, S.; Boggi, U.; Filipponi, F.; Marselli, L.; Masiello, P.; Marchetti, P. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009, 52, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Badaru, A.; Pihoker, C. Type 2 Diabetes in Childhood: Clinical Characteristics and Role of β-Cell Autoimmunity. Curr. Diabetes Rep. 2011, 12, 75–81. [Google Scholar] [CrossRef]
- Cooksey, R.C.; Jouihan, H.A.; Ajioka, R.S.; Hazel, M.W.; Jones, D.L.; Kushner, J.P.; McClain, D.A. Oxidative Stress, β-Cell Apoptosis, and Decreased Insulin Secretory Capacity in Mouse Models of Hemochromatosis. Endocrinology 2004, 145, 5305–5312. [Google Scholar] [CrossRef]
- Sha, W.; Hu, F.; Xi, Y.; Chu, Y.; Bu, S. Mechanism of Ferroptosis and Its Role in Type 2 Diabetes Mellitus. J. Diabetes Res. 2021, 2021, 9999612. [Google Scholar] [CrossRef] [PubMed]
- Jouihan, H.A.; Cobine, P.A.; Cooksey, R.C.; Hoagland, E.A.; Boudina, S.; Abel, E.D.; Winge, D.R.; McClain, D.A. Iron-Mediated Inhibition of Mitochondrial Manganese Uptake Mediates Mitochondrial Dysfunction in a Mouse Model of Hemochromatosis. Mol. Med. 2008, 14, 98–108. [Google Scholar] [CrossRef]
- Perl, S.; Kushner, J.A.; Buchholz, B.A.; Meeker, A.K.; Stein, G.M.; Hsieh, M.; Kirby, M.; Pechhold, S.; Liu, E.H.; Harlan, D.M.; et al. Significant Human β-Cell Turnover Is Limited to the First Three Decades of Life as Determined by in Vivo Thymidine Analog Incorporation and Radiocarbon Dating. J. Clin. Endocrinol. Metab. 2010, 95, E234–E239. [Google Scholar] [CrossRef]
- Cnop, M.; Hughes, S.J.; Igoillo-Esteve, M.; Hoppa, M.B.; Sayyed, F.; Van De Laar, L.; Gunter, J.; De Koning, E.J.P.; Walls, G.V.; Gray, D.W.G.; et al. The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia 2009, 53, 321–330. [Google Scholar] [CrossRef]
- Bouwens, L. Beta Cell Regeneration. Curr. Diabetes Rev. 2006, 2, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Bonner-Weir, S.; Guo, L.; Li, W.-C.; Ouziel-Yahalom, L.; Weir, G.C.; Sharma, A. Islet Neogenesis: A Possible Pathway for Beta-Cell Replenishment. Rev. Diabet. Stud. 2012, 9, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Chera, S.; Herrera, P.L. Regeneration of pancreatic insulin-producing cells by in situ adaptive cell conversion. Curr. Opin. Genet. Dev. 2016, 40, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, S.; Uno, S.; Iwahashi, H.; Fujita, Y.; Yoshikawa, A.; Kozawa, J.; Okita, K.; Takiuchi, D.; Eguchi, H.; Nagano, H.; et al. Predominance of β-Cell Neogenesis Rather Than Replication in Humans with an Impaired Glucose Tolerance and Newly Diagnosed Diabetes. J. Clin. Endocrinol. Metab. 2013, 98, 2053–2061. [Google Scholar] [CrossRef]
- Furuyama, K.; Chera, S.; Van Gurp, L.; Oropeza, D.; Ghila, L.; Damond, N.; Vethe, H.; Paulo, J.A.; Joosten, A.M.; Berney, T.; et al. Diabetes relief in mice by glucose-sensing insulin-secreting human α-cells. Nature 2019, 567, 43–48. [Google Scholar] [CrossRef]
- Lysy, P.A.; Weir, G.C.; Bonner-Weir, S. Making β Cells from Adult Cells within the Pancreas. Curr. Diabetes Rep. 2013, 13, 695–703. [Google Scholar] [CrossRef]
- Masini, M.; Martino, L.; Marselli, L.; Bugliani, M.; Boggi, U.; Filipponi, F.; Marchetti, P.; De Tata, V. Ultrastructural alterations of pancreatic beta cells in human diabetes mellitus. Diabetes/Metabolism Res. Rev. 2017, 33, e2894. [Google Scholar] [CrossRef]
- Mezza, T.; Sorice, G.P.; Conte, C.; Sun, V.A.; Cefalo, C.M.A.; Moffa, S.; Pontecorvi, A.; Mari, A.; Kulkarni, R.N.; Giaccari, A. β-Cell Glucose Sensitivity Is Linked to Insulin/Glucagon Bihormonal Cells in Nondiabetic Humans. J. Clin. Endocrinol. Metab. 2016, 101, 470–475. [Google Scholar] [CrossRef]
- Meier, J.J.; Köhler, C.U.; Alkhatib, B.; Sergi, C.; Junker, T.; Klein, H.H.; Schmidt, W.E.; Fritsch, H. β-cell development and turnover during prenatal life in humans. Eur. J. Endocrinol. 2010, 162, 559–568. [Google Scholar] [CrossRef]
- Ng, K.Y.; Ma, M.T.; Leung, K.K.; Leung, P.S. Vitamin D and vitamin A receptor expression and the proliferative effects of ligand activation of these receptors on the development of pancreatic progenitor cells derived from human fetal pancreas. Stem Cell Rev. Rep. 2011, 7, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Öström, M.; Loffler, K.A.; Edfalk, S.; Selander, L.; Dahl, U.; Ricordi, C.; Jeon, J.; Correa-Medina, M.; Diez, J.; Edlund, H. Retinoic acid promotes the generation of pancreatic endocrine progenitor cells and their further differentiation into beta-cells. PLoS ONE 2008, 3, e2841. [Google Scholar] [CrossRef] [PubMed]
- Masini, M.; Marselli, L.; Himpe, E.; Martino, L.; Bugliani, M.; Suleiman, M.; Boggi, U.; Filipponi, F.; Occhipinti, M.; Bouwens, L.; et al. Co-localization of acinar markers and insulin in pancreatic cells of subjects with type 2 diabetes. PLoS ONE 2017, 12, e0179398. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, L.; Bouwens, L. Can β-cells be derived from exocrine pancreas? Diabetes Obes. Metab. 2008, 10, S170–S178. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Pechberty, S.; Bellini, L.; Göpel, S.O.; Campana, M.; Rouch, C.; Dairou, J.; Cosentino, C.; Fantuzzi, F.; Toivonen, S.; et al. Stearoyl CoA desaturase is a gatekeeper that protects human beta cells against lipotoxicity and maintains their identity. Diabetologia 2019, 63, 395–409. [Google Scholar] [CrossRef]
- Bensellam, M.; Jonas, J.-C.; Laybutt, D.R. Mechanisms of β-cell dedifferentiation in diabetes: Recent findings and future research directions. J. Endocrinol. 2018, 236, R109–R143. [Google Scholar] [CrossRef]
- Cunha, D.A.; Hekerman, P.; Ladrière, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic β-cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef]
- Wang, Z.; York, N.W.; Nichols, C.G.; Remedi, M.S. Pancreatic β-cell Dedifferentiation in Diabetes and Re-differentiation following Insulin Therapy. Cell Metab. 2014, 19, 872–882. [Google Scholar] [CrossRef]
- Nordmann, T.M.; Dror, E.; Schulze, F.; Traub, S.; Berishvili, E.; Barbieux, C.; Böni-Schnetzler, M.; Donath, M.Y. The Role of Inflammation in β-cell Dedifferentiation. Sci. Rep. 2017, 7, 6285. [Google Scholar] [CrossRef]
- Diedisheim, M.; Oshima, M.; Albagli, O.; Huldt, C.W.; Ahlstedt, I.; Clausen, M.; Menon, S.; Aivazidis, A.; Andreasson, A.C.; Haynes, W.G.; et al. Modeling human pancreatic beta cell dedifferentiation. Mol. Metab. 2018, 10, 74–86. [Google Scholar] [CrossRef]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of β-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef]
- Spijker, H.S.; Song, H.; Ellenbroek, J.H.; Roefs, M.M.; Engelse, M.A.; Bos, E.; Koster, A.J.; Rabelink, T.J.; Hansen, B.C.; Clark, A.; et al. Loss of β-Cell Identity Occurs in Type 2 Diabetes and Is Associated With Islet Amyloid Deposits. Diabetes 2015, 64, 2928–2938. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Ni, Q.; Sun, J.; Xie, J.; Liu, J.; Ning, G.; Wang, W.; Wang, Q. Aging Impairs Adaptive Unfolded Protein Response and Drives Beta Cell De-differentiation in Humans. J. Clin. Endocrinol. Metab. 2022, 107, 3231–3241. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, P.V.; Speckmann, T.; Lynn, F.C. Friend and foe: β-cell Ca2+ signaling and the development of diabetes. Mol. Metab. 2019, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Banaei-Bouchareb, L.; Peuchmaur, M.; Czernichow, P.; Polak, M. A transient microenvironment loaded mainly with macrophages in the early developing human pancreas. J. Endocrinol. 2006, 188, 467–480. [Google Scholar] [CrossRef]
- Dalmas, E. Innate immune priming of insulin secretion. Curr. Opin. Immunol. 2018, 56, 44–49. [Google Scholar] [CrossRef]
- Arous, C.; Ferreira, P.G.; Dermitzakis, E.T.; Halban, P.A. Short Term Exposure of Beta Cells to Low Concentrations of Interleukin-1β Improves Insulin Secretion through Focal Adhesion and Actin Remodeling and Regulation of Gene Expression. J. Biol. Chem. 2015, 290, 6653–6669. [Google Scholar] [CrossRef]
- Marselli, L.; Bugliani, M.; Suleiman, M.; Olimpico, F.; Masini, M.; Petrini, M.; Boggi, U.; Filipponi, F.; Syed, F.; Marchetti, P. β-Cell inflammation in human type 2 diabetes and the role of autophagy. Diabetes, Obes. Metab. 2013, 15, 130–136. [Google Scholar] [CrossRef]
- Ying, W.; Fu, W.; Lee, Y.S.; Olefsky, J.M. The role of macrophages in obesity-associated islet inflammation and β-cell abnormalities. Nat. Rev. Endocrinol. 2019, 16, 81–90. [Google Scholar] [CrossRef]
- Donath, M.Y.; Dalmas, É.; Sauter, N.S.; Böni-Schnetzler, M. Inflammation in Obesity and Diabetes: Islet Dysfunction and Therapeutic Opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef]
- Cuenco, J.; Dalmas, E. Islet Inflammation and β Cell Dysfunction in Type 2 Diabetes. In From Obesity to Diabetes. Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2022; Volume 274, pp. 227–251. [Google Scholar] [CrossRef]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated fatty acid and TLR signaling link β cell dysfunction and islet inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef]
- Cucak, H.; Grunnet, L.G.; Rosendahl, A. Accumulation of M1-like macrophages in type 2 diabetic islets is followed by a systemic shift in macrophage polarization. J. Leukoc. Biol. 2013, 95, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, T. Inflammatory markers in children and adolescents with type 2 diabetes mellitus. Clin. Chim. Acta 2019, 496, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Gokulakrishnan, K.; Amutha, A.; Ranjani, H.; Bibin, S.Y.; Balakumar, M.; Pandey, G.K.; Anjana, R.M.; Ali, M.K.; Narayan, K.V.; Mohan, V. Relationship of Adipokines and Proinflammatory Cytokines Among Asian Indians with Obesity and Youth Onset Type 2 Diabetes. Endocr. Pract. 2015, 21, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Su, S.-C.; Pei, D.; Hsieh, C.-H.; Hsiao, F.-C.; Wu, C.-Z.; Hung, Y.-J. Circulating pro-inflammatory cytokines and adiponectin in young men with type 2 diabetes. Acta Diabetol. 2010, 48, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, T.; Karges, B.; Meissner, T.; Wiegand, S.; Stoffel-Wagner, B.; Holl, R.W.; Woelfle, J. Inflammatory Markers in Obese Adolescents with Type 2 Diabetes and Their Relationship to Hepatokines and Adipokines. J. Pediatr. 2016, 173, 131–135. [Google Scholar] [CrossRef]
- Reinehr, T.; Karges, B.; Meissner, T.; Wiegand, S.; Fritsch, M.; Holl, R.W.; Woelfle, J. Fibroblast growth factor 21 and fetuin-A in obese adolescents with and without type 2 diabetes. J. Clin. Endocrinol. Metab. 2015, 100, 3004–3010. [Google Scholar] [CrossRef]
- Karin, A.; Jon, E.; Martin, A.; Lena, B.; Martin, L.; Naveed, S.; Marcus, L.; Maria, Å.; Annika, R. Body mass index in adolescence, risk of type 2 diabetes and associated complications: A nationwide cohort study of men. Eclinicalmedicine 2022, 46, 101356. [Google Scholar] [CrossRef]
- Twig, G.; Zucker, I.; Afek, A.; Cukierman-Yaffe, T.; Bendor, C.D.; Derazne, E.; Lutski, M.; Shohat, T.; Mosenzon, O.; Tzur, D.; et al. Adolescent Obesity and Early-Onset Type 2 Diabetes. Diabetes Care 2020, 43, 1487–1495. [Google Scholar] [CrossRef]
- Wolden-Kirk, H.; Rondas, D.; Bugliani, M.; Korf, H.; Van Lommel, L.; Brusgaard, K.; Christesen, H.T.; Schuit, F.; Proost, P.; Masini, M.; et al. Discovery of Molecular Pathways Mediating 1,25-Dihydroxyvitamin D3 Protection Against Cytokine-Induced Inflammation and Damage of Human and Male Mouse Islets of Langerhans. Endocrinology 2014, 155, 736–747. [Google Scholar] [CrossRef]
- Sadek, K.; Shaheen, H. Biochemical efficacy of vitamin D in ameliorating endocrine and metabolic disorders in diabetic rats. Pharm. Biol. 2013, 52, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Sergeev, I.N. 1,25-Dihydroxyvitamin D3 and type 2 diabetes: Ca2+-dependent molecular mechanisms and the role of vitamin D status. Horm. Mol. Biol. Clin. Investig. 2016, 26, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Vecoli, C.; Neglia, D.; Tavazzi, B.; Lazzarino, G.; Novelli, M.; Masiello, P.; Wang, Y.-T.; Puri, N.; Paolocci, N.; et al. Cobalt-Protoporphyrin Improves Heart Function by Blunting Oxidative Stress and Restoring NO Synthase Equilibrium in an Animal Model of Experimental Diabetes. Front. Physiol. 2012, 3, 160. [Google Scholar] [CrossRef]
- Cruz, K.J.C.; de Oliveira, A.R.S.; do Nascimento Marreiro, D. Antioxidant role of zinc in diabetes mellitus. World J. Diabetes 2015, 6, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Kloubert, V.; Rink, L. Zinc as a micronutrient and its preventive role of oxidative damage in cells. Food Funct. 2015, 6, 3195–3204. [Google Scholar] [CrossRef]
- Nazem, M.R.; Asadi, M.; Adelipour, M.; Jabbari, N.; Allameh, A. Zinc supplementation ameliorates type 2 diabetes markers through the enhancement of total antioxidant capacity in overweight patients. Postgrad. Med. J. 2022. [Google Scholar] [CrossRef]
- Aly, H.; Mantawy, M.M. Comparative effects of zinc, selenium and vitamin E or their combination on carbohydrate metabolizing enzymes and oxidative stress in streptozotocin induced-diabetic rats. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 66–78. [Google Scholar]
- Liu, M.; Jeong, E.-M.; Liu, H.; Xie, A.; So, E.Y.; Shi, G.; Jeong, G.E.; Zhou, A.; Dudley, S.C. Magnesium supplementation improves diabetic mitochondrial and cardiac diastolic function. J. Clin. Investig. 2019, 4, 123182. [Google Scholar] [CrossRef]
- Hwang, D.; Seo, S.; Kim, Y.; Kim, C.; Shim, S.; Jee, S.; Lee, S.; Jang, M.; Kim, M.; Yim, S.; et al. Selenium acts as an insulin-like molecule for the down-regulation of diabetic symptoms via endoplasmic reticulum stress and insulin signalling proteins in diabetes-induced non-obese diabetic mice. J. Biosci. 2007, 32, 723–735. [Google Scholar] [CrossRef]
- González de Vega, R.; Fernández-Sánchez, M.L.; Fernández, J.C.; Álvarez Menéndez, F.V.; Sanz-Medel, A. Selenium levels and Glu-tathione peroxidase activity in the plasma of patients with type II diabetes mellitus. J. Trace Elem. Med. Biol. 2016, 3, 44–49. [Google Scholar] [CrossRef]
- Li, X.C.; Zhuo, J.L. Current Insights and New Perspectives on the Roles of Hyperglucagonemia in Non-Insulin–Dependent Type 2 Diabetes. Curr. Hypertens. Rep. 2013, 15, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.; Bagger, J.I.; Christensen, M.; Knop, F.K.; Vilsbøll, T. Glucagon and Type 2 Diabetes: The Return of the Alpha Cell. Curr. Diabetes Rep. 2014, 14, 555. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Berglund, E.D.; Wang, M.-Y.; Fu, X.; Yu, X.; Charron, M.J.; Burgess, S.C.; Unger, R.H. Metabolic manifestations of insulin deficiency do not occur without glucagon action. Proc. Natl. Acad. Sci. USA 2012, 109, 14972–14976. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, M.E.; Wait, J.B.; Koech, J.; Gordon, A.N.; Coch, R.W.; Svendsen, B.; Finan, B.; D’alessio, D.A.; Campbell, J.E. Glucagon lowers glycemia when β cells are active. J. Clin. Investig. 2019, 4, e129954. [Google Scholar] [CrossRef] [PubMed]
- Huising, M.O. Paracrine regulation of insulin secretion. Diabetologia 2020, 63, 2057–2063. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Rorsman, P. KATP channels and islet hormone secretion: New insights and controversies. Nat. Rev. Endocrinol. 2013, 9, 660–669. [Google Scholar] [CrossRef]
- Weiss, R.; D’Adamo, E.; Santoro, N.; Hershkop, K.; Caprio, S. Basal α-Cell Up-Regulation in Obese Insulin-Resistant Adolescents. J. Clin. Endocrinol. Metab. 2011, 96, 91–97. [Google Scholar] [CrossRef]
- Kahn, S.E.; Mather, K.J.; Arslanian, S.A.; Barengolts, E.; Buchanan, T.A.; Caprio, S.; Ehrmann, D.A.; Hannon, T.S.; Marcovina, S.; Nadeau, K.J.; et al. Hyperglucagonemia Does Not Explain the β-Cell Hyperresponsiveness and Insulin Resistance in Dysglycemic Youth Compared With Adults: Lessons From the RISE Study. Diabetes Care 2021, 44, 1961–1969. [Google Scholar] [CrossRef]
- Hartter, E.; Svoboda, T.; Ludvik, B.; Schuller, M.; Lell, B.; Kuenburg, E.; Brunnbauer, M.; Woloszczuk, W.; Prager, R. Basal and stimulated plasma levels of pancreatic amylin indicate its co-secretion with insulin in humans. Diabetologia 1991, 34, 52–54. [Google Scholar] [CrossRef]
- Westermark, P.; Wilander, E. The influence of amyloid deposits on the islet volume in maturity onset diabetes mellitus. Diabetologia 1978, 15, 417–421. [Google Scholar] [CrossRef]
- Clark, A.; Saad, M.F.; Nezzer, T.; Uren, C.; Knowler, W.C.; Bennett, P.H.; Turner, R.C. Islet amyloid polypeptide in diabetic and non-diabetic Pima Indians. Diabetologia 1990, 33, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Paulsson, J.F.; Westermark, G.T. Aberrant Processing of Human Proislet Amyloid Polypeptide Results in Increased Amyloid Formation. Diabetes 2005, 54, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Gurlo, T.; Huang, C.J.; Butler, P.C. Islet Amyloid in Type 2 Diabetes, and the Toxic Oligomer Hypothesis. Endocr. Rev. 2008, 29, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Visa, M.; Alcarraz-Vizán, G.; Montane, J.; Cadavez, L.; Castaño, C.; Villanueva-Peñacarrillo, M.L.; Servitja, J.; Novials, A. Islet amyloid polypeptide exerts a novel autocrine action in β-cell signaling and proliferation. FASEB J. 2015, 29, 2970–2979. [Google Scholar] [CrossRef] [PubMed]
- Mather, K.J.; Paradisi, G.; Leaming, R.; Hook, G.; Steinberg, H.O.; Fineberg, N.; Hanley, R.; Baron, A.D. Role of amylin in insulin secretion and action in humans: Antagonist studies across the spectrum of insulin sensitivity. Diabetes Metab. Res. Rev. 2002, 18, 118–126. [Google Scholar] [CrossRef]
- Hull, R.L.; Westermark, G.T.; Westermark, P.; Kahn, S.E. Islet Amyloid: A Critical Entity in the Pathogenesis of Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 3629–3643. [Google Scholar] [CrossRef] [PubMed]
- Kiriyama, Y.; Nochi, H. Role and Cytotoxicity of Amylin and Protection of Pancreatic Islet β-Cells from Amylin Cytotoxicity. Cells 2018, 7, 95. [Google Scholar] [CrossRef]
- Sakagashira, S.; Sanke, T.; Hanabusa, T.; Shimomura, H.; Ohagi, S.; Kumagaye, K.Y.; Nakajima, K.; Nanjo, K. Missense mutation of amylin gene (S20G) in Japanese NIDDM patients. Diabetes 1996, 45, 1279–1281. [Google Scholar] [CrossRef]
- Fukunaka, A.; Shimura, M.; Ichinose, T.; Pereye, O.B.; Nakagawa, Y.; Tamura, Y.; Mizutani, W.; Inoue, R.; Inoue, T.; Tanaka, Y.; et al. Zinc and iron dynamics in human islet amyloid polypeptide-induced diabetes mouse model. Sci. Rep. 2023, 13, 3484. [Google Scholar] [CrossRef]
- Mukherjee, S.; Dey, S.G. Heme Bound Amylin: Spectroscopic Characterization, Reactivity, and Relevance to Type 2 Diabetes. Inorg. Chem. 2013, 52, 5226–5235. [Google Scholar] [CrossRef]
- Seal, M.; Mukherjee, S.; Dey, S.G. Fe–oxy adducts of heme–Aβ and heme–hIAPP complexes: Intermediates in ROS generation. Metallomics 2016, 8, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Du, X.; Guo, P.; Huang, L.; Qi, P.; Gong, Q. Ascorbic acid supplementation in type 2 diabetes mellitus: A protocol for systematic review and meta-analysis. Medicine 2020, 99, e23125. [Google Scholar] [CrossRef] [PubMed]
- Ashor, A.; Werner, A.D.; Lara, J.; Willis, N.D.; Mathers, J.C.; Siervo, M. Effects of vitamin C supplementation on glycaemic control: A systematic review and meta-analysis of randomised controlled trials. Eur. J. Clin. Nutr. 2017, 71, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Gillani, S.W.; Sulaiman, S.A.S.; Abdul, M.I.M.; Baig, M.R. Combined effect of metformin with ascorbic acid versus acetyl salicylic acid on diabetes-related cardiovascular complication; a 12-month single blind multicenter randomized control trial. Cardiovasc. Diabetol. 2017, 16, 103. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Xu, Q.; Park, Y.; Hollenbeck, A.; Schatzkin, A.; Chen, H. Multivitamins, Individual Vitamin and Mineral Supplements, and Risk of Diabetes Among Older U.S. Adults. Diabetes Care 2010, 34, 108–114. [Google Scholar] [CrossRef]
- Sims, E.K.; Lakhter, A.J.; Anderson-Baucum, E.; Kono, T.; Tong, X.; Evans-Molina, C. MicroRNA 21 targets BCL2 mRNA to increase apoptosis in rat and human beta cells. Diabetologia 2017, 60, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Roggli, E.; Gattesco, S.; Caille, D.; Briet, C.; Boitard, C.; Meda, P.; Regazzi, R. Changes in MicroRNA Expression Contribute to Pancreatic β-Cell Dysfunction in Prediabetic NOD Mice. Diabetes 2012, 61, 1742–1751. [Google Scholar] [CrossRef] [PubMed]
- Belgardt, B.-F.; Ahmed, K.; Spranger, M.; Latreille, M.; Denzler, R.; Kondratiuk, N.; von Meyenn, F.; Villena, F.N.; Herrmanns, K.; Bosco, D.; et al. The microRNA-200 family regulates pancreatic beta cell survival in type 2 diabetes. Nat. Med. 2015, 21, 619–627. [Google Scholar] [CrossRef]
- Latreille, M.; Hausser, J.; Stützer, I.; Zhang, Q.; Hastoy, B.; Gargani, S.; Kerr-Conte, J.; Pattou, F.; Zavolan, M.; Esguerra, J.L.; et al. MicroRNA-7a regulates pancreatic β cell function. J. Clin. Investig. 2014, 124, 2722–2735. [Google Scholar] [CrossRef]
- Poy, M.N.; Hausser, J.; Trajkovski, M.; Braun, M.; Collins, S.; Rorsman, P.; Zavolan, M.; Stoffel, M. miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc. Natl. Acad. Sci. USA 2009, 106, 5813–5818. [Google Scholar] [CrossRef]
- He, Y.; Ding, Y.; Liang, B.; Lin, J.; Kim, T.-K.; Yu, H.; Hang, H.; Wang, K. A Systematic Study of Dysregulated MicroRNA in Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2017, 18, 456. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, O.; Fabris, G.; Pisani, D.F.; Casamento, V.; Gautier, N.; Hinault, C.; Lebrun, P.; Duranton, C.; Tauc, M.; Dalle, S.; et al. microRNA-375 regulates glucose metabolism-related signaling for insulin secretion. J. Endocrinol. 2020, 244, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Erener, S.; Mojibian, M.; Fox, J.K.; Denroche, H.; Kieffer, T.J. Circulating miR-375 as a Biomarker of β-Cell Death and Diabetes in Mice. Endocrinology 2013, 154, 603–608. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Man, B.; Li, D. Assessing MicroRNA-375 Levels in Type 2 Diabetes Mellitus (T2DM) Patients and Their First-Degree Relatives with T2DM. Diabetes Metab. Syndr. Obes. 2021, 14, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Schoenborn, J.; Wang, Z.; Chen, X.; Matson, K.; Mohan, R.; Zhang, S.; Tang, X.; Arunagiri, A.; Arvan, P.; et al. Transgenic overexpression of microRNA-30d in pancreatic beta-cells progressively regulates beta-cell function and identity. Sci. Rep. 2022, 12, 11969. [Google Scholar] [CrossRef]
- Ferrero, G.; Carpi, S.; Polini, B.; Pardini, B.; Nieri, P.; Impeduglia, A.; Grioni, S.; Tarallo, S.; Naccarati, A. Intake of natural compounds and circulating microrna expression levels: Their relationship investigated in healthy subjects with different dietary habits. Front. Pharmacol. 2021, 11, 2214–2225. [Google Scholar] [CrossRef]
- Karavanaki, K.; Paschou, S.A.; Tentolouris, N.; Karachaliou, F.; Soldatou, A. Type 2 diabetes in children and adolescents: Distinct characteristics and evidence-based management. Endocrine 2022, 78, 280–295. [Google Scholar] [CrossRef]
- Bjornstad, P.; Chao, L.C.; Cree-Green, M.; Dart, A.B.; King, M.; Looker, H.C.; Magliano, D.J.; Nadeau, K.J.; Pinhas-Hamiel, O.; Shah, A.S.; et al. Youth-onset type 2 diabetes mellitus: An urgent challenge. Nat. Rev. Nephrol. 2022, 19, 168–184. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Adult-Onset T2D | Additional Factors Related to Youth-Onset T2D | |
|---|---|---|
| Genetic traits | PPARγ, KCNJ11, TCF7L2 variants from earlier studies, >700 loci from GWAS studies | No studies linking genetic traits with T2D specifically in youth populations |
| Early life and epigenetics | Intrauterine diabetic environment (mother’s gestational diabetes) | Corroborated by data from the TODAY study on youth populations |
| Prenatal nutrient insufficiency leading to SGA | ||
| Decreased secretory rate by the individual β-cells | Present already at the time of diagnosis (possibly due to several mechanisms) | Identified in youth studies together with a much more rapid deterioration in β-cell function |
| Defective first and second phase insulin secretion | ||
| Impaired insulin processing | Increased proinsulin to insulin ratio | Identified in youth studies too |
| Reduced β-cell mass | Both islet volume β-cell density and total β-cell mass decreased at the beginning and gradually progressed | No studies specifically in youth populations |
| Increased β-cell death and lower regeneration rate | Increased apoptosis due to several mechanisms (glucotoxicity, lipotoxicity, ER stress, oxidative stress etc.) | No studies specifically in youth populations |
| Increase autophagy | ||
| Defective regeneration | ||
| Trans- and de-differentiation of β-cells | Defective trans-differentiation may play a role. More robust data on increased de-differentiation of β-cells | May be important due to high rates of obesity and increased inflammation in youth with T2D, but no studies have been published |
| Pancreatic islet inflammation | Macrophages and cytokines important under normal conditions for β-cell function, but participate in their malfunction in T2D | Scarce studies mainly about obesity-related inflammatory markers. Must be important in the obesity-driven inflammatory milieu of youth-onset T2D |
| Role of α- and other islet cell dysfunction | Increased α-cell function and hyperglucagonemia has been implicated in T2D pathogenesis | Scarce studies with conflicting results regarding α-cell function |
| Islet amyloid polypeptide accumulation | Mainly IAAP toxic oligomers have been incriminated in defective insulin production | No relevant studies in populations with youth-onset T2D |
| Role of miRNAs dysfunction | Involved in β-cell development, identity preservation, survival, and function but also in their malfunction and apoptotic death in T2D (e.g., miR-375) | No relevant studies in populations with youth-onset T2D |
| Factors Identified to Have a Protective Role | Factors Identified to Have a Harmful Effect | Comments | |
|---|---|---|---|
| Vitamin D | Vitamin D supplementation associated with improved glucose metabolism | Low vitamin D levels as well as specific VDR polymorphisms associated with increased T2D risk | Higher levels and supplementation seem to play a protective role only in subjects at risk for T2D, possibly through decreased inflammation |
| Vitamin A | Experimental animals fed vitamin A-poor diets showed increased β-cell apoptosis, decreased β-cell mass, and increased α-cells | Essential in maintaining adequate β-cell differentiation and mass in experimental conditions | |
| Vitamin C | Some studies have linked vitamin C supplementation with decreased fasting blood glucose levels and glycosylated hemoglobin | May have a protective role against amylin formation | |
| Calcium | Calcium supplementation associated with improved glucose metabolism | Low calcium levels or decreased dairy product intake | Conflicting results from some studies showing that higher calcium levels increase T2D risk |
| Iron | Higher levels are independently associated with impaired glucose metabolism and T2D. Possibly lower levels too | Not clear if β-cell function is mostly affected, peripheral insulin sensitivity, or both | |
| Magnesium | Intake linked with decreased T2D risk | Possibly acts as an antioxidant. Not clear if β-cell function is improved, peripheral insulin sensitivity, or both | |
| Selenium | Some studies have linked selenium intake with lower T2D risk | Others have associated higher levels or increased intake with higher T2D risk | Role not clear, possibly acts as an antioxidant when in the right concentration |
| Zinc | Zinc intake seems to have a mildly protective role against T2D and better glycemic control in women, patients with T2D, and in experimental animals | Specific SLC30A8 gene polymorphisms (codes for ZnT8 protein) have been linked with higher T2D risk | Its action may be influenced by several factors such as obesity and specific genetic traits. Important role in insulin hexamers. Possibly acts through decreased inflammation |
| Chromium | Chromium supplementation improves glucose metabolism in people with T2D | Not clear if β-cell function is improved, peripheral insulin sensitivity, or both |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serbis, A.; Giapros, V.; Tsamis, K.; Balomenou, F.; Galli-Tsinopoulou, A.; Siomou, E. Beta Cell Dysfunction in Youth- and Adult-Onset Type 2 Diabetes: An Extensive Narrative Review with a Special Focus on the Role of Nutrients. Nutrients 2023, 15, 2217. https://doi.org/10.3390/nu15092217
Serbis A, Giapros V, Tsamis K, Balomenou F, Galli-Tsinopoulou A, Siomou E. Beta Cell Dysfunction in Youth- and Adult-Onset Type 2 Diabetes: An Extensive Narrative Review with a Special Focus on the Role of Nutrients. Nutrients. 2023; 15(9):2217. https://doi.org/10.3390/nu15092217
Chicago/Turabian StyleSerbis, Anastasios, Vasileios Giapros, Konstantinos Tsamis, Foteini Balomenou, Assimina Galli-Tsinopoulou, and Ekaterini Siomou. 2023. "Beta Cell Dysfunction in Youth- and Adult-Onset Type 2 Diabetes: An Extensive Narrative Review with a Special Focus on the Role of Nutrients" Nutrients 15, no. 9: 2217. https://doi.org/10.3390/nu15092217