
Hydrocarbon Sorption in Flexible MOFs—Part I: Thermodynamic Analysis with the Dubinin-Based Universal Adsorption Theory (D-UAT)



Abstract
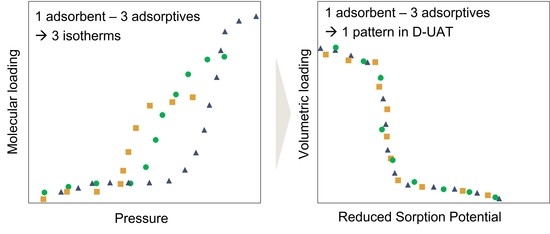
:
1. Introduction
- Applying the D-UAT on an empirical sorption data set with several adsorbents, adsorptives, and temperatures, and evaluate the practical abilities of the theory for visualization, analysis, and prediction of sorption isotherms for both rigid and flexible materials
- Revisit the D-UAT from a purely theoretical point of view and find mathematical proof of its applicability for visualization, analysis, and prediction
2. Materials and Methods
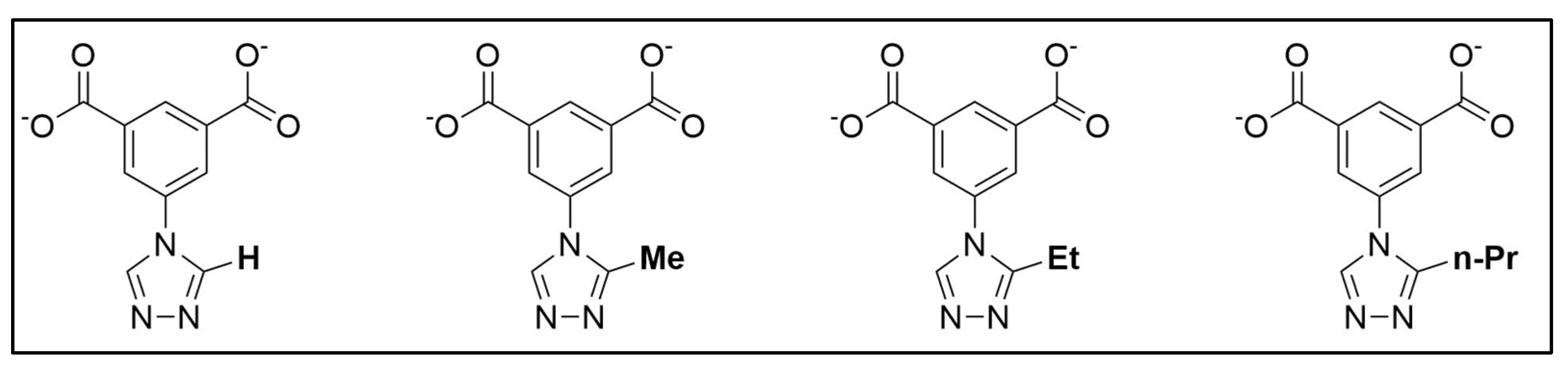
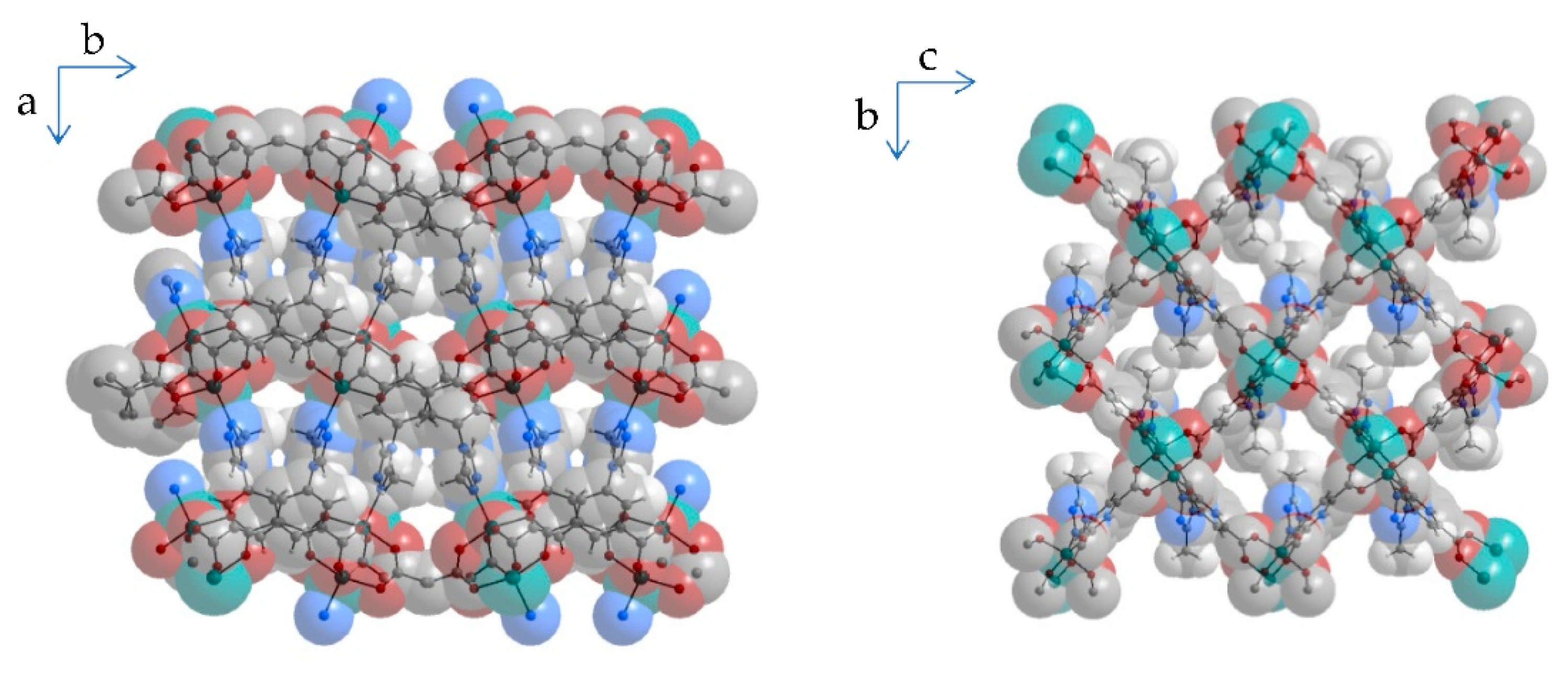
2.1. Materials
2.2. Methods
2.2.1. Sorption Isotherms
2.2.2. Adsorption Enthalpies
2.2.3. Dubinin-Based Universal Adsorption Theory (D-UAT)
3. Results
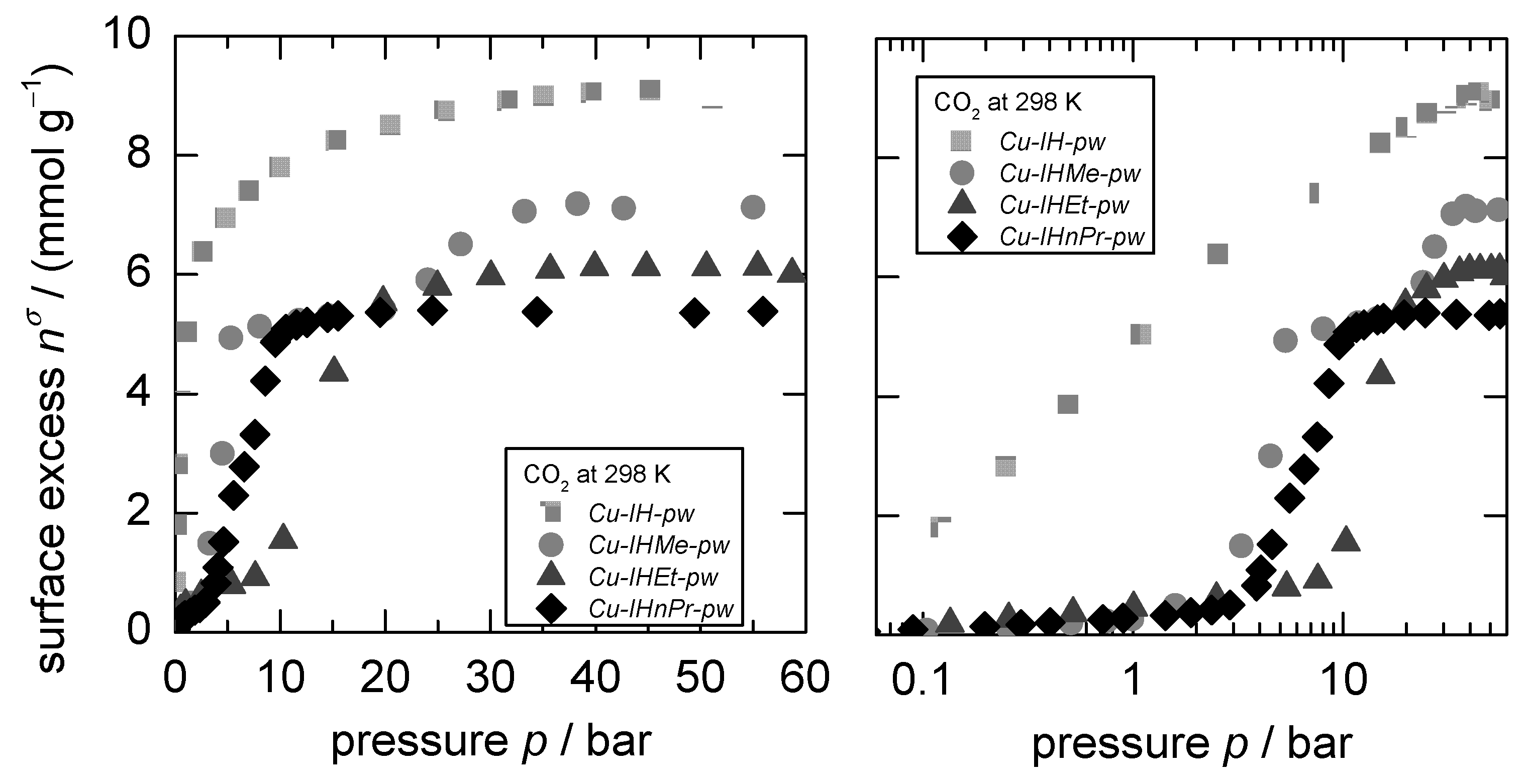
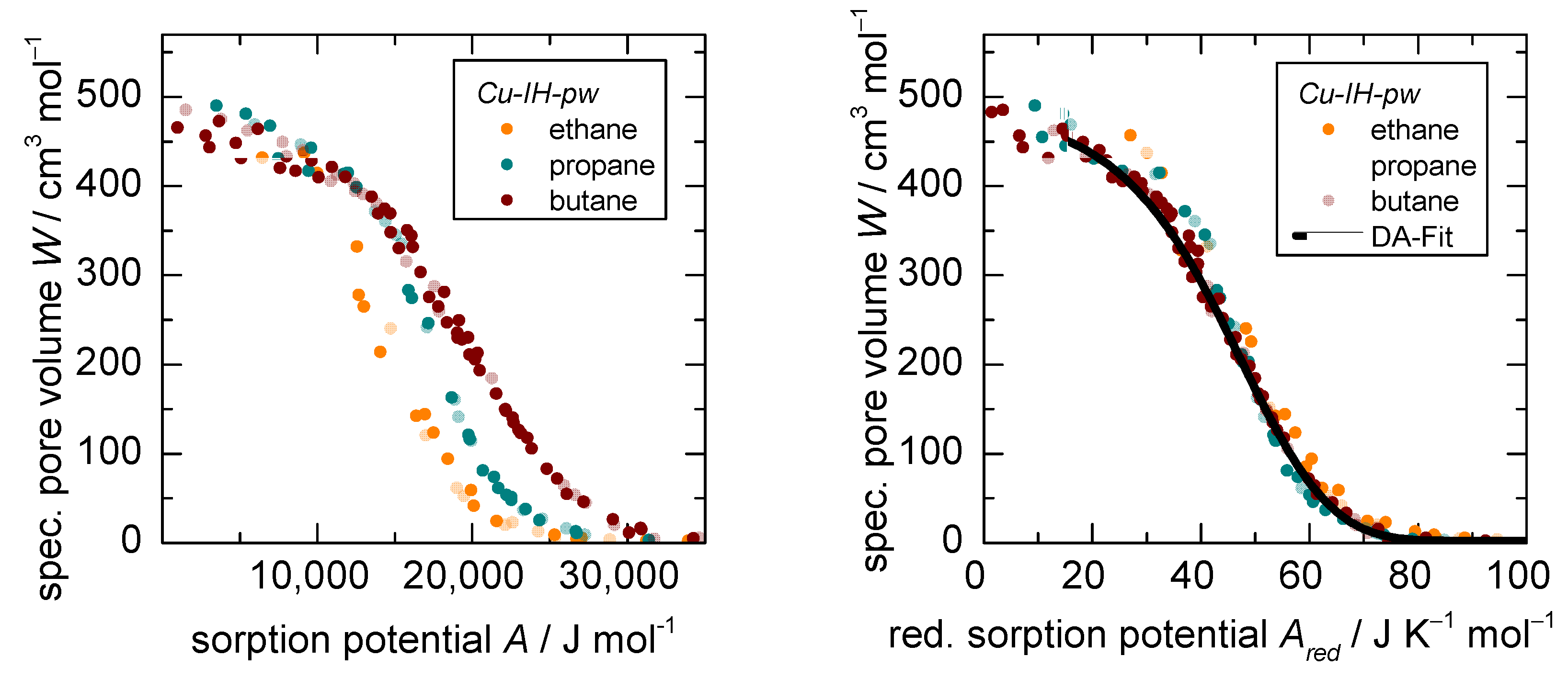
3.1. D-UAT on the Rigid MOF Cu-IH-pw
- The accessible pore volume (derived from the modelling parameter in Equation (3)) and
- The reduced adsorption enthalpy (derived from the modelling parameters and in Equation (3)).
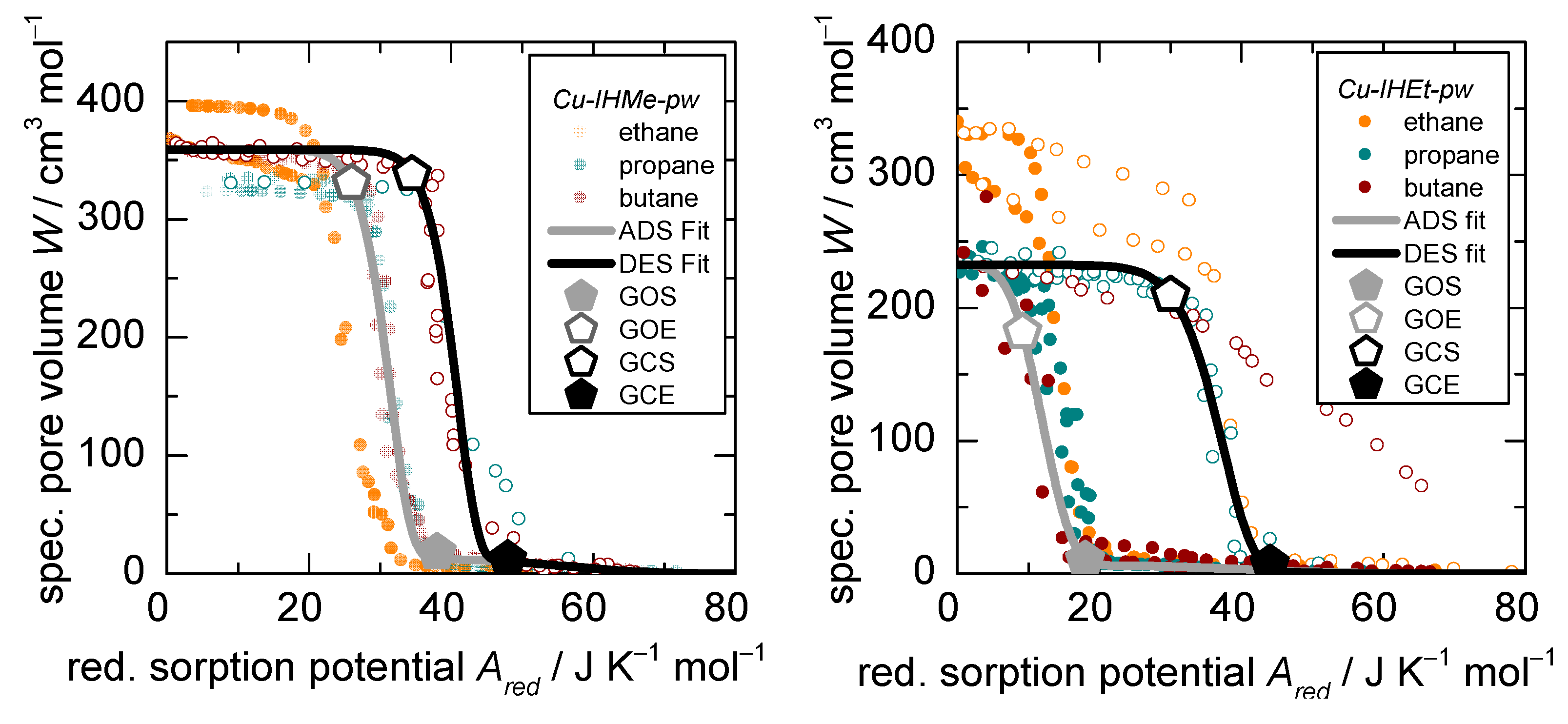
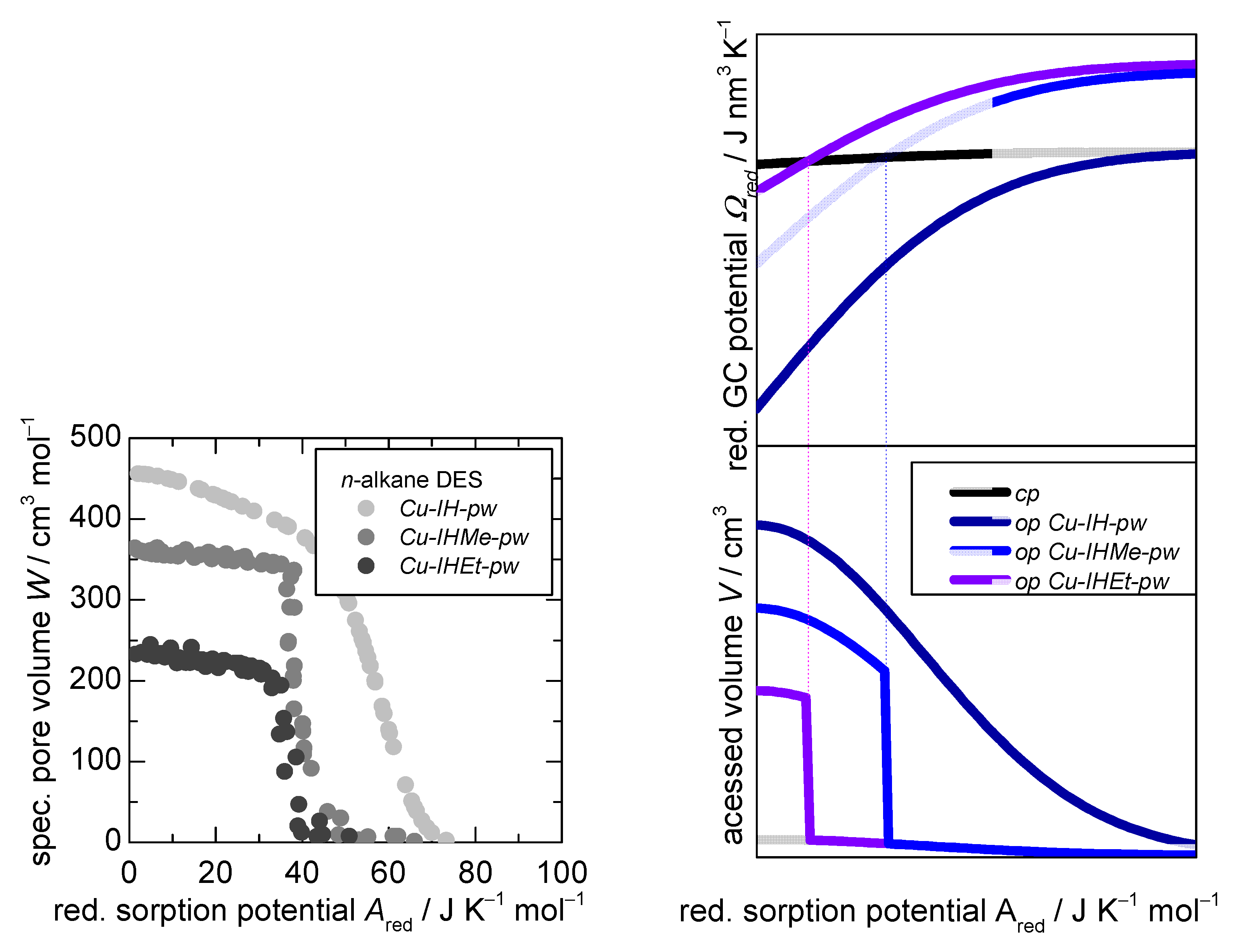
3.2. D-UAT on the Flexible MOFs Cu-IHMe-pw, Cu-IHEt-pw, and Cu-IHnPr-pw
- The accessible pore volumes for (a) open and (b) closed form (derived from the modelling parameters and within Equation (3)),
- The reduced adsorption enthalpies for (a) open and (b) closed form (derived from the modelling parameters / and /) within Equation (3) and
- The resulting reduced energetic offset between the opened and closed structures as indicated by the calculated (, derived from all fitting parameters)
3.3. D-UAT Simulation and Sensitivity Analysis of Governing Parameters , and
- -
- Investigate a potential np phase for the “rigid” Cu-IH-pw under vacuum conditions with in situ PXRD
- -
- Investigate whether ethane is able to open Cu-IHEt-pw to the lp phase rather than the mp phase utilizing in situ PXRD
- -
- Further investigate the kinetic hindrance during the gate-opening and gate-closing processes in dependence of MOF, adsorptive, temperature, and pressure jumps
- -
- Investigate whether the affinity of the MOF series towards olefins can be utilized for alkane–alkene separation processes.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henke, S.; Schneemann, A.; Fischer, R.A. Massive anisotropic thermal expansion and thermo-responsive breathing in metal-organic frameworks modulated by linker functionalization. Adv. Funct. Mater. 2013, 23, 5990–5996. [Google Scholar] [CrossRef]
- Rodriguez, J.; Beurroies, I.; Coulet, M.-V.; Fabry, P.; Devic, T.; Serre, C.; Denoyel, R.; Llewellyn, P.L. Thermodynamics of the structural transition in metal–organic frameworks. Dalton Trans. 2016, 45, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Ghoufi, A.; Benhamed, K.; Boukli-Hacene, L.; Maurin, G. Electrically induced breathing of the MIL-53(Cr) metal-organic framework. ACS Cent. Sci. 2017, 3, 394–398. [Google Scholar] [CrossRef]
- Schmid, R. An electric field induced breath for metal-organic frameworks. ACS Cent. Sci. 2017, 3, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Serre, C.; Millange, F.; Thouvenot, C.; Noguès, M.; Marsolier, G.; Louër, D.; Férey, G. Very large breathing effect in the first nanoporous Chromium(III)-based solids. J. Am. Chem. Soc. 2002, 124, 13519–13526. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.J.; Thomas, K.M.; Rosseinsky, M.J. Flexibility in metal-organic framework materials: Impact on sorption properties. J. Solid State Chem. 2005, 178, 2491–2510. [Google Scholar] [CrossRef]
- Coudert, F.-X.; Jeffroy, M.; Fuchs, A.H.; Boutin, A.; Mellot-Draznieks, C. Thermodynamics of guest-induced structural transitions in hybrid organic-inorganic frameworks. J. Am. Chem. Soc. 2008, 130, 14294–14302. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, Y.; Li, D.-S.; Bu, X.; Feng, P. Metal-organic frameworks for separation. Adv. Mater. 2018, 30, 1705189–1705223. [Google Scholar] [CrossRef]
- Herm, Z.R.; Bloch, E.D.; Long, J.R. Hydrocarbon separations in metal–organic frameworks. Chem. Mater. 2014, 26, 323–338. [Google Scholar] [CrossRef]
- Mason, J.A.; Oktawiec, J.; Taylor, M.K.; Hudson, M.R.; Rodriguez, J.; Bachman, J.E.; Gonzalez, M.I.; Cervellino, A.; Guagliardi, A.; Brown, C.M.; et al. Methane storage in flexible metal-organic frameworks with intrinsic thermal management. Nature 2015, 527, 357–361. [Google Scholar] [CrossRef]
- Bozbiyik, B.; Lannoeye, J.; de Vos, D.E.; Baron, G.V.; Denayer, J.F.M. Shape selective properties of the Al-fumarate metal-organic framework in the adsorption and separation of n-alkanes, iso-alkanes, cyclo-alkanes and aromatic hydrocarbons. Phys. Chem. Chem. Phys. 2016, 18, 3294–3301. [Google Scholar] [CrossRef] [PubMed]
- Farrusseng, D.; Aguado, S.; Pinel, C. Metal-organic frameworks: Opportunities for catalysis. Angew. Chem. Int. Ed. 2009, 48, 7502–7513. [Google Scholar] [CrossRef] [PubMed]
- Stassen, I.; Burtch, N.; Talin, A.; Falcaro, P.; Allendorf, M.; Ameloot, R. An updated roadmap for the integration of metal-organic frameworks with electronic devices and chemical sensors. Chem. Soc. Rev. 2017, 46, 3185–3241. [Google Scholar] [CrossRef]
- Horcajada, P.; Serre, C.; Maurin, G.; Ramsahye, N.A.; Balas, F.; Vallet-Regí, M.; Sebban, M.; Taulelle, F.; Férey, G. Flexible porous metal-organic frameworks for a controlled drug delivery. J. Am. Chem. Soc. 2008, 130, 6774–6780. [Google Scholar] [CrossRef]
- The Cambridge Structural Database (CSD)—The Cambridge Crystallographic Data Centre (CCDC). Available online: https://www.ccdc.cam.ac.uk/solutions/csd-system/components/csd/ (accessed on 22 September 2019).
- Wilmer, C.E.; Farha, O.K.; Bae, Y.-S.; Hupp, J.T.; Snurr, R.Q. Structure–property relationships of porous materials for carbon dioxide separation and capture. Energy Environ. Sci. 2012, 5, 9849. [Google Scholar] [CrossRef]
- Wilmer, C.E.; Leaf, M.; Lee, C.Y.; Farha, O.K.; Hauser, B.G.; Hupp, J.T.; Snurr, R.Q. Large-scale screening of hypothetical metal-organic frameworks. Nat. Chem. 2011, 4, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Miyahara, M.T. Free energy calculations for adsorption-induced deformation of flexible metal–organic frameworks. Curr. Opin. Chem. Eng. 2019, 24, 19–25. [Google Scholar] [CrossRef]
- Vanduyfhuys, L.; Rogge, S.M.J.; Wieme, J.; Vandenbrande, S.; Maurin, G.; Waroquier, M.; van Speybroeck, V. Thermodynamic insight into stimuli-responsive behaviour of soft porous crystals. Nat. Commun. 2018, 9, 204. [Google Scholar] [CrossRef]
- Ghysels, A.; Vanduyfhuys, L.; Vandichel, M.; Waroquier, M.; van Speybroeck, V.; Smit, B. On the thermodynamics of framework breathing: A Free Energy Model for Gas Adsorption in MIL-53. J. Phys. Chem. C 2013, 117, 11540–11554. [Google Scholar] [CrossRef]
- Fraux, G.; Coudert, F.-X. Recent advances in the computational chemistry of soft porous crystals. Chem. Commun. 2017, 53, 7211–7221. [Google Scholar] [CrossRef]
- Yamazaki, T.; Takahashi, Y.; Yoshida, D. Adsorption of several gases on flexible metal organic framework Cu(dhbc)2(4,4′-bpy)·H2O. J. Colloid Interface Sci. 2011, 362, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Sircar, S.; Pramanik, S.; Li, J.; Cole, M.W.; Lueking, A.D. Corresponding states interpretation of adsorption in gate-opening metal-organic framework Cu(dhbc)2(4,4′-bpy). J. Colloid Interface Sci. 2015, 446, 177–184. [Google Scholar] [CrossRef]
- Quinn, D.F. Supercritical adsorption of ‘permanent’ gases under corresponding states on various carbons. Carbon 2002, 40, 2767–2773. [Google Scholar] [CrossRef]
- Angela, D. Lueking. A corresponding states principle for physisorption and deviations for quantum fluids. Mol. Phys. 2008, 106, 1579–1585. [Google Scholar]
- Kobalz, M.; Lincke, J.; Kobalz, K.; Erhart, O.; Bergmann, J.; Lässig, D.; Lange, M.; Möllmer, J.; Gläser, R.; Staudt, R.; et al. Paddle wheel based triazolyl isophthalate MOFs: Impact of linker modification on crystal structure and gas sorption properties. Inorg. Chem. 2016, 55, 3030–3039. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.U.; Staudt, R. Gas Adsorption Equilibria: Experimental Methods and Adsorption Isotherms; Springer: New York, NY, USA, 2004. [Google Scholar]
- Dubinin, M.M. The potential theory of adsorption of gases and vapors for adsorbents with energetically nonuniform surfaces. Chem. Rev. 1960, 60, 235–242. [Google Scholar] [CrossRef]
- Johnston, D.C. Thermodynamic properties of the van der Waals fluid. In Advances in Thermodynamics of the van der Waals Fluid; Morgan&Claypool: San Rafael, CA, USA, 2014. [Google Scholar]
- Burevski, D. The application of the Dubinin-Astakhov equation to the characterization of microporous carbons. Colloid Polym. Sci. 1982, 260, 623–627. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Do, D.D. Adsorption Analysis: Equilibria and Kinetics; Imperial College Press: London, UK, 1998; ISBN 978-1-86094-130-6. [Google Scholar]
- Numaguchi, R.; Tanaka, H.; Hiraide, S.; Miyahara, M.T. Potential theory for gate adsorption on soft porous crystals. Mol. Simul. 2015, 41, 1329–1338. [Google Scholar] [CrossRef]
- Adolphs, J. Excess surface work?: A modelless way of getting surface energies and specific surface areas directly from sorption isotherms. App. Surf. Sci. 2007, 253, 5645–5649. [Google Scholar] [CrossRef]
- Couck, S.; van Assche, T.R.C.; Liu, Y.-Y.; Baron, G.V.; van der Voort, P.; Denayer, J.F.M. Adsorption and separation of small hydrocarbons on the flexible, vanadium-containing MOF, COMOC-2. Langmuir 2015, 31, 5063–5070. [Google Scholar] [CrossRef] [PubMed]
- Hähnel, T.; Kalies, G.; Krishna, R.; Möllmer, J.; Hofmann, J.; Kobalz, M.; Krautscheid, H. Adsorptive separation of C2/C3/C4-hydrocarbons on a flexible Cu-MOF: The influence of temperature, chain length and bonding character. Micropor. Mesopor. Mat. 2016, 224, 392–399. [Google Scholar] [CrossRef]
- Sircar, S.; Wu, H.; Li, J.; Lueking, A.D. Effect of time, temperature, and kinetics on the hysteretic adsorption-desorption of H2, Ar, and N2 in the metal-organic framework Zn2(bpdc)2(bpee). Langmuir 2011, 27, 14169–14179. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, D.; Coudert, F.-X.; Fossati, A.G.J.; Neimark, A.V.; Fuchs, A.H.; Boutin, A. Adsorption induced transitions in soft porous crystals: An osmotic potential approach to multistability and intermediate structures. J. Chem. Phys. 2013, 138, 174706. [Google Scholar] [CrossRef]
- Boutin, A.; Springuel-Huet, M.-A.; Nossov, A.; Gédéon, A.; Loiseau, T.; Volkringer, C.; Férey, G.; Coudert, F.-X.; Fuchs, A.H. Breathing transitions in MIL-53(Al) metal-organic framework upon xenon adsorption. Angew. Chem. Int. Ed. 2009, 48, 8314–8317. [Google Scholar] [CrossRef]
- de Toni, M.; Pullumbi, P.; Coudert, F.-X.; Fuchs, A.H. Understanding the effect of confinement on the liquid–gas transition: A study of adsorption isotherms in a family of metal–organic frameworks. J. Phys. Chem. C 2010, 114, 21631–21637. [Google Scholar] [CrossRef]
- Ortiz, A.U.; Springuel-Huet, M.-A.; Coudert, F.-X.; Fuchs, A.H.; Boutin, A. Predicting mixture coadsorption in soft porous crystals: Experimental and theoretical Study of CO2/CH4 in MIL-53(Al). Langmuir 2012, 28, 494–498. [Google Scholar] [CrossRef]
- Sircar, S.; Myers, A.L. Characteristic adsorption isotherm for adsorption of vapors on heterogeneous adsorbents. AIChE J. 1986, 32, 650–656. [Google Scholar] [CrossRef]
- Myers, A.L.; Prausnitz, J.M. Prediction of the adsorption isotherm by the principle of corresponding states. Chem. Eng. Sci. 1965, 20, 549–556. [Google Scholar] [CrossRef]
- Myers, A.L. Thermodynamics of adsorption in porous materials. AIChE J. 2002, 48, 145–160. [Google Scholar] [CrossRef]
- Numaguchi, R.; Tanaka, H.; Watanabe, S.; Miyahara, M.T. Simulation study for adsorption-induced structural transition in stacked-layer porous coordination polymers: Equilibrium and hysteretic adsorption behaviors. J. Chem. Phys. 2013, 138, 54708. [Google Scholar] [CrossRef] [PubMed]
- Kobalz, M. Metal-Organic Frameworks Basierend auf 1,2,4-Triazolylisophtalaten und -benzoaten: Einfluss von Liganden- und Metallionensubstitution auf Struktur und Adsorptionseigenschaften. Ph.D. Thesis, Universität Leipzig, Leipzig, Germany, 24 August 2016. [Google Scholar]
- Lange, M.; Kobalz, M.; Bergmann, J.; Lässig, D.; Lincke, J.; Möllmer, J.; Möller, A.; Hofmann, J.; Krautscheid, H.; Staudt, R.; et al. Structural flexibility of a copper-based metal–organic framework: Sorption of C4-hydrocarbons and in situ XRD. J. EIMater. Chem. A 2014, 2, 8075. [Google Scholar] [CrossRef]
- Coelho, A. TOPAS; Bruker AXS GmbH: Karlsruhe, Germany, 2014. [Google Scholar]
- Larson, A.C.; Von Dreele, R.B. General Structure Analysis System (GSAS); Los Alamos National Laboratory Report LAUR 86-748; The Regents of the University of California: Los Alamos, NM, USA, 2004. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Unit | Cu-IHMe-pw | Cu-IHEt-pw |
|---|---|---|---|
| 38 | 19 | ||
| 26 | 11 | ||
| 30 | 14 | ||
| 35 | 30 | ||
| 44 | 42 | ||
| 41 | 37 | ||
| 3.8 | 4.3 | ||
| 3.9 | 4.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Preißler-Kurzhöfer, H.; Lange, M.; Kolesnikov, A.; Möllmer, J.; Erhart, O.; Kobalz, M.; Krautscheid, H.; Gläser, R. Hydrocarbon Sorption in Flexible MOFs—Part I: Thermodynamic Analysis with the Dubinin-Based Universal Adsorption Theory (D-UAT). Nanomaterials 2022, 12, 2415. https://doi.org/10.3390/nano12142415
Preißler-Kurzhöfer H, Lange M, Kolesnikov A, Möllmer J, Erhart O, Kobalz M, Krautscheid H, Gläser R. Hydrocarbon Sorption in Flexible MOFs—Part I: Thermodynamic Analysis with the Dubinin-Based Universal Adsorption Theory (D-UAT). Nanomaterials. 2022; 12(14):2415. https://doi.org/10.3390/nano12142415
Chicago/Turabian StylePreißler-Kurzhöfer, Hannes, Marcus Lange, Andrei Kolesnikov, Jens Möllmer, Oliver Erhart, Merten Kobalz, Harald Krautscheid, and Roger Gläser. 2022. "Hydrocarbon Sorption in Flexible MOFs—Part I: Thermodynamic Analysis with the Dubinin-Based Universal Adsorption Theory (D-UAT)" Nanomaterials 12, no. 14: 2415. https://doi.org/10.3390/nano12142415