1. Introduction
Continuously adjusting the band gap of semiconductors is expected to revolutionize semiconductor technology [
1,
2]. The charge current flowing through semiconductor devices such as p-n junctions, field-effect transistors, and bipolar junction transistors are related to the energy barrier across the transition region between semiconductor interfaces [
2]. Recently, carbon nanotube (CNT) transistors outperform some high-class silicon-based transistors, and [
3] this remarkable performance deserves attention. If the band gap of a CNT can be tuned continuously, more robust CNT transistors will debut and it will diversify the applications of CNT transistors. However, tuning the band gap of semiconductors continuously [
1], especially for carbon nanotubes, is an uphill challenge. The band gap of carbon nanotubes depends on discrete chirality numbers [
1]. In order to get out of this dilemma, a continuously tunable length-dependent direct band gap of carbyne (in form of a linear carbon chain LCC) has been proposed for fine-tuning the energy barriers across semiconductor junctions [
1,
4]. Carbyne-based transistors have been demonstrated to have an outstanding current–voltage IV characteristic experimentally and the IV performance always depends on the applied strain or the size of carbyne [
5], in which the band gap of carbyne can also be widely tuned by applying less than 10% strain [
6]. The strain or length-triggered continuously tunable band gap of the carbyne inside a CNT may give a solution to widening the practical uses of CNT-based transistors. Unfortunately, the length of the carbyne during sample fabrication is unpredictable even though advanced manufacturing techniques are applied. There is no systematic routine that provides the theoretical input for optimizing the chain length of the carbyne, which hinders experimental bottom-up studies of the continuously tunable band gap.
The preparation of carbon chains with a length in the order of 20 atoms has been a difficult problem in the last decade [
7,
8]. To extend the chain length, Shi et al. encapsulated the carbyne with carbon nanotubes (CNTs) as a nanoreactor, resulting in carbon chains with up to 6000 atoms [
9]. Based on the analysis of the carbyne@nanotube, it was argued that the van der Waals (VDW) force emanating from the carbon nanotube plays an important role in stabilizing the internal carbyne [
4,
9]. Inspired by the work of Shi et al., Chi Ho Wong et al. developed a Monte Carlo method (linear carbon chain (LCC) model) to study the stability of free-standing carbon chains laterally coupled by VDW forces at finite temperatures [
10]. In the absence of carbon nanotubes, the direct action of the environment on the carbyne can be detrimental to the development of long chain lengths, but the kink structure [
11,
12] in a short carbyne can induce strong ferromagnetism [
13]. One year later, Chi Ho Wong et al. succeeded in fabricating a ferromagnetic VDW-coupled free-standing carbon chain array with a length of ~100 atoms at room temperature assisted by the LCC model [
13], where its ferromagnetism survives above 400 K. This observation is consistent with the argument of Shi et al. that the VDW interaction can be used to stabilize carbyne. On the other hand, several dopants have been proposed to enhance the magnetism of carbyne-based semiconductors with a local magnetic moment above 1.7 µB [
13,
14].
Since the discovery of the 400 K ferromagnetism in carbyne, carbyne may become a potential material to study the magnetic interface/edge problems for the development of room-temperature spintronic devices. To take spintronic nanodevices to the next level, edge magnetism [
15] of semiconductors is the most promising route. The monoatomic structure makes carbyne an ideal candidate for studying the physics of edge magnetism, as it is much thinner than any unit cell thick nanowire or ultrathin zigzag nanoribbons of graphene [
16,
17]. From a scientific point of view, the spin–spin interaction of a monoatomic chain contributes to one of the types of boundary magnetism in a higher-dimensional Bravais lattice. If the chain length of a magnetic carbyne can be properly tuned [
11,
12,
13], the length-dependent spin–spin interaction can be experimentally investigated down to the monoatomic scale, providing not only valuable scientific information for monitoring the interfaces of magnetic compounds for spintronics [
18], but also insights into the interface problems of magnetic heterostructures [
19] on an industrial scale.
In spite of technological and scientific advances, a tiny change in experimental parameters can fragmentize carbyne during sample fabrications. Theoretical guidelines are needed to control the carbyne length within a targeted range. However, the LCC model can only compare the stability of fixed-length carbyne [
10], which is not designed for a variable-length condition. The kink structuring of a carbyne chain simulated by the LCC model remains continuous no matter how long the chain is. A similar problem can be noticed in DFT software. Chain breakage is not observed in an isolated carbyne even though the carbyne is very long, and the entire isolated chain remains linear, surprisingly, after applying geometric optimizations. Worse still, the DFT study of finite-length carbyne is limited to only ~100–200 atoms due to prohibitive computational cost [
20]. For example, a ~6000-atom-long carbyne surrounded by a CNT (over ~100,000 atoms per unit cell) may not be a realistic task for DFT software [
21]. To speed up the computational progress, using DFT software to calculate an infinitely long LCC with few atoms per unit cell is inevitably a popular option [
14,
22]. DFT methods are ideal for situations at 0 K [
23]. DFT results are usually representative unless the materials are extremely unstable at finite temperatures. If a long linear carbon chain (LCC) dissociates at room temperature, a transfer function should be imposed on DFT methods to align theoretical results with experimental observations. Better software is needed to monitor carbyne in variable-length conditions. Otherwise, it is unclear how the sample’s quality affects the growth of internal LCC, and how the fragmentation of LCC takes place.
In view of the above problems, we developed a Monte Carlo algorithm to predict the stability of carbyne@nanotubes at finite temperatures with reasonable computational effort. A stochastic process is monitored to predict the stability of the carbyne@nanotubes up to 300 K. Parametric studies of the composite focus on the external parameters (e.g., pressure, temperature) and the internal parameters (chain length of carbyne, chirality, porosity of the carbon nanotube, etc.). Molecular modeling for the formation of fragmentized carbyne will debut in this work. After the Monte Carlo algorithm is introduced, we will propose a potential LCC@CNT composite to vary the band gap continuously in the CNT-based transistors.
3. Results
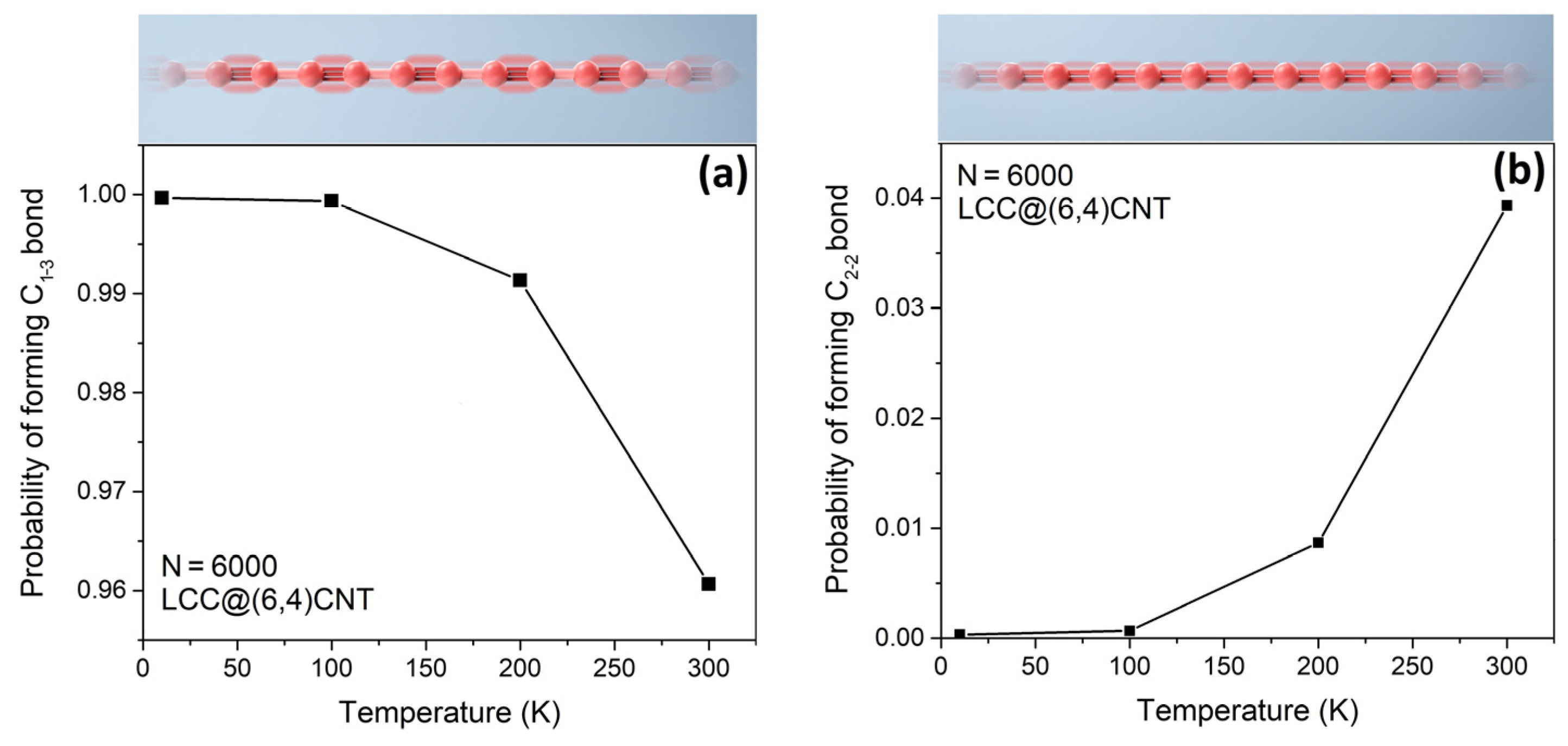
Thermal excitation from 0 K to 300 K has changed the proportion of polyyne and cumulene phases in
Figure 2. We found that the probability of forming a C
1–3 bond (polyyne phase) in the internal LCC decreases from 1.00 to 0.96 upon heating. Due to the growth of the polyyne phase, the probability of forming a C
2–2 bond (cumulene) correspondingly increases to ~0.04. The formation of C
1–2 ([
] or [
]) and C
1–1 ([
]) bonds is rare. The most energetically favorable phase of the internal LCC is still polyyne, even when the temperature is 300 K [
9]. After the pre-calibration process at T~0 K, the average bond length of the internal LCC is ~1.3 Å and the bond length alternation is above 99.9%.
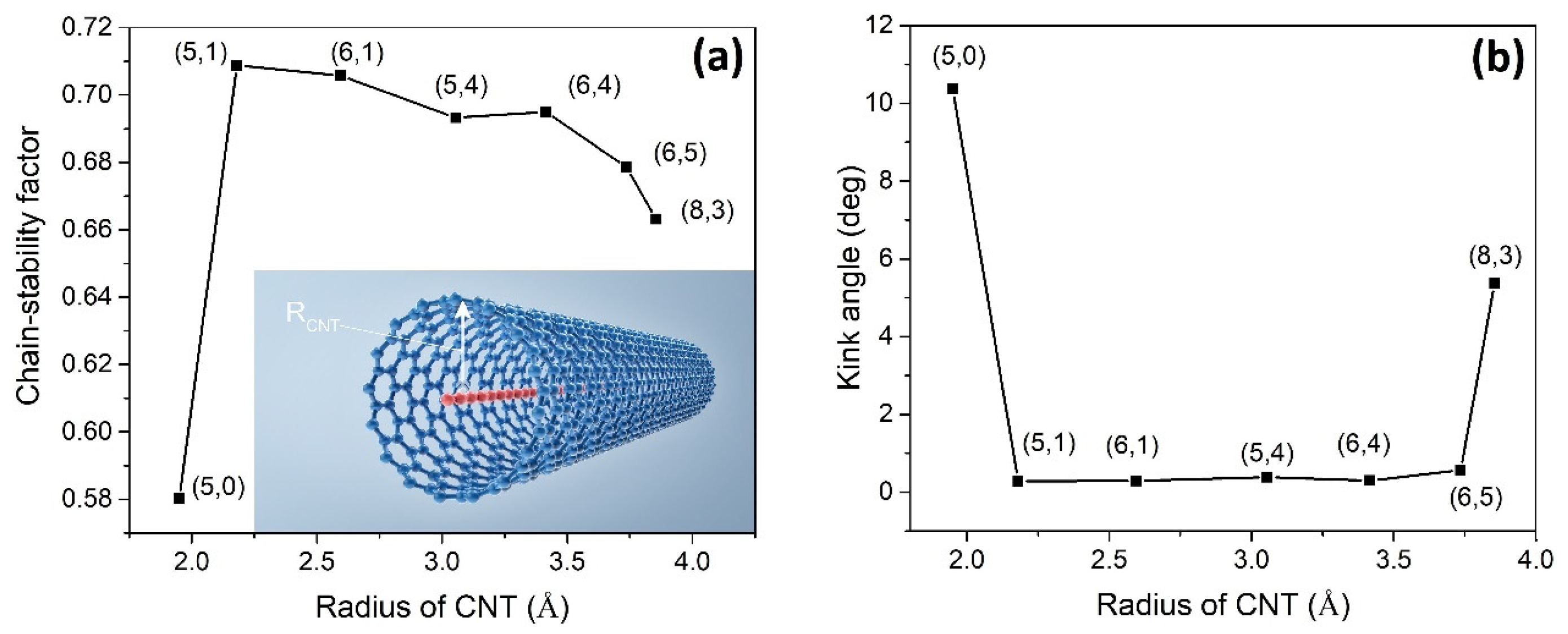
As illustrated in
Figure 3a, the stability of the carbyne@nanotube depends on the radius of the CNT associated with its chirality (N
c,M
c) [
4,
9]. In the carbyne@nanotube experiment [
9], (6,4) and (6,5) CNTs were the most abundant components. At a constant temperature of 300 K, our simulation data show that the (6,4)CNT should have a longer LCC than (6,5)CNT. Although the experiment [
9] does not contain (5,0), (5,1), (6,1), and (5,4)CNTs, we used these samples in our simulation. The LCC@(5,1)CNT has the highest chain stability factor among the others, whereas the chain stability of LCC@(5,0)CNT is the worst.
Figure 3b demonstrates how the kink angles of the internal LCC are affected by the geometry of the CNTs. The average kink angle of LCC@(5,0)CNT is much higher than the others. It can be observed that the higher the chain stability factor, the lower the average kink angle of the LCC.
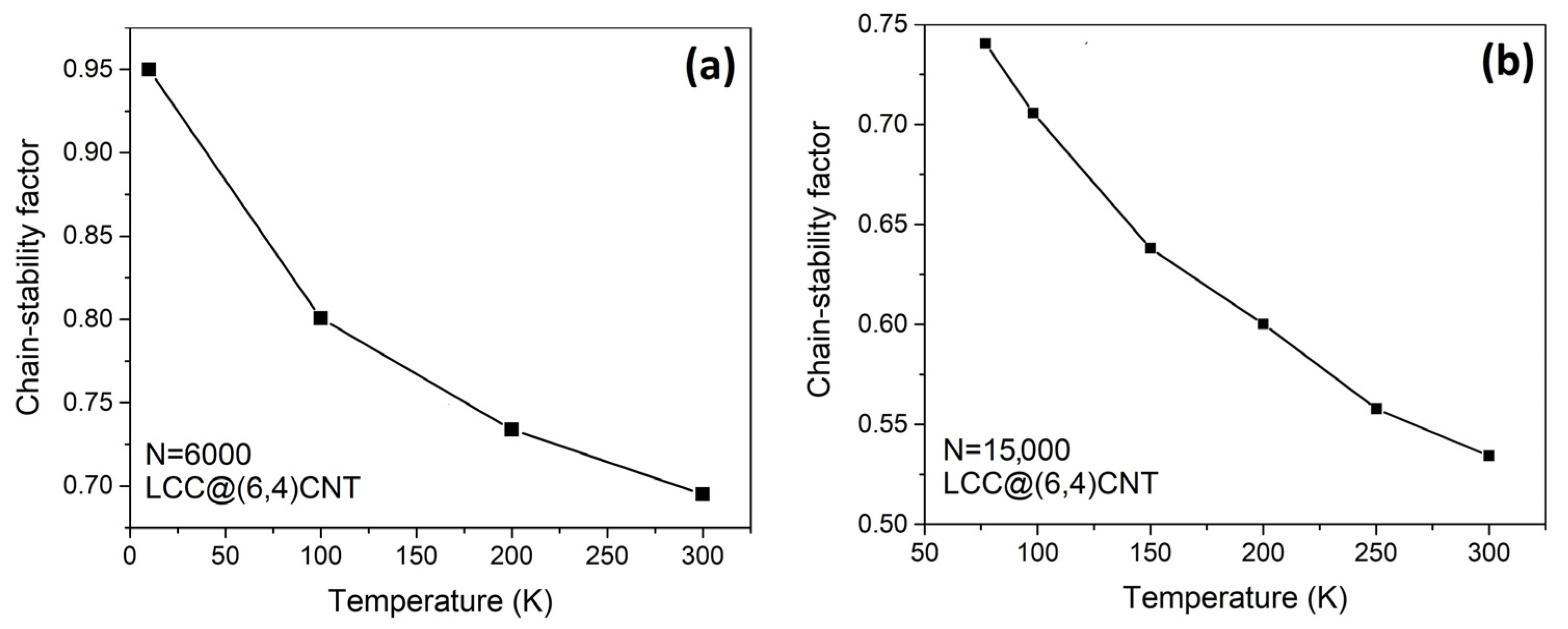
The chain stability of the internal LCC consisting of 6000 atoms and 15,000 atoms is reduced by thermal excitation, as demonstrated in
Figure 4. The LCC with 6000 atoms in length has a higher chain stability factor
than the LCC with 15,000 atoms in length, regardless of the temperature. Comparing the chain stabilities in both cases, it can be interpreted that the atomic fluctuations of the 6000-atom-long LCC are always lower at the same temperature. The chain stability of the carbyne@nanotube in different lengths is compared in
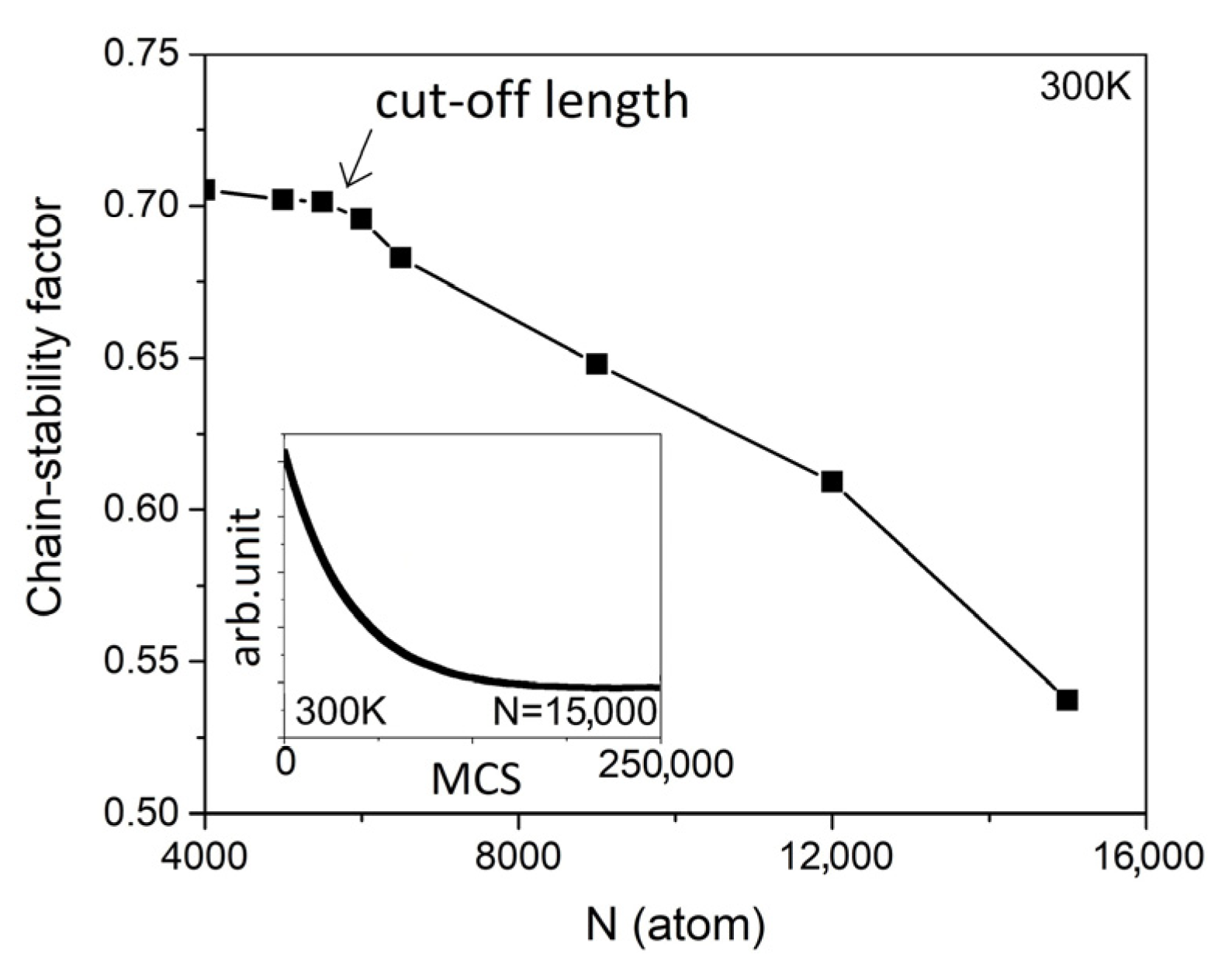
Figure 5. At T = 300 K, the chain stability factor gradually decreases when N is less than 5500. However, at N > 6000, a rapid reduction in the chain stability factor is observed when we halve the cut-off length of the LCC to N = 5750. The inset in
Figure 5 refers to the average energy of the internal LCC as a function of the Monte Carlo iterations.
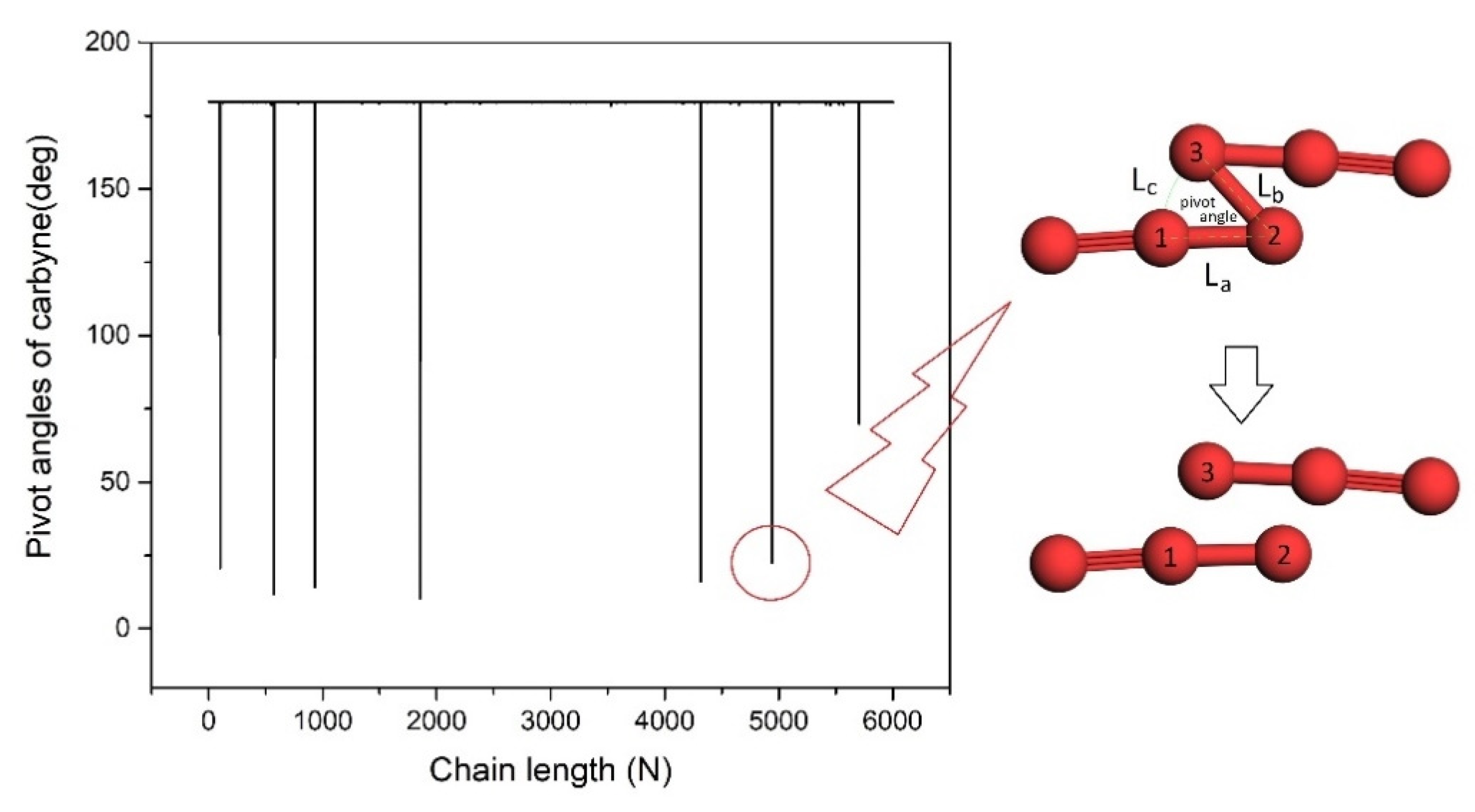
A compressive strain in the radial direction to the LCC@(6,4)CNT deteriorates the chain stability. The chain stability factor drops from 0.69 to 0.68 at a compressive strain of 2%. However, when the compressive strain reaches 4% in test 1 (
Table 2), the formation of seven extremely sharp pivot angles within ~10
o < pivot angle < ~60° at n = 103rd, 576th, 932nd, 1853rd, 4318th, 4943rd, and 5705th atom are observed in
Figure 6. The critical kink structure around n = 4943rd (marked by a red circle) is drawn. The carbon–carbon distances nearby the sharp kink points always reach ~1.73 Å. The carbon atoms at the sharp kink points are mostly connected by
. More samplings are shown in
Table 2 where we repeat the studies of 4% radial compression in test 2 and test 3.
The porosity of the (6,4)CNT harms the chain stability of the internal LCC, as demonstrated in
Table 3. The introduction of a tiny amount of vacancy defects is sufficient to significantly shorten the cut-off length to N~5600. In
Table 4, the cut-off length of the LCC@(N
c,M
c)CNT in various chiralities at 300 K is investigated. The internal LCC in the (5,1)CNT is the longest. The internal LCC in the (6,4)CNT is ~10% longer than that in a (6,5)CNT. Based on the data in
Table 5, it is possible to extend the internal LCC to a length of 15,000 atoms. However, the sample should be cooled to T~120 K. Our model expects that the internal LCC in the (6,4)CNT should increase by less than 10% at 273 K.
4. Discussion
Although the cumulene phase should dominate in an isolated LCC [
22], the polyyne phase is energetically more favorable in the LCC@(N
c,M
c)CNT [
9]. According to
Figure 2, the polyyne phase fades upon heating because the atomic fluctuations are more severe at high temperatures. This process can be controlled by the Boltzmann factor [
26], which more readily accepts the trial type of covalent bonds at high temperatures [
10]. The formation of [
], [
], and [
] is rare, confirming that our Monte Carlo simulation effectively monitors the process of energy minimization. For instance, the energy difference between [
] and [
] is |348 + 839 − (614 + 614)| = 41 kJ/mol per site. However, the energy difference (per site) between [
] and [
], and the energy difference (per site) between [
] and [
] are 225 kJ/mol and 491 kJ/mol per site, respectively. Hence, when the sample heats up, the LCC prefers to accept the trial state [
] rather than [
], [
], and [
].
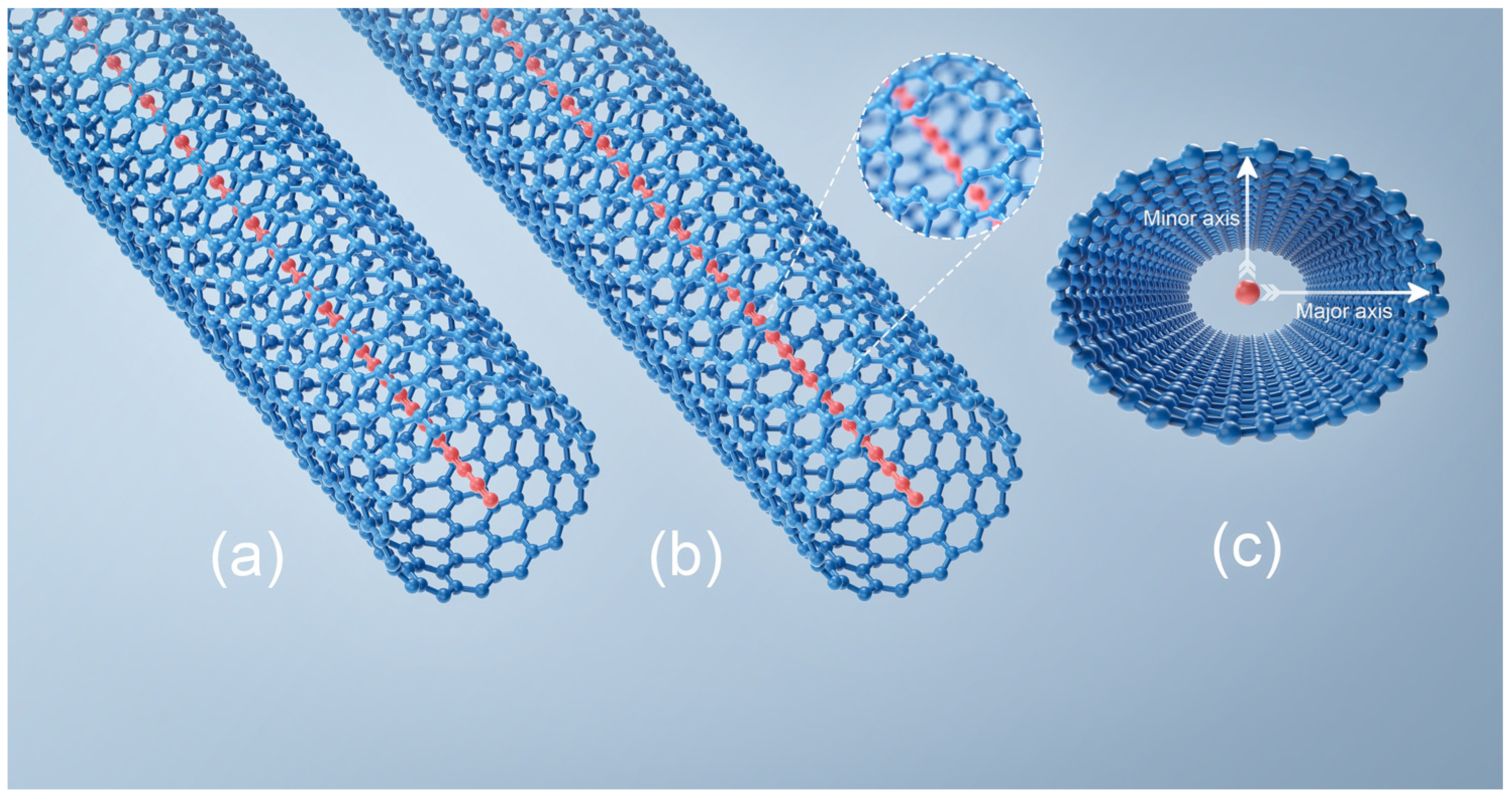
We study the stability of the internal LCC as a function of R
CNT in
Figure 3a. While the chain length is fixed, the optimization process focuses only on the radial direction of the CNT. An isolated long LCC is always unstable in a room temperature environment due to strong atomic fluctuations [
5,
6,
7,
30,
31]. The kink angle is a measure of the atomic fluctuations in the LCC. The kink angle of the LCC in
Figure 3b can be used to judge whether the CNT provides appropriate VDW protection or not. The average kink angle of the internal LCC in (5,1)CNT is only 0.3 degrees (or pivot angle = 179.7 degrees) in
Figure 3b. This explains why the LCC surrounded by (5,1)CNT has the highest chain stability factor where self-dissociation or chain breakage is less likely. Our simulation result is consistent with the experimental results that the internal LCC in a (6,4)CNT is longer than that in a (6,5)CNT [
9].
The chain stability factor of the LCC@(6,4)CNT increases upon cooling (see
Figure 4). At low temperatures, the movement of carbon atoms is inactive (i.e., the amplitude of atomic vibration is small). The carbon atoms in the LCC tend to remain close to their equilibrium positions due to reduced atomic fluctuations at lower temperatures. (i.e.,
~
) that increases the chain stability factor
. At high temperatures, the strength of the covalent bond between carbon atoms may not be sufficient enough to resist atomic fluctuations, leading to chain breakage or self-dissociation of the LCC. In other words, the minimization of atomic fluctuations in the LCC is a way to maintain the effectiveness of covalent bonds in order to avoid chain breakage or self-dissociation. Therefore, it is feasible to extend the length of the LCC beyond N~6000, but precautions must be taken to minimize atomic fluctuations, such as implementing a cooling mechanism. A cooler environment can effectively limit the atomic fluctuations, reinforcing the covalent bond strength and reducing the risk of chain breakage or self-dissociation. As atomic fluctuations can be influenced by temperature continuously, and since the maximum length of the LCC is dependent on these atomic fluctuations, it may build a relationship between the maximum LCC length and temperature.
If all covalent bonds in the LCC are stable, the LCC with a length of 15,000 atoms should have weaker atomic fluctuations than the LCC with a length of 6000 atoms. However, extensive studies of an LCC show that the elongation of an LCC always leads to chain termination or self-dissociation [
4,
5,
6,
7,
9,
30,
31,
32,
33]. So far, the formation of all stable covalent bonds in an extremely long LCC has not been observed experimentally (48 carbon atoms at most). To make a longer LCC unstable in our model, Hooke’s factor is used to tune the trial range of atomic fluctuations. When our previous LCC model is run this time, the maximum bond length of an isolated LCC calculated by the thermal expansion coefficient being set to
~1.56 Å manually. In this case, all covalent bonds are stable and eventually
is shorter in a longer LCC.
The accuracy of the thermal expansion coefficient in our model was justified by Costa et al. [
29]. After the substitution of
into the pre-calibration process, the trial Hooke’s factor increases for a longer LCC. By setting the maximum bond length in this carbyne@nanotube model to ~1.73 Å [
28], we impose a boundary condition that chain breaking is not allowed even if two carbon atoms are ~1.73 Å apart. Then, our Monte Carlo system can force the atoms to form unstable covalent bonds at any chain length L in excited states. The unstable covalent bonds associated with large atomic fluctuations outweigh the effect of stabilization toward the bulk state. Therefore, the LCC with a length of 15,000 atoms (
Figure 4b) has a lower chain stability factor than the LCC with a length of 6000 atoms (
Figure 4a). Without this boundary condition, we cannot adequately compare the stability of the different LCC@(Nc,Mc)CNT types at the same length.
Although Hooke’s factor is designed to be semi-empirical, our simulation can mimic the stability of a long LCC that is consistent with experimental observations, with the cut-off length (5750-atom-long) in
Figure 5 being comparable to the experimental data (~6000-atom-long) [
9]. Indeed, Hooke’s factor is just a ratio of velocity, because any elastic force multiplied by
gives a unit of velocity [
27]. Therefore,
can be read as an adjustment of the trial velocity. If a carbon chain is going to break [
28], Hooke’s factor in a local region approaches 1, so using the free-particle velocity as the maximum velocity at the trial state would be appropriate [
27]. The random number
refines the spatial fluctuations to aid the process of energy minimization.
In our simulation, a single-walled CNT is used instead of a double-walled CNT to protect the LCC. A double-walled CNT mainly exerts influence on the internal LCC via the inner wall and the influence of the outer wall is very small, which was observed in the measurement of radial breathing modes [
9]. Lei Shi et al. conducted the analysis of radial breathing modes (RBM) of the double-walled CNT experimentally, where only the RBM peaks corresponding to the inner CNT are split and enhanced significantly [
9]. This indicates that the inner CNT is a dominant source interacting with the internal LCC [
9]. Therefore, our simulations may not need to take the outer wall into account which mainly prevents the inner wall from collapsing (or maintaining a good shape) under perturbations only [
4,
9]. Our DFT simulation runs a parallel array of LCC@(N
c,M
c)CNT with a lateral spacing of 0.5 nm, and the lateral wall-to-wall interaction is enough to stabilize the single-walled CNT in a good shape. The cut-off length of the LCC@(6,4)CNT is slightly underestimated in our model. There are some sources of error. The
in Hooke’s factor is obtained from the pre-calibration process for N > 4000, but the
is the spring constant of an infinitely long LCC. Since the computational cost of a DFT simulation may not be realistic when the size exceeds 4000 atoms per unit cell [
21], our model assumes that the mechanical properties of the LCC consisting of more than 4000 atoms are likely to move toward the bulk state [
34,
35]. Moreover, the iterative process of our Monte Carlo simulation is not applicable to the atoms in CNTs, as experimental results show that the atomic vibrations of CNTs are much weaker than those of the internal LCCs. Based on these experimental findings, we can assume that the atoms in the CNTs are relatively at rest. Although the cut-off length is slightly underestimated by these approximations, the computational cost becomes affordable. The inset in
Figure 5 confirms that 250,000 Monte Carlo steps are sufficient to drive the system to equilibrium, even if the longest sample is chosen.
The shape of the CNT plays an important role in stabilizing the carbyne@nanotube. The compressive strain changes the CNT from a circular to an elliptical shape. Although the elliptical CNT has a slightly more negative VDW interaction along the minor axis (shorter axis), the VDW interaction along the major axis (longer axis) is much weaker. This is the reason why radial compression destabilizes the LCC@(6,4)CNT. On the other hand, a stronger spatial fluctuation in the internal LCC is observed in porous CNTs. If the selected atom in the internal LCC sums up the negative VDW terms along the angular plane ϕ, the missing carbon atoms in the porous CNT cannot turn the VDW force more negatively, and thus maintaining a good sample quality of a CNT is important to prolong the internal LCC.
Our Monte Carlo results are consistent with several experimental observations. (1) The LCC in a (6,4)CNT is more stable than the LCC in a (6,5)CNT [
9]. (2) The quality of the CNT affects the stability of the internal LCC [
4,
9]. (3) The cut-off length of the LCC@(6,4)CNT is about ~6000 atoms long [
9]. (4) Polyyne is the dominant phase in LCC@(N
c,M
c)CNT at room temperature [
9]. These Monte Carlo results encourage us to predict the cut-off length of the internal LCC surrounded by different CNT types in
Table 4. Based on our Monte Carlo model, the LCC surrounded by a (5,1)CNT should be the longest among the others because the atomic fluctuation is the weakest. The effect of free radical electrons [
27] in the LCC is ignored in our model since the probability of forming a polyyne phase is close to 1. When the trial kink angle is large, the selected carbon atom experiences a non-uniform VDW interaction along the angular ϕ plane. The non-uniform VDW interaction makes the Hamiltonian more positive, which is consistent with the data in the compressed CNT. For example, the selected carbon atom in the LCC orthogonally shifted by 0.01 nm within the (5,4)CNT, increasing the VDW energy by 2% more positive.
Our model assists in the exploration of the chain-breaking process and the growth mechanism of carbyne. An overlength carbyne (exceeding the cut-off length) always breaks into fragments during sample fabrication due to chemical instability [
33]. What happens to an overlength carbyne before it breaks naturally? To answer this question, an overlength carbyne needs to be generated in our simulation model to “visualize” the instantaneous atomic fluctuations right before it breaks. A carbon chain made up of 5500–6000 atoms (~780 nm) ought to be stable inside a (6,4)CNT. When the LCC@(6,4)CNT is compressed by ~4% (
Figure 6), the ~780 nm long internal carbyne becomes overlengthened which may be fragmentized. Nair et al. [
34] justified that a mechanical strain of at least 5% is needed to break a finite-length carbyne. In other words, the carbon chain should break when the
bond length reaches 1.5 Å× (1 + 5%) ~1.6 Å. After the 6000-atom-long LCC@(6,4)CNT is compressed, the seven sharp kink structures are always connected by the
bond lengths above 1.6 Å, which may initiate chain breakage. Hence, an effective covalent bond between sites ‘2’ and ‘3’ (
Figure 6) should vanish. The reestablishment of an effective covalent bond between sites ‘1’ and ‘3’ is unlikely to occur because our ab-initio calculation shows that the carbon chain with a pivot angle around 20 degrees generates a local electrostatic repulsion at least two times stronger than the triple-bond energy. In addition, for the case of a pivot angle ~50 deg (
Figure 6), the carbon atom in site ‘3’ should be unable to form an effective covalent bond with the CNT wall because the radial separation between the carbon atom in ‘site 3’ and the CNT wall is still as large as 1.96 Å. As a result, the LCC in the compressed (6,4)CNT is probably fragmentized into eight pieces during test 1, where the average size of the carbyne fragment is 780 nm/(7 + 1) = 97 nm. The eight short carbyne chains should consist of 103 − 1=102 atoms, 576 − 103 = 473 atoms, 932 − 576 = 356 atoms, 1853 − 932=921 atoms, 4318 − 1853 = 2465 atoms, 4943 − 4318 = 625 atoms, 7505 − 4943 = 762 atoms, and 6000 − 5705 = 295 atoms, respectively. Our model can be used to forecast where the long carbon chain should break at finite temperatures [
33] and the instantaneous atomic configuration of the carbyne right before it breaks. Our model does not probe the further atomic movement of carbyne fragments after the long chain breaks. Based on Ref. [
34], the mechanical properties of carbyne show a size effect up to N~20. If the carbyne fragment is less than 20 atoms long, the atomic spring constant in Hooke’s factor should be revised. However, the shortest carbyne fragment formed by compression still contains over 100 atoms and, hence, Hooke’s factor is still applicable to monitor the fragmentation of the carbyne.
As an example, we investigate if scientists want to use LCC@(5,5)CNT in the transistor for an emerging continuously tunable band gap, where the radius of (5,5)CNT of 0.339 nm is comparable to the radius of (6,4)CNT of 0.341 nm. A metallic (5,5)CNT exists [
36] and the precise control of chiral angles in a CNT enters a new era [
37]. When LCC@(5,5)CNT is unstrained, the cut-off length of the LCC is ~800 nm. After LCC@(5,5)CNT is strained by 1% longitudinally, the cut-off length drops to ~750 nm. To ensure the internal carbyne is stable during the operation, the maximum length of the unstrained LCC@(5,5)CNT should not exceed ~750 nm. The ~750 nm long carbyne in the (5,5)CNT is stable at 1% strain. When the LCC@(5,5)CNT is unstrained, the stability of the LCC is even stronger, based on the analysis in
Figure 5. Hence, neither the 1% strain nor the unstrained condition destroys the internal carbyne, and more encouragingly, the 1% strain should change the band gap of the carbyne by ~15% continuously [
6], which provides precise control in the energy barriers in the transistors.
The low computational cost of our Monte Carlo model is credited to two approximations. The first approximation is that the atomic positions of the nanoreactor (finite-length CNT) in our Monte Carlo model are obtained by scaling the repeating unit in the ab-initio calculations. This speeds up the computational progress because a time-consuming geometric optimization process of a big supercell (over 100,000 atoms in a non-repeating unit) is avoidable. To validate this approximation, we consider the length dependence of the Raman spectrum of a finite-length CNT where the size effect starts to pale for a CNT length longer than ~200 nm [
38]. As the shape of the Raman spectrum is closely related to the atomic positions of materials and the minimum length of the nanoreactor CNT in our model is far above 200 nm, it is arguable that the first approximation will not create a large error in Monte Carlo results. The second approximation is that the Monte Carlo iterations are conducted to the internal chain only. This approximation can be validated by the experimental fact that the atomic fluctuations of the CNT are relatively at rest when compared to the internal chain [
9]. With the second approximation, the size of a one-dimensional array in our Monte Carlo simulation is 4000–15,000 only, which reduces the computational time massively without creating a large error in Monte Carlo results. However, the appropriateness of using our Monte Carlo simulation for modeling carbyne is dependent on the particular scenario. For example, our Monte Carlo simulation may need modifications if it is asked to accurately predict the length of the LCC inside a very short CNT. In contrast, DFT software does not suffer from this issue because any super-cell function in DFT software is designed for studying nanomaterials approaching the size of quantum dots.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}