
In Situ Monitoring of Non-Thermal Plasma Cleaning of Surfactant Encapsulated Nanoparticles
 and
and
Abstract
:1. Introduction
2. Materials and Methods
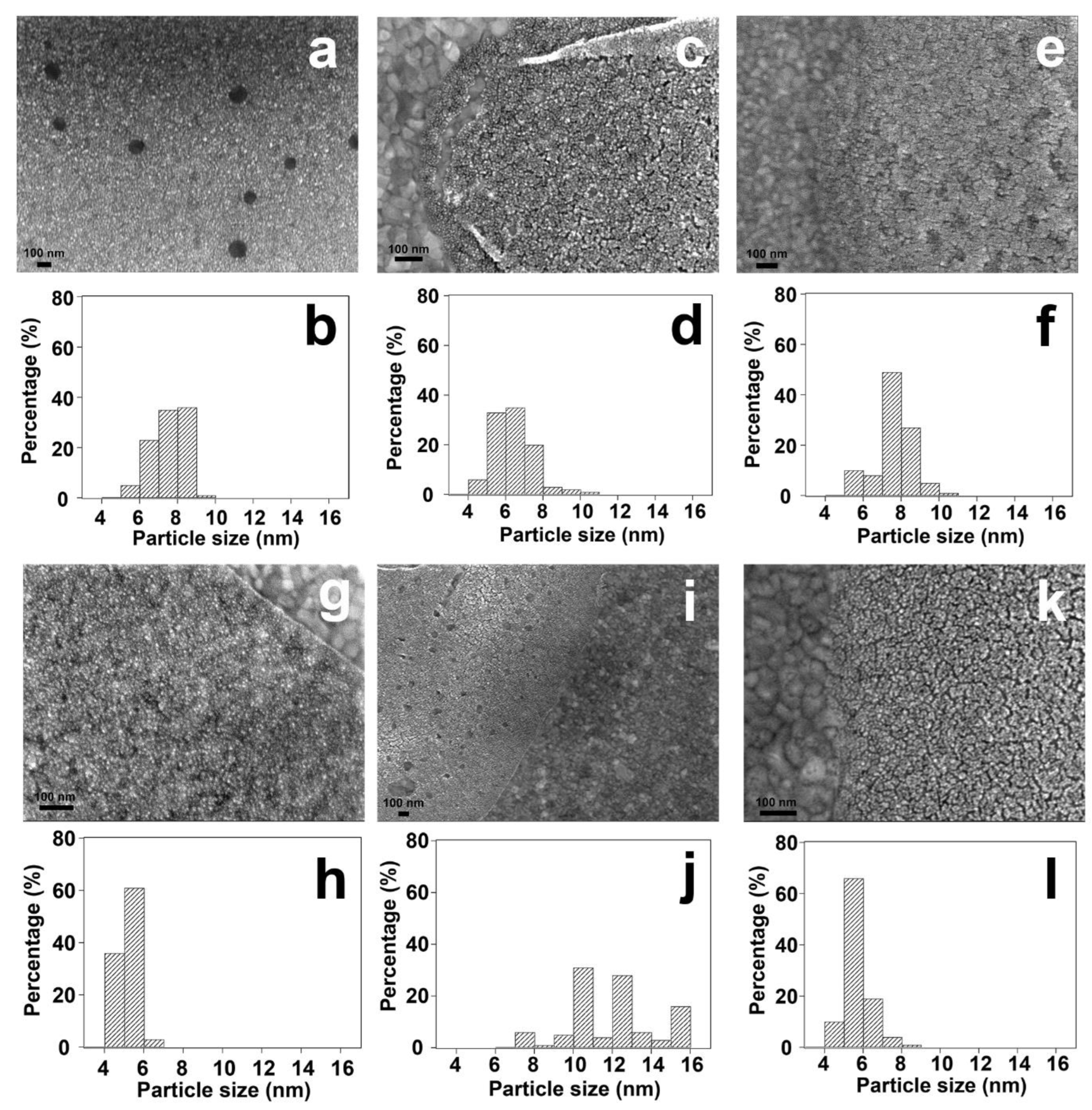
2.1. Materials Synthesis
2.2. Structural Characterization
2.3. Plasma and UV-Ozone Treatments
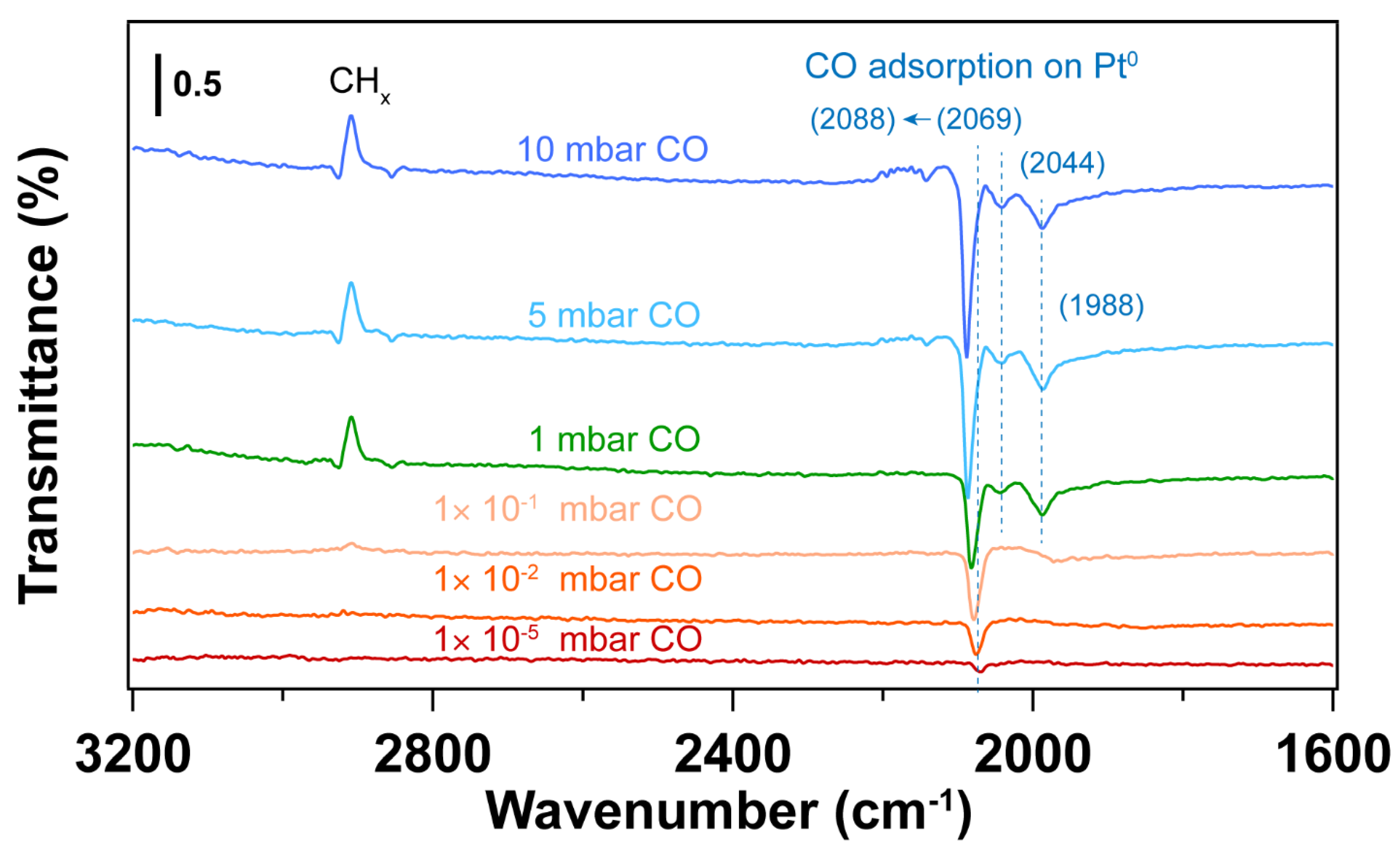
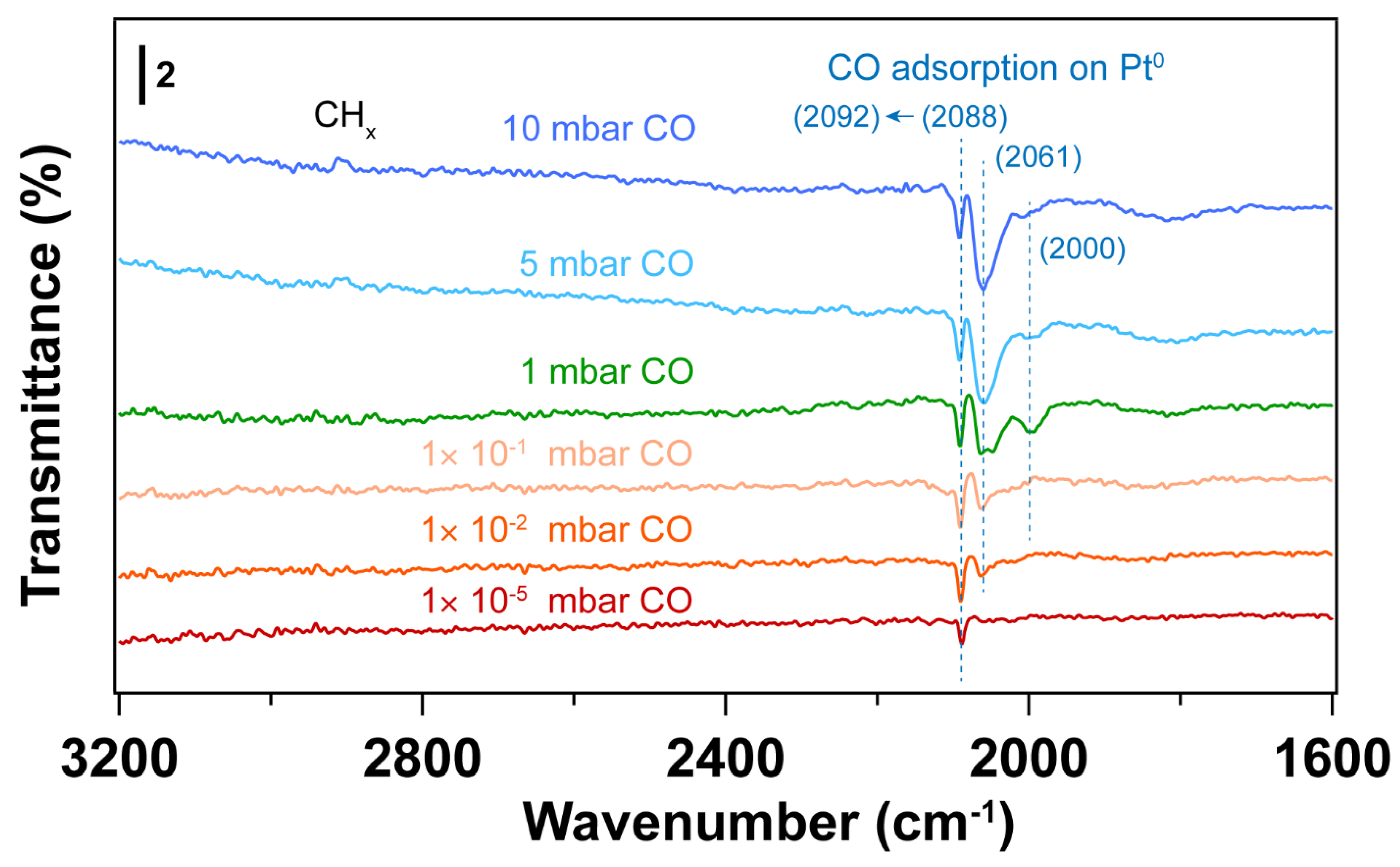
3. Results and Discussion
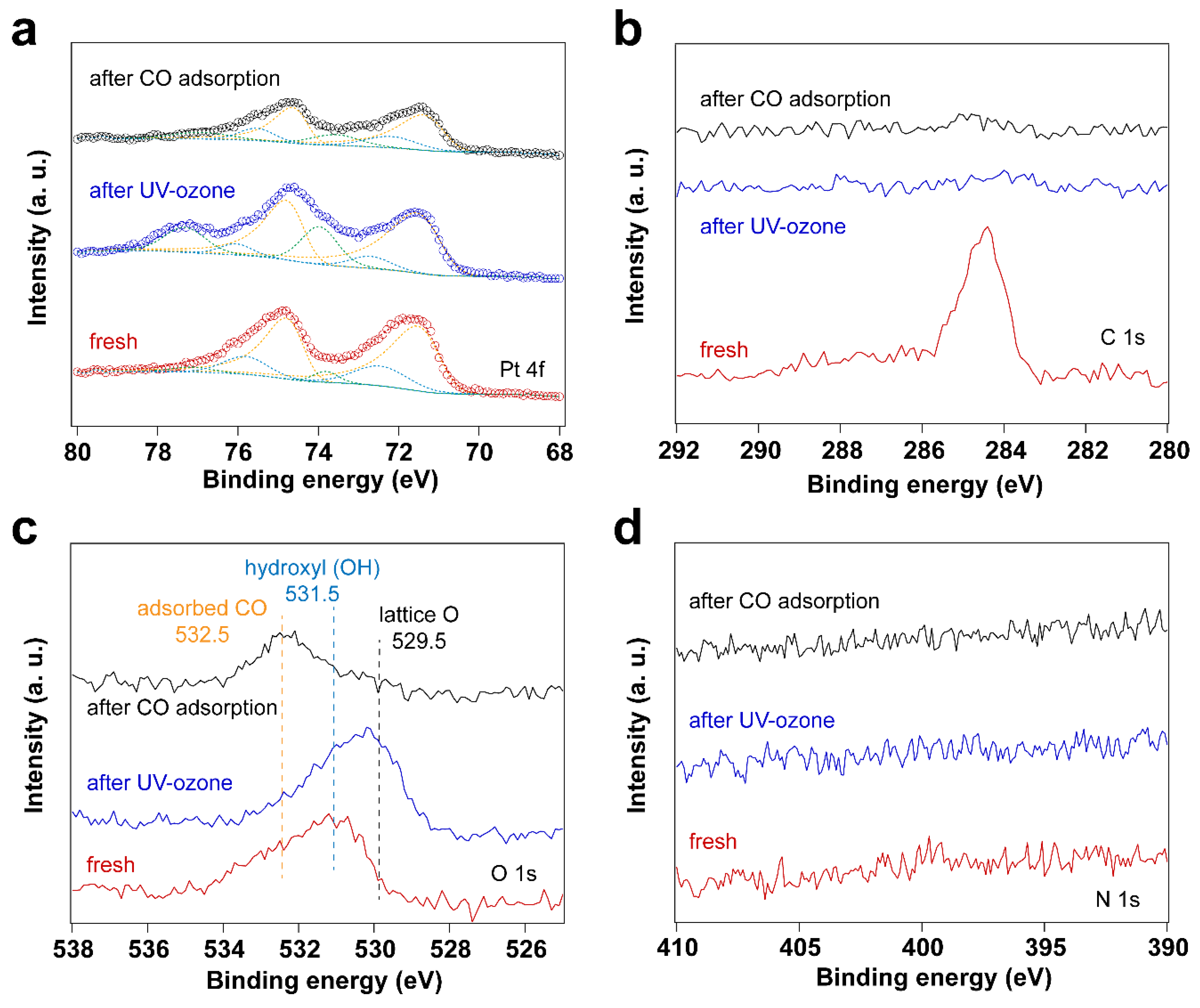
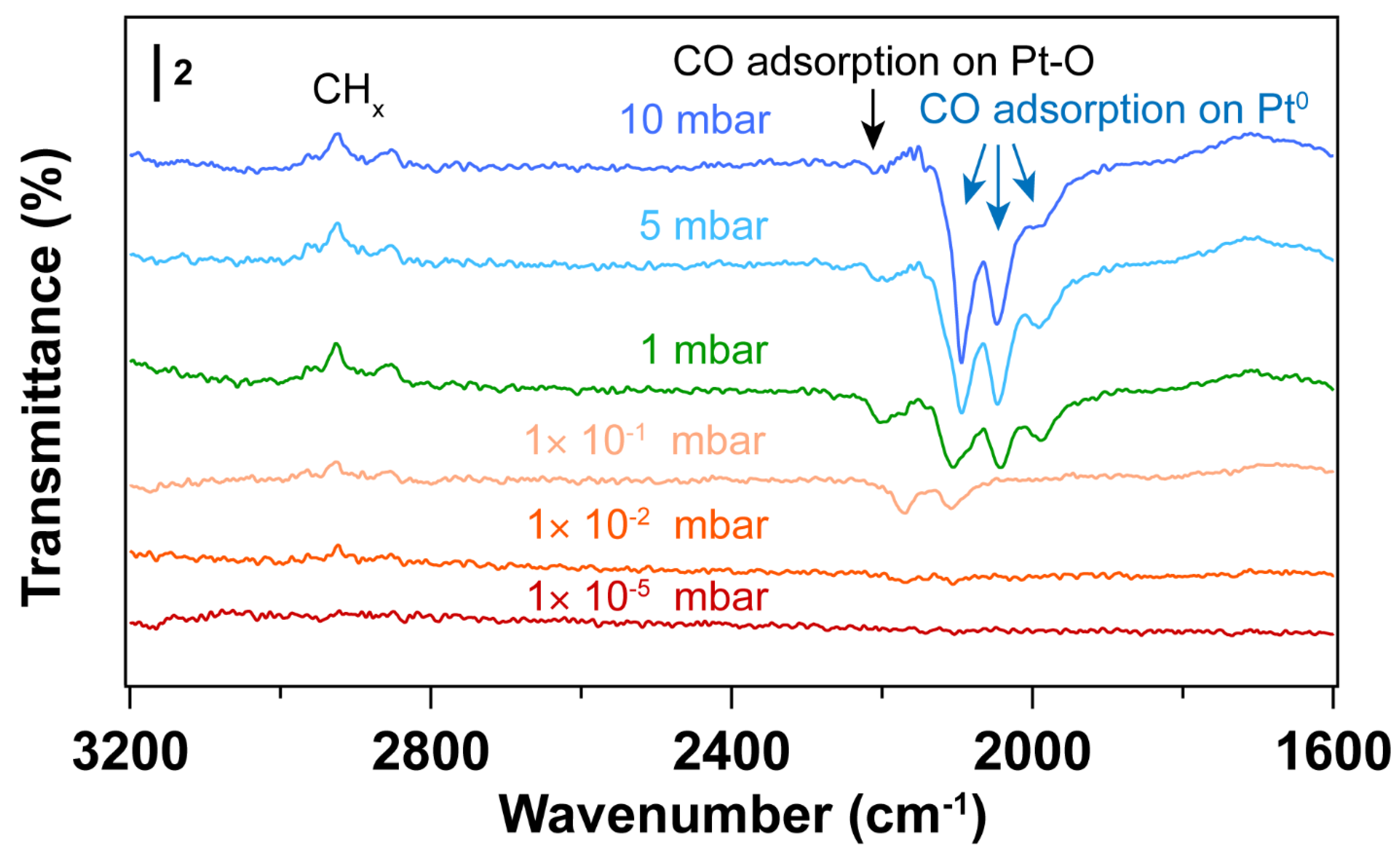
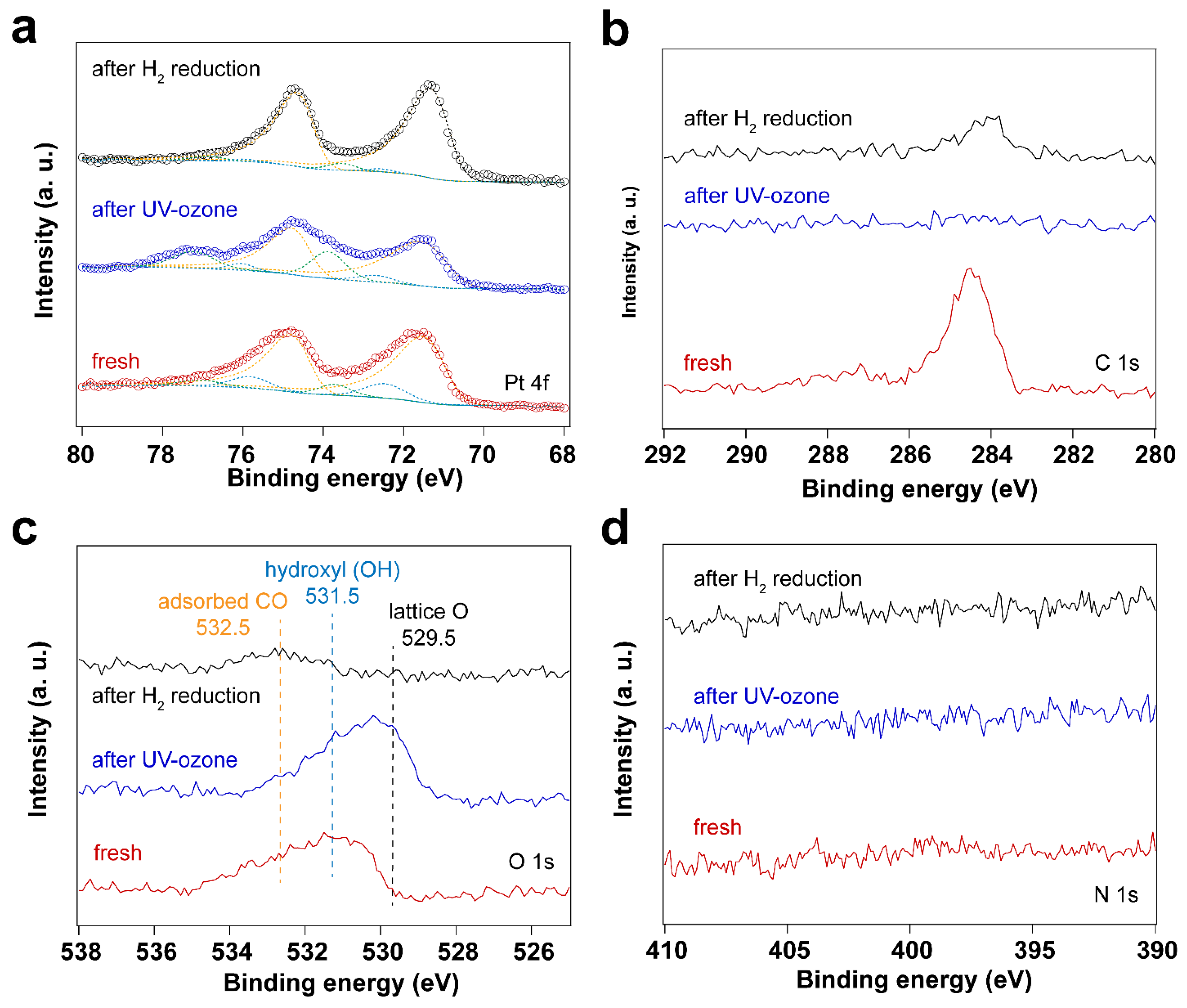
3.1. Removal of Surfactant by UV-Ozone Treatment
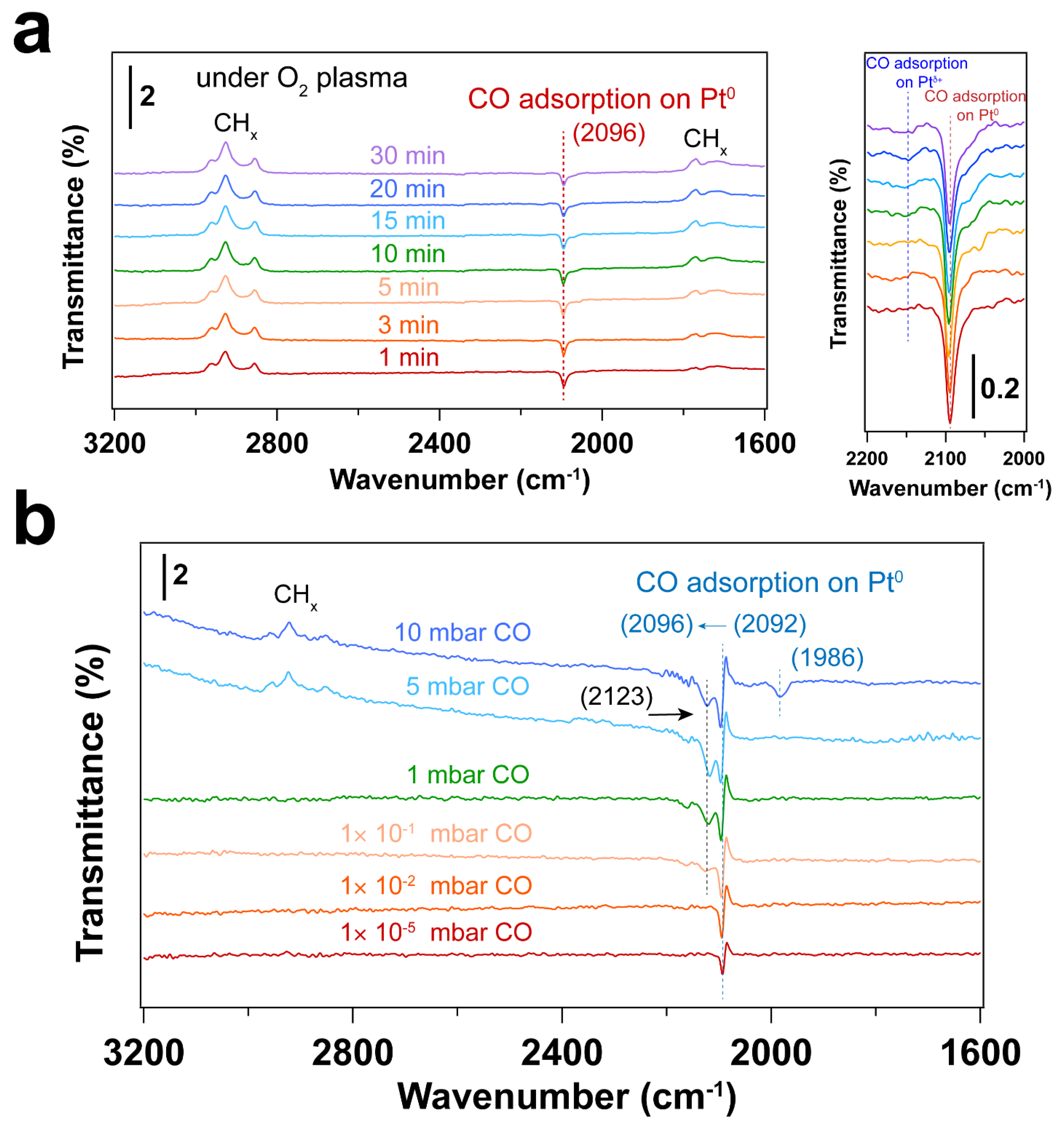
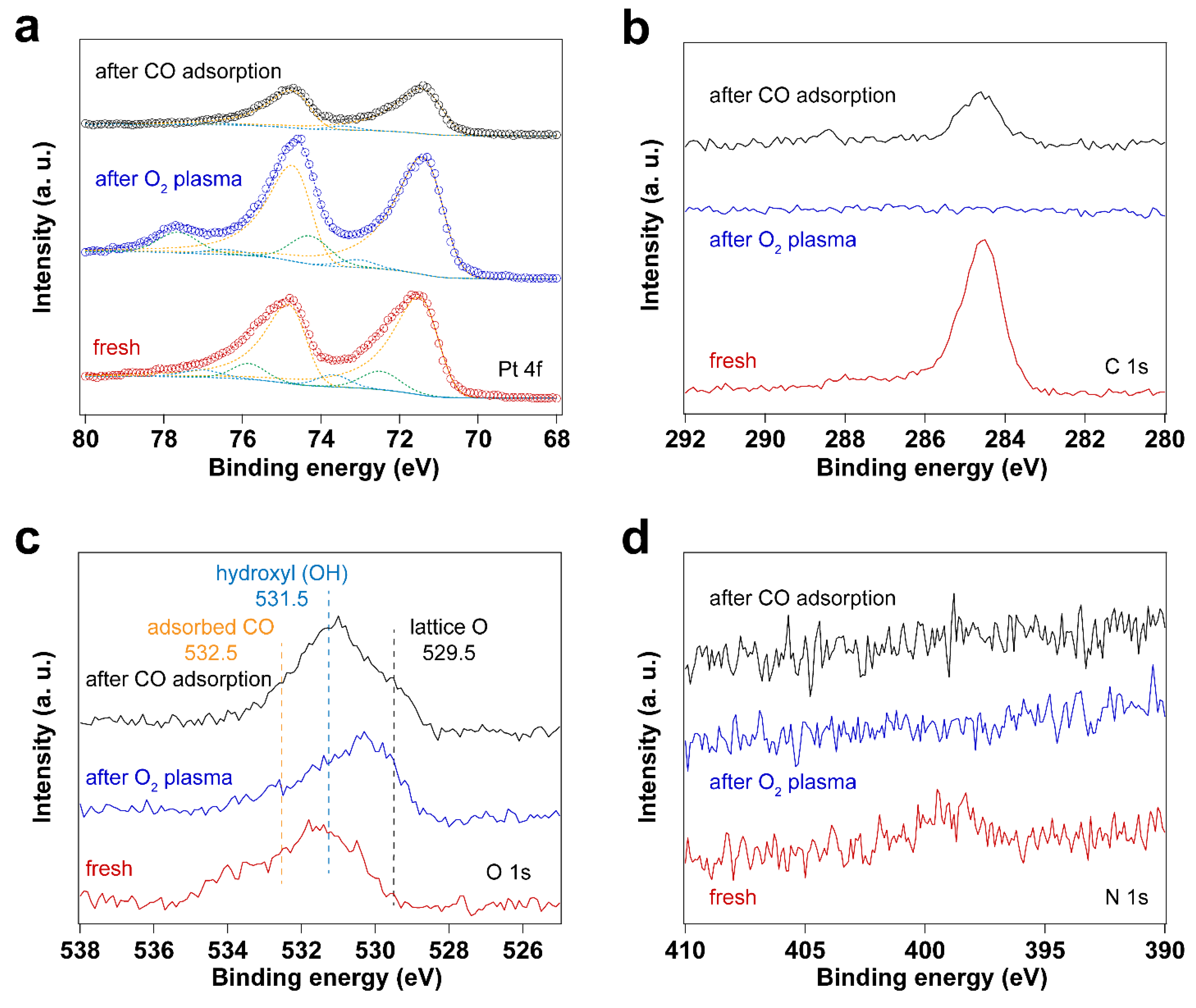
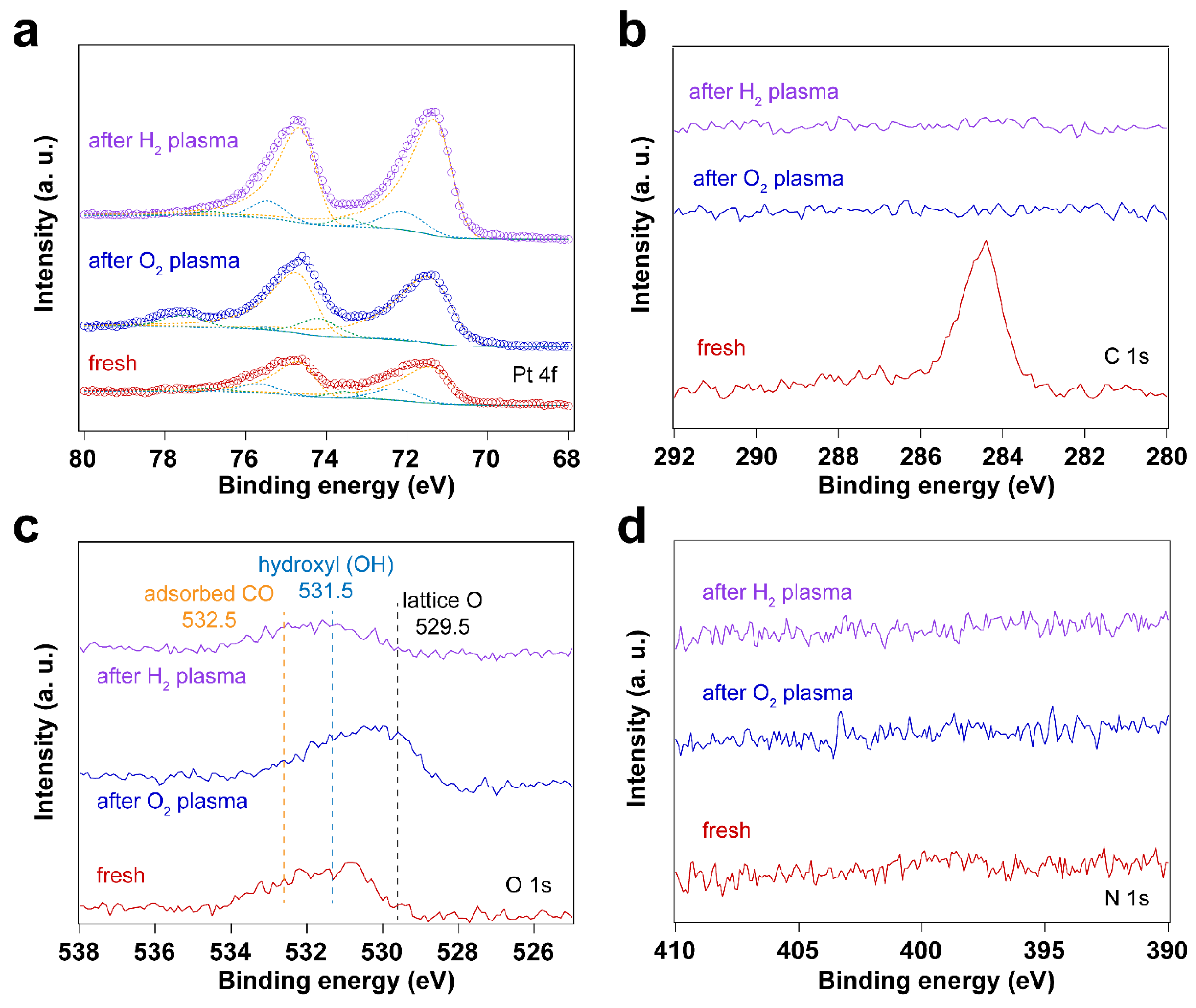
3.2. Removal of Surfactant by O2 Plasma Treatment
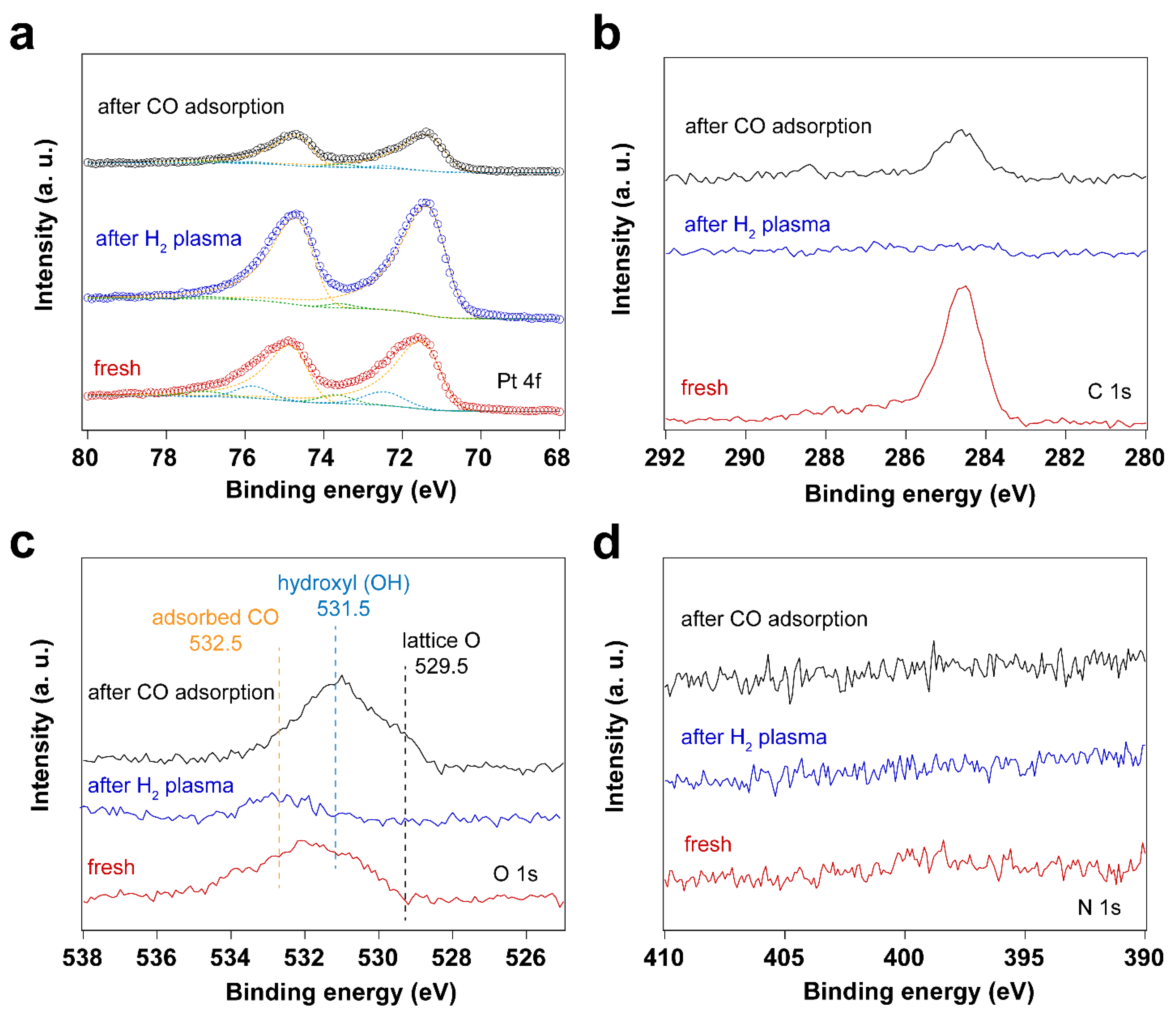
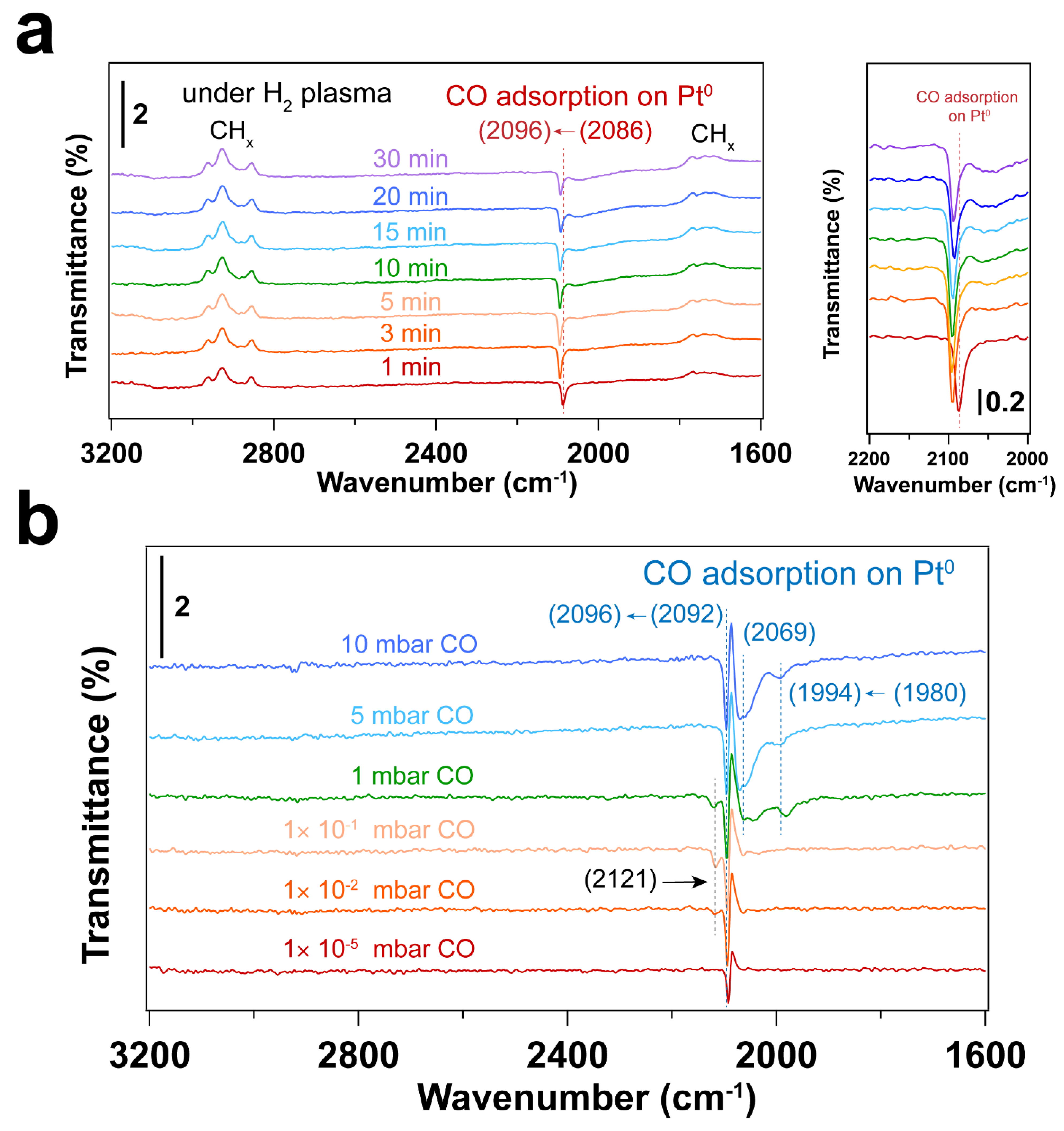
3.3. Removal of Surfactant by H2 Plasma Treatment
3.4. Using H2 Plasma to Reduce Pt Nanoparticles Treated by O2 Sources
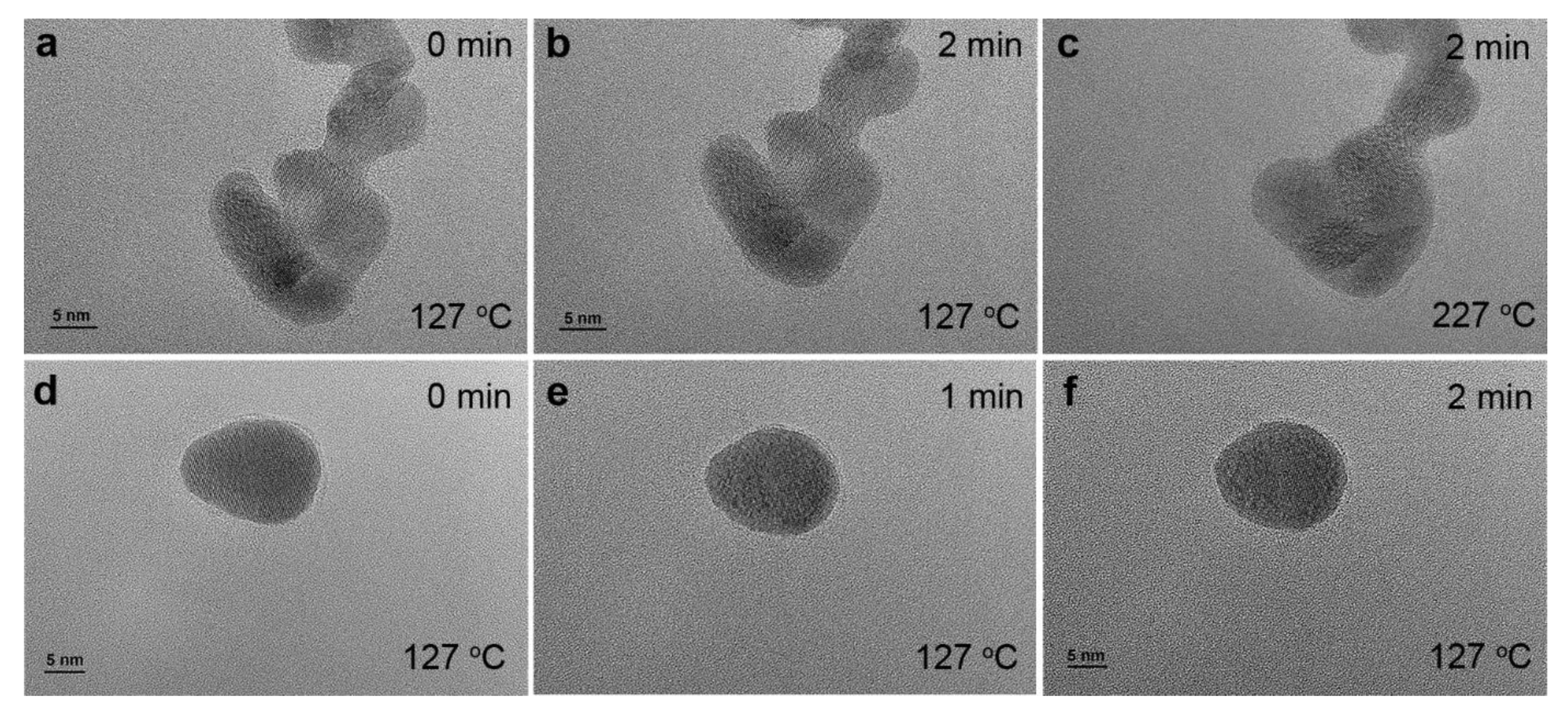
3.5. Practical Applications of Plasma Cleaning in Advanced Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zhang, J.; Yang, H.; Fang, J.; Zou, S. Synthesis and oxygen reduction activity of shape-controlled Pt3Ni nanopolyhedra. Nano Lett. 2010, 10, 638–644. [Google Scholar] [CrossRef]
- Puntes, V.F.; Krishnan, K.M.; Alivisatos, A.P. Colloidal nanocrystal shape and size control: The case of cobalt. Science 2001, 291, 2115–2117. [Google Scholar] [CrossRef]
- Wang, C.; van der Vliet, D.; Chang, K.-C.; You, H.; Strmcnik, D.; Schlueter, J.A.; Markovic, N.M.; Stamenkovic, V.R. Monodisperse Pt3Co nanoparticles as a catalyst for the oxygen reduction reaction: Size-dependent activity. J. Phys. Chem. C 2009, 113, 19365–19368. [Google Scholar] [CrossRef]
- Sun, S.; Zeng, H.; Robinson, D.B.; Raoux, S.; Rice, P.M.; Wang, S.X.; Li, G. Monodisperse MFe2O4 (M = Fe, Co, Mn) nanoparticles. J. Am. Chem. Soc. 2004, 126, 273–279. [Google Scholar] [CrossRef]
- Tsung, C.-K.; Kuhn, J.N.; Huang, W.; Aliaga, C.; Hung, L.-I.; Somorjai, G.A.; Yang, P. Sub-10 nm platinum nanocrystals with size and shape control: Catalytic study for ethylene and pyrrole hydrogenation. J. Am. Chem. Soc. 2009, 131, 5816–5822. [Google Scholar] [CrossRef]
- Heinz, H.; Pramanik, C.; Heinz, O.; Ding, Y.; Mishra, R.K.; Marchon, D.; Flatt, R.J.; Estrela-Lopis, I.; Llop, J.; Moya, S. Nanoparticle decoration with surfactants: Molecular interactions, assembly, and applications. Surf. Sci. Rep. 2017, 72, 1–58. [Google Scholar] [CrossRef]
- Zeng, Z.; Chen, Y.; Zhu, X.; Yu, L. Polyaniline-supported nano metal-catalyzed coupling reactions: Opportunities and challenges. Chin. Chem. Lett. 2023, 34, 107728. [Google Scholar] [CrossRef]
- Darmadi, I.; Piella, J.; Stolas, A.; Andersson, C.; Tiburski, C.; Moth-Poulsen, K.; Langhammer, C. Plasma Cleaning of Cationic Surfactants from Pd Nanoparticle Surfaces: Implications for Hydrogen Sorption. ACS Appl. Nano Mater. 2023, 6, 8168–8177. [Google Scholar] [CrossRef]
- Saman, N.M.; Zakaria, I.H.; Ahmad, M.H.; Abdul-Malek, Z. Effects of plasma treated alumina nanoparticles on breakdown strength, partial discharge resistance, and thermophysical properties of mineral oil-based nanofluids. Materials 2021, 14, 3610. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Orozco, C.; Urban, C.; Knight, M.W.; Urban, A.S.; Neumann, O.; Bishnoi, S.W.; Mukherjee, S.; Goodman, A.M.; Charron, H.; Mitchell, T. Au nanomatryoshkas as efficient near-infrared photothermal transducers for cancer treatment: Benchmarking against nanoshells. ACS Nano 2014, 8, 6372–6381. [Google Scholar] [CrossRef]
- Dreaden, E.C.; Alkilany, A.M.; Huang, X.; Murphy, C.J.; El-Sayed, M.A. The golden age: Gold nanoparticles for biomedicine. Chem. Soc. Rev. 2012, 41, 2740–2779. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.H.; Kamat, P.V. Quantum dot sensitized solar cells. A tale of two semiconductor nanocrystals: CdSe and CdTe. ACS Nano 2009, 3, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Jariwala, D.; Sangwan, V.K.; Lauhon, L.J.; Marks, T.J.; Hersam, M.C. Emerging device applications for semiconducting two-dimensional transition metal dichalcogenides. ACS Nano 2014, 8, 1102–1120. [Google Scholar] [CrossRef] [PubMed]
- Radisavljevic, B.; Radenovic, A.; Brivio, J.; Giacometti, V.; Kis, A. Single-layer MoS2 transistors. Nat. Nanotechnol. 2011, 6, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Chen, T.; Chen, W.; Wang, Z.; Chang, K.; Ma, L.; Huang, F.; Chen, D.; Lee, J.Y. Graphene-Like MoS2/Graphene Composites: Cationic Surfactant-Assisted Hydrothermal Synthesis and Electrochemical Reversible Storage of Lithium. Small 2013, 9, 3693–3703. [Google Scholar] [CrossRef]
- Wang, C.; Van Der Vliet, D.; More, K.L.; Zaluzec, N.J.; Peng, S.; Sun, S.; Daimon, H.; Wang, G.; Greeley, J.; Pearson, J. Multimetallic Au/FePt3 nanoparticles as highly durable electrocatalyst. Nano Lett. 2011, 11, 919–926. [Google Scholar] [CrossRef]
- Chen, W.; Kim, J.; Sun, S.; Chen, S. Electrocatalytic reduction of oxygen by FePt alloy nanoparticles. J. Phys. Chem. C 2008, 112, 3891–3898. [Google Scholar] [CrossRef]
- Swafford, L.A.; Weigand, L.A.; Bowers, M.J.; McBride, J.R.; Rapaport, J.L.; Watt, T.L.; Dixit, S.K.; Feldman, L.C.; Rosenthal, S.J. Homogeneously alloyed CdSxSe1-x nanocrystals: Synthesis, characterization, and composition/size-dependent band gap. J. Am. Chem. Soc. 2006, 128, 12299–12306. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xia, Y. Shape-controlled synthesis of gold and silver nanoparticles. Science 2002, 298, 2176–2179. [Google Scholar] [CrossRef]
- Hrelescu, C.; Sau, T.K.; Rogach, A.L.; Ja, F.; Laurent, G.; Douillard, L.; Charra, F. Selective excitation of individual plasmonic hotspots at the tips of single gold nanostars. Nano Lett. 2011, 11, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Zheng, S.; Wang, D.; Liu, X.; Li, H.; Han, S.; Chen, J.; Tang, Z.; Xu, G. Selective synthesis of single-crystalline rhombic dodecahedral, octahedral, and cubic gold nanocrystals. J. Am. Chem. Soc. 2009, 131, 697–703. [Google Scholar] [CrossRef]
- Huang, X.; Neretina, S.; El-Sayed, M.A. Gold nanorods: From synthesis and properties to biological and biomedical applications. Adv. Mater. 2009, 21, 4880–4910. [Google Scholar] [CrossRef]
- Martinsson, E.; Shahjamali, M.M.; Enander, K.; Boey, F.; Xue, C.; Aili, D.; Liedberg, B. Local refractive index sensing based on edge gold-coated silver nanoprisms. J. Phys. Chem. C 2013, 117, 23148–23154. [Google Scholar] [CrossRef]
- Jing, H.; Zhang, Q.; Large, N.; Yu, C.; Blom, D.A.; Nordlander, P.; Wang, H. Tunable plasmonic nanoparticles with catalytically active high-index facets. Nano Lett. 2014, 14, 3674–3682. [Google Scholar] [CrossRef]
- Zhang, Q.; Large, N.; Wang, H. Gold nanoparticles with tipped surface structures as substrates for single-particle surface-enhanced Raman spectroscopy: Concave nanocubes, nanotrisoctahedra, and nanostars. ACS Appl. Mater. Interfaces 2014, 6, 17255–17267. [Google Scholar] [CrossRef] [PubMed]
- Martinsson, E.; Shahjamali, M.M.; Large, N.; Zaraee, N.; Zhou, Y.; Schatz, G.C.; Mirkin, C.A.; Aili, D. Influence of surfactant bilayers on the refractive index sensitivity and catalytic properties of anisotropic gold nanoparticles. Small 2016, 12, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, R.J.; Jones, M.R.; Lee, B.; Auyeung, E.; Mirkin, C.A. Topotactic interconversion of nanoparticle superlattices. Science 2013, 341, 1222–1225. [Google Scholar] [CrossRef] [PubMed]
- Alkilany, A.M.; Thompson, L.B.; Boulos, S.P.; Sisco, P.N.; Murphy, C.J. Gold nanorods: Their potential for photothermal therapeutics and drug delivery, tempered by the complexity of their biological interactions. Adv. Drug Deliv. Rev. 2012, 64, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Hauck, T.S.; Ghazani, A.A.; Chan, W.C. Assessing the effect of surface chemistry on gold nanorod uptake, toxicity, and gene expression in mammalian cells. Small 2008, 4, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, C.; Tripkovic, D.; Sun, S.; Markovic, N.M.; Stamenkovic, V.R. Surfactant removal for colloidal nanoparticles from solution synthesis: The effect on catalytic performance. ACS Catal. 2012, 2, 1358–1362. [Google Scholar] [CrossRef]
- Lu, L.; Zou, S.; Fang, B. The critical impacts of ligands on heterogeneous nanocatalysis: A review. ACS Catal. 2021, 11, 6020–6058. [Google Scholar] [CrossRef]
- Aliaga, C.; Park, J.Y.; Yamada, Y.; Lee, H.S.; Tsung, C.-K.; Yang, P.; Somorjai, G.A. Sum frequency generation and catalytic reaction studies of the removal of organic capping agents from Pt nanoparticles by UV—Ozone treatment. J. Phys. Chem. C 2009, 113, 6150–6155. [Google Scholar] [CrossRef]
- Kim, G.W.; Ha, J.W. Single-particle study: Effects of oxygen plasma treatment on structural and spectral changes of anisotropic gold nanorods. Phys. Chem. Chem. Phys. 2020, 22, 11767–11770. [Google Scholar] [CrossRef]
- Choi, K.; Ghosh, S.; Lim, J.; Lee, C. Removal efficiency of organic contaminants on Si wafer by dry cleaning using UV/O3 and ECR plasma. Appl. Surf. Sci. 2003, 206, 355–364. [Google Scholar] [CrossRef]
- Gehl, B.; Frömsdorf, A.; Aleksandrovic, V.; Schmidt, T.; Pretorius, A.; Flege, J.I.; Bernstorff, S.; Rosenauer, A.; Falta, J.; Weller, H. Structural and Chemical Effects of Plasma Treatment on Close-Packed Colloidal Nanoparticle Layers. Adv. Funct. Mater. 2008, 18, 2398–2410. [Google Scholar] [CrossRef]
- Nguyen, D.K.; Vargheese, V.; Liao, V.; Dimitrakellis, P.; Sourav, S.; Zheng, W.; Vlachos, D.G. Plasma-Enabled Ligand Removal for Improved Catalysis: Furfural Conversion on Pd/SiO2. ACS Nano 2023, 17, 21480–21492. [Google Scholar] [CrossRef]
- Han, G.; Wang, K.; Elkins, K.E.; Qiu, Z.; Timmons, R.B.; Savage, C.R.; Kang, S.; Liu, J.P. Cold plasma reduction of surface carbon on SmCo5 nano-flakes prepared by surfactant-assisted ball-milling. J. Magn. Magn. Mater. 2019, 471, 250–254. [Google Scholar] [CrossRef]
- de Mello, M.D.; Ahmad, M.; Lee, D.T.; Dimitrakellis, P.; Miao, Y.; Zheng, W.; Nykypanchuk, D.; Vlachos, D.G.; Tsapatsis, M.; Boscoboinik, J.A. In Situ Tracking of Nonthermal Plasma Etching of ZIF-8 Films. ACS Appl. Mater. Interfaces 2022, 14, 19023–19030. [Google Scholar] [CrossRef]
- Yamada, Y.; Tsung, C.-K.; Huang, W.; Huo, Z.; Habas, S.E.; Soejima, T.; Aliaga, C.E.; Somorjai, G.A.; Yang, P. Nanocrystal bilayer for tandem catalysis. Nat. Chem. 2011, 3, 372–376. [Google Scholar] [CrossRef]
- Li, G.; Marinkovic, N.; Wang, B.; Komarneni, M.R.; Resasco, D.E. Manipulating the Microenvironment of Surfactant-Encapsulated Pt Nanoparticles to Promote Activity and Selectivity. ACS Catal. 2022, 12, 13930–13940. [Google Scholar] [CrossRef]
- Vovk, E.I.; Kalinkin, A.V.; Smirnov, M.Y.; Klembovskii, I.O.; Bukhtiyarov, V.I. XPS study of stability and reactivity of oxidized Pt nanoparticles supported on TiO2. J. Phys. Chem. C 2017, 121, 17297–17304. [Google Scholar] [CrossRef]
- Li, G.; Li, L.; Wu, B.; Li, J.; Yuan, Y.; Shi, J. Controlled one-step synthesis of Pt decorated octahedral Fe3O4 and its excellent catalytic performance for CO oxidation. Nanoscale 2015, 7, 17855–17860. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, C.; Walker, M.; McConville, C. Reaction of atomic oxygen with a Pt (111) surface: Chemical and structural determination using XPS, CAICISS and LEED. Surf. Sci. 2003, 545, 19–33. [Google Scholar] [CrossRef]
- Motin, A.M.; Haunold, T.; Bukhtiyarov, A.V.; Bera, A.; Rameshan, C.; Rupprechter, G. Surface science approach to Pt/carbon model catalysts: XPS, STM and microreactor studies. Appl. Surf. Sci. 2018, 440, 680–687. [Google Scholar] [CrossRef]
- Johansson, N.; Andersen, M.; Monya, Y.; Andersen, J.N.; Kondoh, H.; Schnadt, J.; Knudsen, J. Ambient pressure phase transitions over Ir (111): At the onset of CO oxidation. J. Phys. Condens. Matter 2017, 29, 444002. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, F.; García-Fernández, C.; Simonovis, J.P.; Hunt, A.; Walter, A.; Waluyo, I.; Bertram, F.; Merte, L.R.; Shipilin, M.; Pfaff, S. Catalytic Oxidation of CO on a Curved Pt (111) Surface: Simultaneous Ignition at All Facets through a Transient CO-O Complex. Angew. Chem. 2020, 132, 20212–20218. [Google Scholar] [CrossRef]
- Ding, K.; Gulec, A.; Johnson, A.M.; Schweitzer, N.M.; Stucky, G.D.; Marks, L.D.; Stair, P.C. Identification of active sites in CO oxidation and water-gas shift over supported Pt catalysts. Science 2015, 350, 189–192. [Google Scholar] [CrossRef]
- Ivanova, E.; Mihaylov, M.; Thibault-Starzyk, F.; Daturi, M.; Hadjiivanov, K. FTIR spectroscopy study of CO and NO adsorption and co-adsorption on Pt/TiO2. J. Mol. Catal. A Chem. 2007, 274, 179–184. [Google Scholar] [CrossRef]
- Crossley, A.; King, D.A. Infrared spectra for co isotopes chemisorbed on Pt “111”: Evidence for strong absorbate coupling interactions. Surf. Sci. 1977, 68, 528–538. [Google Scholar] [CrossRef]
- Olsen, C.; Masel, R. An infrared study of CO adsorption on Pt (111). Surf. Sci. 1988, 201, 444–460. [Google Scholar] [CrossRef]
- Allian, A.D.; Takanabe, K.; Fujdala, K.L.; Hao, X.; Truex, T.J.; Cai, J.; Buda, C.; Neurock, M.; Iglesia, E. Chemisorption of CO and mechanism of CO oxidation on supported platinum nanoclusters. J. Am. Chem. Soc. 2011, 133, 4498–4517. [Google Scholar] [CrossRef] [PubMed]
- Bordenyuk, A.N.; Weeraman, C.; Yatawara, A.; Jayathilake, H.D.; Stiopkin, I.; Liu, Y.; Benderskii, A.V. Vibrational sum frequency generation spectroscopy of dodecanethiol on metal nanoparticles. J. Phys. Chem. C 2007, 111, 8925–8933. [Google Scholar] [CrossRef]
- Borodko, Y.; Habas, S.E.; Koebel, M.; Yang, P.; Frei, H.; Somorjai, G.A. Probing the Interaction of Poly (vinylpyrrolidone) with Platinum Nanocrystals by UV− Raman and FTIR. J. Phys. Chem. B 2006, 110, 23052–23059. [Google Scholar] [CrossRef] [PubMed]
- Michalka, J.R.; Latham, A.P.; Gezelter, J.D. CO-induced restructuring on stepped Pt surfaces: A molecular dynamics study. J. Phys. Chem. C 2016, 120, 18180–18190. [Google Scholar] [CrossRef]
- Zabidi, N.; Zaaba, S.; Sut, K.E.; Mohamad, C.; Masiman, R. A Brief Review on Atmospheric Air Plasma. J. Phys. Conf. Ser. 2021, 2071, 012004. [Google Scholar] [CrossRef]
- Škoro, N.; Puač, N.; Lazović, S.; Cvelbar, U.; Kokkoris, G.; Gogolides, E. Characterization and global modelling of low-pressure hydrogen-based RF plasmas suitable for surface cleaning processes. J. Phys. D Appl. Phys. 2013, 46, 475206. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Fresh | After UV-Ozone Treatment | After CO Adsorption | |
|---|---|---|---|---|
| Peak position (eV) | Pt0 4f7/2 | 71.17 | 71.16 | 71.04 |
| Pt0 4f5/2 | 74.52 | 74.51 | 74.39 | |
| Pt–O(I) 4f7/2 | 72.45 | 72.70 | 72.18 | |
| Pt–O(I) 4f5/2 | 75.80 | 76.05 | 75.53 | |
| Pt–O(II) 4f7/2 | 73.79 | 73.98 | 73.57 | |
| Pt–O(II) 4f5/2 | 77.14 | 77.33 | 76.92 | |
| Peak area ratio | Pt–O(I)/Pt0 | 0.28 | 0.13 | 0.30 |
| Pt–O(II)/Pt0 | 0.07 | 0.37 | 0.27 | |
| Sample | Fresh | After O2 Plasma Treatment | After CO Adsorption | |
|---|---|---|---|---|
| Peak position (eV) | Pt0 4f7/2 | 71.16 | 71.05 | 71.07 |
| Pt0 4f5/2 | 74.51 | 74.40 | 74.42 | |
| Pt–O(I) 4f7/2 | 73.69 | 73.06 | 73.43 | |
| Pt–O(I) 4f5/2 | 77.04 | 76.41 | 76.77 | |
| Pt–O(II) 4f7/2 | 72.48 | 74.29 | 74.80 | |
| Pt–O(II) 4f5/2 | 75.83 | 77.64 | 78.15 | |
| Peak area ratio | Pt–O(I)/Pt0 | 0.13 | 0.04 | 0.04 |
| Pt–O(II)/Pt0 | 0.07 | 0.17 | <0.001 | |
| Sample | Fresh | After H2 Plasma Treatment | After CO Adsorption | |
|---|---|---|---|---|
| Peak position (eV) | Pt0 4f7/2 | 71.22 | 71.05 | 71.07 |
| Pt0 4f5/2 | 74.57 | 74.40 | 74.42 | |
| Pt–O(I) 4f7/2 | 72.50 | 72.05 | 72.46 | |
| Pt–O(I) 4f5/2 | 75.85 | 75.40 | 75.81 | |
| Pt–O(II) 4f7/2 | 74.60 | 73.58 | 73.54 | |
| Pt–O(II) 4f5/2 | 77.95 | 76.93 | 76.89 | |
| Peak area ratio | Pt–O(I)/Pt0 | 0.15 | 0.03 | 0.03 |
| Pt–O(II)/Pt0 | 0.08 | 0.02 | 0.02 | |
| Sample | Fresh | After UV-Ozone Treatment | After H2 Reduction | |
|---|---|---|---|---|
| Peak position (eV) | Pt0 4f7/2 | 71.14 | 71.13 | 71.02 |
| Pt0 4f5/2 | 74.49 | 74.48 | 74.37 | |
| Pt–O(I) 4f7/2 | 72.47 | 72.70 | 72.58 | |
| Pt–O(I) 4f5/2 | 75.82 | 76.05 | 75.93 | |
| Pt–O(II) 4f7/2 | 73.72 | 73.88 | 73.54 | |
| Pt–O(II) 4f5/2 | 77.07 | 77.23 | 76.89 | |
| Peak area ratio | Pt–O(I)/Pt0 | 0.15 | 0.07 | 0.02 |
| Pt–O(II)/Pt0 | 0.09 | 0.30 | 0.04 | |
| Sample | Fresh | After O2 Plasma Treatment | After H2 Plasma Treatment | |
|---|---|---|---|---|
| Peak position (eV) | Pt0 4f7/2 | 71.07 | 71.08 | 71.03 |
| Pt0 4f5/2 | 74.42 | 74.43 | 74.38 | |
| Pt–O(I) 4f7/2 | 72.31 | 72.15 | 72.10 | |
| Pt–O(I) 4f5/2 | 75.66 | 75.50 | 75.45 | |
| Pt–O(II) 4f7/2 | 73.56 | 74.19 | 73.52 | |
| Pt–O(II) 4f5/2 | 76.91 | 77.54 | 76.87 | |
| Peak area ratio | Pt–O(I)/Pt0 | 0.22 | 0.01 | 0.13 |
| Pt–O(II)/Pt0 | 0.08 | 0.15 | 0.04 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Zakharov, D.N.; Sikder, S.; Xu, Y.; Tong, X.; Dimitrakellis, P.; Boscoboinik, J.A. In Situ Monitoring of Non-Thermal Plasma Cleaning of Surfactant Encapsulated Nanoparticles. Nanomaterials 2024, 14, 290. https://doi.org/10.3390/nano14030290
Li G, Zakharov DN, Sikder S, Xu Y, Tong X, Dimitrakellis P, Boscoboinik JA. In Situ Monitoring of Non-Thermal Plasma Cleaning of Surfactant Encapsulated Nanoparticles. Nanomaterials. 2024; 14(3):290. https://doi.org/10.3390/nano14030290
Chicago/Turabian StyleLi, Gengnan, Dmitri N. Zakharov, Sayantani Sikder, Yixin Xu, Xiao Tong, Panagiotis Dimitrakellis, and Jorge Anibal Boscoboinik. 2024. "In Situ Monitoring of Non-Thermal Plasma Cleaning of Surfactant Encapsulated Nanoparticles" Nanomaterials 14, no. 3: 290. https://doi.org/10.3390/nano14030290