
Ketogenic Diet Treatment of Defects in the Mitochondrial Malate Aspartate Shuttle and Pyruvate Carrier
 , , , , , , , , , , , , , , , , , , , and add
Show full author list
, , , , , , , , , , , , , , , , , , , and add
Show full author list
Abstract
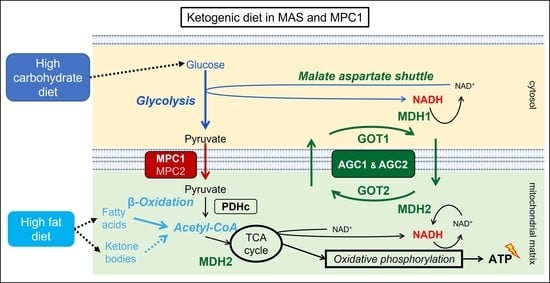
:
1. Introduction
1.1. The Ketogenic Diet
1.2. The Mitochondrial Malate Aspartate Shuttle (MAS)
1.3. The Mitochondrial Pyruvate Carriers 1 and 2
1.4. Ketogenic Diet for MAS-Defects and MPC1-Deficiency in the Literature
2. Materials and Methods
3. Results
3.1. AGC1-Deficiency (SLC25A12)
3.1.1. Presentation before Initiation of KD
3.1.2. Follow Up during KD
3.2. MDH1-Deficiency (MDH1)
3.3. MDH2-Deficiency (MDH2)
3.3.1. Presentation before Initiation of KD
3.3.2. Follow Up during KD
3.4. GOT2-Deficiency (GOT2)
Treatment
3.5. MPC1-Deficiency (MPC1)
3.5.1. Presentation before Initiation of KD
3.5.2. Follow-Up during KD
3.6. Citrin Deficiency (Also Known as AGC2-Deficiency, SLC25A13)
3.7. Genotypes
3.8. Functional Investigations
4. Discussion
4.1. Phenotype
4.2. Effect of KD on Neurological MAS-Defects and MPC1-Deficiency
4.3. Serine Supplementation
4.4. Citrin Deficiency
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wortmann, S.B.; Mayr, J.A.; Nuoffer, J.M.; Prokisch, H.; Sperl, W. A Guideline for the Diagnosis of Pediatric Mitochondrial Disease: The Value of Muscle and Skin Biopsies in the Genetics Era. Neuropediatrics 2017, 48, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.L.; Prokisch, H. Genetics of Mitochondrial Diseases: Identifying Mutations to Help Diagnosis. EBioMedicine 2020, 56, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.; Thomas, R.H. The Mitochondrial Epilepsies. Eur. J. Paediatr. Neurol. 2020, 24, 47–52. [Google Scholar] [CrossRef]
- Repp, B.M.; Mastantuono, E.; Alston, C.L.; Schiff, M.; Haack, T.B.; Rötig, A.; Ardissone, A.; Lombès, A.; Catarino, C.B.; Diodato, D.; et al. Clinical, Biochemical and Genetic Spectrum of 70 Patients with ACAD9 Deficiency: Is Riboflavin Supplementation Effective? Orphanet J. Rare Dis. 2018, 13, 1–10. [Google Scholar] [CrossRef]
- Hoytema van Konijnenburg, E.M.M.; Wortmann, S.B.; Koelewijn, M.J.; Tseng, L.A.; Houben, R.; Stöckler-Ipsiroglu, S.; Ferreira, C.R.; van Karnebeek, C.D.M. Treatable Inherited Metabolic Disorders Causing Intellectual Disability: 2021 Review and Digital App. Orphanet J. Rare Dis. 2021, 16, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.; Jackson, C.F.; Levy, R.G.; Cooper, P.N. Ketogenic Diet and Other Dietary Treatments for Epilepsy. Cochrane Database Syst. Rev. 2016, 2, 1–26. [Google Scholar] [CrossRef]
- Sofou, K.; Dahlin, M.; Hallböök, T.; Lindefeldt, M.; Viggedal, G.; Darin, N. Ketogenic Diet in Pyruvate Dehydrogenase Complex Deficiency: Short- and Long-Term Outcomes. J. Inherit. Metab. Dis. 2017, 40, 237–245. [Google Scholar] [CrossRef]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of Action for the Medium-Chain Triglyceride Ketogenic Diet in Neurological and Metabolic Disorders. Lancet. Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Zweers, H.; van Wegberg, A.M.J.; Janssen, M.C.H.; Wortmann, S.B. Ketogenic Diet for Mitochondrial Disease: A Systematic Review on Efficacy and Safety. Orphanet J. Rare Dis. 2021, 16, 295. [Google Scholar] [CrossRef]
- Broeks, M.H.; van Karnebeek, C.D.M.; Wanders, R.J.A.; Jans, J.J.M.; Verhoeven-Duif, N.M. Inborn Disorders of the Malate Aspartate Shuttle. J. Inherit. Metab. Dis. 2021, 44, 792. [Google Scholar] [CrossRef]
- Saheki, T.; Song, Y. Citrin Deficiency-PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/20301360/ (accessed on 7 July 2022).
- Tavoulari, S.; Lacabanne, D.; Thangaratnarajah, C.; Kunji, E.R.S. Pathogenic Variants of the Mitochondrial Aspartate/Glutamate Carrier Causing Citrin Deficiency. Trends Endocrinol. Metab. 2022, 33, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.B.; Kobayashi, K.; Ushikai, M.; Tabata, A.; Iijima, M.; Li, M.X.; Lei, L.; Kawabe, K.; Taura, S.; Yang, Y.; et al. Frequency and Distribution in East Asia of 12 Mutations Identified in the SLC25A13 Gene of Japanese Patients with Citrin Deficiency. J. Hum. Genet. 2005, 50, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Hutchin, T.; Preece, M.A.; Hendriksz, C.; Chakrapani, A.; McClelland, V.; Okumura, F.; Song, Y.Z.; Iijima, M.; Kobayashi, K.; Saheki, T.; et al. Neonatal Intrahepatic Cholestasis Caused by Citrin Deficiency (NICCD) as a Cause of Liver Disease in Infants in the UK. J. Inherit. Metab. Dis. 2009, 32, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Buffet, A.; Morin, A.; Castro-Vega, L.-J.; Habarou, F.; Lussey-Lepoutre, C.; Letouzé, E.; Lefebvre, H.; Guilhem, I.; Haissaguerre, M.; Raingeard, I.; et al. Germline Mutations in the Mitochondrial 2-Oxoglutarate/Malate Carrier SLC25A11 Gene Confer a Predisposition to Metastatic Paragangliomas. Cancer Res. 2018, 78, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, M.; Martin, D.A.; Hedlund, Z.; Jonsson, M.; Von Döbeln, U.; Wedell, A. The Ketogenic Diet Compensates for AGC1 Deficiency and Improves Myelination. Epilepsia 2015, 56, e176–e181. [Google Scholar] [CrossRef]
- Pfeiffer, B.; Sen, K.; Kaur, S.; Pappas, K. Expanding Phenotypic Spectrum of Cerebral Aspartate-Glutamate Carrier Isoform 1 (AGC1) Deficiency. Neuropediatrics 2020, 51, 160–163. [Google Scholar] [CrossRef]
- Pérez-Liébana, I.; Casarejos, M.J.; Alcaide, A.; Herrada-Soler, E.; Llorente-Folch, I.; Contreras, L.; Satrústegui, J.; Pardo, B. ΒOHB Protective Pathways in Aralar-KO Neurons and Brain: An Alternative to Ketogenic Diet. J. Neurosci. 2020, 40, 9293–9305. [Google Scholar] [CrossRef]
- Brivet, M.; Garcia-Cazorla, A.; Lyonnet, S.; Dumez, Y.; Nassogne, M.C.; Slama, A.; Boutron, A.; Touati, G.; Legrand, A.; Saudubray, J.M. Impaired Mitochondrial Pyruvate Importation in a Patient and a Fetus at Risk. Mol. Genet. Metab. 2003, 78, 186–192. [Google Scholar] [CrossRef]
- Vanderperre, B.; Herzig, S.; Krznar, P.; Hörl, M.; Ammar, Z.; Montessuit, S.; Pierredon, S.; Zamboni, N.; Martinou, J.C. Embryonic Lethality of Mitochondrial Pyruvate Carrier 1 Deficient Mouse Can Be Rescued by a Ketogenic Diet. PLoS Genet. 2016, 12, e1006056. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Kido, J.; Häberle, J.; Sugawara, K.; Tanaka, T.; Nagao, M.; Sawada, T.; Wada, Y.; Numakura, C.; Murayama, K.; Watanabe, Y.; et al. Clinical Manifestation and Long-Term Outcome of Citrin Deficiency: Report from a Nationwide Study in Japan. J. Inherit. Metab. Dis. 2022, 45, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Wibom, R.; Lasorsa, F.M.; Töhönen, V.; Barbaro, M.; Sterky, F.H.; Kucinski, T.; Naess, K.; Jonsson, M.; Pierri, C.L.; Palmieri, F.; et al. AGC1 Deficiency Associated with Global Cerebral Hypomyelination. N. Engl. J. Med. 2009, 361, 489–495. [Google Scholar] [CrossRef]
- Falk, M.J.; Li, D.; Gai, X.; McCormick, E.; Place, E.; Lasorsa, F.M.; Otieno, F.G.; Hou, C.; Kim, C.E.; Abdel-Magid, N.; et al. AGC1 Deficiency Causes Infantile Epilepsy, Abnormal Myelination, and Reduced N-Acetylaspartate. JIMD Rep. 2014, 14, 77–85. [Google Scholar] [CrossRef]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Ciara, E.; Trubicka, J.; Rokicki, D.; Karkucinska-Wieckowska, A.; Pajdowska, M.; Jurkiewicz, E.; Halat, P.; Kosinska, J.; et al. New Perspective in Diagnostics of Mitochondrial Disorders: Two Years’ Experience with Whole-Exome Sequencing at a National Paediatric Centre. J. Transl. Med. 2016, 14, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, B.C.; Warren, E.B.; Baytas, O.; Schmidt, M.; Merck, D.; Buch, K.; Liu, J.S.; Phornphutkul, C.; Caruso, P.; Morrow, E.M. Longitudinal MRI Findings in Patient with SLC25A12 Pathogenic Variants Inform Disease Progression and Classification. Am. J. Med. Genet. A 2019, 179, 2284–2291. [Google Scholar] [CrossRef]
- Saleh, M.; Helmi, M.; Yacop, B. A Novel Nonsense Gene Variant Responsible for Early Infantile Epileptic Encephalopathy Type 39: Case Report. Pakistan J. Biol. Sci. PJBS 2020, 23, 973–976. [Google Scholar] [CrossRef]
- Kose, M.; Isik, E.; Aykut, A.; Durmaz, A.; Kose, E.; Ersoy, M.; Diniz, G.; Adebali, O.; Ünalp, A.; Yilmaz, Ü.; et al. The Utility of Next-Generation Sequencing Technologies in Diagnosis of Mendelian Mitochondrial Diseases and Reflections on Clinical Spectrum. J. Pediatr. Endocrinol. Metab. 2021, 34, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Broeks, M.H.; Shamseldin, H.E.; Alhashem, A.; Hashem, M.; Abdulwahab, F.; Alshedi, T.; Alobaid, I.; Zwartkruis, F.; Westland, D.; Fuchs, S.; et al. MDH1 Deficiency Is a Metabolic Disorder of the Malate-Aspartate Shuttle Associated with Early Onset Severe Encephalopathy. Hum. Genet. 2019, 138, 1247–1257. [Google Scholar] [CrossRef]
- Ait-El-Mkadem, S.; Dayem-Quere, M.; Gusic, M.; Chaussenot, A.; Bannwarth, S.; François, B.; Genin, E.C.; Fragaki, K.; Volker-Touw, C.L.M.; Vasnier, C.; et al. Mutations in MDH2, Encoding a Krebs Cycle Enzyme, Cause Early-Onset Severe Encephalopathy. Am. J. Hum. Genet. 2017, 100, 151–159. [Google Scholar] [CrossRef]
- Laemmle, A.; Steck, A.L.; Schaller, A.; Kurth, S.; Perret Hoigné, E.; Felser, A.D.; Slavova, N.; Salvisberg, C.; Atencio, M.; Mochel, F.; et al. Triheptanoin-Novel Therapeutic Approach for the Ultra-Rare Disease Mitochondrial Malate Dehydrogenase Deficiency. Mol. Genet. Metab. Rep. 2021, 29, 100814. [Google Scholar] [CrossRef]
- van Karnebeek, C.D.M.; Ramos, R.J.; Wen, X.Y.; Tarailo-Graovac, M.; Gleeson, J.G.; Skrypnyk, C.; Brand-Arzamendi, K.; Karbassi, F.; Issa, M.Y.; van der Lee, R.; et al. Bi-Allelic GOT2 Mutations Cause a Treatable Malate-Aspartate Shuttle-Related Encephalopathy. Am. J. Hum. Genet. 2019, 105, 534–548. [Google Scholar] [CrossRef]
- Bricker, D.K.; Taylor, E.B.; Schell, J.C.; Orsak, T.; Boutron, A.; Chen, Y.C.; Cox, J.E.; Cardon, C.M.; Van Vranken, J.G.; Dephoure, N.; et al. A Mitochondrial Pyruvate Carrier Required for Pyruvate Uptake in Yeast, Drosophila, and Humans. Science 2012, 337, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Oonthonpan, L.; Rauckhorst, A.J.; Gray, L.R.; Boutron, A.C.; Taylor, E.B. Two Human Patient Mitochondrial Pyruvate Carrier Mutations Reveal Distinct Molecular Mechanisms of Dysfunction. JCI Insight 2019, 5, 1–14. [Google Scholar] [CrossRef]
- Tabata, A.; Sheng, J.S.; Ushikai, M.; Song, Y.Z.; Gao, H.Z.; Lu, Y.B.; Okumura, F.; Iijima, M.; Mutoh, K.; Kishida, S.; et al. Identification of 13 Novel Mutations Including a Retrotransposal Insertion in SLC25A13 Gene and Frequency of 30 Mutations Found in Patients with Citrin Deficiency. J. Hum. Genet. 2008, 53, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Yamaguchi, N.; Kobayashi, K.; Nishi, I.; Horinouchi, H.; Jalil, M.A.; Li, M.X.; Ushikai, M.; Iijima, M.; Kondo, I.; et al. Identification of Two Novel Mutations in the SLC25A13 Gene and Detection of Seven Mutations in 102 Patients with Adult-Onset Type II Citrullinemia. Hum. Genet. 2000, 107, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Pouwels, P.J.W.; Brockmann, K.; Kruse, B.; Wilken, B.; Wick, M.; Hanefeld, F.; Frahm, J. Regional Age Dependence of Human Brain Metabolites from Infancy to Adulthood as Detected by Quantitative Localized Proton MRS. Pediatr. Res. 1999, 46, 474. [Google Scholar] [CrossRef]
- Palmieri, F. Mitochondrial Carrier Proteins. FEBS Lett. 1994, 346, 48–54. [Google Scholar] [CrossRef]
- Monné, M.; Palmieri, F.; Kunji, E.R.S. The Substrate Specificity of Mitochondrial Carriers: Mutagenesis Revisited. Mol. Membr. Biol. 2013, 30, 149–159. [Google Scholar] [CrossRef]
- Palmieri, L.; Pardo, B.; Lasorsa, F.M.; Del Arco, A.; Kobayashi, K.; Iijima, M.; Runswick, M.J.; Walker, J.E.; Saheki, T.; Satrústegui, J.; et al. Citrin and Aralar1 Are Ca(2+)-Stimulated Aspartate/Glutamate Transporters in Mitochondria. EMBO J. 2001, 20, 5060–5069. [Google Scholar] [CrossRef]
- Wolf, N.I.; van der Knaap, M.S. AGC1 Deficiency and Cerebral Hypomyelination. N. Engl. J. Med. 2009, 361, 1997–1998. [Google Scholar] [CrossRef] [PubMed]
- van der Knaap, M.S.; Bugiani, M. Leukodystrophies: A Proposed Classification System Based on Pathological Changes and Pathogenetic Mechanisms. Acta Neuropathol. 2017, 134, 351–382. [Google Scholar] [CrossRef]
- Jalil, M.A.; Begumi, L.; Contreras, L.; Pardo, B.; Iijima, M.; Li, M.X.; Ramos, M.; Marmol, P.; Horiuchi, M.; Shimotsu, K.; et al. Reduced N-Acetylaspartate Levels in Mice Lacking Aralar, a Brain- and Muscle-Type Mitochondrial Aspartate-Glutamate Carrier. J. Biol. Chem. 2005, 280, 31333–31339. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Rodrigues, T.B.; Contreras, L.; Garzón, M.; Llorente-Folch, I.; Kobayashi, K.; Saheki, T.; Cerdan, S.; Satrüstegui, J. Brain Glutamine Synthesis Requires Neuronal-Born Aspartate as Amino Donor for Glial Glutamate Formation. J. Cereb. Blood Flow Metab. 2011, 31, 90–101. [Google Scholar] [CrossRef]
- Amaral, A.I. Effects of Hypoglycaemia on Neuronal Metabolism in the Adult Brain: Role of Alternative Substrates to Glucose. J. Inherit. Metab. Dis. 2013, 36, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Vigueira, P.A.; McCommis, K.S.; Schweitzer, G.G.; Remedi, M.S.; Chambers, K.T.; Fu, X.; McDonald, W.G.; Cole, S.L.; Colca, J.R.; Kletzien, R.F.; et al. Mitochondrial Pyruvate Carrier 2 Hypomorphism in Mice Leads to Defects in Glucose-Stimulated Insulin Secretion. Cell Rep. 2014, 7, 2042–2053. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.N.; Cousteils, K.; Lou, J.W.; Fox, J.E.M.; MacDonald, P.E.; Joseph, J.W. Mitochondrial Metabolism of Pyruvate Is Essential for Regulating Glucose-Stimulated Insulin Secretion. J. Biol. Chem. 2014, 289, 13335–13346. [Google Scholar] [CrossRef]
- Buchanan, J.L.; Taylor, E.B. Mitochondrial Pyruvate Carrier Function in Health and Disease across the Lifespan. Biomolecules 2020, 10, 1162. [Google Scholar] [CrossRef] [PubMed]
- Kossoff, E.H.; Zupec-Kania, B.A.; Auvin, S.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Cross, J.H.; Dahlin, M.G.; et al. Optimal Clinical Management of Children Receiving Dietary Therapies for Epilepsy: Updated Recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 2018, 3, 175–192. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Kumar, I.; Verma, N.; Aggarwal, P.; Ojha, R. Magnetic Resonance Spectroscopy-Revisiting the Biochemical and Molecular Milieu of Brain Tumors. BBA Clin. 2016, 5, 170–178. [Google Scholar] [CrossRef]
- Provencher, S.W. Estimation of Metabolite Concentrations from Localized in Vivo Proton NMR Spectra. Magn. Reson. Med. 1993, 30, 672–679. [Google Scholar] [CrossRef]
- Juaristi, I.; García-Martín, M.L.; Rodrigues, T.B.; Satrústegui, J.; Llorente-Folch, I.; Pardo, B. ARALAR/AGC1 Deficiency, a Neurodevelopmental Disorder with Severe Impairment of Neuronal Mitochondrial Respiration, Does Not Produce a Primary Increase in Brain Lactate. J. Neurochem. 2017, 142, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Maugard, M.; Vigneron, P.A.; Bolaños, J.P.; Bonvento, G. L-Serine Links Metabolism with Neurotransmission. Prog. Neurobiol. 2021, 197, 1–11. [Google Scholar] [CrossRef]
- Porcelli, V.; Longo, A.; Palmieri, L.; Closs, E.I.; Palmieri, F. Asymmetric Dimethylarginine Is Transported by the Mitochondrial Carrier SLC25A2. Amino Acids 2016, 48, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Profilo, E.; Peña-Altamira, L.E.; Corricelli, M.; Castegna, A.; Danese, A.; Agrimi, G.; Petralla, S.; Giannuzzi, G.; Porcelli, V.; Sbano, L.; et al. Down-Regulation of the Mitochondrial Aspartate-Glutamate Carrier Isoform 1 AGC1 Inhibits Proliferation and N-Acetylaspartate Synthesis in Neuro2A Cells. Biochim. Biophys. Acta Mol. basis Dis. 2017, 1863, 1422–1435. [Google Scholar] [CrossRef] [PubMed]
- Feichtinger, R.G.; Zimmermann, F.; Mayr, J.A.; Neureiter, D.; Hauser-Kronberger, C.; Schilling, F.H.; Jones, N.; Sperl, W.; Kofler, B. Low Aerobic Mitochondrial Energy Metabolism in Poorly- or Undifferentiated Neuroblastoma. BMC Cancer 2010, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Rustin, P.; Chretien, D.; Bourgeron, T.; Gérard, B.; Rötig, A.; Saudubray, J.M.; Munnich, A. Biochemical and Molecular Investigations in Respiratory Chain Deficiencies. Clin. Chim. Acta. 1994, 228, 35–51. [Google Scholar] [CrossRef]
- Sperl, W.; Trijbels, J.M.F.; Ruitenbeek, W.; Van Laack, H.L.J.M.; Janssen, A.J.M.; Kerkhof, C.M.C.; Sengers, R.C.A. Measurement of Totally Activated Pyruvate Dehydrogenase Complex Activity in Human Muscle: Evaluation of a Useful Assay. Enzyme Protein 1993, 47, 37–46. [Google Scholar] [CrossRef]
- Kušíková, K.; Feichtinger, R.G.; Csillag, B.; Kalev, O.K.; Weis, S.; Duba, H.C.; Mayr, J.A.; Weis, D. Case Report and Review of the Literature: A New and a Recurrent Variant in the VARS2 Gene Are Associated With Isolated Lethal Hypertrophic Cardiomyopathy, Hyperlactatemia, and Pulmonary Hypertension in Early Infancy. Front. Pediatr. 2021, 9, 1–12. [Google Scholar] [CrossRef]
- Bookelman, H.; Trijbels, J.M.F.; Sengers, R.C.A.; Janssen, A.J.M.; Veerkamp, J.H.; Stadhouders, A.M. Pyruvate Oxidation in Rat and Human Skeletal Muscle Mitochondria. Biochem. Med. 1978, 20, 395–403. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a: Phenotype of individuals with AGC1-defect without/before ketogenic diet | ||||||||||||||
| #, Sex | Pregnancy & Birth | Age of Onset (m); Presenting Findings | Onset of Seizures (m) | Last FU (y, m) | Start KD (y, m) | Development At Start KD (Age If No/Before KD) | Developmental Course | Muscle Tone at Start KD (Last FU If No KD) | Movement Disorder | Seizure Semiology | ASM Before KD | Microcephaly | Other Findings Before Start KD | Reference |
| AGC1-1, m | AT, polyhydr- amnios | birth; NRDS, muscle tone ↓ | 4 | 4 y 3 m | 1 y 9 m | no social interaction, movements ↓, tube fed | sec DI upon SO | ↓ | choreo- athetotic | focal clonic; (multi)focal with apnea; tonic eye deviation & laughter, +/− motor | LEV, OxCBZ, Pb, LTG, CLB, VGB, TPM, Pred, STM, RUF, STP | yes, sec | GOR/vomiting, FTT, feeding difficulties, LFT ↑; non-epileptic apnea, dysm, muscle weakness, tongue fibrillation | this paper |
| AGC1-2, m | AT | 5 m; seizures | 5 | 7 y 8 m (6 m after stopp KD) | 5 y 8 m | non-verbal, spoon-fed, no sitting | sec DI upon SO | ↓ | no | focal clonic | LEV | yes | FTT | this paper |
| AGC1-3, f | AT, CS | 5 m; muscle tone ↓ | 7 | 3 y 11 m (2 w after stopp KD) | 1 y 4 m | no visual fixation | sec DI, 3 m before SO | ↓ | no | focal with starring, motor arrest, cyanosis | LEV | no, p3-10 | growth retardation | this paper |
| AGC1-4, f | AT; foetal move-ments ↓ | birth; lethargic, muscle tone ↓ | 5 | 7 y (off) | 3 y (1 m KD) | non-verbal, no motor milestones, tube fed | sec DI, 1 m after SO | ↓ | dystonic- spastic (7y) | n/r | Pb, LEV, STM, OxCBZ. LAC, (7y): OxCBZ | yes, sec | GOR/vomiting, FTT | this paper |
| AGC1-5, m | AT | birth; lethargic, breast-feeding difficulties | 7 | 4 y 6 m | 2.5 | no sitting, no crawling, non-verbal, interaction ↓, eye contact ↓, tube fed | prim DI, worse- ning upon SO | ↓ | hypo-tonic- dys-kinetic | tonic | LEV, TPM, esliCBZ | yes, sec | feeding difficulties | this paper |
| AGC1-6, m | AT | 6 m; seizures | 6 | 1 y 5 m | 1 y 2 m | babbeling | global DI | ↓ | no | focal with apnea | LEV, TPM | n/r | n/r | this paper |
| AGC1-7, f | preg normal | 5 m; DI | 7 | 7 y 8 m | 6 | no head control, rolling, grasping, eye contact ↓, smiling response | sec DI, stag- nation after SO | ↓ | n/r | apneas, tonic, focal clonic, myoclonic, lip smacking, chewing, laughter | LEV, CBZ, OxCBZ | no | n/r | [16,23] |
| AGC1-8, f | PT (36 wg) | birth; hypogly-cemia (7 d NICU), muscle tone ↓ | 10 | 6 y 8 m | n/a | non-verbal, smiling, sitting assisted, drooling | sec DI, regr after SE | ↓ | no | focal clonic, gen tonic-clonic | TPM, Pb, ZNS | yes, sec | dysm, short stature | [24] |
| AGC1-9, m | PT, CS (33 wg) | birth; polyhydr- amnios | 10 | 1 y 1 m | n/a | bearing weight, sitting, rolling over, smiling | n/r | ↓ | no | focal, clonic, sec bilateral | LEV | no | dysm, jejunal atresia | [24] |
| AGC1-10, m | AT, preg normal | 7 m; febrile seizures | 7 | >2 y 1 m (4 m KD) | > 1 y 9 m | sitting assisted, monosyllables | sec DI upon SO | ↓ | n/r | tonic-clonic, myoclonic | n/r | n/r | not dysm | [17] |
| AGC1-11, n/r | CS | birth | 8 | n/r | n/a | n/r | n/r | ↓ | n/r | n/r | n/r | n/r | ptosis | [25] |
| AGC1-12, m | AT, delayed transition | birth; oxy-gen (8 d) | 3 | 11 y | n/a | sitting with support, smiling, vocalization, understanding some language, tube fed | prim DI, regr upon SO | ↑ | dystonic-spastic (11y) | focal eye/head deviation, control at 3 y. At 12y relapse, starring spells | OxCBZ, LEV, TPM, Pb | yes | GOR, ear infections, blepharitis, osteopenia, scoliosis, optic neuropathy, CVI, short stature | [26] |
| AGC1-13, f | n/r | 1 m; seizures | 1 | 7 y (†) | n/a | severe DI, non-verbal | n/r | ↑ | n/r | n/r | n/r | n/r | subtle dysm | [27] |
| AGC1-14, f | n/r | 3 m; n/r | n/r | 1 y 4 m | n/a | n/r | n/r | n/r | n/r | n/r | n/r | n/r | optic atrophy | [28] |
| b: Phenotype of individuals with MDH1/2 defect without/before ketogenic diet. | ||||||||||||||
| #, Sex | Pregnancy & Birth | Age of Onset (m); Presenting Findings | Onset of Seizures (m) | Last FU (y, m) | Start KD (y, m) | Development At Start KD (Age If No/Before KD) | Developmental Course | Muscle Tone at Start KD (Last FU If No KD) | Movement Disorder | Seizure Semiology | ASM Before KD | Microcephaly | Other Findings Before Start KD | Reference |
| MDH1-1, m | PT (32 wg), preg normal | 3 m; micro-cephaly, dysm | 13 | 2 y 6 m | n/a | DI, (13 m: rolling over, some babbling) | n/r | ↑ | n/r | hypsarrhyth-mia (infantile spasms) | TPM, CLN | yes, prog | growth retardation, strabism | [29] |
| MDH1-2, f | n/r | n/r | n/r | 4 y | n/a | DI, (walking 3 y, a few words 4 y) | n/r | ↑ | n/r | n/r | n/r | yes | dysm | [29] |
| MDH2-1, m | AT, preg normal | 5 m; muscle tone ↓, no head control | 7 | 4½ y (5 y alive) | 3 y | DI, 18 m sitting, crawling, good eye contact, averbal, tube fed | n/r | ↓ | dys-kinetic / dystonic | myoclonic | n/r | yes | FTT, constipation, strabism | [30] |
| MDH2-2, m | preg normal | birth; constipation | 2 | 1 y 8 m (†) | 1 y 6 m | DI, 1y: no sitting, no crawling; good eye contact, babbling | n/r | ↓ | no | gen tonic-clonic, spasms | n/r | no | strabism | [30] |
| MDH2-3, m | AT | birth; muscle tone ↓, macro-cephaly/-somia, two add. nipples | (?) | 7 ½ y (12 y alive) | 3 y | no crawling; no good eye contact; no language, tube fed | n/r | ↓ | dystonic | myoclonic, gen tonic | n/r | no | FTT, loss of vision, von Willebrand disease, congenital cystic adenomatoid malformation | [30] |
| MDH2-4, f | AT, preg normal | 1st m of life; febrile seizures | 1st m | 4 y | 3 y (Tri- hepta-noin) | DI | n/r | ↓ | dys-kinetic / choreatic | gen, absences | LTG, LEV | yes | 1st year: FTT. 18 m met stroke; episodes of met decompensation | [31] |
| c: Phenotype of individuals with GOT2 defect without ketogenic diet. | ||||||||||||||
| #, Sex | Pregnancy & Birth | Age of Onset (m); Presenting Findings | Onset of Seizures (m) | Last FU (y, m) | Start KD (y, m) | Development At Start KD (Age If No/Before KD) | Developmental Course | Muscle Tone at Start KD (Last FU If No KD) | Movement Disorder | Seizure Semiology | ASM Before KD | Microcephaly | Other Findings Before Start KD | Reference |
| GOT2-1, m | AT, CS | 1 m; DI, muscle tone ↓, abdominal spasms, feeding difficulties | 9 | 7 y 10 m | n/a | profound DI, no words (6 m: no head control, no sitting, no visual fixation) | n/r | ↑ | n/r | upward gaze, clonic seizures (left/right) | VPA, Pb, LEV, CBZ, LTG, TPM | yes | frequent infections, sleep disturbance | [32] |
| GOT2-2, f | PT (32 wg) | birth; DI, drooling, feeding difficulties | 7 | 10 y | n/a | severe DI, <10 words, following objects, smiling | n/r | ↑ | n/r | myoclonic, gen tonic-clonic, tonic | LTG, VPA | yes | frequent infections, acrocyanosis and chillblains | [32] |
| GOT2-3, f | AT | birth; DI, drooling, feeding difficulties | 6 | 8 y | n/a | severe DI, sitting, using hands, following objects, smiling, vocalize | n/r | ↑ | n/r | myoclonic, gen tonic-clonic | VPA, LTG, LEV | yes | frequent infections, acrocyanosis and chillblains | [32] |
| GOT2-4, m | AT | birth; feeding difficulties | 4 | 4 y | n/a | profound DI, non-verbal, unable to follow objects, abnormal eye movements | n/r | ↑ | n/r | myoclonic, tonic (upward gaze) | VPA, LEV, VGB, CLN | yes | frequent infections | [32] |
| d: Phenotype of individuals with MPC1-defect without/before KD. | ||||||||||||||
| #, Sex | Pregnancy & Birth | Age of Onset (m); Presenting Findings | Onset of Seizures (m) | Last FU (y, m) | Start KD (y, m) | Development At Start KD (Age If No/Before KD) | Developmental Course | Muscle Tone at Start KD (Last FU If No KD) | Movement Disorder | Seizure Semiology | ASM Before KD | Microcephaly | Other Findings Before Start KD | Reference |
| MPC1-1, m | AT, CS, breech, foetal move-ments ↓ | birth; lactate ↑, resp distress | 2.5 | 1 y 7 m | 1 y 3 m | profound DI, no change of position, no grasping, eye contact/fixation ↓, non-verbal | prim DI (regr at 2 m) | ↓ | no | 2 ½ m: myoclonia; 8 m: infantile spasms | LEV, CLB, OXC, LCS, VGB, RUF, LTG, BRV | yes, sec | muscle weakness | this paper |
| MPC1-2, m | AT | 6 y (?); seizures | 72 (?) | 12 y | n/a | IQ 56 | sec DI | normal | no | gen tonic clonic | VPA | yes | diabetes mellitus, splenomegaly, growth retardation (12 y p < 2), fractures | this paper |
| MPC1-3, f | AT | birth; truncal tone ↓ | 4 | 11 y | n/a | short sentences, counting to 5, walking | prim DI | ↓ | no | zyanotic, tonic, tonic-clonic | LTG | no | no | this paper |
| MPC1-4, m | AT | birth; truncal tone ↓ | no | 6 y | n/a | some words (sitting > 1 y, walking 4 y) | prim DI | ↓ | no | no seizures | n/a | no | no | this paper |
| MPC1-5, f | AT; induced delivery (mother) | birth; muscle tone ↓, dysm, hepato- megaly, resp distress | n/r | 1 y 7 m | neo | 1st m: rotatory nystagmus, poor visual contact, worsening over time | prim DI, sec regr | ↓ | n/r | n/r (no seizures?) | n/a | yes, prog | congenital heart defect, mild renal insufficiency, growth failure | [19,33,34] |
| MPC1-6, m | n/r | n/r | n/r | ~20 y | n/r | mild DI (last FU) | primDI | ↓ | n/r | n/r (seizures) | n/r | n/r | n/r | [33,34] |
| MPC1-7, m | n/r | n/r | n/r | ~17 y | n/r | severe DI (last FU) | n/r | n/r | n/r | n/r (no seizures?) | n/a | n/r | peripheral neuropathy, visual impairment | [33,34] |
| MPC1-8, f | n/r | n/r | n/r | ~12 y | n/r | mild DI (last FU) | n/r | n/r | n/r | n/r (no seizures?) | n/a | n/r | peripheral neuropathy | [33,34] |
| #, Sex | Country of Origin | Pregnancy, Delivery, Postnatal Course; Past History | Reason for Referral (Age) | Max. Blood Citrulline (μmol/L) | NH3 [Ref < 50 µmol/L] | Serum Galactose [Ref <20 mg/dl] | Other Lab Values | Hypo- gly- cemia | Ultrasound Liver | Treatment | Outcome/ Course |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AGC2-1, m | Austria | unremark | hepatomegaly, -pathy (4 m) | 104.8 (10–36) | normal | in urine ↑ | liver transaminases ↑, blood threonine ↑, methionine ↑, ferritin ↑; prothrombin time ↓ | no | mildly ↑ echogenicity of liver parenchyma | CH 40–45%, MCT 15–20% | lab normalised within weeks, thrives and develops age-adequate (2.4 y) |
| AGC2-2, m | Austria | n/a; epilepsy since adolescence | ↑ NH3, seizures, vomiting (35 y) | 571 (<50) | 151 | n/a | urinary arginino-succinic acid ↑ | no | n/a | glu inf, prot restr, ammonia scavengers | death (35 y) |
| AGC2-3, f | Austria | SGA, T21, neonatal bilirubin ↑ | NBS: citrulline ↑ (5 w) | 940 (12.6–58) | normal | normal (NBS), ↑ to max 25 (blood) | threonine ↑, methionine ↑, tyrosine ↑, threonine/serine ratio ↑; hyperbilirubi-nemia/ cholestasis, liver transaminases ↑, disturbed clotting | no | mildly ↑ echogenicity of liver parenchyma | CH 40–45%, MCT 15–20% | lab normalised within weeks, thrives and develops age-adequate (4½ y) |
| AGC2-4, m | Austria | SGA | cholestatic jaundice (4 w), positive family history | 523 (12.6–58) | normal | 73.9 (<15) | unremark serum amino acids, liver transaminases, clotting parameters | no | unremark | lab normalised within 10 d, thrives and development age-adequate (2.2 y) | |
| AGC2-5, m | Syria | neonatal hyperbili- rubinemia | suspected abdominal neoplasia (7 m) | 28 (5–24) | 79–182 | n/a | tyrosine ↑, cholestasis, hemolytic anemia, absent succinylacetone in urine/dried blood spot | no | n/a | glu inf, prot restr, nitisinone | death within 7 d (7 m) |
| AGC2-6, f | Syria | unremark | positive family history, (diagnosis from cord blood on d 12) | 764 (5–24) (45 d) | normal | normal (NBS), upon diagnosis ↑ (7542 (<800) μmol/L) | plasma threonine ↑ | no | n/a | lab normalised within 7 d, thrives and development age-adequate (22 m) | |
| AGC2-7, m | Syria | SGA | FTT (6 m) | 465 (4–65) | normal | ↑ | cholestasis, triglycerides ↑ | no | unremark | CH 26%, lactose and galactose free, MCT fat-enriched 61% | FTT improved, hepatopathy (5 y) |
| AGC2-8, f | Austria | uneventful | NBS: citrulli- ne ↑ (25 d) | 860 | 103 | 4.8 → ↑ 90 | cholestasis | no | mildly ↑ echogenicity of liver parenchyma | CH restr | normal growth and laboratory values (4½ y) |
| Patient ID | Gene | RefSeq | Variant | Predicted Change | HGMD | ACMG Classification | Reference |
|---|---|---|---|---|---|---|---|
| GOT2-1 | GOT2 | NM_002080.4 | c.617_619delTTC | p.Leu209del | DM | PTH | [32] |
| GOT2-2, GOT-3 | GOT2 | NM_002080.4 | c.784C > G | p.Arg262Gly | DM | PTH | [32] |
| GOT2-1 | GOT2 | NM_002080.4 | c.1009C > G | p.Arg337Gly | DM | PTH | [32] |
| GOT2-4 | GOT2 | NM_002080.4 | c.1097G > T | p.Gly366Val | DM | PTH | [32] |
| MDH1-1, MDH1-2 | MDH1 | NM_001199111.1 | c.413C > T | p.Ala138Val | DM | LPTH | [29] |
| MDH2-3 | MDH2 | NM_005918.4 | c.109G > A | p.Gly37Arg | DM | PTH | [30] |
| MDH2-1, MDH2-2, MDH2-3, MDH2-4 | MDH2 | NM_005918.4 | c.398C > T | p.Pro133Leu | DM | PTH | [30] |
| MDH2-4 | MDH2 | NM_005918.4 | c.445delinsACA | p.Pro149Hisfs*22 | DM | PTH | [31] |
| MDH2-2 | MDH2 | NM_005918.4 | c.596delG | p.Gly199Alafs*10 | DM | PTH | [30] |
| MDH2-1 | MDH2 | NM_005918.4 | c.620C > T | p.Pro207Leu | DM | LPTH | [30] |
| MPC1-2 | MPC1 | NM_016098.4 | c.95C > G | p.Ala32Gly | not listed | LPTH | this paper |
| MPC1-1 | MPC1 | NM_016098.4 | c.214A > G | p.Lys72Glu | not listed | LPTH | this paper |
| MPC1-3, MPC1-4, MPC1-6, MPC1-7, MPC1-8 | MPC1 | NM_016098.4 | c.236T > A | p.Leu79His | DM | LPTH | [33,34] |
| MPC1-5 | MPC1 | NM_016098.4 | c.289C > T | p.Arg97Trp | DM | LPTH | [33,34] |
| AGC1-14 | SLC25A12 | NM_003705.5 | c.125G > C | p.Arg42Pro | not listed | LPTH | [28] |
| AGC1-5 | SLC25A12 | NM_003705.5 | c.225del | p.Glu76Serfs*17 | not listed | PTH | this paper |
| AGC1-13 | SLC25A12 | NM_003705.5 | c.400C > T | p.Arg134* | not listed | PTH | [27] |
| AGC1-4 | SLC25A12 | NM_003705.5 | c.810_811insA | p.Leu271Thrfs*9 | not listed | PTH | this paper |
| AGC1-8, AGC1-9 | SLC25A12 | NM_003705.5 | c.1058G > A | p.Arg353Gln | DM | LPTH | [24] |
| AGC1-12 | SLC25A12 | NM_003705.5 | c.1295C > T | p.Ala432Val | DM | PTH | [26] |
| AGC1-10 | SLC25A12 | NM_003705.5 | c.1331C > T | p.Thr444Ile | not listed | LPTH | [17] |
| AGC1-6, AGC1-11 | SLC25A12 | NM_003705.5 | c.1335C > A | p.Asn445Lys | DM | LPTH | [25] |
| AGC1-12 | SLC25A12 | NM_003705.5 | c.1447-2_1447-1delAG | p.? | not listed | PTH | [26] |
| AGC1-1 | SLC25A12 | NM_003705.5 | c.1586-?_1835 + ?del (deletion exon 16 and 17, exact break-point not determined) | p.? | not listed | PTH | this paper |
| AGC1-2, AGC1-3 | SLC25A12 | NM_003705.5 | c.1618G > A | p.Asp540Asn | not listed | LPTH | this paper |
| AGC1-5 | SLC25A12 | NM_003705.5 | c.1747C > A | p.Arg583Arg | not listed | LPTH | this paper |
| AGC1-7 | SLC25A12 | NM_003705.5 | c.1769A > G | p.Gln590Arg | DM | LPTH | [16,23] |
| AGC2-7 | SLC25A13 | NM_014251.3 | c.173_174delTG | p.Val58Glyfs*24 | not listed | PTH | this paper |
| AGC2-1, AGC2-8 | SLC25A13 | NM_014251.3 | c.848 + 1G > T | p.? | not listed | PTH | this paper |
| AGC2-2, AGC2-3, AGC2-4 | SLC25A13 | NM_014251.3 | c.1078C > T | p.Arg360* | DM | PTH | [35] |
| AGC2-1 | SLC25A13 | NM_014251.3 | c.1173T > G | p.Tyr391* | not listed | LPTH | this paper |
| AGC2-2, AGC2-8 | SLC25A13 | NM_014251.3 | c.1307_1308delinsAA | p.Gly436Glu | not listed | LPTH | this paper |
| AGC2-5, AGC2-6 | SLC25A13 | NM_014251.3 | c.1629dup | p.Ile544Tyrfs*24 | not listed | PTH | this paper |
| AGC2-7 | SLC25A13 | NM_014251.3 | c.1813C > T | p.Arg605* | DM | PTH | [36] |
| #, sex | Start (y) | Duration (m) | Last FU (y) | Composition of KD | BHB (mmol/L) | General Effect | Effect on Epilepsy | Effect on EEG | Effect on Motor Abilities | Muscle Tone & Head Control | Effect on Communication | Effect on Thriving/Feeding | Effect on MRI | Effect on MRS | Effect on Lab |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AGC1-1, m | 1.75 | 30 | 4.25 | 3:1 → 4:1 | 2.5–3.5 | ++ | ++ | + | ++ | ↑ | + | + | ++ | + | LFT ↓ L (n) |
| AGC1-2, m | 5.67 | 18 | 7.67 | 2:1 | n/r | + | (+) | + | 0 | 0 | 0 | 0 | n/a | n/a | n/r |
| AGC1-3, m | 1.3 | 31 | 3.92 | 2:1 | 0.4–4.0 | + | + | + | ++ | ↑ | + | 0 | n/a | n/a | L ↓ |
| AGC1-4, m | 3 | 1 | 3.08 | 3:1 | n/r | 0 | 0 | n/r | 0 | 0 | 0 | 0 | n/a | n/a | n/r |
| AGC1-5, m | 2.5 | 12 | 4.5 | 3:1 | 3–4 | ++ | ++ | + | ++ | ↑ | + | + | n/a | n/a | n/r |
| AGC1-6, m | 1.17 | 3 | 1.42 | 3:1 | n/r | + | ++ | n/a | + | ↑ | n/r | n/r | n/a | n/a | n/r |
| AGC1-7, f | 6 | 20 | 7.67 | 3:1→4:1 | 5.5–6.8 | ++ | ++ | + | ++ | ↑ | + | n/r | ++ | + | 0 |
| AGC1-10, m | 1.75 | 4 | n/r | 4:1 | n/r | ++ | ++ | n/r | n/r | ↑ | n/r | n/r | n/a | n/a | n/r |
| MDH2-1, m | 3 | 18 | 4.5 | n/r | n/r | + | + | n/r | n/r | n/r | n/r | n/r | n/r | n/r | n/r |
| MDH2-2, m | 1.5 | 2 | 1.66 | n/r | n/r | + | + | n/r | n/r | n/r | n/r | n/r | n/r | n/r | n/r |
| MDH2-3, m | 3 | 54 | 7.5 | n/r | n/r | + | ? | n/r | n/r | n/r | n/r | n/r | n/r | n/r | n/r |
| MPC1-1, m | 1.25 | 36 | 4.25 | 2.5:1→4:1 | 2–3 | + | ++ | ++ | + | ↑ | + | n/r | n/a | n/a | L ↓ |
| MPC1-5, f | neo | 19 | † | n/r | <1.5; 0.3–3.7 (>16 m) | 0 | n/r | n/r | - | 0 | - | n/r | n/a | n/a | 0 |
| No KD | KD | No KD | KD | |||||
|---|---|---|---|---|---|---|---|---|
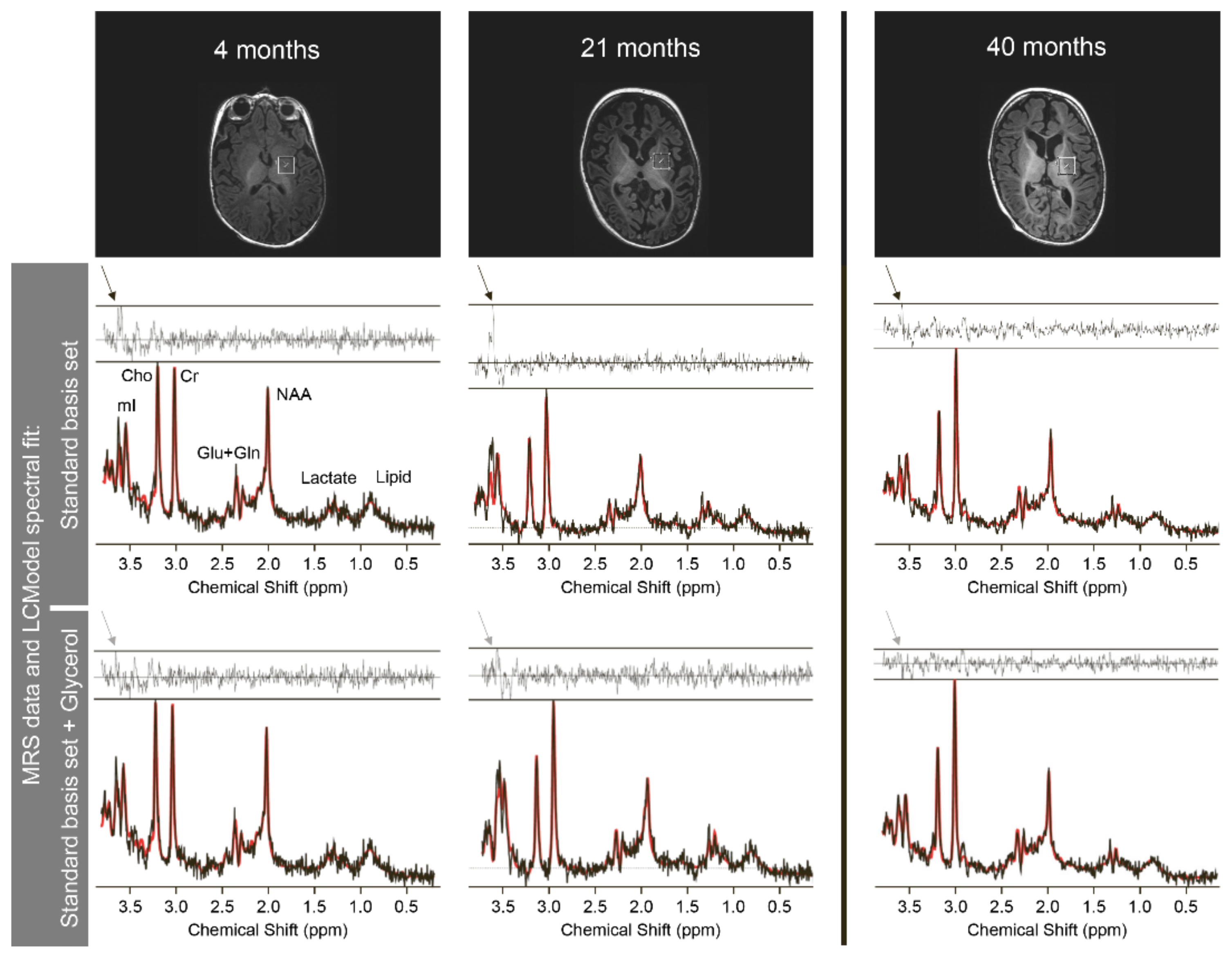
| Age | 4 m | 7 m | 21 m | 40 m | 4 m | 7 m | 21 m | 40 m |
| white matter concentrations | basal ganglia concentrations | |||||||
| Lac (mM) | 0.73 | 2.01 | 2.53 | 0.98 | 0.73 | 0 | 2.57 | 1.54 |
| mI (mM) | 6.42 | 6.14 | 10.1 | 4.66 | 4.7 | 10.67 | 6.81 | 4.84 |
| Asp (mM) * | 0 | 0 | 0 | 0.68 | 0 | 0.16 | 0.21 | 1.11 |
| NAA (mM) | 3.67 | 2.35 | 2.07 | 3.08 | 3.63 | 3.47 | 3.19 | 3.42 |
| white matter NAA/Cr-ratio | basal ganglia NAA/Cr-ratio | |||||||
| NAA/Cr | 0.45 | 0.46 | 0.32 | 0.38 | 0.7 | 0.34 | 0.37 | 0.4 |
| white matter concentrations | basal ganglia concentrations | |||||||
| Glyc (aU) * | 5.73 | 33.44 | 20.92 | 6.4 | 13.04 | 20.64 | 23.88 | 0 |
| Abbreviations: Lac = lactate, mI = myo-inositol, Asp = aspartate, NAA= N-acetyl-aspartate, Cr = creatinine, Glyc = glycerol, aU = arbitrary unit (scaling factor based on a phantom of known glycerol-3-phosphate concentration), m = month(s) * = no reference values available. Means and SD of mI/NAA-concentrations are age-dependent and were taken from Pouwels et al. [37] (cf. their tables 3 & 6) | ||||||||
| Color codes: | normal (for Lac <2mM; for mI: within mean ± 2.5 SD [37]) | |||||||
| increased (for Lac >2mM; for mI: above mean + 2.5 SD [37]) | ||||||||
| reduced (for NAA: below mean−2.5 SD [37]; NAA/Cr: <1) | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bölsterli, B.K.; Boltshauser, E.; Palmieri, L.; Spenger, J.; Brunner-Krainz, M.; Distelmaier, F.; Freisinger, P.; Geis, T.; Gropman, A.L.; Häberle, J.; et al. Ketogenic Diet Treatment of Defects in the Mitochondrial Malate Aspartate Shuttle and Pyruvate Carrier. Nutrients 2022, 14, 3605. https://doi.org/10.3390/nu14173605
Bölsterli BK, Boltshauser E, Palmieri L, Spenger J, Brunner-Krainz M, Distelmaier F, Freisinger P, Geis T, Gropman AL, Häberle J, et al. Ketogenic Diet Treatment of Defects in the Mitochondrial Malate Aspartate Shuttle and Pyruvate Carrier. Nutrients. 2022; 14(17):3605. https://doi.org/10.3390/nu14173605
Chicago/Turabian StyleBölsterli, Bigna K., Eugen Boltshauser, Luigi Palmieri, Johannes Spenger, Michaela Brunner-Krainz, Felix Distelmaier, Peter Freisinger, Tobias Geis, Andrea L. Gropman, Johannes Häberle, and et al. 2022. "Ketogenic Diet Treatment of Defects in the Mitochondrial Malate Aspartate Shuttle and Pyruvate Carrier" Nutrients 14, no. 17: 3605. https://doi.org/10.3390/nu14173605