
Synthetic Routes and Clinical Application of Representative Small-Molecule EGFR Inhibitors for Cancer Therapy

1
First People’s Hospital of Shangqiu, Shangqiu 476100, China
2
Key Laboratory of Natural Medicines of the Changbai Mountain, Ministry of Education, College of Pharmacy, Yanbian University, Yanji 133002, China
3
College of Chemistry and Chemical Engineering, Zhengzhou Normal University, Zhengzhou 450044, China
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2024, 29(7), 1448; https://doi.org/10.3390/molecules29071448
Submission received: 7 December 2023
/
Revised: 20 March 2024
/
Accepted: 21 March 2024
/
Published: 23 March 2024
(This article belongs to the Special Issue Synthesis and Evaluation of Bioactivity of Enzyme Inhibitors)
Abstract
:The epidermal growth factor receptor (EGFR) plays a pivotal role in cancer therapeutics, with small-molecule EGFR inhibitors emerging as significant agents in combating this disease. This review explores the synthesis and clinical utilization of EGFR inhibitors, starting with the indispensable role of EGFR in oncogenesis and emphasizing the intricate molecular aspects of the EGFR-signaling pathway. It subsequently provides information on the structural characteristics of representative small-molecule EGFR inhibitors in the clinic. The synthetic methods and associated challenges pertaining to these compounds are thoroughly examined, along with innovative strategies to overcome these obstacles. Furthermore, the review discusses the clinical applications of FDA-approved EGFR inhibitors such as erlotinib, gefitinib, afatinib, and osimertinib across various cancer types and their corresponding clinical outcomes. Additionally, it addresses the emergence of resistance mechanisms and potential counterstrategies. Taken together, this review aims to provide valuable insights for researchers, clinicians, and pharmaceutical scientists interested in comprehending the current landscape of small-molecule EGFR inhibitors.
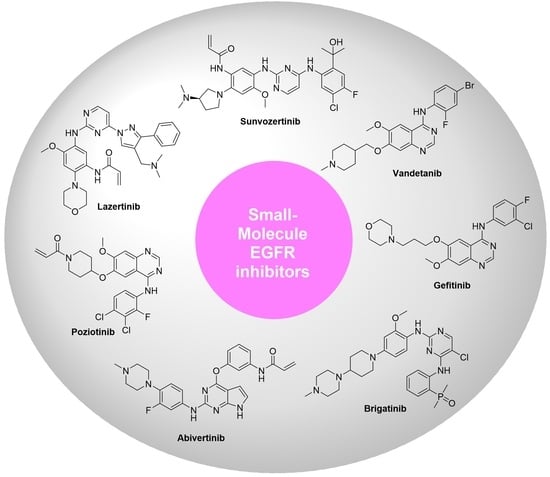
1. Introduction
EGFR, also known as ErbB1 or HER1, belongs to the ErbB family of receptor tyrosine kinases (RTKs). Structurally, EGFR consists of an extracellular ligand-binding domain, a single transmembrane domain, an intracellular tyrosine kinase domain, and a C-terminal tail [1]. Upon ligand binding, EGFR undergoes conformational changes that lead to dimerization and activation of its intrinsic kinase activity, resulting in autophosphorylation of specific tyrosine residues within the receptor’s intracellular domain. This phosphorylation event initiates downstream signaling cascades involved in cell proliferation, survival, and differentiation. Crystallographic studies have provided detailed insights into the structure of EGFR and its interactions with ligands and inhibitors [2]. The crystal structure of the EGFR kinase domain complexed with inhibitors such as gefitinib and erlotinib has been elucidated, revealing the binding mode of these small molecules within the ATP-binding pocket of the kinase domain [3,4]. Additionally, structural studies have elucidated the mechanisms of resistance mutations, such as the T790M gatekeeper mutation, which affects the binding affinity of certain EGFR inhibitors [5,6,7]. The identification and advancement of small-molecule inhibitors aimed at the EGFR have significantly transformed the therapeutic paradigm across multiple cancer types, notably within non-small-cell lung cancer (NSCLC) [8,9,10]. EGFR inhibitors have demonstrated remarkable clinical efficacy, significantly improving patient outcomes. This review delves into the synthesis and application of representative small-molecule EGFR inhibitors, shedding light on the historical evolution, mechanisms of action, pharmacological properties, and therapeutic implications of these agents.
The journey of EGFR inhibitors began with the identification of EGFR as a key player in cancer progression. Subsequent research efforts led to the discovery of small molecules capable of inhibiting EGFR signaling. Significant strides in the advancement of EGFR inhibitors encompass pivotal landmarks marked by the discovery of activating mutations, notably the L858R substitution and Exon 19 deletions, which render tumors highly responsive to these agents [11]. The advent of initial EGFR inhibitors such as gefitinib and erlotinib represented a pivotal advancement, presenting a promising prospect for individuals afflicted with EGFR-mutated NSCLC [12]. EGFR inhibitors exert their primary mechanism through the inhibition of the tyrosine kinase activity inherent in EGFR. This action effectively hampers the progression of downstream signaling cascades crucially implicated in processes related to cell proliferation, survival, and the regulation of angiogenesis [13]. The interaction between these diminutive compounds and the intracellular kinase domain of EGFR obstructs the process of autophosphorylation, consequently impeding the activation of subsequent cascades, including the Raf/Ras/ERK/MEK and PI3K/Akt/mTOR pathways [14]. Additionally, third-generation EGFR inhibitors like osimertinib exhibit unique mechanisms of action, effectively targeting T790M resistance mutations in EGFR [15].
Understanding the pharmacological properties of EGFR inhibitors is crucial for optimizing their clinical use. Factors like pharmacokinetics, bioavailability, and drug–drug interactions influence dosing regimens and treatment outcomes. Some EGFR inhibitors, like afatinib, are irreversible binders, while others, like osimertinib, are additionally highly selective for mutant EGFR variants, minimizing off-target effects [16]. These agents exhibit varying pharmacokinetic profiles, which impact their efficacy and tolerability [17].
The advent of clinically endorsed EGFR inhibitors has revolutionized the landscape of cancer treatment, yielding significant enhancements in both progression-free survival and overall survival among individuals afflicted with EGFR-mutated tumors [18]. These therapies have become standard-of-care for EGFR-positive NSCLC and are also being explored in other malignancies with EGFR alterations. Moreover, recent advances in liquid biopsies and patient stratification enable precise identification of EGFR mutations, facilitating personalized treatment approaches [19]. The synthesis and application of representative small-molecule EGFR inhibitors are regarded as a remarkable achievement in the field of oncology (Table 1 and Figure 1). These agents have redefined cancer treatment paradigms and significantly improved patient outcomes. Understanding their historical evolution, mechanisms of action, pharmacological properties, and therapeutic implications is essential for optimizing their use and driving further advancements in precision medicine.
Among the current tyrosine kinase inhibitors (TKIs) targeting EGFR, the majority were developed with the aid of structure-based drug-discovery techniques. Specifically, gefitinib, erlotinib, afatinib, and osimertinib are prominent examples of TKIs that benefited from structure-based drug-design approaches [1,17,20,21]. Furthermore, a significant portion of these TKI drugs were optimized from lead compounds using structure-based docking and analysis methods. For instance, afatinib and osimertinib underwent optimization through structure-based drug-design strategies, leveraging molecular docking and structure-activity relationship studies to enhance their potency and selectivity against EGFR mutations [17,21]. These examples underscore the pivotal role of structure-based drug discovery in both the development and optimization of TKI drugs targeting EGFR, contributing to their clinical efficacy and therapeutic success.
2. EGFR and Its Signal Transduction
EGFR signaling is a complex process involving a series of phosphorylation events and downstream signaling pathways. Here is a simplified overview of the signal transduction process—Ligand Binding (Figure 2): Upon ligand engagement, such as the union of EGF to the extracellular section of EGFR, a consequential conformational alteration in the receptor is initiated. Dimerization: The induced conformational alteration upon ligand binding to the extracellular domain of EGFR triggers and facilitates the dimerization process of the receptor, thereby effectively aligning the intracellular kinase domains in close spatial proximity. Auto-phosphorylation: The intracellular kinase modules inherent in EGFR catalyze the phosphorylation of distinct tyrosine residues present on the receptor itself. These phosphorylated tyrosine moieties function as pivotal sites for the recruitment and interaction with downstream signaling proteins [22]. Activation of downstream signaling: The activation of EGFR through phosphorylation of its tyrosine residues facilitates the recruitment of diverse adaptor proteins and enzymes. This assemblage notably encompasses pivotal components such as Grb2, Shc, and the consequential engagement of the Raf–Ras–MEK–MAPK pathway. Such interactions orchestrate intricate signaling cascades integral to the regulation of cellular growth and proliferation. Additionally, PI3K–Akt and STAT pathways are activated, influencing cell survival and gene expression [13]. Cellular responses: The varied cellular responses triggered by the activation of downstream signaling pathways following EGFR activation are indeed dependent on various factors, including the specific ligand, the context of cellular signaling, and the interplay between different signaling pathways. Ligand specificity: Different ligands binding to EGFR can activate distinct downstream signaling pathways. For example, while EGF primarily activates the MAPK pathway, other ligands like transforming growth factor alpha (TGF-α) may preferentially activate the PI3K–Akt pathway. This ligand specificity can lead to different cellular responses based on the particular ligand bound to EGFR. The cellular context, including the presence of other growth factors, cytokines, or environmental cues, can modulate the downstream signaling responses initiated by EGFR activation. For instance, the presence of certain growth factors may synergistically enhance or inhibit the effects of EGFR signaling, leading to context-dependent cellular responses. Interaction between pathways like MAPK and PI3K-Akt can modify cellular responses initiated by EGFR activation. Responses vary based on cell type and differentiation status, with EGFR signaling promoting proliferation in cancer cells but regulating normal cell growth and repair. In some cases, EGFR signaling can also influence cell migration and invasion [23].
3. Clinically Approved Representative Small-Molecule TKIs of EGFR
3.1. Sunvozertinib
Sunvozertinib, a novel TKI manufactured by Dizal Pharmaceuticals, represents an advancement arising from the need to overcome resistance mechanisms and thereby replaces previous generations of EGFR inhibitors. As marketed under the proprietary name DZD9008, this therapeutic entity embodies an innovative modality aimed at addressing NSCLC characterized by distinct mutations within the EGFR gene [24]. The pharmacological efficacy of sunvozertinib is predicated upon its specific capacity to inhibit EGFR with Exon 20 mutations, alongside targeting human epidermal growth factor receptor 2 (HER2) with Exon 20 insertions. This targeted inhibition is of significance due to the diminished responsiveness of cancer cells with these mutations to earlier generations of EGFR inhibitors. By competitively obstructing the ATP-binding site of these mutated tyrosine kinases, sunvozertinib effectively hinders the proliferative signaling pathways, exhibiting potent anti-tumor activity (ClinicalTrials.gov Identifier: NCT03974022). Preclinical models have demonstrated sunvozertinib’s capacity to inhibit tumor growth, especially in NSCLC models harboring Exon 20 insertions. Moreover, its substantiated ability to traverse the blood–brain barrier has provided favorable support for its prospective efficacy in managing central nervous system metastases. In clinical settings, sunvozertinib has shown promising efficacy, with ongoing trials further assessing its utility as a targeted intervention for individuals presenting specific EGFR mutations. The toxicity profile has been comparable to other EGFR inhibitors, with manageable side effects that do not significantly diminish its therapeutic value [25].
The preparation of sunvozertinib is shown in Scheme 1 [26]. SUNV-001 and SUNV-002 engage in nucleophilic substitution reactions under alkaline conditions, yielding the formation of SUNV-003 through a subsequent nucleophilic substitution process involving SUNV-004, ultimately leading to the generation of SUNV-005. SUNV-005 and SUNV-006 consecutively engage in nucleophilic substitution reactions, culminating in the formation of SUNV-007. The nitro moiety present in SUNV-007 undergoes a reduction process to yield an amino functional group, through the utilization of hydrogen gas as the reducing agent and platinum carbon as the catalytic mediator, ultimately affording the formation of SUNV-008. The amino moiety present in SUNV-008 and the acyl chloride functionality of SUNV-009 engage in a condensation reaction, leading to the formation of the amide compound SUNV-010. SUNV-010 is subjected to an elimination reaction in alkaline environments, leading to the formation of sunvozertinib.
3.2. Mobocertinib Succinate
Mobocertinib succinate, developed by Takeda Oncology, represents an orally administered TKI strategically engineered to selectively act upon EGFR with Exon 20 insertion mutations identified in NSCLC. Marketed under the brand name Exkivity, it was developed in response to the unmet medical need for effective treatments for NSCLC patients harboring these specific mutations [27]. Mobocertinib exhibits its therapeutic activity by selectively inhibiting the EGFR Exon 20 insertions, thereby obstructing aberrant signaling pathways that promote cancer cell proliferation. Its unique structure allows for targeted activity, minimizing the interaction with wild-type EGFR and reducing off-target effects [28]. Preclinical studies have shown that mobocertinib possessed significant antitumor efficacy in cell lines and animal models specifically engineered to express EGFR Exon 20 insertions, indicating its robust preclinical pharmacodynamics [29]. Clinically, mobocertinib has exhibited notable efficacy by eliciting tumor reduction in a distinct subgroup of individuals afflicted with advanced NSCLC featuring these infrequent genetic mutations. The endorsement by the U.S. Food and Drug Administration (FDA) stemmed from the outcomes observed in a comprehensive phase 1/2 clinical trial, emphasizing a noteworthy response rate and sustained response durability as pivotal determinants for approval [30]. Side effects noted include diarrhea, rash, and paronychia, which are consistent with the known toxicity profile of EGFR inhibitors.
The synthetic method of mobocertinib succinate is shown in Scheme 2 [31]. The compounds MOBO-001 and MOBO-002 are subjected to a nucleophilic substitution reaction, resulting in the formation of MOBO-003. Subsequently, MOBO-003 engages in a nucleophilic substitution reaction with MOBO-004, yielding the synthesis of MOBO-005. The nitro moieties within MOBO-005 undergo hydrogenation and subsequent reduction to amino functionalities employing palladium catalysis, yielding the compound denoted as MOBO-006. The condensation reaction between the amino group and carboxylic acid functional moieties, specifically denoted as MOBO-007 and MOBO-006, respectively, culminates in the formation of MOBO-008. MOBO-008 undergoes a process of elimination when exposed to robust alkaline conditions, resulting in the formation of MOBO-009. The compound MOBO-009 was finally converted to the corresponding succinate.
3.3. Alflutinib
Alflutinib, also known as AST2818, represents a third-generation EGFR TKI strategically formulated for the therapeutic intervention in NSCLC. It is tailored to selectively address both the prevalent EGFR-sensitive mutations and the challenging T790M resistance mutation, acknowledged as pivotal pathogenic mechanisms in NSCLC progression [32]. Alflutinib was engineered to selectively bind to mutant forms of EGFR, sparing the wild-type receptor. This selectivity intends to optimize its anticancer efficacy by minimizing adverse effects attributed to the inhibition of wild-type EGFR in non-cancerous cells. In preclinical studies, alflutinib demonstrated potent activity against NSCLC cell lines harboring EGFR mutations, indicating promising preclinical pharmacodynamics. In diverse in vitro and in vivo models, the drug demonstrated efficacy, resulting in the attenuation of neoplastic proliferation. Clinical trials have reported that alflutinib exhibits substantial efficacy in individuals afflicted with NSCLC, particularly those carrying the EGFR T790M mutation, which is associated with resistance to earlier-generation EGFR inhibitors [33,34]. However, as with many TKIs, alflutinib has been associated with adverse events, including diarrhea and rash, though its toxicity profile is considered manageable with appropriate supportive care.
The synthetic approach utilized for the synthesis of alflutinib is elucidated within the framework of Scheme 3 [35]. ALFT-001 reacts with ALFT-002, in the presence of sodium hydride dissolved in tetrahydrofuran (THF), culminating in the formation of ALFT-003. Afterward, the nitro moiety within ALFT-003 undergoes reduction, leading to the generation of ALFT-004. ALFT-004 undergoes a chemical transformation upon reaction with acetyl chloride under the catalytic influence of N,N-diisopropylethylamine (DIPEA) in dichloromethane (DCM), affording the formation of ALFT-005. Subsequent nitration of ALFT-005 using nitric acid and trifluoroacetic acid anhydride (TFAA) leads to the synthesis of ALFT-006. Subsequently, the compound ALFT-006 engages in a chemical transformation with ALFT-007, yielding the product ALFT-008. A subsequent conversion of ALFT-008 into ALFT-009 is accomplished through treatment with methanol in the presence of hydrochloric acid (HCl). Next, the synthesis of ALFT-011 is accomplished through the chemical transformation involving the reaction of ALFT-009 with ALFT-010. The nitro moiety of Compound ALFT-011 was reduced using hydrogen in the presence of platinum oxide as a catalyst to result in ALFT-012. In the final step of the synthetic pathway, ALFT-012 undergoes a chemical transformation through its reaction with acryloyl chloride under the influence of triethylamine (TEA) within DCM, culminating in the emergence of alflutinib.
3.4. Lazertinib
Lazertinib, marketed as Leclaza, is a third-generation EGFR inhibitor specifically designed to target EGFR mutations, with a particular focus on the T790M mutation. It was developed to combat resistance mechanisms by selectively inhibiting both EGFR mutations and sparing wild-type EGFR to reduce off-target effects [36]. Mechanistically, lazertinib works by binding to the ATP-binding site of mutant EGFRs, preventing autophosphorylation and subsequent activation of downstream pro-survival signaling pathways. Preclinically, lazertinib showed a potent inhibition of tumor growth in cell lines and animal models possessing EGFR mutations, demonstrating effective pharmacodynamics. This drug demonstrated significant efficacy in clinical trials, particularly showing a positive response in NSCLC patients with EGFR mutations, specifically those who acquired T790M mutations after prior treatment with initial or second-generation EGFR inhibitors [37]. The toxicity profile of lazertinib has been noted to include manageable side effects typical of EGFR inhibitors, such as rash and diarrhea, without the severe cardiotoxicity or interstitial lung disease often associated with this drug class.
The synthetic strategy employed for the preparation of lazertinib is outlined in Scheme 4 [38]. LAZE-001 undergoes nucleophilic substitution by LAZE-002, leading to the formation of LAZE-003. Subsequently, the amino moiety within LAZE-003 undergoes formaldehyde-mediated transformation, yielding LAZE-004. A reduction of the nitro group of LAZE-004 via hydrogenation using Pd/C as a catalyst provides the corresponding aminoderivative LAZE-005. Following the condensation process, the compound LAZE-005 and the acyl chloride LAZE-006 experience elimination reactions when subjected to alkaline conditions, ultimately yielding the formation of LAZE-007. In the context of alkaline conditions, the compounds LAZE-007 and LAZE-008 engage in a condensation reaction, resulting in the generation of LAZE-009. Subsequently, the aldehyde moiety within the molecular structure of LAZE-009 is subjected to reductive amination, resulting in the synthesis of lazertinib.
3.5. Dacomitinib
Dacomitinib, developed by Pfizer, represents a second-generation EGFR TKI. The mechanism of action for dacomitinib differs from that of first-generation TKIs as it forms an irreversible bond with the ATP binding site of the EGFR, resulting in a sustained inhibition of kinase activity. This irreversible binding mechanism is ascribed to the formation of a covalent bond with the cysteine residue situated within the ATP-binding pocket of EGFR, as well as its homologous family members HER2 and HER4 [39]. The FDA awarded dacomitinib (VIZIMPRO) with orphan drug designation in 2018. This endorsement specifically pertains to its primary application as a therapeutic option for individuals diagnosed with metastatic NSCLC featuring EGFR Exon 19 deletion or Exon 21 L858R substitution mutations [40]. Preclinical studies of dacomitinib showed potent activity against cancer cells harboring EGFR mutations and provided insight into its pharmacodynamics, indicating a longer duration of receptor downregulation compared to the first-generation TKIs [41]. Clinically, dacomitinib has exhibited efficacy in enhancing the duration of progression-free survival among individuals diagnosed with untreated NSCLC possessing EGFR mutations, surpassing the effects of gefitinib. In the ARCHER 1050 study, dacomitinib exhibited a notable extension in the median duration of progression-free survival [42]. However, dacomitinib’s toxicity profile includes diarrhea, dermatologic adverse effects, and mucosal inflammation, which are consistent with the inhibition of EGFR in normal tissues. These toxicities often require dose reductions and management with supportive care measures [43].
The synthetic pathway utilized for the synthesis of dacomitinib is depicted in Scheme 5 [44]. In the context of 2-methoxyethanol as the reaction medium, the chemical transformation involving 2-amino-4-fluoro-benzoic acid (referred to as DACT-001) with DACT-002 culminates in the generation of a novel chemical entity denoted as DACT-003. DACT-003 undergoes nitration using nitric acid to produce DACT-004. Subsequently, DACT-004 is treated with thionyl chloride (SOCl2) in the presence of dimethylformamide (DMF) to produce DACT-005 through chlorination. Following this, DACT-005 engages in a substitution process with 3-chloro-4-fluoroaniline (DACT-006), facilitated by TEA within an isopropanol solvent milieu, ultimately yielding the compound designated as DACT-007. Upon subjecting DACT-007 to methanol in the presence of sodium hydride, DACT-008 is synthesized. A subsequent reduction of the nitro moiety within DACT-008, utilizing a Raney nickel catalyst, yields the DACT-009. Subsequent to this reduction, DACT-009 undergoes a condensation reaction with DACT-010, leading to the synthesis of DACT-011. DACT-011 undergoes a chemical reaction with piperidine (DACT-012), leading to the synthesis of the desired molecular entity, namely dacomitinib.
3.6. Pyrotinib Maleate
Pyrotinib maleate, sold under the brand name Hengrui, is a novel oral TKI designed to selectively target the EGFR and HER2. Developed by Jiangsu Hengrui Medicine Co., Ltd., it emerged from the need to treat HER2-positive breast cancers, which are often more aggressive and less responsive to hormonal therapy [45]. The drug acts by inhibiting the intracellular phosphorylation of HER2, leading to blockade of downstream MAPK and PI3K–Akt-signaling pathways, thereby impeding tumor cell growth and proliferation. Pyrotinib has displayed a broad range of inhibitory activity against HER2-overexpressing tumor cells in preclinical studies, with promising pharmacodynamic properties [46]. Clinically, pyrotinib has shown significant efficacy among individuals diagnosed with HER2-positive breast cancer. In a Phase II clinical investigation, it notably enhanced the objective response rate and median progression-free survival when contrasted against the combination of lapatinib and capecitabine in female patients grappling with advanced breast cancer [47]. The drug’s toxicity profile includes diarrhea, hand–foot syndrome, and hematologic toxicity. Despite these adverse effects, pyrotinib is generally well-tolerated when administered with appropriate supportive measures.
The synthesis of pyrotinib maleate is depicted in Scheme 6 [48]. The condensation reaction of the aminoderivative PYRO-001 and diethylphosphonoacetate leads to the formation of PYRO-002. Subsequently, PYRO-002 and PYRO-003 undergo a Wittig reaction, resulting in the synthesis of PYRO-004. The conversion of PYRO-004 into pyrotinib maleate is achieved through the intervention of maleic acid.
3.7. Neratinib
Neratinib, branded as Nerlynx and developed by Puma Biotechnology, is an oral TKI targeting HER2 and EGFR. This drug was developed following the recognition of HER2’s pivotal involvement in the aggressive forms of breast cancer and the need for improved therapeutic options [49]. Neratinib irreversibly engages the ATP-binding cavity within these receptor proteins, resulting in the cessation of their kinase activity. Through the obstruction of signal transduction cascades, it interferes with cellular proliferation and viability, manifesting robust antineoplastic properties in preclinical model systems [50]. The drug showed clinical efficacy in the ExteNET study, a Phase III clinical trial where it significantly improved disease-free survival among early-stage patients afflicted with HER2-positive breast cancer who completed adjuvant trastuzumab therapy [51]. Toxicity profiles of neratinib include diarrhea, which is the most common adverse event, along with hepatotoxicity and rash. Effective management strategies, including a prophylactic use of anti-diarrheal medications, have been developed to mitigate these side effects.
The synthetic methodology employed for the preparation of neratinib is disclosed in Scheme 7. The synthesis of neratinib is initiated through the reduction of NERA-001 employing iron as the reducing agent, yielding the production of NERA-002. Following this, NERA-002 undergoes a condensation reaction with NERA-003, resulting in the synthesis of NERA-004. The chloride moiety within the molecular structure of NERA-004 undergoes displacement by NERA-005, culminating in the synthesis of neratinib [52].
3.8. Brigatinib
Brigatinib, commercially known as Alunbrig, was developed by Ariad Pharmaceuticals (now part of Takeda Oncology) and is a subsequent-generation inhibitor of anaplastic lymphoma kinase (ALK) [53]. The development of the compound was initiated in response to the emergence of resistance encountered with preceding ALK inhibitors within NSCLC [54]. Mechanistically, brigatinib exhibits selective binding affinity towards the ATP-binding domain of the ALK receptor tyrosine kinase, thereby inducing inhibition of ALK-mediated signaling pathways, consequently resulting in the demise of tumor cells. The drug is renowned for its ability to maintain efficacy against various ALK mutations associated with resistance to crizotinib, which belongs to the first generation of ALK inhibitors [55]. Preclinical studies demonstrated brigatinib’s potent activity against a range of ALK-positive tumor models, highlighting its potential pharmacodynamic benefits [56]. Clinically, discernible efficacy has been observed in individuals diagnosed with ALK-positive metastatic NSCLC, notably encompassing cases involving central nervous system (CNS) metastases [57]. The toxicity profile of brigatinib includes side effects such as elevated blood pressure, gastrointestinal disturbances, and the more severe, albeit less common, interstitial lung disease or pneumonitis. These adverse events necessitate careful patient monitoring [58].
The synthetic route utilized for the production of brigatinib is outlined in Scheme 8 [59]. The synthesis of BRIT-002 entails the condensation of 2-iodoaniline (BRIT-001) with dimethylphosphine oxide, employing an adapted Hirao method featuring Xantphos as the ligand and palladium acetate as the catalyst. Subsequent to this stage, a nucleophilic aromatic substitution process occurs, characterized by the interaction between BRIT-002 and trichloropyrimidine (BRIT-003), resulting in the synthesis of BRIT-004. The synthesis of aniline BRIT-005 involves a nucleophilic aromatic substitution (SNAr) process, wherein piperidine BRIT-007 displaces the fluoronitroarene BRIT-006. This reaction pathway results in the formation of aniline BRIT-005. Subsequently, a catalyzed hydrogenative reduction of the nitro functional group is conducted, with Pd/C serving as the catalyst for this transformative chemical reaction. The ensuing transformation involves the reaction of BRIT-004 with BRIT-005, culminating in the chemical synthesis of brigatinib.
3.9. Olmutinib
Olmutinib (brand name: Olita) was developed by Hanmi Pharmaceutical Co., Ltd. and later licensed to Boehringer Ingelheim for its potential in targeting EGFR mutations in NSCLC, particularly those patients with the T790M mutation, which induces resistance against initial-generation EGFR TKIs [60]. This mutation hinders the binding of earlier TKIs, prompting the need for development of third-generation inhibitors like olmutinib. The mechanism of action of Olmutinib involves irreversible binding to specific mutant EGFR, thereby inhibiting autophosphorylation and subsequent downstream signaling pathways. This ultimately leads to the inhibition of tumor cell proliferation and survival [61]. The efficacy of olmutinib against cell lines harboring the T790M mutation has been demonstrated in preclinical studies. The results of clinical trials demonstrated that olmutinib exhibited significant efficacy in patients diagnosed with T790M-positive NSCLC who experienced disease progression following prior TKI therapy, thereby providing a novel treatment option [62]. However, the clinical development of the drug faced challenges due to reports of serious adverse events such as severe skin toxicity and the rare but serious side effect known as Stevens–Johnson syndrome [63]. The development and distribution of the drug have been consequently limited, reflecting the paramount importance of safety in the risk–benefit assessment of novel cancer therapies.
A synthetic approach for the production of olmutinib is delineated within Scheme 9 [64]. The nucleophilic addition reaction of 2,4-dichloro-thieno[3,2-d]pyrimidine (OLMT-001) with N-(3-hydroxyphenyl)-2-propenamide (OLMT-002) is conducted employing potassium carbonate (K2CO3) as a base in dimethyl sulfoxide (DMSO), yielding the diaryl ether OLMT-003. In accordance with specialized reaction conditions characterized by an acid-mediated heating process, the process of introducing piperazinyl aniline OLMT-004 into the synthesis pathway transpires. This reaction necessitates the concurrent presence of dimethyl adipate, isopropanol, and trifluoroacetic acid. This precisely orchestrated sequence culminates in the successful synthesis of the principal compound, known as olmutinib.
3.10. Osimertinib Mesylate
Osimertinib, commercially known as Tagrisso, is a meticulously engineered third-generation covalent EGFR inhibitor developed by AstraZeneca. It has been specifically designed to selectively target the prevalent EGFR T790M resistance mutation commonly encountered in NSCLC [65]. The development of osimertinib was undertaken as part of an endeavor to tackle the challenge posed by resistance observed in initial and subsequent generations of EGFR inhibitors. Osimertinib functions through selective inhibition, specifically targeting EGFR-TKI-sensitizing and EGFR T790M-resistance mutations. Its impact on wild-type EGFR demonstrates reduced activity, contributing to a discernibly diminished toxicity profile [17]. The preclinical studies demonstrated potent and selective pharmacodynamic activity against mutant EGFR forms, including the T790M mutation, in tumor models [66]. Osimertinib has exhibited a notable response rate in clinical settings for individuals suffering from NSCLC who have developed resistance to previous EGFR TKIs due to the T790M mutation. Additionally, it has demonstrated remarkable efficacy as a first-line treatment, significantly enhancing progression-free survival [67]. The safety profile of osimertinib is considered favorable, but it includes some risks such as cardiotoxicity, interstitial lung disease, and dermal side effects, although these are generally manageable [68].
Scheme 10 delineates a demonstrative synthetic method employed in the production of osimertinib mesylate, offering an illustrative framework for its synthesis [69]. The Friedel–Crafts arylation reaction involving N-methylindole (referred to as OSIM-001) and dichloropyrimidine (designated as OSIM-002) results in the formation of 3-pyrazinyl indole, denoted as OSIM-003. Subsequently, an SNAr reaction with nitroaniline (identified as OSIM-004) is executed, affording the final product, aminopyrazine OSIM-005. The consecutive SNAr reaction involving OSIM-005 and N,N,N′-trimethylated ethylenediamine (OSIN-006) results in the formation of OSIM-007. Subsequently, OSIM-007 undergoes a nitro reduction process facilitated by iron in an acidic milieu, ultimately yielding the triaminated arene compound recognized as OSIM-008. OSIM-008 undergoes transformation into osimertinib through acylation utilizing 3-chloropropanoyl chloride (OSIM-009), followed by elimination in the presence of TEA within an acetonitrile milieu. Ultimately, the transformation of osimertinib is executed through a methanesulfonic acid-mediated treatment, denoted as OSIM-011, yielding the resultant product identified as osimertinib mesylate.
3.11. Afatinib Dimaleate
Afatinib, marketed as Gilotrif, is a second-generation TKI, developed by Boehringer Ingelheim, primarily for the treatment of NSCLC. Afatinib irreversibly inhibits the tyrosine kinases within the ErbB family, encompassing EGFR (ErbB1), HER2 (ErbB2), and ErbB4, and it also inhibits transphosphorylation of ErbB3 [21]. The objective behind the development of afatinib was to enhance outcomes for patients diagnosed with NSCLC with EGFR mutations. The inhibitory efficacy of afatinib was observed in preclinical investigations, effectively impeding signaling pathways arising from both homodimers and heterodimers across the ErbB family constellation. The observed phenomenon ultimately resulted in a clear inhibitory effect on the proliferation of tumor cells [5]. In clinical settings, afatinib has demonstrated efficacy as a viable first-line intervention for NSCLC patients harboring EGFR mutations, leading to improved progression-free survival compared to standard chemotherapy [70]. However, it is associated with side effects such as diarrhea, rash, and paronychia. Despite these toxicities, afatinib has been approved for use in several countries and is generally well tolerated with appropriate supportive care [71].
The synthetic process for the production of afatinib dimaleate commences with the initial reaction involving 2-amino-4-chlorobenzoic acid (designated as AFAT-001) and formamidine acetate, thereby affording the intermediate compound denominated as AFAT-002 (Scheme 11) [72]. AFAT-002 undergoes nitration, yielding the nitroquinazolinone AFAT-003. An ensuing chlorination reaction, employing phosphorus oxychloride (POCl3), is employed to effectuate the transformation of AFAT-003 into the compound denominated as AFAT-004. AFAT-006 is subsequently acquired through the chemical transformation of AFAT-004 via a reaction involving the commercially accessible 3-chloro-4-fluoroaniline (AFAT-005), succeeded by sulfonylation using sodium benzenesulfonate. AFAT-006 engages in a chemical reaction with (S)-tetrahydrofuran-3-ol, designated as AFAT-007, yielding the compound AFAT-008. Subsequently, a Raney–Nickel (Raney–Ni) catalyzed reduction of the nitro functional group ensues, leading to the generation of the aniline derivative denoted as AFAT-009. The treatment of AFAT-009 with a combination of AFAT-010 and N,N′-carbonyldiimidazole results in the generation of AFAT-011. Subsequently, employing the Horner–Wadsworth–Emmons homologation reaction scheme facilitates the transformation, ultimately affording the (E)-olefinic afatinib hydrate compound. Ultimately, the final step involves the treatment of afatinib hydrate with maleic acid, leading to the proficient synthesis of the desired compound, namely afatinib dimaleate.
3.12. Vandetanib
Vandetanib, commercially known as Caprelsa, is a TKI developed by AstraZeneca. Its FDA approval in 2011 specifically pertains to its application in the management of advanced medullary thyroid cancer (MTC). Vandetanib targets multiple tyrosine kinases, including EGFR, vascular endothelial growth factor receptor (VEGFR), and the rearranged during transfection (RET) proto-oncogene [73]. The principal mode of operation for vandetanib primarily revolves around the suppression of these kinases, which holds significant significance in the context of tumor angiogenesis and cellular proliferation. By blocking these pathways, vandetanib suppresses tumor growth and the formation of new blood vessels [74]. The preclinical pharmacodynamic studies demonstrated the potent anti-angiogenic and antitumor effects of vandetanib in various cancer models [75]. The clinical trials of vandetanib demonstrated promising outcomes in patients with advanced MTC, leading to its approval by the FDA. However, the use of this medication is associated with a range of adverse effects that necessitate careful monitoring and management, including diarrhea, rash, hypertension, and cardiac toxicity [76]. While vandetanib represents a significant advancement in the treatment of advanced MTC, its use is limited to this specific indication. It highlights the significance of targeted therapies and individualized medicine in oncology, where drugs like vandetanib can provide substantial clinical benefit in well-defined patient populations.
Scheme 12 outlines the preparation methodology employed for the synthesis of vandetanib [77]. VAND-001 and VAND-002 engage in a Mitsunobu reaction, yielding VAND-003 as the resultant product. Subsequently, VAND-003 undergoes a deprotection process involving the removal of the Boc protective group from amino moieties, executed under acidic conditions, thereby affording the formation of VAND-004. The amino moiety residing within VAND-004 is subjected to a reduction amination process catalyzed by formaldehyde, yielding the formation of VAND-005. VAND-005 facilitates the deprotection of the POM protective group through a reaction conducted in the presence of methanolic ammonia. This process yields VAND-006 as the resultant product. VAND-006 undergoes a chlorination process catalyzed by thionyl chloride, resulting in the formation of VAND-007. VAND-007 and VAND-008 engage in a nucleophilic substitution reaction, resulting in the formation of VAND-009. Subsequently, VAND-009 undergoes an alkaline treatment process, ultimately affording the synthesis of vandetanib.
3.13. Lapatinib Ditosylate
Lapatinib ditosylate, recognized by the brand Tykerb, represents a TKI developed by GlaxoSmithKline (GSK). In 2007, it obtained authorization from the FDA for managing advanced or metastatic breast malignancy [78]. The drug primarily targets HER2 and EGFR, both of which are overexpressed or mutated in certain types of breast cancer. The mechanism of action of lapatinib involves the inhibition of receptor phosphorylation, thereby obstructing downstream signaling pathways implicated in cellular proliferation and survival. The implementation of this targeted approach effectively regulates the proliferation of cancer cells [79]. The preclinical pharmacodynamic studies demonstrated that lapatinib exhibited potent inhibition of HER2 and EGFR signaling, resulting in a significant reduction in tumor growth in breast cancer models [80]. The efficacy of this treatment in managing HER2-positive metastatic breast cancer has been demonstrated through clinical trials, both as a standalone therapeutic regimen and when used in conjunction with adjunctive treatments. However, like many cancer drugs, lapatinib is associated with potential side effects, including diarrhea, skin rash, and cardiotoxicity, which require monitoring and management during treatment [81]. The development and approval of lapatinib represent a significant milestone in personalized medicine, as it specifically targets HER2-positive breast cancer. It underscores the importance of identifying and targeting specific molecular markers in cancer therapy, leading to more effective treatments for patients.
The initiation of lapatinib ditosylate synthesis is instigated through a Suzuki coupling reaction involving the precursors LAPT-001 and LAPT-002. This catalytic event engenders the formation of an intermediate compound LAPT-003, as comprehensively explicated within Scheme 13. Subsequent to the preceding step, the chlorination of LAPT-003 is conducted employing a reagent system composed of SOCl2 and DMF within a refluxing acetonitrile medium. This procedure yields the compound LAPT-004 [82]. Subsequent to the initial step, the chlorinated functional group residing in LAPT-004 is subjected to a substitution process with LAPT-005, resulting in the formation of LAPT-006. The ultimate stage in the synthetic route entails the reductive amination process of LAPT-006 and LAPT-007, subsequently subjecting the resultant compound to treatment with para-toluenesulfonic acid (p-TsOH), leading to the conclusive formation of lapatinib ditosylate.
3.14. Erlotinib Hydrochloride
Erlotinib, marketed as Tarceva, is a TKI that has made a significant impact on cancer therapy. Developed by OSI Pharmaceuticals, it received FDA approval in 2004 for the management of NSCLC and subsequently expanded its therapeutic indications to include pancreatic cancer [83]. Erlotinib primarily targets the inhibition of EGFR, a pivotal cell surface receptor critically involved in regulating neoplastic cell proliferation and growth. Its mechanism of action involves inhibiting the activation of EGFR, thereby blocking downstream signaling pathways that are essential for cell division and survival. This targeted approach renders it highly effective against EGFR-mutated NSCLC and pancreatic cancer [3]. Preclinical pharmacodynamic studies have demonstrated the effective inhibition of EGFR signaling by erlotinib, resulting in reduced tumor growth in animal models. In clinical investigations, it has exhibited notable efficacy in improving overall survival and progression-free survival among individuals with advanced NSCLC and pancreatic malignancies, particularly those harboring distinct EGFR mutations [84]. Adverse effects associated with erlotinib include dermatological manifestations such as cutaneous rash, gastrointestinal disturbances, notably diarrhea, and fatigue, which can generally be managed with appropriate medical care. Severe adverse events like interstitial lung disease and liver toxicity are rare but necessitate close monitoring during treatment [85]. The development and clinical success of erlotinib underscores the significance of targeted therapies in oncology, where personalized treatments can offer substantial benefits for patients with specific genetic mutations. This has spurred the advancement of additional therapeutic modalities aimed at EGFR in the context of oncological interventions.
In Scheme 14, a detailed delineation of the synthetic pathway for the production of erlotinib hydrochloride is meticulously expounded and presented [86]. ERLT-001 undergoes a chemical transformation upon interaction with bromoethyl methyl ether (ERLT-002). In this synthetic procedure, potassium carbonate is used as the base, while tetrabutyl ammonium iodide (TBAI) functions as a catalytic agent. The result of this process yields ERLT-003, which subsequently undergoes a nitration step involving nitric acid, yielding the final product ERLT-004. The reduction of ERLT-004 in an ethanol medium, catalyzed by PtO2/H2O under a hydrogen atmosphere, affords ERLT-005. ERLT-005 experiences a cyclization process when subjected to specific reaction conditions comprising formamide and ammonium formate. This transformative reaction eventuates in the generation of the quinazolone derivative denominated as ERLT-006. Subsequently, the compound ERLT-006 engages in a chemical transformation through its reaction with oxalyl chloride, yielding the ERLT-007. This intermediate undergoes a subsequent reaction with 3-ethynyl aniline, denoted as ERLT-008, facilitated by pyridine as a catalyst within an isopropanol environment. The resulting base was converted to the corresponding hydrochloride to result in erlotinib hydrochloride.
3.15. Gefitinib
The tyrosine kinase inhibitor Gefitinib, marketed as Iressa, has significantly impacted the therapeutic landscape of NSCLC. Originally developed by AstraZeneca, it received its first FDA approval in 2003 [87]. Gefitinib exerts its function through the inhibition of EGFR tyrosine kinase, thereby impeding downstream signaling pathways responsible for the proliferation of cancer cells [88]. Preclinical pharmacodynamic studies demonstrated the ability of gefitinib to effectively block EGFR signaling and inhibit tumor growth in animal models [20]. The efficacy of gefitinib has been further validated through clinical trials, particularly in individuals diagnosed with NSCLC carrying specific EGFR mutations. These mutations enhance the responsiveness of tumor cells to EGFR inhibitors [89]. Adverse events associated with gefitinib commonly manifest as dermatologic reactions, gastrointestinal disturbances such as diarrhea, and mild gastrointestinal discomfort. However, infrequent but critical incidences of interstitial lung disease and hepatic toxicity necessitate vigilant monitoring throughout the treatment regimen due to their severity. The development and clinical success of Gefitinib represent a significant advancement in targeted cancer therapy, showcasing the potential for personalized treatments based on genetic mutations. It has acted as a catalyst for further advancements in EGFR-focused therapeutic modalities within the field of oncology.
The synthetic procedure initiates by the selective removal of the methyl group from the 6-methoxy group of GEFT-001 by means of GEFT-002 in methanesulphonic acid (Scheme 15) [90]. This orchestrated reaction pathway culminates in the generation of GEFT-003. Subsequent to the initial step, GEFT-003 undergoes a chemical transformation involving acetic anhydride in the presence of pyridine, leading to the production of GEFT-004. Subsequently, this intermediate, GEFT-004, is subjected to a reaction with SOCl2 in the solvent DMF, ultimately affording the desired compound GEFT-005. The chlorine functional group present in GEFT-005 undergoes substitution with GEFT-006, leading to the generation of GEFT-007. Subsequently, a hydrolytic transformation of GEFT-007 into GEFT-008 is achieved in the presence of ammonium hydroxide (NH4OH) under refluxing conditions in methanol. The interaction between GEFT-008 and GEFT-009, facilitated by K2CO3 as the base in the solvent DMF, affords the successful synthesis of compound gefitinib.
3.16. Abivertinib
The novel TKI Abivertinib, marketed as Aociga, has been developed by Spectrum Pharmaceuticals and exhibits promising potential as a targeted therapy for various solid tumors, particularly NSCLC [91]. The primary target of the compound is located within EGFR and HER2. By inhibiting these receptor tyrosine kinases, abivertinib disrupts the intricate signaling cascades that facilitate cancer cell growth and proliferation [92]. Preclinical studies have demonstrated the potent inhibitory effects of abivertinib on EGFR and HER2 activation, leading to significant suppression of tumor proliferation in animal models. Ongoing clinical trials are currently underway to evaluate the compound’s safety profile and efficacy in oncology patients [93]. While abivertinib shows promise as a therapeutic option, similar to other targeted treatments, it may be associated with certain toxicities. Common adverse effects observed in clinical trials include skin rash, diarrhea, and fatigue. In line with other EGFR inhibitors, abivertinib potentially poses heightened concerns regarding severe adverse reactions such as interstitial lung disease and hepatotoxicity. The development and evaluation of abivertinib exemplify the ongoing efforts to discover and refine targeted therapies for cancer treatment. As research progresses and clinical trials continue, abivertinib holds potential to provide renewed hope for patients with EGFR- and HER2-driven cancers.
The preparation procedure for abivertinib is presented in Scheme 16 [94]. ABIV-001 is subjected to a safeguarding process employing SEM protective group under conditions of strong alkalinity, leading to the generation of ABIV-002. ABIV-002 and ABIV-003 partake in a nucleophilic substitution process, resulting in the generation of ABIV-004. Subsequently, ABIV-004 experiences a nucleophilic substitution reaction with ABIV-005, affording the ABIV-006. ABIV-006 undergoes reduction to form ABIV-007, which then reacts with acryloyl chloride ABIV-008 through condensation to produce ABIV-009. The deprotection of the SEM safeguarding moiety in ABIV-009 is achieved via acidic conditions, thereby affording the formation of hydroxyl functionalities, thereby yielding ABIV-010. ABIV-010 undergoes a hydroxymethyl group elimination process mediated by ammonia, ultimately leading to the successful synthesis of abivertinib.
3.17. Poziotinib
Poziotinib is an orally administered TKI that has demonstrated significant potential in the therapeutic management of NSCLC. It is developed by Hanmi Pharmaceutical Co., Ltd. Specifically targeting EGFR mutations, including those associated with resistance to other EGFR inhibitors, Poziotinib offers a valuable therapeutic option [95]. The drug’s mechanism of action involves the inhibition of EGFR and other kinases responsible for signaling pathways pivotal to cellular proliferation and growth. What sets poziotinib apart from other EGFR inhibitors is its unique ability to selectively target a diverse range of EGFR mutations, including Exon 20 insertions [96]. Preclinical studies have shown that poziotinib exhibits potent activity against various EGFR mutations, resulting in tumor regression in lung cancer models. Clinical trials have shown its efficacy in NSCLC patients with EGFR mutations, particularly those patients with Exon 20 insertions [97]. Like many targeted therapies, poziotinib is associated with side effects, including skin rash, diarrhea, and hepatotoxicity, which require careful monitoring and management during treatment. However, its potential to address EGFR-mutant NSCLC represents a significant advancement in personalized medicine for lung cancer patients. In summary, the development of poziotinib marks a crucial milestone in the treatment of EGFR-mutant NSCLC, particularly those with Exon 20 insertions. Its ability to target a broader spectrum of EGFR mutations offers renewed hope to patients who have previously encountered limited therapeutic options.
The synthetic strategy employed for the preparation of poziotinib is outlined in Scheme 17 [98]. POZI-001 undergoes a chlorination reaction, catalyzed by the reagent POCl3, subsequently proceeding through a nucleophilic substitution reaction with POZI-002, ultimately yielding the compound POZI-003. Following this, POZI-003 experiences deacetylation in an alkaline environment, resulting in its transformation into POZI-004. The phenolic hydroxyl moiety residing within the molecular structure of POZI-004 actively engages in a nucleophilic substitution reaction with POZI-005, ultimately culminating in the synthesis of poziotinib.
4. Challenges and Opportunities
The advent of EGFR inhibitors has revolutionized the therapeutic landscape for various cancers, particularly in the case of NSCLC. While these therapies offer substantial advantages, they also present distinctive challenges and opportunities. This discussion delves into the fundamental aspects of EGFR inhibitors in cancer treatment.
Challenges: Resistance Mechanisms: Despite initial responses, resistance to EGFR inhibitors inevitably develops. Acquired resistance mechanisms, such as secondary mutations (e.g., T790M) and bypass signaling pathways, limit the duration of response to first- and second-generation EGFR inhibitors [99]. Toxicities: skin rash, diarrhea, and hepatotoxicity are common side effects associated with EGFR inhibitors. These toxicities can impact patients’ quality of life and may necessitate dose adjustments or treatment interruptions [100]. Intratumoral Heterogeneity: Tumors often exhibit intratumoral heterogeneity, with different regions harboring distinct EGFR mutations. Targeting all subclones effectively remains a challenge [101]. CNS Metastases: EGFR-mutant NSCLC patients are prone to central nervous system (CNS) metastases. EGFR inhibitors have variable efficacy in addressing brain lesions owing to poor blood–brain barrier penetration [102].
Opportunities: Third-Generation EGFR Inhibitors: Third-generation EGFR inhibitors, exemplified by osimertinib, have shown efficacy against T790M resistance mutations. These agents extend progression-free survival and improve outcomes in NSCLC patients [67]. Combination Therapies: Combining EGFR inhibitors with other targeted agents or immunotherapies holds promise. Combinations like osimertinib plus savolitinib have demonstrated potential in addressing resistance mechanisms [103]. Next-Generation EGFR Inhibitors: Ongoing research is focused on developing next-generation EGFR inhibitors with enhanced efficacy and reduced toxicities. These agents aim to address existing challenges and improve patient outcomes. Liquid Biopsies: Liquid biopsies afford a non-invasive avenue for the surveillance of EGFR mutations and the identification of resistance mechanisms. An early detection of resistance can guide treatment decisions [19]. Patient Stratification: Advancements in molecular profiling enable precise patient stratification. Identifying specific EGFR mutations and resistance mechanisms helps tailor treatment strategies. Centralized CNS Therapy: Developing EGFR inhibitors with improved blood–brain barrier penetration can better manage CNS metastases, enhancing the overall effectiveness of these therapies [66]. EGFR inhibitors have transformed cancer treatment by targeting specific molecular alterations. While challenges like resistance and toxicities persist, ongoing research and the development of next-generation inhibitors and combination therapies provide opportunities to further improve patient outcomes. Patient stratification, liquid biopsies, and centralized CNS therapy are vital components of a comprehensive approach to addressing these challenges.
5. Conclusions
The synthesis and utilization of clinically approved small-molecule EGFR inhibitors in the field of cancer therapy represent a significant advancement towards precision medicine. These inhibitors have revolutionized oncology, providing targeted treatments for individuals affected by EGFR-driven malignancies, such as NSCLC and head and neck cancer. Despite substantial progress, persistent challenges remain in this field that require continued exploration and innovation. These challenges include the emergence of resistance mechanisms, potential off-target effects, synthetic complexity, patient stratification, and the need for effective combination therapies. Addressing these challenges necessitates rigorous scientific inquiry and clinical collaboration. Overcoming them requires the development of successive iterations of inhibitors with improved selectivity, better strategies to manage resistance, and enhanced patient-stratification techniques. The opportunities ahead are promising as personalized medicine gains momentum through advances in genomic profiling and non-invasive diagnostics enabling precise patient stratification. Next-generation EGFR inhibitors continue to evolve offering improved therapeutic options. Additionally, exploring combination therapies, including immunotherapies, holds immense potential for enhancing patient outcomes. As we navigate this dynamic landscape, it is essential for researchers, clinicians, and pharmaceutical scientists to work synergistically, leveraging their collective expertise to tackle challenges while maximizing opportunities. Regulatory entities play a crucial role in evaluating and authorizing these therapeutic interventions, prioritizing patient safety along with confirming efficacy. In summary, the synthesis and application of clinically approved small-molecule EGFR inhibitors signify a pivotal chapter in the ongoing battle against cancer. The journey is far from over, but the strides made in understanding EGFR-driven cancers and developing targeted therapies provide hope for improved patient outcomes and a future where precision medicine becomes the standard of care in oncology. Through continued dedication to research and innovation, we aim to better equip ourselves in the fight against this formidable disease and offer more effective treatment options to those in need.
Author Contributions
Conceptualization, Y.-T.W. and J.-F.S.; resources, Y.-T.W. and P.-C.Y.; data curation, J.-Y.Z.; writing—original draft preparation, Y.-T.W., P.-C.Y. and J.-Y.Z.; writing—review and editing, Y.-T.W. and J.-F.S.; supervision, Y.-T.W. and J.-F.S.; project administration, Y.-T.W.; funding acquisition, J.-F.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by National Natural Science Foundation of China (No. 82360805).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declared no financial interests.
References
- Yun, C.H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef]
- Moyer, J.D.; Barbacci, E.G.; Iwata, K.K.; Arnold, L.; Boman, B.; Cunningham, A.; DiOrio, C.; Doty, J.; Morin, M.J.; Moyer, M.P.; et al. Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res. 1997, 57, 4838–4848. [Google Scholar]
- Louie, G.V.; Yang, W.; Bowman, M.E.; Choe, S. Crystal structure of the complex of diphtheria toxin with an extracellular fragment of its receptor. Mol. Cell 1997, 1, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.R.; Padera, R.F.; Shapiro, G.I.; Baum, A.; Himmelsbach, F.; et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef] [PubMed]
- Gajiwala, K.S.; Feng, J.; Ferre, R.; Ryan, K.; Brodsky, O.; Weinrich, S.; Kath, J.C.; Stewart, A. Insights into the aberrant activity of mutant EGFR kinase domain and drug recognition. Structure 2013, 21, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Heppner, D.E.; Wittlinger, F.; Beyett, T.S.; Shaurova, T.; Urul, D.A.; Buckley, B.; Pham, C.D.; Schaeffner, I.K.; Yang, B.; Ogboo, B.C.; et al. Structural basis for inhibition of mutant EGFR with lazertinib (YH25448). ACS Med. Chem. Lett. 2022, 13, 1856–1863. [Google Scholar] [CrossRef]
- Sequist, L.V.; Yang, J.C.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef]
- Lan, C.C.; Hsieh, P.C.; Huang, C.Y.; Yang, M.C.; Su, W.L.; Wu, C.W.; Wu, Y.K. Review of epidermal growth factor receptor-tyrosine kinase inhibitors administration to non-small-cell lung cancer patients undergoing hemodialysis. World J. Clin. Cases 2022, 10, 6360–6369. [Google Scholar] [CrossRef]
- Cheng, Z.; Cui, H.; Wang, Y.; Yang, J.; Lin, C.; Shi, X.; Zou, Y.; Chen, J.; Jia, X.; Su, L. The advance of the third-generation EGFR-TKI in the treatment of non-small cell lung cancer (Review). Oncol. Rep. 2024, 51, 16. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Pao, W.; Chmielecki, J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat. Rev. Cancer 2010, 10, 760–774. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Wu, Y.L.; Schuler, M.; Sebastian, M.; Popat, S.; Yamamoto, N.; Zhou, C.; Hu, C.P.; O’Byrne, K.; Feng, J.; et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): Analysis of overall survival data from two randomised, phase 3 trials. Lancet. Oncol. 2015, 16, 141–151. [Google Scholar] [CrossRef]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef]
- Cheng, Y.; He, Y.; Li, W.; Zhang, H.L.; Zhou, Q.; Wang, B.; Liu, C.; Walding, A.; Saggese, M.; Huang, X.; et al. Osimertinib versus comparator EGFR TKI as first-line treatment for EGFR-mutated advanced NSCLC: FLAURA China, a randomized study. Target. Oncol. 2021, 16, 165–176. [Google Scholar] [CrossRef]
- Rolfo, C.; Mack, P.C.; Scagliotti, G.V.; Baas, P.; Barlesi, F.; Bivona, T.G.; Herbst, R.S.; Mok, T.S.; Peled, N.; Pirker, R.; et al. Liquid biopsy for advanced non-small cell lung cancer (NSCLC): A statement paper from the IASLC. J. Thorac. Oncol. 2018, 13, 1248–1268. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, A.E.; Guy, S.P.; Woodburn, J.R.; Ashton, S.E.; Curry, B.J.; Barker, A.J.; Gibson, K.H. ZD1839 (Iressa): An orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002, 62, 5749–5754. [Google Scholar] [PubMed]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, J.C.; Mitchell, P.L.; Fang, J.; Camidge, D.R.; Nian, W.; Chiu, C.H.; Zhou, J.; Zhao, Y.; Su, W.C.; et al. Sunvozertinib, a selective EGFR inhibitor for previously treated non-small cell lung cancer with EGFR exon 20 insertion mutations. Cancer Discov. 2022, 12, 1676–1689. [Google Scholar] [CrossRef]
- Wang, M.; Fan, Y.; Sun, M.; Wang, Y.; Zhao, Y.; Jin, B.; Hu, Y.; Han, Z.; Song, X.; Liu, A.; et al. Sunvozertinib for the treatment of NSCLC with EGFR Exon20 insertion mutations: The first pivotal study results. J. Clin. Oncol. 2023, 41, 9002. [Google Scholar] [CrossRef]
- Zheng, J.; Jiang, J.; Guo, Q.; Chang, S.; Zeng, Q.; Tsui, H.; Yang, Z.; Zhang, X. Novel Pharmaceutical Salts and Polymorphic Forms of an Erbb and btk Inhibitor. International Patent Application No. WO2023011358A1, 29 July 2022. [Google Scholar]
- Markham, A. Mobocertinib: First approval. Drugs 2021, 81, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, J.M. Mobocertinib: A potential treatment for NSCLC with EGFR exon 20 insertions. Cancer Discov. 2021, 11, 1617–1619. [Google Scholar] [CrossRef]
- Vasconcelos, P.; Kobayashi, I.S.; Kobayashi, S.S.; Costa, D.B. Preclinical characterization of mobocertinib highlights the putative therapeutic window of this novel EGFR inhibitor to EGFR exon 20 insertion mutations. JTO Clin. Res. Rep. 2021, 2, 100105. [Google Scholar] [CrossRef]
- Zhou, C.; Ramalingam, S.S.; Kim, T.M.; Kim, S.W.; Yang, J.C.; Riely, G.J.; Mekhail, T.; Nguyen, D.; Garcia Campelo, M.R.; Felip, E.; et al. Treatment outcomes and safety of mobocertinib in platinum-pretreated patients with EGFR exon 20 insertion-positive metastatic non-small cell lung cancer: A phase 1/2 open-label nonrandomized clinical trial. JAMA Oncol. 2021, 7, e214761. [Google Scholar] [CrossRef]
- Durak, L.; Langston, M.; Sharma, P.; Nguyen, T.; Li, S.; Zhang, X. Pharmaceutical Salts of Pyrimidine Derivatives and Method of Treating Disorders. International Patent Application No. WO2019222093A1, 13 May 2019. [Google Scholar]
- Shi, Y.; Zhang, S.; Hu, X.; Feng, J.; Ma, Z.; Zhou, J.; Yang, N.; Wu, L.; Liao, W.; Zhong, D.; et al. Safety, clinical activity, and pharmacokinetics of alflutinib (AST2818) in patients with advanced NSCLC With EGFR T790M mutation. J. Thorac. Oncol. 2020, 15, 1015–1026. [Google Scholar] [CrossRef]
- Shi, Y.; Hu, X.; Zhang, S.; Yang, N.; Zhang, Y.; Li, W.; Han, X.; Mo, H.; Sun, Y. P2.03-028 third generation EGFR inhibitor AST2818 (alflutinib) in NSCLC patients with EGFR T790M mutation: A phase1/2 multi-center clinical trial. J. Thorac. Oncol. 2017, 12, S2138. [Google Scholar] [CrossRef]
- Dong, R.F.; Zhu, M.L.; Liu, M.M.; Xu, Y.T.; Kong, L.Y. EGFR mutation mediates resistance to EGFR tyrosine kinase inhibitors in NSCLC: From molecular mechanisms to clinical research. Pharmacol. Res. 2021, 167, 105583. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Zhou, H.; Wang, S.; Wu, Y. Pyridylamino Pyrimidine Derivative, Preparation Method Therefor and Application of Pyridylamino Pyrimidine Derivative. Chinese Patent Application No. CN105315259A, 13 May 2019. [Google Scholar]
- Dhillon, S. Lazertinib: First approval. Drugs 2021, 81, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.J.; Han, J.Y.; Lee, K.H.; Kim, S.W.; Kim, D.W.; Lee, Y.G.; Cho, E.K.; Kim, J.H.; Lee, G.W.; Lee, J.S.; et al. Lazertinib in patients with EGFR mutation-positive advanced non-small-cell lung cancer: Results from the dose escalation and dose expansion parts of a first-in-human, open-label, multicentre, phase 1–2 study. Lancet. Oncol. 2019, 20, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Khoo, J.; Lim, J.; Lee, D.; Lee, J.; Lee, J.; Ju, H.; Shin, W.; Jeon, S. Novel Intermediates Useful for the Synthesis of Aminopyrimidine Derivatives, Process for Preparing the Same, and Process for Preparing Aminopyrimidine Derivatives Using the Same. International Patent Application No. WO2019022486A1, 25 July 2018. [Google Scholar]
- Gonzales, A.J.; Hook, K.E.; Althaus, I.W.; Ellis, P.A.; Trachet, E.; Delaney, A.M.; Harvey, P.J.; Ellis, T.A.; Amato, D.M.; Nelson, J.M.; et al. Antitumor activity and pharmacokinetic properties of PF-00299804, a second-generation irreversible pan-erbB receptor tyrosine kinase inhibitor. Mol. Cancer Ther. 2008, 7, 1880–1889. [Google Scholar] [CrossRef]
- Shirley, M. Dacomitinib: First global approval. Drugs 2018, 78, 1947–1953. [Google Scholar] [CrossRef]
- Tan, C.S.; Gilligan, D.; Pacey, S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet. Oncol. 2015, 16, e447–e459. [Google Scholar] [CrossRef]
- Wu, Y.L.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Tsuji, F.; Linke, R.; Rosell, R.; Corral, J.; et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): A randomised, open-label, phase 3 trial. Lancet. Oncol. 2017, 18, 1454–1466. [Google Scholar] [CrossRef]
- Mok, T.S.; Cheng, Y.; Zhou, X.; Lee, K.H.; Nakagawa, K.; Niho, S.; Lee, M.; Linke, R.; Rosell, R.; Corral, J.; et al. Improvement in overall survival in a randomized study that compared dacomitinib with gefitinib in patients with advanced non-small-cell lung cancer and EGFR-activating mutations. J. Clin. Oncol. 2018, 36, 2244–2250. [Google Scholar] [CrossRef]
- Alan, F.S.; Tsenwhei, L.H.; Elizabeth, R.J.; Matthew, S.K.; Elaine, S.K.; Haile, T.; Thomas, W.R. 4-Phenylamino-quinazolin-6-yl-Amides. International Patent Application No. WO2005107758A1, 25 April 2005. [Google Scholar]
- Blair, H.A. Pyrotinib: First global approval. Drugs 2018, 78, 1751–1755. [Google Scholar] [CrossRef]
- Xuhong, J.C.; Qi, X.W.; Zhang, Y.; Jiang, J. Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer. Am. J. Cancer Res. 2019, 9, 2103–2119. [Google Scholar]
- Ma, F.; Ouyang, Q.; Li, W.; Jiang, Z.; Tong, Z.; Liu, Y.; Li, H.; Yu, S.; Feng, J.; Wang, S.; et al. Pyrotinib or lapatinib combined with capecitabine in HER2-positive metastatic breast cancer with prior taxanes, anthracyclines, and/or trastuzumab: A randomized, phase II study. J. Clin. Oncol. 2019, 37, 2610–2619. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, B. Pharmaceutically Acceptable Salt of (e)-n-[4-[[3-chloro-4-(2-pyridylmethoxy)phenyl]amino]-3-cyano-7-ethoxy-6-quinolyl]-3-[(2r)-1-methylpyrrolidin-2-yl]prop-2-enamide, Preparation Method Thereof, and Medical Use Thereof. U.S. Patent Application No. US20130338190A1, 2 October 2012. [Google Scholar]
- Echavarria, I.; López-Tarruella, S.; Márquez-Rodas, I.; Jerez, Y.; Martin, M. Neratinib for the treatment of HER2-positive early stage breast cancer. Expert. Rev. Anticancer. Ther. 2017, 17, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004, 64, 3958–3965. [Google Scholar] [CrossRef]
- Chan, A.; Delaloge, S.; Holmes, F.A.; Moy, B.; Iwata, H.; Harvey, V.J.; Robert, N.J.; Silovski, T.; Gokmen, E.; von Minckwitz, G.; et al. Neratinib after trastuzumab-based adjuvant therapy in patients with HER2-positive breast cancer (ExteNET): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Oncol. 2016, 17, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Tsou, H.R.; Overbeek-Klumpers, E.G.; Hallett, W.A.; Reich, M.F.; Floyd, M.B.; Johnson, B.D.; Michalak, R.S.; Nilakantan, R.; Discafani, C.; Golas, J.; et al. Optimization of 6,7-disubstituted-4-(arylamino)quinoline-3-carbonitriles as orally active, irreversible inhibitors of human epidermal growth factor receptor-2 kinase activity. J. Med. Chem. 2005, 48, 1107–1131. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.N.; Bazhenova, L.A.; Langer, C.J.; Salgia, R.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Tugnait, M.; Narasimhan, N.I.; et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: A single-arm, open-label, phase 1/2 trial. Lancet. Oncol. 2016, 17, 1683–1696. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Brigatinib: First global approval. Drugs 2017, 77, 1131–1135. [Google Scholar] [CrossRef]
- Kim, D.W.; Tiseo, M.; Ahn, M.J.; Reckamp, K.L.; Hansen, K.H.; Kim, S.W.; Huber, R.M.; West, H.L.; Groen, H.J.M.; Hochmair, M.J.; et al. Brigatinib in patients with crizotinib-refractory anaplastic lymphoma kinase-positive non-small-cell lung cancer: A randomized, multicenter phase II trial. J. Clin. Oncol. 2017, 35, 2490–2498. [Google Scholar] [CrossRef]
- Siaw, J.T.; Wan, H.; Pfeifer, K.; Rivera, V.M.; Guan, J.; Palmer, R.H.; Hallberg, B. Brigatinib, an anaplastic lymphoma kinase inhibitor, abrogates activity and growth in ALK-positive neuroblastoma cells, Drosophila and mice. Oncotarget 2016, 7, 29011–29022. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Kim, H.R.; Ahn, M.J.; Yang, J.C.; Han, J.Y.; Lee, J.S.; Hochmair, M.J.; Li, J.Y.; Chang, G.C.; Lee, K.H.; et al. Brigatinib versus crizotinib in ALK-positive non-small-cell lung cancer. N. Engl. J. Med. 2018, 379, 2027–2039. [Google Scholar] [CrossRef]
- Sabari, J.K.; Santini, F.C.; Schram, A.M.; Bergagnini, I.; Chen, R.; Mrad, C.; Lai, W.V.; Arbour, K.C.; Drilon, A. The activity, safety, and evolving role of brigatinib in patients with ALK-rearranged non-small cell lung cancers. Onco Targets Ther. 2017, 10, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.S.; Liu, S.; Zou, D.; Thomas, M.; Wang, Y.; Zhou, T.; Romero, J.; Kohlmann, A.; Li, F.; Qi, J.; et al. Discovery of brigatinib (AP26113), a phosphine oxide-containing, potent, orally active inhibitor of anaplastic lymphoma kinase. J. Med. Chem. 2016, 59, 4948–4964. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S. Olmutinib: First global approval. Drugs 2016, 76, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.O.; Cha, M.Y.; Kim, M.; Song, J.Y.; Son, J. Abstract LB-100: Discovery of HM61713 as an orally available and mutant EGFR selective inhibitor. Cancer Res. 2014, 74, LB–100. [Google Scholar] [CrossRef]
- Park, K.; Jänne, P.A.; Kim, D.W.; Han, J.Y.; Wu, M.F.; Lee, J.S.; Kang, J.H.; Lee, D.H.; Cho, B.C.; Yu, C.J.; et al. Olmutinib in T790M-positive non-small cell lung cancer after failure of first-line epidermal growth factor receptor-tyrosine kinase inhibitor therapy: A global, phase 2 study. Cancer 2021, 127, 1407–1416. [Google Scholar] [CrossRef]
- Noh, Y.S.; Yoon, S.; Kim, S.R.; Lee, K.T.; Jang, I.J. A safety, pharmacokinetic, pharmacogenomic and population pharmacokinetic analysis of the third-generation EGFR TKI, olmutinib (HM61713), after single oral administration in healthy volunteers. Basic Clin. Pharmacol. Toxicol. 2019, 125, 370–381. [Google Scholar] [CrossRef]
- Wook, J.; Young Ho, M.; Tae Hee, H.; Kwee Hyun, S. Novel Process for Preparing Thienopyrimidine Compound and Intermediates Used Therein. International Patent Application No. WO2017074147A1, 27 August 2019. [Google Scholar]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Ballard, P.; Yates, J.W.; Yang, Z.; Kim, D.W.; Yang, J.C.; Cantarini, M.; Pickup, K.; Jordan, A.; Hickey, M.; Grist, M.; et al. Preclinical comparison of osimertinib with other EGFR-TKIs in EGFR-mutant NSCLC brain metastases models, and early evidence of clinical brain metastases activity. Clin. Cancer Res. 2016, 22, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef]
- Wu, Y.L.; Tsuboi, M.; He, J.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Laktionov, K.; Kim, S.W.; Kato, T.; et al. Osimertinib in resected EGFR-mutated non-small-cell lung cancer. N. Engl. J. Med. 2020, 383, 1711–1723. [Google Scholar] [CrossRef]
- Sam, B.; Verschoyle, F.M.R.; Andrew, W.R.; Krishna, K.V. 2-(2,4,5-substituted-anilino)Pyrimidine Derivatives as EGFR Modulators Useful for Treating Cancer. International Patent Application No. WO2013014448A1, 25 July 2012. [Google Scholar]
- Harvey, R.D.; Adams, V.R.; Beardslee, T.; Medina, P. Afatinib for the treatment of EGFR mutation-positive NSCLC: A review of clinical findings. J. Oncol. Pharm. Pract. 2020, 26, 1461–1474. [Google Scholar] [CrossRef]
- Sartori, G.; Belluomini, L.; Lombardo, F.; Avancini, A.; Trestini, I.; Vita, E.; Tregnago, D.; Menis, J.; Bria, E.; Milella, M.; et al. Efficacy and safety of afatinib for non-small-cell lung cancer: State-of-the-art and future perspectives. Expert. Rev. Anticancer. Ther. 2020, 20, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Juergen, S.; Georg, D.; Thomas, F.; Burkhard, J.; Carsten, R.; Svenja, R. Process for Preparing Aminocrotonylamino-Substituted Quinazoline Derivatives. International Patent Application No. WO2007085638A1, 25 January 2007. [Google Scholar]
- Carlomagno, F.; Vitagliano, D.; Guida, T.; Ciardiello, F.; Tortora, G.; Vecchio, G.; Ryan, A.J.; Fontanini, G.; Fusco, A.; Santoro, M. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002, 62, 7284–7290. [Google Scholar] [PubMed]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef]
- Ciardiello, F.; Tortora, G. EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.J.; Seib, C.D.; Gosnell, J. Vandetanib and the management of advanced medullary thyroid cancer. Curr. Opin. Oncol. 2013, 25, 39–43. [Google Scholar] [CrossRef]
- Hennequin, L.F.; Stokes, E.S.; Thomas, A.P.; Johnstone, C.; Plé, P.A.; Ogilvie, D.J.; Dukes, M.; Wedge, S.R.; Kendrew, J.; Curwen, J.O. Novel 4-anilinoquinazolines with C-7 basic side chains: Design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors. J. Med. Chem. 2002, 45, 1300–1312. [Google Scholar] [CrossRef]
- Geyer, C.E.; Forster, J.; Lindquist, D.; Chan, S.; Romieu, C.G.; Pienkowski, T.; Jagiello-Gruszfeld, A.; Crown, J.; Chan, A.; Kaufman, B.; et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2006, 355, 2733–2743. [Google Scholar] [CrossRef]
- Scaltriti, M.; Verma, C.; Guzman, M.; Jimenez, J.; Parra, J.L.; Pedersen, K.; Smith, D.J.; Landolfi, S.; Ramon y Cajal, S.; Arribas, J.; et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene 2009, 28, 803–814. [Google Scholar] [CrossRef]
- Bilancia, D.; Rosati, G.; Dinota, A.; Germano, D.; Romano, R.; Manzione, L. Lapatinib in breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2007, 18 (Suppl. S6), vi26–vi30. [Google Scholar] [CrossRef]
- Spector, N.L.; Xia, W.; Burris, H., 3rd; Hurwitz, H.; Dees, E.C.; Dowlati, A.; O’Neil, B.; Overmoyer, B.; Marcom, P.K.; Blackwell, K.L.; et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 2502–2512. [Google Scholar] [CrossRef]
- Chen, Y.; Henschke, J.P.; Liu, Y.; Chu, G.; Zhang, X. Process and Intermediates for Preparing Lapatinib. International Patent Application No. WO2011116634A1, 23 March 2010. [Google Scholar]
- Kobayashi, K.; Hagiwara, K. Epidermal growth factor receptor (EGFR) mutation and personalized therapy in advanced nonsmall cell lung cancer (NSCLC). Target. Oncol. 2013, 8, 27–33. [Google Scholar] [CrossRef]
- Brower, J.V.; Robins, H.I. Erlotinib for the treatment of brain metastases in non-small cell lung cancer. Expert. Opin. Pharmacother. 2016, 17, 1013–1021. [Google Scholar] [CrossRef]
- Abdelgalil, A.A.; Al-Kahtani, H.M.; Al-Jenoobi, F.I. Erlotinib. Profiles Drug Subst. Excip. Relat. Methodol. 2020, 45, 93–117. [Google Scholar] [CrossRef] [PubMed]
- Schnur, R.C.; Arnold, L.D. Quinazoline Derivatives. International Patent Application No. WO1996030347A1, 6 June 1995. [Google Scholar]
- Rawluk, J.; Waller, C.F. Gefitinib. Recent Results Cancer Res. 2018, 211, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.W.; Pedersen, N.; Ottesen, L.H.; Poulsen, H.S. Differential response to gefitinib of cells expressing normal EGFR and the mutant EGFRvIII. Br. J. Cancer 2005, 93, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Kris, M.G.; Natale, R.B.; Herbst, R.S.; Lynch, T.J., Jr.; Prager, D.; Belani, C.P.; Schiller, J.H.; Kelly, K.; Spiridonidis, H.; Sandler, A.; et al. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: A randomized trial. JAMA 2003, 290, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Barker, A.J.; Gibson, K.H.; Grundy, W.; Godfrey, A.A.; Barlow, J.J.; Healy, M.P.; Woodburn, J.R.; Ashton, S.E.; Curry, B.J.; Scarlett, L.; et al. Studies leading to the identification of ZD1839 (IRESSA): An orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorganic Med. Chem. Lett. 2001, 11, 1911–1914. [Google Scholar] [CrossRef]
- Wang, H.; Pan, R.; Zhang, X.; Si, X.; Wang, M.; Zhang, L. Abivertinib in patients with T790M-positive advanced NSCLC and its subsequent treatment with osimertinib. Thorac. Cancer 2020, 11, 594–602. [Google Scholar] [CrossRef]
- He, J.; Huang, Z.; Han, L.; Gong, Y.; Xie, C. Mechanisms and management of 3rd-generation EGFR-TKI resistance in advanced non-small cell lung cancer (Review). Int. J. Oncol. 2021, 59, 90. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Chen, Z.H.; Zhang, X.C.; Xu, C.R.; Yan, H.H.; Xie, Z.; Chuai, S.K.; Ye, J.Y.; Han-Zhang, H.; Zhang, Z.; et al. Analysis of resistance mechanisms to abivertinib, a third-generation EGFR tyrosine kinase inhibitor, in patients with EGFR T790M-positive non-small cell lung cancer from a phase I trial. EBioMedicine 2019, 43, 180–187. [Google Scholar] [CrossRef]
- Xu, X.; Wang, X.; Mao, L.; Zhao, L.; Xi, B. Novel pyrrolopyrimidine compounds as inhibitors of protein kinases. US2015210702A1.
- Rosell, R.; Cardona Zorrilla, A.F. Poziotinib treatment in intractable NSCLC: Epidermal growth factor receptor and human epidermal growth factor receptor 2 exon 20 insertion mutation disease. Eur. J. Cancer 2021, 149, 233–234. [Google Scholar] [CrossRef]
- Robichaux, J.P.; Elamin, Y.Y.; Tan, Z.; Carter, B.W.; Zhang, S.; Liu, S.; Li, S.; Chen, T.; Poteete, A.; Estrada-Bernal, A.; et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20-selective kinase inhibitor in non-small cell lung cancer. Nat. Med. 2018, 24, 638–646. [Google Scholar] [CrossRef]
- Elamin, Y.Y.; Robichaux, J.P.; Carter, B.W.; Altan, M.; Tran, H.; Gibbons, D.L.; Heeke, S.; Fossella, F.V.; Lam, V.K.; Le, X.; et al. Poziotinib for EGFR exon 20-mutant NSCLC: Clinical efficacy, resistance mechanisms, and impact of insertion location on drug sensitivity. Cancer Cell 2022, 40, 754–767.e756. [Google Scholar] [CrossRef]
- Bang, K.C.; Jung, J.H.; Moon, Y.H. Method for Preparing 1-(4-(4-(3,4-dichloro-2-fluorophenylamino)-7-methoxyquinazolin-6-yloxy)piperidin-1-yl)prop-2-en-1-one. U.S. Patent Application No. US20150344458A1, 13 December 2016. [Google Scholar]
- Piotrowska, Z.; Niederst, M.J.; Karlovich, C.A.; Wakelee, H.A.; Neal, J.W.; Mino-Kenudson, M.; Fulton, L.; Hata, A.N.; Lockerman, E.L.; Kalsy, A.; et al. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov. 2015, 5, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Lacouture, M.E.; Anadkat, M.J.; Bensadoun, R.J.; Bryce, J.; Chan, A.; Epstein, J.B.; Eaby-Sandy, B.; Murphy, B.A. Clinical practice guidelines for the prevention and treatment of EGFR inhibitor-associated dermatologic toxicities. Support. Care Cancer 2011, 19, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, W.J.; Lester-Coll, N.H.; Wu, A.J.; Yang, T.J.; Lockney, N.A.; Gerber, N.K.; Beal, K.; Amini, A.; Patil, T.; Kavanagh, B.D.; et al. Management of brain metastases in tyrosine kinase inhibitor-naïve epidermal growth factor receptor-mutant non-small-cell lung cancer: A retrospective multi-institutional analysis. J. Clin. Oncol. 2017, 35, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.; Lee, J.S.; Su, W.C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet. Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Chemical structures of small-molecule EGFR inhibitors.


Figure 2.
EGFR signal pathway.

Scheme 1.
Synthesis of sunvozertinib.

Scheme 2.
Synthesis of mobocertinib succinate.

Scheme 3.
Synthesis of alflutinib.

Scheme 4.
Synthesis of lazertinib.

Scheme 5.
Synthesis of dacomitinib.

Scheme 6.
Synthesis of pyrotinib maleate.

Scheme 7.
Synthesis of neratinib.

Scheme 8.
Synthesis of brigatinib.

Scheme 9.
Synthesis of olmutinib.

Scheme 10.
Synthesis of osimertinib mesylate.

Scheme 11.
Synthesis of afatinib dimaleate.

Scheme 12.
Synthesis of vandetanib.

Scheme 13.
Synthesis of lapatinib ditosylate.

Scheme 14.
Synthesis of erlotinib hydrochloride.

Scheme 15.
Synthesis of gefitinib.

Scheme 16.
Synthesis of abivertinib.

Scheme 17.
Synthesis of poziotinib.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Small-molecule EGFR inhibitors for cancer therapy a.
| NO | Drug | Company | Indications | Target | Status |
|---|---|---|---|---|---|
| 1 | Sunvozertinib | Dizal Pharmaceuticals (Shanghai, China) | NSCLC | EGFR; HER2; | Approved (2023) |
| 2 | Mobocertinib succinate | Takeda Oncology (Cambridge, MA, USA) | NSCLC | EGFR exon 20 insertion mutations; | Approved (2021) |
| 3 | Alflutinib | Shanghai Allist Pharmaceuticals Co., Ltd. (Shanghai, China) | NSCLC | EGFR T790M; | Approved (2021) |
| 4 | Lazertinib | Yuhan (Seoul, Republic of Korea) and Janssen Biotech (Titusville, NJ, USA) | NSCLC | EGFR; EGFR T790M; EGFR L858R; EGFR 19del; | Approved (2021) |
| 5 | Dacomitinib | Pfizer (New York, NY, USA) | NSCLC | HER2; HER4; EGFR; EGFR 19del; EGFR L858R; | Approved (2018) |
| 6 | Pyrotinib maleate | Jiangsu Hengrui Medicine Co., Ltd. (Lianyungang, China) | Breast cancer | HER2; HER4; EGFR; | Approved (2018) |
| 7 | Neratinib | Puma Biotechnology (Los Angeles, CA, USA) | Breast cancer | HER2; HER4; EGFR; | Approved (2017) |
| 8 | Brigatinib | Ariad Pharmaceuticals (San Diego, CA, USA) | NSCLC | EGFR; IGF1R; ALK; ROS; FLT3; | Approved (2017) |
| 9 | Olmutinib | Hanmi Pharmaceutical Co., Ltd. (Hwaseong-si, Gyeonggi-do, Republic of Korea) | NSCLC | EGFR T790M; | Approved (2016) |
| 10 | Osimertinib mesylate | AstraZeneca (London, UK) | NSCLC | EGFR; HER2; HER3; HER4; EGFR T790M; EGFR L858R; EGFR 19del; | Approved (2015) |
| 11 | Afatinib dimaleate | Boehringer Ingelheim (Dortmund, Germany) | NSCLC | HER4; HER2; EGFR; EGFR L858R; EGFR 19del; | Approved (2013) |
| 12 | Vandetanib | AstraZeneca (London, UK) | MTC | VEGFR2; RET; EGFR T790M; | Approved (2011) |
| 13 | Lapatinib ditosylate | GlaxoSmithKline (London, UK) | breast cancer | EGFR; HER2; | Approved (2007) |
| 14 | Erlotinib hydrochloride | Genentech (San Francisco, CA, USA) | NSCLC | EGFR; EGFR 19del; EGFR L858R; | Approved (2004) |
| 15 | Gefitinib | AstraZeneca (London, UK) | NSCLC | EGFR; EGFR 19del; EGFR L858R; | Approved (2002) |
| 16 | Abivertinib | Spectrum Pharmaceuticals (Irvine, CA, USA) | Acute respiratory distress syndrome | BTK; EGFR T790M; | Investigational |
| 17 | Poziotinib | Hanmi Pharmaceutical Co., Ltd. (Hwaseong-si, Gyeonggi-do, Republic of Korea) | NSCLC | HER2; HER4; EGFR; | Investigational |
a Company: Original company responsible for the development and marketing of the drug; indications: primary clinical indications; target: target of the drug’s action; status: the most recent updates regarding the research and development status of the drug, with the year provided indicating the approval date.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, Y.-T.; Yang, P.-C.; Zhang, J.-Y.; Sun, J.-F. Synthetic Routes and Clinical Application of Representative Small-Molecule EGFR Inhibitors for Cancer Therapy. Molecules 2024, 29, 1448. https://doi.org/10.3390/molecules29071448
AMA Style
Wang Y-T, Yang P-C, Zhang J-Y, Sun J-F. Synthetic Routes and Clinical Application of Representative Small-Molecule EGFR Inhibitors for Cancer Therapy. Molecules. 2024; 29(7):1448. https://doi.org/10.3390/molecules29071448
Chicago/Turabian StyleWang, Ya-Tao, Peng-Cheng Yang, Jing-Yi Zhang, and Jin-Feng Sun. 2024. "Synthetic Routes and Clinical Application of Representative Small-Molecule EGFR Inhibitors for Cancer Therapy" Molecules 29, no. 7: 1448. https://doi.org/10.3390/molecules29071448