The Effects of Endoplasmic-Reticulum-Resident Selenoproteins in a Nonalcoholic Fatty Liver Disease Pig Model Induced by a High-Fat Diet



Abstract
:1. Introduction
2. Methods
2.1. Treatment of Experimental Animals
2.2. Growth Performance
2.3. Serum Enzyme Activity Assays
2.4. Determination of Liver Biochemical Indexes
2.5. Histological Examinations
2.6. Ultrastructural Observation
2.7. Gene Expression by Real-Time PCR
2.8. Measurement of Apoptosis
2.9. Statistical Analysis
3. Results
3.1. Growth Performance
3.2. Serum ALT and AST Analysis
3.3. Antioxidant Enzyme Activity
3.4. Concentrations of TC and TG
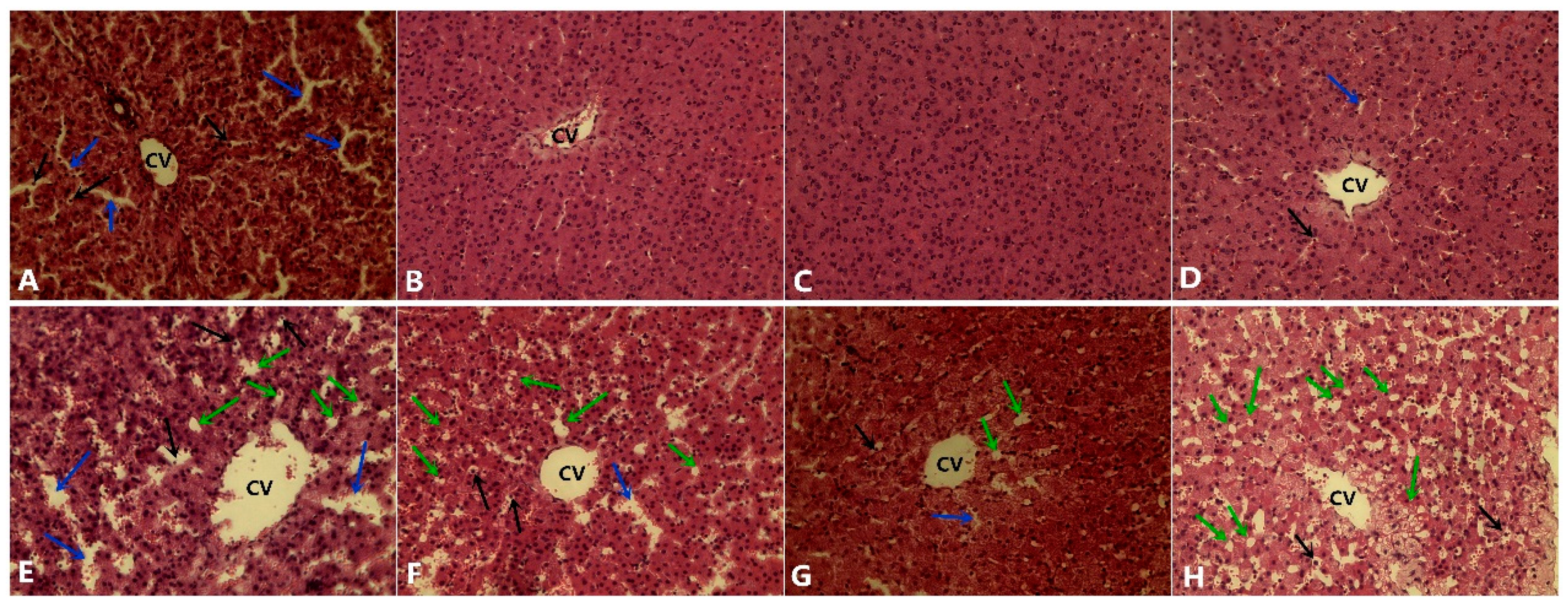
3.5. Histological Analysis
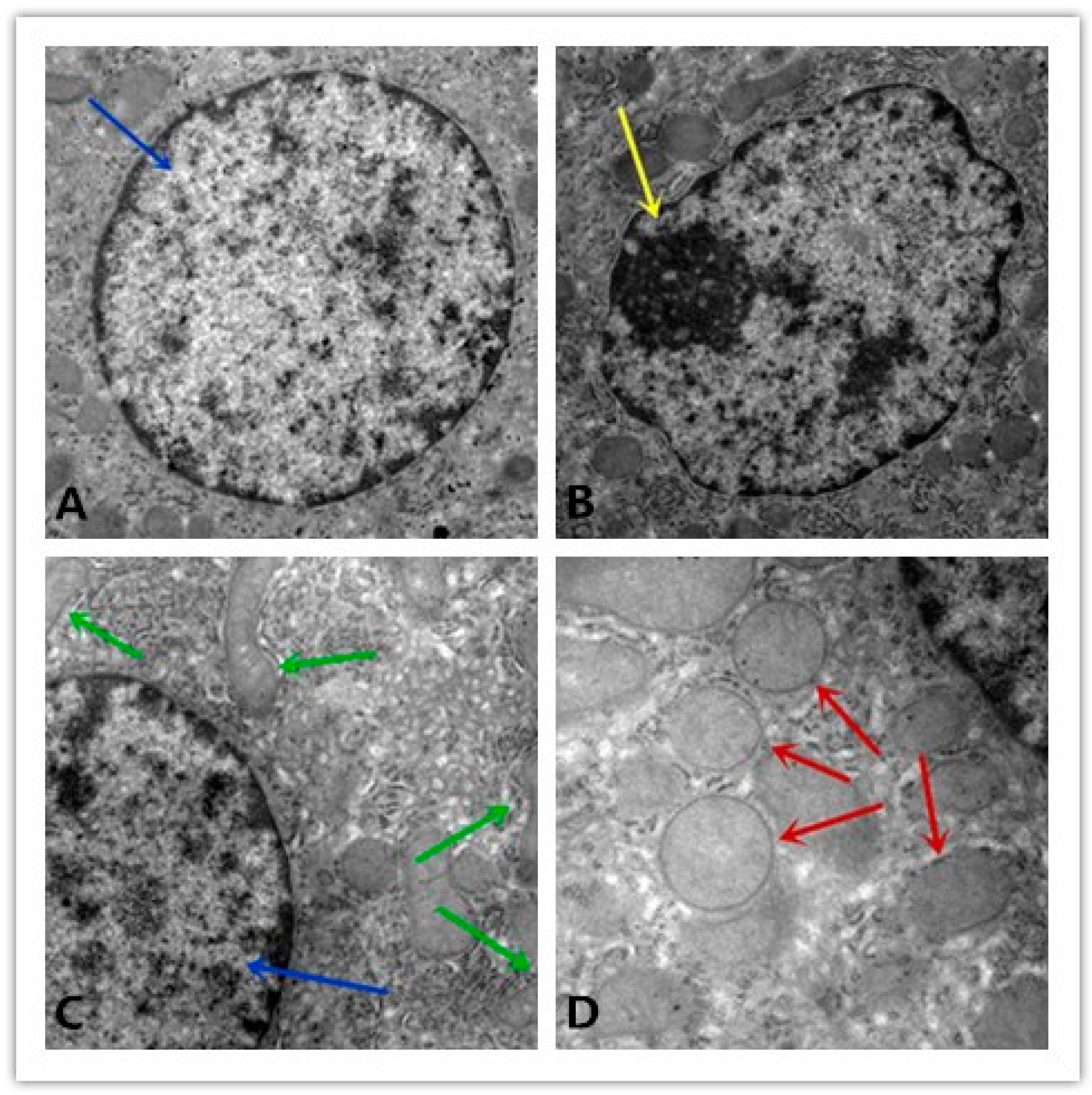
3.6. Ultrastructural Alterations
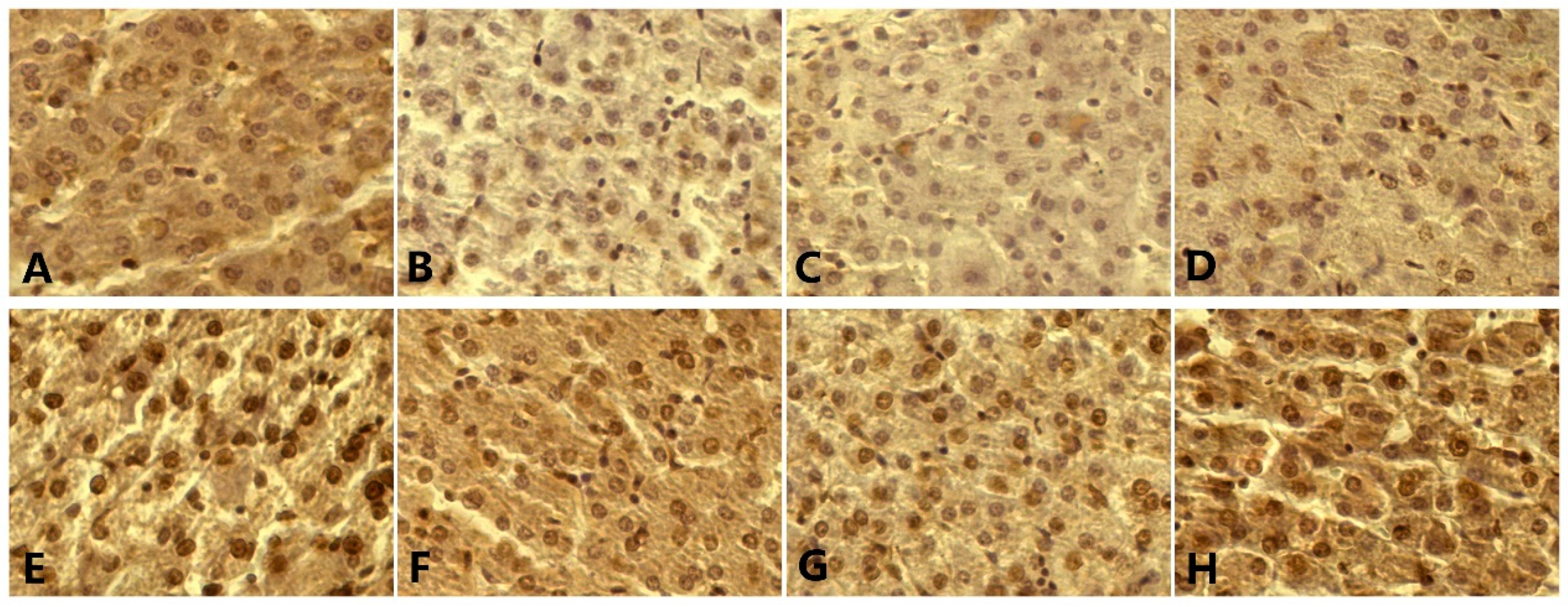
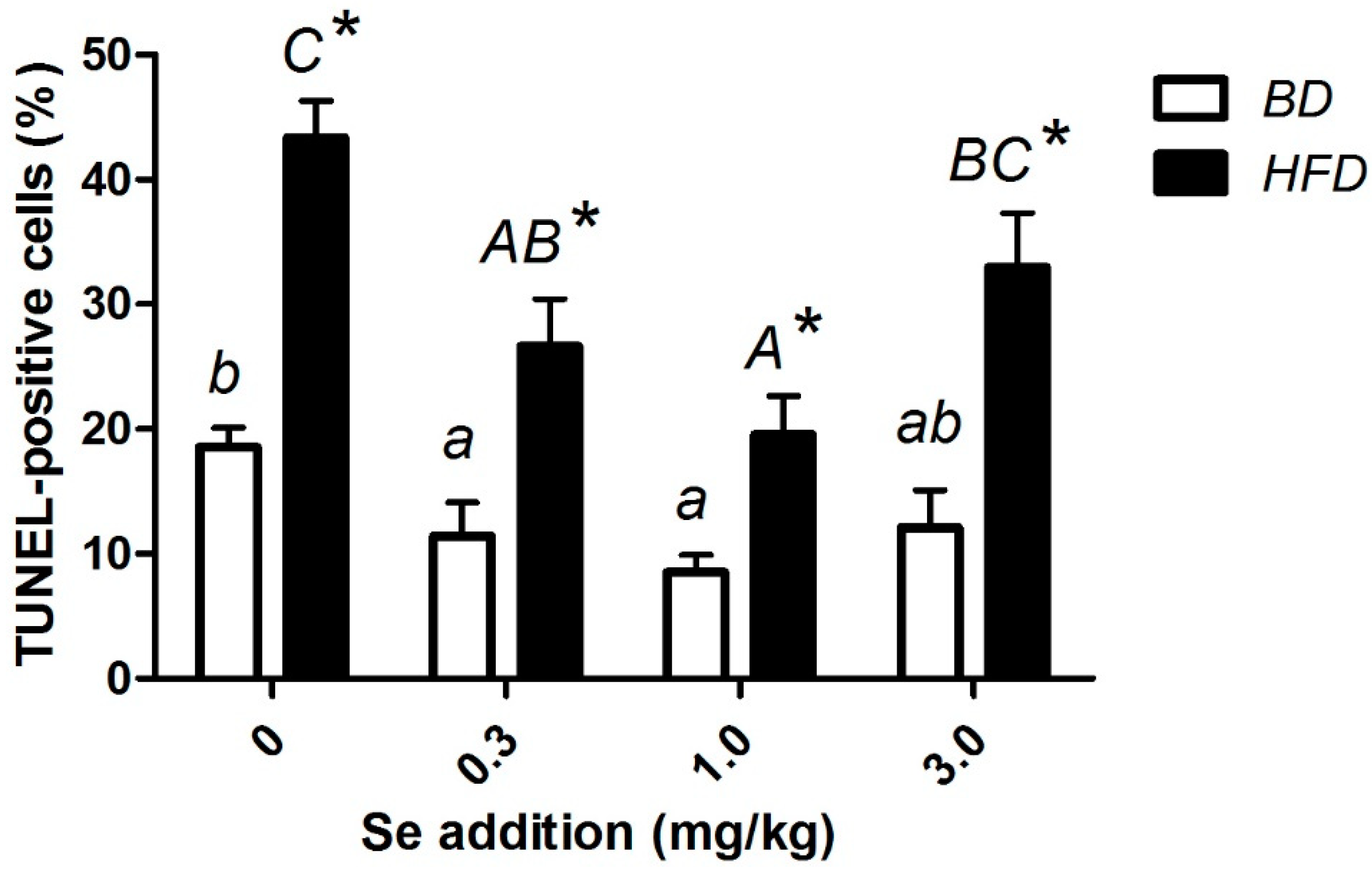
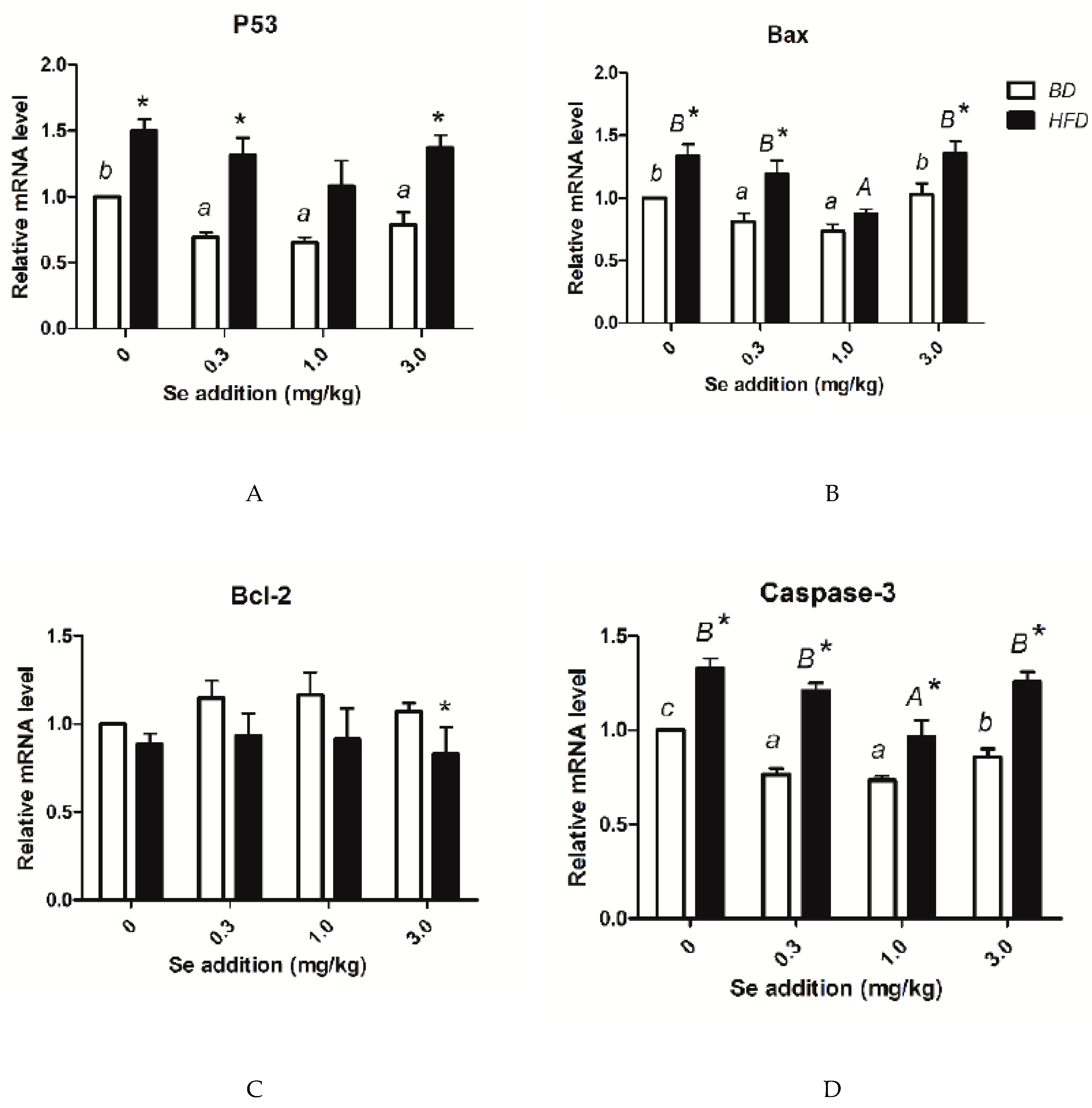
3.7. p53, Bax, Bcl-2, Caspase-3 Level, and Apoptosis
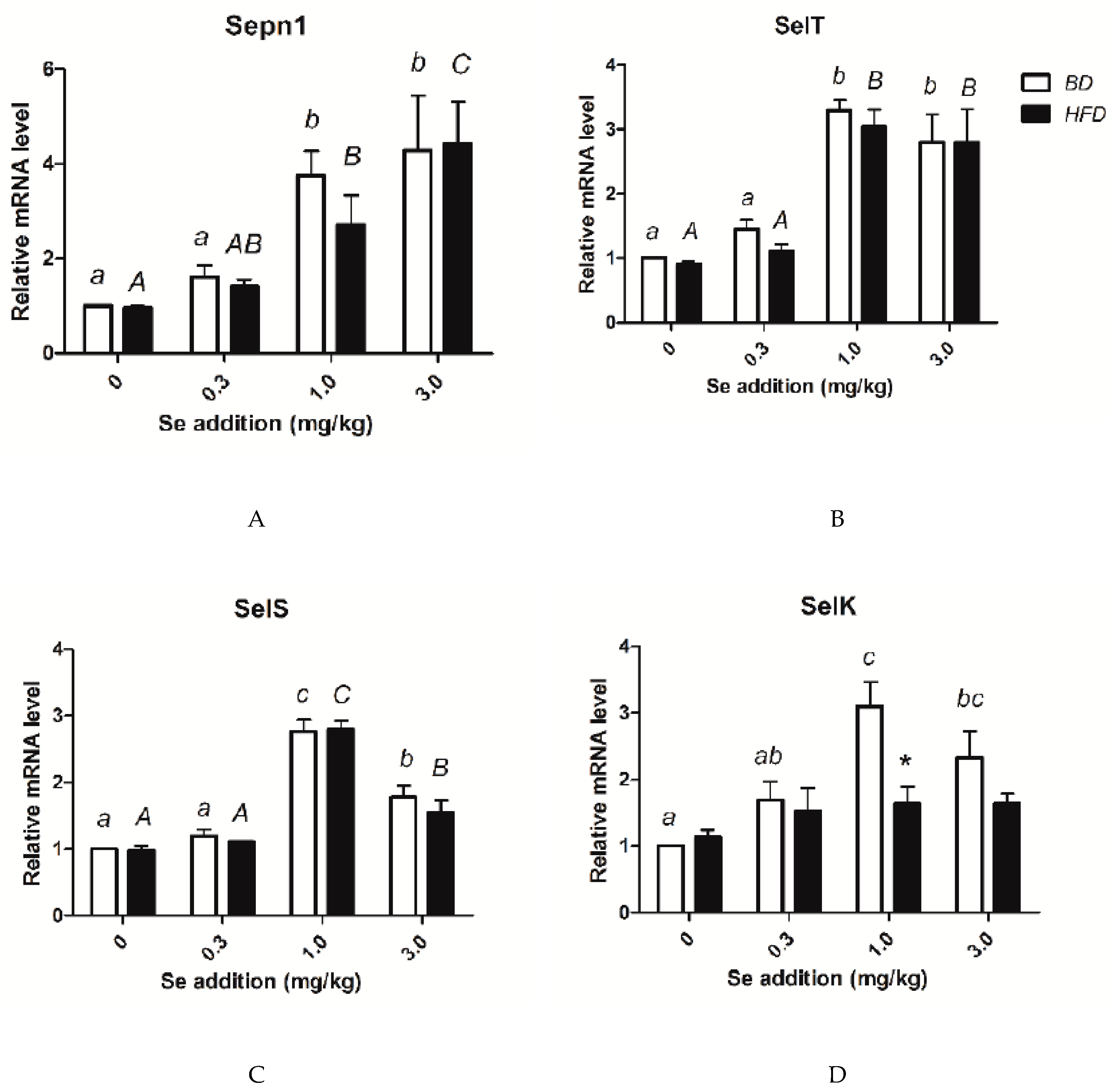
3.8. Relative mRNA Amounts of Selenoprotein Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ong, J.P.; Younossi, Z.M. Epidemiology and natural history of NAFLD and NASH. Clin. Liver Dis. 2007, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Yeh, M.M.; Brunt, E.M. Pathological features of fatty liver disease. Gastroenterology 2014, 147, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Styskal, J.; Van Remmen, H.; Richardson, A.; Salmon, A.B. Oxidative stress and diabetes: What can we learn about insulin resistance from antioxidant mutant mouse models? Free Radic. Biol. Med. 2012, 52, 46–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Yue, W.; Zhang, C.; Ren, Y.; Zhu, X.; Wang, Q.; Shi, L.; Lei, F. Effects of maternal and dietary selenium (Se-enriched yeast) on oxidative status in testis and apoptosis of germ cells during spermatogenesis of their offspring in goats. Anim. Reprod. Sci. 2010, 119, 212–218. [Google Scholar] [CrossRef]
- Ranawat, P.; Bansal, M.P. Apoptosis induced by modulation in selenium status involves p38 MAPK and ROS: Implications in spermatogenesis. Mol. Cell. Biochem. 2009, 330, 83–95. [Google Scholar] [CrossRef]
- Franco, R.; Sanchez-Olea, R.; Reyes-Reyes, E.M.; Panayiotidis, M.I. Environmental toxicity, oxidative stress and apoptosis: Menage a Trois. Mutat. Res. 2008, 674, 3–22. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and Fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Ribeiro, P.S.; Cortez-Pinto, H.; Sola, S.; Castro, R.E.; Ramalho, R.M.; Baptista, A.; Moura, M.C.; Camilo, M.E.; Rodrigues, C.M. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappa B in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am. J. Gastroenterol. 2004, 99, 1708–1717. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Y.; Luo, K.; Liu, Y.; Zhou, M.; Yan, S.; Shi, H.; Cai, Y. The protective effects of selenium on cadmium-induced oxidative stress and apoptosis via mitochondria pathway in mice kidney. Food Chem. Toxicol. 2013, 58, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Spallholz, J.E.; Hoffman, D.J. Selenium toxicity: Cause and effects in aquatic birds. Aquat. Toxicol. 2002, 57, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Andrade, K.Q.; Moura, F.A.; Dos, S.J.; de Araujo, O.R.; de Farias, S.J.; Goulart, M.O. Oxidative stress and inflammation in hepatic diseases: Therapeutic possibilities of N-Acetylcysteine. Int. J. Mol. Sci. 2015, 16, 30269–30308. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. The argument for increasing selenium intake. Proc. Nutr. Soc. 2002, 61, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, T.C. Selenium biochemistry: Mammalian selenoenzymes. Ann. N. Y. Acad. Sci. 2000, 899, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Driscoll, D.M.; Copeland, P.R. Mechanism and regulation of selenoprotein synthesis. Annu. Rev. Nutr. 2003, 23, 17–40. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Gao, Y.; Walder, K.; Collier, G.R.; Skelton, J.; Kissebah, A.H. SEPS1 protects RAW264.7 cells from pharmacological ER stress agent-induced apoptosis. Biochem. Biophys. Res. Commun. 2007, 354, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Du, S.; Zhou, J.; Jia, Y.; Huang, K. SelK is a novel ER stress-regulated protein and protects HepG2 cells from ER stress agent-induced apoptosis. Arch. Biochem. Biophys. 2010, 502, 137–143. [Google Scholar] [CrossRef]
- Dikiy, A.; Novoselov, S.V.; Fomenko, D.E.; Sengupta, A.; Carlson, B.A.; Cerny, R.L.; Ginalski, K.; Grishin, N.V.; Hatfield, D.L.; Gladyshev, V.N. SelT, SelW, SelH, and Rdx12: Genomics and molecular insights into the functions of selenoproteins of a novel thioredoxin-like family. Biochemistry 2007, 46, 6871–6882. [Google Scholar] [CrossRef]
- Petit, N.; Lescure, A.; Rederstorff, M.; Krol, A.; Moghadaszadeh, B.; Wewer, U.M.; Guicheney, P. Selenoprotein N: An endoplasmic reticulum glycoprotein with an early developmental expression pattern. Hum. Mol. Genet. 2003, 12, 1045–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Qiu, F.; Zhou, H.; Peng, Y.; Hao, W.; Xu, J.; Yuan, J.; Wang, S.; Qiang, B.; Xu, C. Identification and characterization of selenoprotein K: An antioxidant in cardiomyocytes. FEBS Lett. 2006, 580, 5189–5197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grumolato, L.; Ghzili, H.; Montero-Hadjadje M Gasman, S.; Lesage, J.; Tanguy, Y.; Galas, L.; Ait-Ali, D.; Leprince, J.; Guérineau, N.C. Selenoprotein T is a PACAP-regulated gene involved in intracellular Ca(2+) mobilization and neuroendocrine secretion. FASEB J. 2008, 22, 1756–1768. [Google Scholar] [CrossRef] [PubMed]
- Ezaki, O. The insulin-like effects of selenate in rat adipocytes. J. Biol. Chem. 1990, 265, 1124–1128. [Google Scholar] [PubMed]
- Mueller, A.S.; Pallauf, J. Compendium of the antidiabetic effects of supranutritional selenate doses. In vivo and in vitro investigations with type II diabetic db/db mice. J. Nutr. Biochem. 2006, 17, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, H. Interference of selenium and selenoproteins with the insulin-regulated carbohydrate and lipid metabolism. Free Radic. Biol. Med. 2013, 65, 1538–1547. [Google Scholar] [CrossRef]
- Choi, H.Y.; Hwang, S.Y.; Lee, C.H.; Hong, H.C.; Yang, S.J.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; et al. Increased selenoprotein p levels in subjects with visceral obesity and nonalcoholic fatty liver disease. Diabetes Metab. J. 2013, 37, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Baumgardner, J.N.; Shankar, K.; Hennings, L.; Badger, T.M.; Ronis, M.J. A new model for nonalcoholic steatohepatitis in the rat utilizing total enteral nutrition to overfeed a high-polyunsaturated fat diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, 27–38. [Google Scholar] [CrossRef]
- Lieber, C.S.; Leo, M.A.; Mak, K.M.; Xu, Y.; Cao, Q.; Ren, C.; Ponomarenko, A.; DeCarli, L.M. Model of nonalcoholic steatohepatitis. Am. J. Clin. Nutr. 2004, 79, 502–509. [Google Scholar] [CrossRef] [Green Version]
- Patterson, J.K.; Lei, X.G.; Miller, D.D. The pig as an experimental model for elucidating the mechanisms governing dietary influence on mineral absorption. Exp. Biol. Med. 2008, 233, 651–664. [Google Scholar] [CrossRef]
- Reitman, S.; Frankel, S. A colorimetric method for the determination of serum glutamic oxalacetic and glutamic pyruvic transaminases. Am. J. Clin. Pathol. 1957, 28, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; RANDALL, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US) Panel on Dietary Antioxidants and Related Compounds. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids; National Academies Press: Washington, DC, USA, 2000. [Google Scholar]
- Bailey, R.L.; Gahche, J.J.; Lentino, C.V.; Dwyer, J.T.; Engel, J.S.; Thomas, P.R.; Betz, J.M.; Sempos, C.T.; Picciano, M.F. Dietary supplement use in the United States, 2003–2006. J. Nutr. 2011, 141, 261–266. [Google Scholar] [CrossRef] [Green Version]
- Burk, R.F.; Norsworthy, B.K.; Hill, K.E.; Motley, A.K.; Byrne, D.W. Effects of chemical form of selenium on plasma biomarkers in a high-dose human supplementation trial. Cancer Epidemiol. Biomark. Prev. 2006, 15, 804–810. [Google Scholar] [CrossRef] [Green Version]
- Adams, L.A.; Angulo, P. Recent concepts in non-alcoholic fatty liver disease. Diabet. Med. 2005, 22, 1129–1133. [Google Scholar] [CrossRef]
- Mulligan, F.J.; Doherty, M.L. Production diseases of the transition cow. Vet. J. 2008, 176, 3–9. [Google Scholar] [CrossRef]
- Pan, D.; Huang, H. Hair selenium levels in hepatic steatosis patients. Biol. Trace Elem. Res. 2013, 152, 305–309. [Google Scholar] [CrossRef]
- Pinto, A.; Juniper, D.T.; Sanil, M.; Morgan, L.; Clark, L.; Sies, H.; Rayman, M.P.; Steinbrenner, H. Supranutritional selenium induces alterations in molecular targets related to energy metabolism in skeletal muscle and visceral adipose tissue of pigs. J. Inorg. Biochem. 2012, 114, 47–54. [Google Scholar] [CrossRef]
- Zeng, M.S.; Li, X.; Liu, Y.; Zhao, H.; Zhou, J.C.; Li, K.; Huang, J.Q.; Sun, L.H.; Tang, J.Y.; Xia, X.J.; et al. A high-selenium diet induces insulin resistance in gestating rats and their offspring. Free Radic. Biol. Med. 2012, 52, 1335–1342. [Google Scholar] [CrossRef] [Green Version]
- Utzschneider, K.M.; Kahn, S.E. Review: The role of insulin resistance in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ausman, L.M.; Russell, R.M.; Greenberg, A.S.; Wang, X.D. Increased apoptosis in high-fat diet-induced nonalcoholic steatohepatitis in rats is associated with c-Jun NH2-terminal kinase activation and elevated proapoptotic Bax. J. Nutr. 2008, 138, 1866–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Sekiya, M.; Najima, Y.; Okazaki, S.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Inoue, N.; et al. P53 involvement in the pathogenesis of fatty liver disease. J. Biol. Chem. 2004, 279, 20571–20575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, G.C.; Larter, C.Z.; Hou, J.Y.; Zhang, R.H.; Yeh, M.M.; Williams, J.; dela Pena, A.; Francisco, R.; Osvath, S.R.; Brooling, J.; et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J. Gastroenterol. Hepatol. 2009, 24, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Panasiuk, A.; Dzieciol, J.; Panasiuk, B.; Prokopowicz, D. Expression of p53, Bax and Bcl-2 proteins in hepatocytes in non-alcoholic fatty liver disease. World J. Gastroenterol. 2006, 12, 6198–6202. [Google Scholar] [CrossRef] [PubMed]
- Pariza, M.; Park, Y.; Cook, M.; Albright, K.; Liu, W. Conjugated linoleic acid (CLA) reduces body fat. FASEB J. 1996, 10, 3227. [Google Scholar]
- Tsuboyama-Kasaoka, N.; Takahashi, M.; Tanemura, K.; Kim, H.J.; Tange, T.; Okuyama, H.; Kasai, M.; Ikemoto, S.; Ezaki, O. Conjugated linoleic acid supplementation reduces adipose tissue by apoptosis and develops lipodystrophy in mice. Diabetes 2000, 49, 1534–1542. [Google Scholar] [CrossRef] [Green Version]
- Korsmeyer, S.J. Bcl-2 initiates a new category of oncogenes - regulators of cell-death. Blood 1992, 80, 879–886. [Google Scholar] [CrossRef] [Green Version]
- Reed, J.C. Regulation of apoptosis by bcl-2 family proteins and its role in cancer and chemoresistance. Curr. Opin. Oncol. 1995, 7, 541–546. [Google Scholar] [CrossRef]
- Miyashita, T.; Krajewski, S.; Krajewska, M.; Wang, H.G.; Lin, H.K.; Liebermann, D.A.; Hoffman, B.; Reed, J.C. Tumor-suppressor p53 is a regulator of Bcl-2 and bax gene-expression in-vitro and in-vivo. Oncogene 1994, 9, 1799–1805. [Google Scholar]
- Wolter, K.G.; Hsu, Y.T.; Smith, C.L.; Nechushtan, A.; Xi, X.G.; Youle, R.J. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell. Biol. 1997, 139, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Broekemeier, K.M.; Pfeiffer, D.R. Recent progress on regulation of the mitochondrial permeability transition pore—A cyclosporine-sensitive pore in the inner mitochondrial-membrane. J. Bioenerg. Biomembr. 1994, 26, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Kluck, R.M.; BossyWetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Liu, X.S.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T.I.; Jones, D.P.; Wang, X. Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef]
- Zimmermann, K.C.; Green, D.R. How cells die: Apoptosis pathways. J. Allergy Clin. Immunol. 2001, 108, S99–S103. [Google Scholar] [CrossRef]
- Porter, A.G.; Janicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.Y.; Leopold, J.A.; Loscalzo, J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 2009, 284, 11913–11921. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.Q.; Li, D.L.; Zhao, H.; Sun, L.H.; Xia, X.J.; Wang, K.N.; Luo, X.; Lei, X.G. The selenium deficiency disease exudative diathesis in chicks is associated with downregulation of seven common selenoprotein genes in liver and muscle. J. Nutr. 2011, 141, 1605–1610. [Google Scholar] [CrossRef]
- Yao, H.D.; Wu, Q.; Zhang, Z.W.; Zhang, J.L.; Li, S.; Huang, J.Q.; Ren, F.Z.; Xu, S.W.; Wang, X.L.; Lei, X.G. Gene expression of endoplasmic reticulum resident selenoproteins correlates with apoptosis in various muscles of se-deficient chicks. J. Nutr. 2013, 143, 613–619. [Google Scholar] [CrossRef]
- Morozova, N.; Forry, E.P.; Shahid E Zavacki, A.M.; Harney, J.W.; Kraytsberg, Y.; Berry, M.J. Antioxidant function of a novel selenoprotein in Drosophila melanogaster. Genes Cells 2003, 8, 963–971. [Google Scholar] [CrossRef]
- Meng, X.L.; Zhang, H.L.; Feng, L.L.; Chen, M.L.; Liu, Y.Y.; Yu, X.; Huan, F.N.; Lu, J.; Wang, D.; Liu, H.S.; et al. Selenoprotein SelK increases the secretion of insulin from MIN6 b cells. RSC Adv. 2017, 7, 35038–35047. [Google Scholar] [CrossRef] [Green Version]
- Moghadaszadeh, B.; Petit, N.; Jaillard, C.; Brockington, M.; Quijano Roy, S.; Merlini, L.; Romero, N.; Estournet, B.; Desguerre, I.; Chaigne, D.; et al. Mutations in SEPN1 cause congenital muscular dystrophy with spinal rigidity and restrictive respiratory syndrome. Nat. Genet. 2001, 29, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.W.; Arner, E.S.J.; Holmgren, A. Structure and mechanism of mammalian thioredoxin reductase: The active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Natl. Acad. Sci. USA 2000, 97, 5854–5859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korotkov, K.V.; Kumaraswamy, E.; Zhou, Y.; Hatfield, D.L.; Gladyshev, V.N. Association between the 15-kDa selenoprotein and UDP-glucose: Glycoprotein glucosyltransferase in the endoplasmic reticulum of mammalian cells. J. Biol. Chem. 2001, 276, 15330–15336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walder, K.; Kantham, L.; McMillan, J.S.; Trevaskis, J.; Kerr, L.; de Silva, A.; Sunderland, T.; Godde, N.; Gao, Y.; Bishara, N.; et al. Tanis: A link between type 2 diabetes and inflammation? Diabetes 2002, 51, 1859–1866. [Google Scholar] [CrossRef] [Green Version]
- Pahl, H.L.; Baeuerle, P.A. Endoplasmic-reticulum-induced signal transduction and gene expression. Trends Cell Biol. 1997, 7, 50–55. [Google Scholar] [CrossRef]
- Gao, Y.; Feng, H.C.; Walder K Bolton, K.; Sunderland, T.; Bishara, N.; Quick, M.; Kantham, L.; Collier, G.R. Regulation of the selenoprotein Sels by glucose deprivation and endoplasmic reticulum stress - Sels is a novel glucose-regulated protein. FEBS Lett. 2004, 563, 185–190. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BD(g/kg) | HFD(g/kg) | |||||
|---|---|---|---|---|---|---|
| Ingredients | 10–30 kg | 30–60 kg | 60–110 kg | 10–30 kg | 30–60 kg | 60–110 kg |
| Corn grain | 721.59 | 753.64 | 802.36 | 670.88 | 655.14 | 654.9 |
| Soybean meal (43.00% CP) | 220 | 195 | 150 | 231 | 214 | 178 |
| Wheat bran | 20 | 20 | 20 | 20 | 20 | 20 |
| Lard | 0 | 0 | 0 | 30 | 70 | 110 |
| Fish oil | 0 | 0 | 0 | 10 | 10 | 10 |
| L-lysine | 7.24 | 4.11 | 3.9 | 6.95 | 3.62 | 3.15 |
| DL-methionine | 0.86 | 0.15 | 0.04 | 0.89 | 0.24 | 0.12 |
| L-threonine | 0.86 | 0 | 0 | 0.85 | 0 | 0 |
| L-tryptophan | 0.15 | 0 | 0.1 | 0.13 | 0 | 0.03 |
| Salt | 3 | 3 | 3 | 3 | 3 | 3 |
| Limestone | 9.8 | 10.6 | 9.6 | 9.8 | 10 | 9.3 |
| Dicalcium phosphate | 10.5 | 7.5 | 5 | 10.5 | 8 | 5.5 |
| Premix 1 | 6 | 6 | 6 | 6 | 6 | 6 |
| Calculated analysis % | ||||||
| Digestible energy kcal/kg | 3296 | 3294 | 3303 | 3492 | 3688 | 3929 |
| Crude protein | 16.1 | 15 | 13.4 | 16.2 | 15 | 13.4 |
| Crude fat | 3 | 3.1 | 3.2 | 6.8 | 10.7 | 14.6 |
| Crude fiber | 3.1 | 3 | 2.8 | 3 | 2.9 | 2.7 |
| ADF * | 4.14 | 4.04 | 3.82 | 4.08 | 3.90 | 3.61 |
| NDF * | 9.94 | 9.94 | 9.85 | 9.60 | 9.25 | 8.82 |
| Calcium | 0.7 | 0.65 | 0.55 | 0.7 | 0.65 | 0.55 |
| Available phosphorus | 0.3 | 0.25 | 0.21 | 0.3 | 0.25 | 0.21 |
| Lysine | 1.15 | 0.92 | 0.8 | 1.15 | 0.92 | 0.8 |
| Methionine | 0.34 | 0.26 | 0.23 | 0.34 | 0.26 | 0.23 |
| Methionine + Cysteine | 0.63 | 0.54 | 0.49 | 0.63 | 0.54 | 0.48 |
| Threonine | 0.68 | 0.56 | 0.5 | 0.68 | 0.56 | 0.5 |
| Tryptophan | 0.19 | 0.16 | 0.15 | 0.19 | 0.17 | 0.15 |
| Gene | Accession Number | Primer sequence 5′→3′ |
|---|---|---|
| P53 | NM_213824.3 | F: GTCACGAACTGGCTGGATG |
| R: GAAGGGACAAAGGACGACAG | ||
| Bax | XM_003355975.2 | F: TGCTTCAGGGTTTCATCCAG |
| R: GACACTCGCTCAACTTCTTGG | ||
| Bcl-2 | XM_003474076.2 | F: CCTTCTCCGTGGTCATCCT |
| R: AAGTCTGAGCGTCCTGTTCC | ||
| Caspase-3 | NM_214131.1 | F: GTGGGATTGAGACGGACAG |
| R: TTCGCCAGGAATAGTAACCAG | ||
| Selk | DQ372075 | F: CAGGAAACCCCCCTAGAAGAA |
| R: CTCATCCACCGGCCATTG | ||
| Sepn1 | EF113595 | F: ACCTGGTCCCTGGTGAAAGAG |
| R: AGGCCAGCCAGCTTCTTGT | ||
| Sels | GU983865 | F: ACAGGAGGCTTTAGCAGCAG |
| R: CGCTGTCCCATCTTTCAATC | ||
| Selt | NM_001163408 | F: CGCTGCTCAAATTCCAGATA |
| R: CTCTCCTTCAATGCGGATGT | ||
| β-actin | AY550069 | F: CCCAAAGCCAACCGTGAGAA |
| R: CCACGTACATGGCTGGGGTG |
| Items | Growth Performance | ||||
|---|---|---|---|---|---|
| Fat | Se | ADG | ADFI | Carcass Weight | Carcass Length |
| (mg/kg) | (kg/d) | (kg/d) | (kg/d) | (cm) | |
| BD | 0 | 0.73 ± 0.01 c | 1.90 ± 0.21 | 63.66 ± 7.52 | 66.67 ± 2.52 |
| 0.3 | 0.70 ± 0.02 b | 1.85 ± 0.15 | 61.67 ± 6.93 | 70.67 ± 0.58 | |
| 1 | 0.73 ± 0.07 c | 1.93 ± 0.10 | 62.50 ± 5.07 | 68.33 ± 2.08 | |
| 3 | 0.67 ± 0.04 a | 1.84 ± 0.18 | 59.33 ± 5.01 | 70.33 ± 3.61 | |
| HFD | 0 | 0.87 ± 0.11 B* | 2.17 ± 0.12 * | 78.83 ± 7.37 B* | 83.00 ± 6.25 B* |
| 0.3 | 0.86 ± 0.06 B* | 2.14 ± 0.09* | 77.33 ± 1.04 B* | 73.00 ± 1.00 A | |
| 1 | 0.82 ± 0.03 A* | 2.00 ± 0.11 | 74.33 ± 4.65 B* | 72.33 ± 0.58 A | |
| 3 | 0.83 ± 0.08 A* | 2.06 ± 0.10 * | 72.00 ± 2.78 A* | 73.23 ± 5.77 A | |
| p-value | |||||
| Fat | <0.01 | <0.01 | <0.01 | <0.01 | |
| Se | 0.25 | 0.47 | 0.38 | 0.06 | |
| Fat × Se | 0.27 | 0.28 | 0.91 | <0.01 | |
| Items | Aminotransferase | ||
|---|---|---|---|
| Fat | Se (mg/kg) | ALT (U/L) | AST (U/L) |
| BD | 0 | 58.16 ± 11.23 | 129.32 ± 11.31 c |
| 0.3 | 52.76 ± 5.43 | 98.10 ± 6.22 a | |
| 1.0 | 48.08 ± 5.88 | 88.70 ± 3.98 a | |
| 3.0 | 53.58 ± 9.26 | 115.14 ± 10.70 b | |
| HFD | 0 | 96.48 ± 10.39 C* | 188.66 ± 8.35 C* |
| 0.3 | 83.18 ± 7.80 B* | 160.90 ± 8.64 B* | |
| 1.0 | 70.42 ± 3.90 A* | 137.20 ± 7.53 A* | |
| 3.0 | 93.04 ± 4.28 C* | 169.54 ± 23.92 C* | |
| p-value | |||
| Fat | <0.01 | <0.01 | |
| Se | <0.01 | <0.01 | |
| Fat × Se | 0.07 | 0.01 | |
| Items | Antioxidant Index | ||
|---|---|---|---|
| Fat | Se (mg/kg) | GSH-Px (U/mg protein) | MDA (nmol/mg protein) |
| BD | 0 | 23.24 ± 2.66 a | 2.51 ± 0.41 bc |
| 0.3 | 128.11 ± 5.61 c | 2.11 ± 0.26 ab | |
| 1.0 | 148.09 ± 8.20 d | 1.94 ± 0.26 a | |
| 3.0 | 100.66 ± 6.04 b | 2.64 ± 0.35 c | |
| HFD | 0 | 22.71 ± 2.26 A | 3.96 ± 0.70 B* |
| 0.3 | 85.03 ± 3.25 C* | 3.68 ± 0.20 B* | |
| 1.0 | 117.14 ± 5.19 D* | 2.58 ± 0.34 A* | |
| 3.0 | 64.14 ± 4.24 B* | 4.26 ± 1.10 B* | |
| p-value | |||
| Fat | <0.01 | <0.01 | |
| Se | <0.01 | <0.01 | |
| Fat × Se | <0.01 | 0.16 | |
| Items | Antioxidant Index | ||
|---|---|---|---|
| Fat | Se (mg/kg) | TC (mmol/L) | TG (mmol/L) |
| BD | 0 | 1.13 ± 0.09 b | 0.6 ± 0.02 a |
| 0.3 | 1.21 ± 0.07 c | 0.68 ± 0.02 b | |
| 1.0 | 1.07 ± 0.04 a | 0.68 ± 0.02 b | |
| 3.0 | 1.51 ± 0.07 d | 0.94 ± 0.03 c | |
| HFD | 0 | 2.07 ± 0.08 C* | 1.26 ± 0.03 C* |
| 0.3 | 1.85 ± 0.09 B* | 1.08 ± 0.01 B* | |
| 1.0 | 1.75 ± 0.04 A* | 0.90 ± 0.02 A* | |
| 3.0 | 1.83 ± 0.05 B* | 1.32 ± 0.03 C* | |
| p-value | |||
| Fat | <0.01 | <0.01 | |
| Se | <0.01 | <0.01 | |
| Fat × Se | <0.01 | <0.01 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Lu, Z.; He, M.; Shi, B.; Lei, X.; Shan, A. The Effects of Endoplasmic-Reticulum-Resident Selenoproteins in a Nonalcoholic Fatty Liver Disease Pig Model Induced by a High-Fat Diet. Nutrients 2020, 12, 692. https://doi.org/10.3390/nu12030692
Wang P, Lu Z, He M, Shi B, Lei X, Shan A. The Effects of Endoplasmic-Reticulum-Resident Selenoproteins in a Nonalcoholic Fatty Liver Disease Pig Model Induced by a High-Fat Diet. Nutrients. 2020; 12(3):692. https://doi.org/10.3390/nu12030692
Chicago/Turabian StyleWang, Pengzu, Zhuang Lu, Meng He, Baoming Shi, Xingen Lei, and Anshan Shan. 2020. "The Effects of Endoplasmic-Reticulum-Resident Selenoproteins in a Nonalcoholic Fatty Liver Disease Pig Model Induced by a High-Fat Diet" Nutrients 12, no. 3: 692. https://doi.org/10.3390/nu12030692