
Total Synthesis of the Racemate of Laurolitsine
by
Mingyu Cao
1,
Yiming Wang
1,
Yong Zhang
1,
Caiyun Zhang
2,
Niangen Chen
1,* and
Xiaopo Zhang
1,2,* 1
Key Laboratory of Tropical Translational Medicine of Ministry of Education, Hainan Key Laboratory for Research and Development of Tropical Herbs, School of Pharmacy, Hainan Medical University, Haikou 571199, China
2
Research Center for Drug Safety Evaluation of Hainan Province, Hainan Medical University, Haikou 571101, China
*
Authors to whom correspondence should be addressed.
Molecules 2024, 29(3), 745; https://doi.org/10.3390/molecules29030745
Submission received: 14 January 2024
/
Revised: 1 February 2024
/
Accepted: 2 February 2024
/
Published: 5 February 2024
(This article belongs to the Section Medicinal Chemistry)
Abstract
:The total synthesis of laurolitsine was achieved for the first time. This reaction was accomplished in 14 steps with a 2.3% yield (this was calculated using 3-hydroxy-4-methoxybenzaldehyde as the starting material) starting from two simple materials, 3-hydroxy-4-methoxybenzaldehyde and 2-(3-hydroxy-4-methoxyphenyl)acetic acid, and the longest linear sequence consisted of 11 steps. The key steps included an electrophilic addition reaction in which a nitro group was reduced to an amino group using lithium tetrahydroaluminum and a Pd-catalyzed direct biaryl coupling reaction. In this paper, many of the experimental steps were optimized, and an innovative postprocessing method in which 2-(3-(benzyloxy)-4-methoxyphenyl)ethanamine is salted with oxalic acid was proposed.
1. Introduction
Laurolitsine is an alkaloid isolated from natural plants such as Litsea glutinosa that exhibits potent antidiabetic effects and hypoglycemic activity in vivo and has wide application prospects in the clinic [1,2]. Laurolitsine was first discovered and named by Tatsuo Nakasato and Shozo Nomura from the leaves of Neolitsea sericea (Blume) Koidz. in 1959 [3]. Sun et al. obtained laurolitsine by efficient isolation from the chloroform extract of Litsea cubeba [4]. Through a series of experiments, Zhang et al. confirmed that laurolitsine, which is abundant in Litsea glutinosa bark, can exhibit potent antidiabetic effects with hypoglycemic activity in vivo. Laurolitsine improved insulin resistance, glucose tolerance and lipid metabolism; protected liver, renal and pancreatic functions; and promoted weight loss in db/db mice [1].
However, since laurolitsine must be isolated and extracted from plants, the cycle is long, and low yields are obtained; thus, the price of this material is relatively high, which greatly limits the development of related experiments on laurolitsine [4]. At present, there are no reports on the total synthesis of laurolitsine. Therefore, exploring a new synthesis route is imperative, and reasonable and efficient chemical synthesis methods for obtaining laurolitsine quickly and in large quantities are very important for related research.
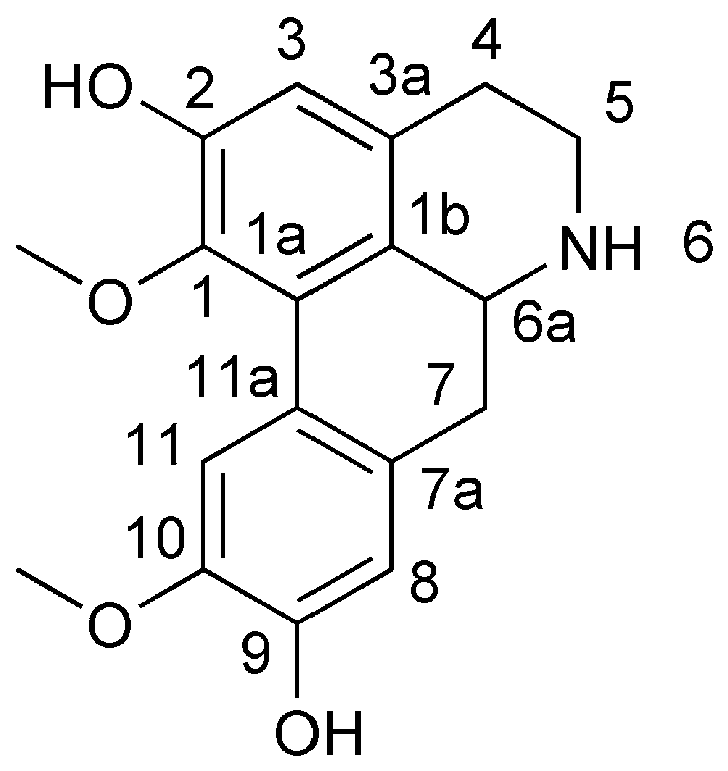
Laurolitsine is an aporphine alkaloid that has a special biphenyl tetracyclic structure with a chiral carbon atom at the 6a position (Figure 1) and a wide range of physiological activities due to its different oxidation states and substituents [5,6,7,8,9,10]. Due to their remarkable pharmacological effects, aporphine alkaloids have attracted widespread attention in organic synthesis. However, directly synthesizing aporphine alkaloids is challenging due to their special benzene ring structure. The 1-benzyl-substituted tetrahydroisoquinoline can be used as a basic skeleton for the biomimetic synthesis of aporphine alkaloids. Therefore, establishing this parent nucleus structure is fundamental to the synthesis of aporphine alkaloids [11].
Lafrance et al. reported that a variety of aporphine alkaloids with 2-position substitutions can be directly synthesized using a Pd-catalyzed arylation reaction [12]. This breakthrough not only highlights the utility of direct arylation in target-oriented synthesis reactions but also allows the synthesis of various compounds with different substituents. Researchers have used the same approach for the enantioselective synthesis of pronuciferine and nuciferin [11]. Gao et al. described a highly efficient and practical multicomponent one-pot reaction. This reaction represents a streamlined pathway for synthesizing functionalized 1,2-dihydroisoquinolines, showcasing the versatility of multicomponent reactions in organic synthesis [13]. Under different reaction conditions, a novel series of aporphine analogs were finally accomplished through intramolecular phenol ortho-arylation using Pd-mediated catalysis [14,15,16,17,18]. However, the operation is complex, and most steps require silica gel column chromatography. The purpose of this study was to synthesize total amounts of laurolitsine for the first time and optimize the reaction steps. An innovative postprocessing method in which 2-(3-(benzyloxy)-4-methoxyphenyl)ethanamine is salted with oxalic acid is also proposed, which greatly reduces the time cost and increases the yield of intermediates.
2. Results and Discussion
2.1. Retrosynthetic Analysis of Laurolitsine
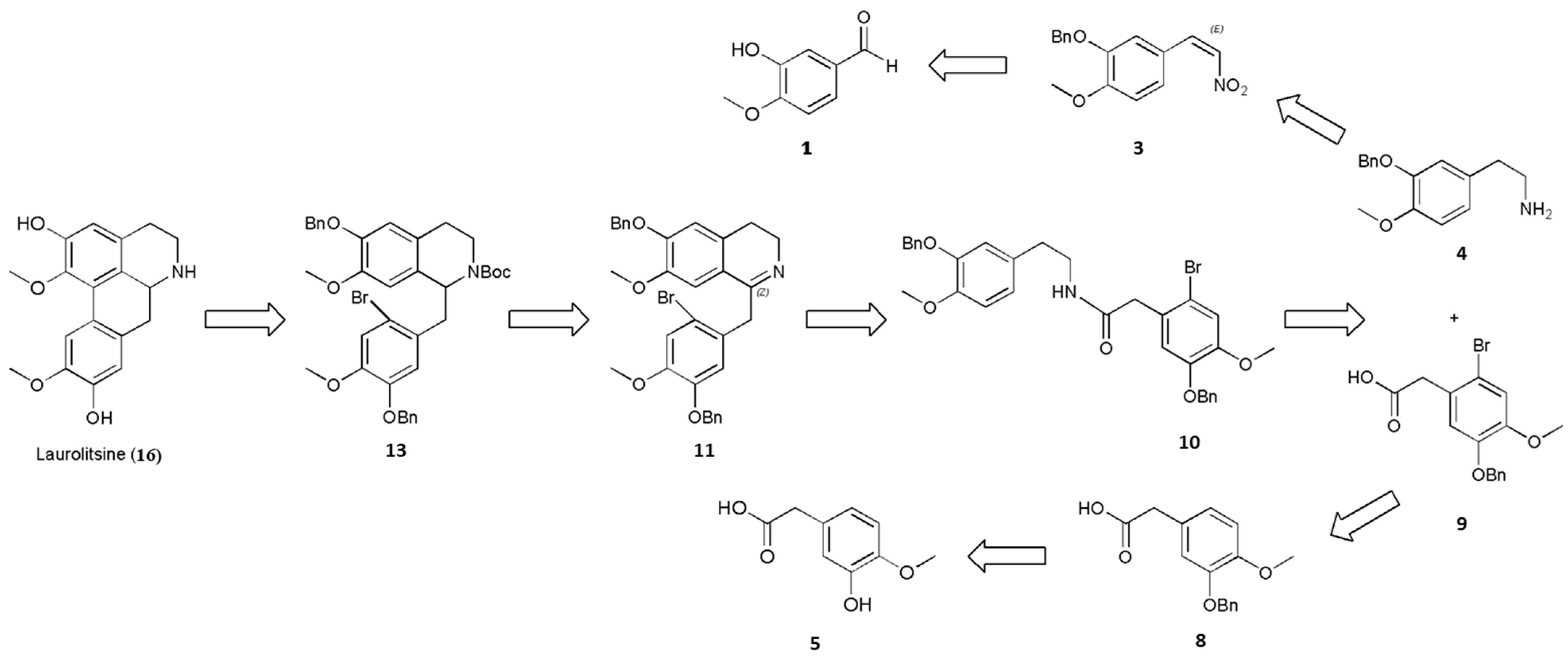
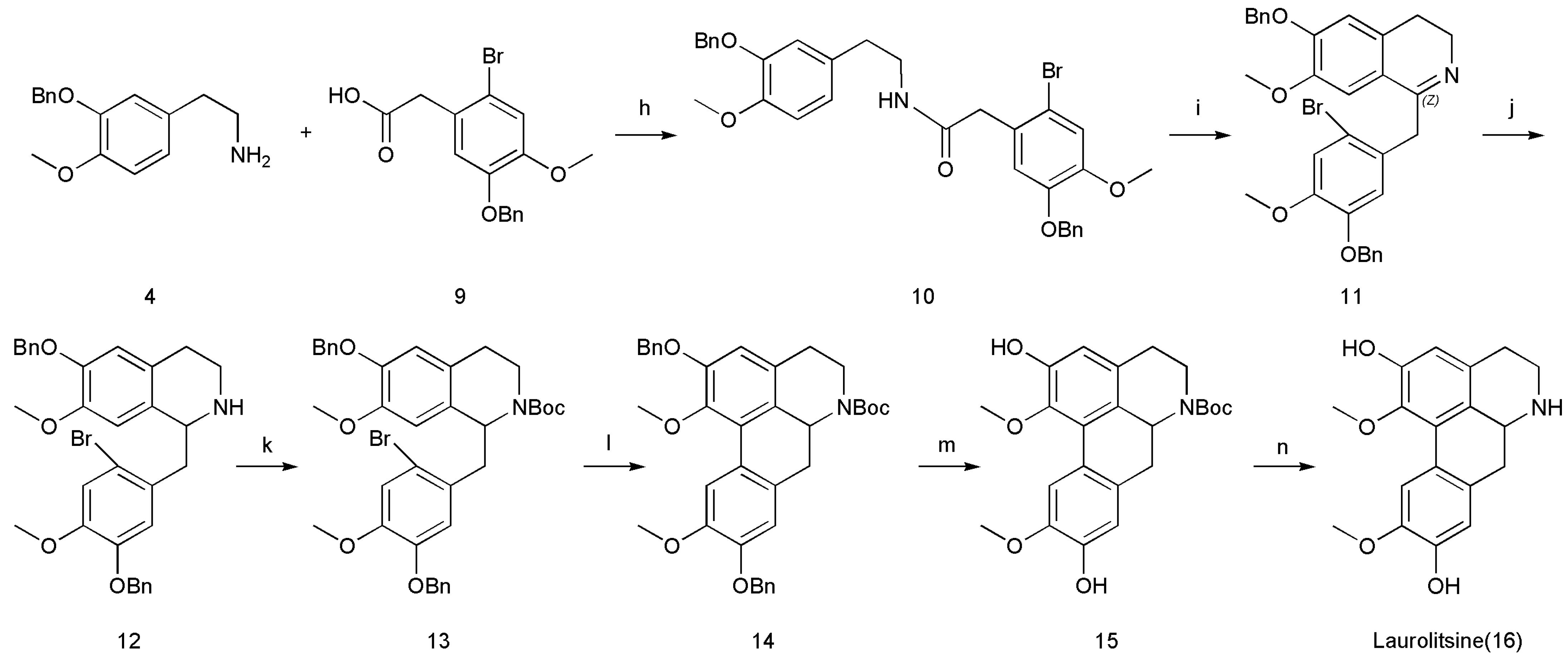
As shown in Scheme 1, the steps for the synthesis of laurolitsine (16) begins with the N-t-butyloxy carbonyl (Boc)-protected carbamate 13 and uses Pd-catalyzed direct arylation and deprotection reaction. N-Boc-protected carbamate 13 is then generated from cyclized imine 11 through a reduction reaction and protection with a Boc group. Cylized imine 11 is generated from amide 10 through the Bischler–Napieralski reaction under acidic conditions. Eventually, amide 10 could be prepared from 2-(3-(benzyloxy)-4-methoxyphenyl)ethanamine (4) coupled with 2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetic acid (9) under standard peptide-coupling conditions.
Compounds 4 and 9 were synthesized by two different routes. Compound 3 was synthesized through a benzylation reaction and a nitration reaction, and 4 was subsequently produced by the reduction of 3. In relation to another reaction route, 9 was produced from 8 through substitution with Br2. Finally, 8 was obtained by esterification, benzylation, and hydrolysis.
2.2. Facile Construction of the Laurolitsine Skeleton
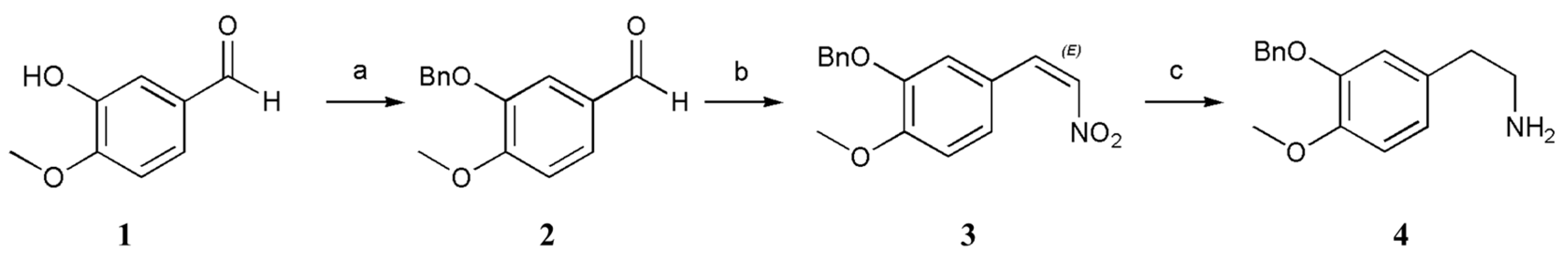
The protection of 1 with a benzyl (Bn) group furnished 2. Then, 3 was prepared via the Henry reaction and elimination reaction by adding 2, ammonium acetate, and nitromethane to an acetic acid solvent at 118 °C [19,20,21]. First, we wanted to complete the reaction by increasing the amount of nitromethane and ammonium acetate, but despite our efforts to perform many experiments, the effect was not good, the product was sticky and difficult to separate and filter, and the yield and purity were poor. After many experimental alterations, we finally determined the reaction conditions (CH3NO2 (4 equiv.), NH4OAc (1.3 equiv.), HOAc, 118 °C). The viscosity of the product may be related to the amount of ammonium acetate. If the amount of ammonium acetate is too high, side reactions increase and intermediate 3 becomes viscous and difficult to filter. A small amount of ammonium acetate causes the reaction to be incomplete. Regarding the next step of reduction, we attempted to reduce Pd/C, iron powder, sodium sulfide and sodium borohydride and did not achieve good results; subsequently, we succeeded by adding the strong reductant LiAlH4, which was converted to substituted phenyl ethanamine 4 via reduction. LiAlH4 was selected to reduce the double bond and nitro group simultaneously. A much lower yield was obtained when LiAlH4 was added at ambient temperature due to the generation of impurities including two compounds that only reduced the nitro group but not the double bond and only reduced the double bond but not the nitro group. Purifying the product was difficult because reduction of the double bond and nitro group was incomplete, and the polarity difference between the two impurities was very small [19,20,21]. In terms of the feeding temperature, reaction temperature, and reductant ratio, we performed a large number of experiments and ultimately determined the most suitable reaction conditions (LiAlH4 (4 equiv.), THF, 0 → 35 °C, THF = tetrahydrofuran). A method involving salt formation between oxalic acid and amino groups was used for purification. Compound 4 was combined with oxalic acid in methanol to form oxalate, after which the salt was hydrolyzed with sodium hydroxide solution, which achieved high purity and reduced the number of tedious purification steps. The specific method is described in detail later (Scheme 2).
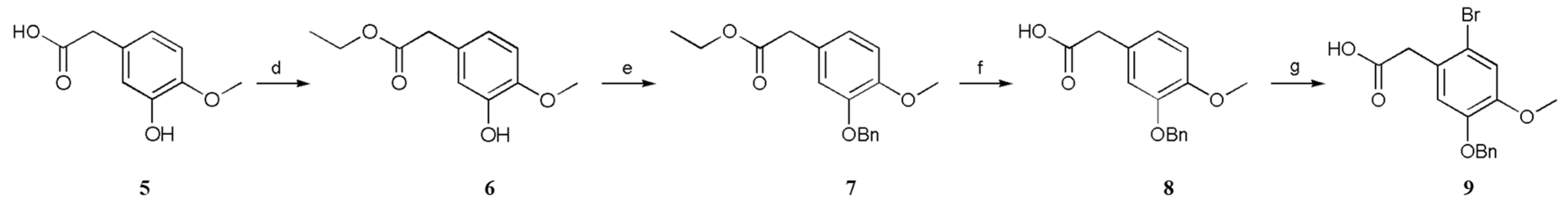
First, 5 was esterified with ethanol to furnish 6, and then 7 was formed by a benzylation reaction. Next, 8 was obtained by hydrolysis with sodium hydroxide solution, and 9 was obtained by reaction with Br2, acetic acid and sodium acetate [15]. Notably, this reaction requires Br2 to be slowly added to the system at the end; otherwise, the reaction will not be complete (Scheme 3).
We thus attempted to repeat the work reported by Sharma et al. [18] by coupling 2-(3-(benzyloxy)-4-methoxyphenyl)ethanamine (4) with 2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetic acid (9) under standard peptide-coupling conditions to afford amide 10 (1,1′-carbonyldiimidazole, THF, RT, 20 h). Unfortunately, we could not observe any reactions between 4 and 9. We varied the concentration of the reaction mixture and the number of equivalents of each reagent, and no good effects were observed. Considering that 4 contains a benzyl group and has a large steric hindrance, we subsequently changed the solvent to xylene and dehydrated and condensed the mixture with an oil–water separator at 135 °C; however, no satisfactory results were obtained. Finally, we tried to obtain 10 successfully by using DMF, HOBt and EDC. The use of the dehydrating condensation agent EDC results in mild reaction conditions and an easy process.
Amide 10 was subjected to the Bischler–Napieralski reaction [22] to afford cyclized imine 11, which was subjected to NaBH4-mediated reduction without further purification to furnish secondary amine 12. The protection of secondary amine 12 with a tert-butoxycarbonyl (Boc) group furnished N-Boc-protected 13. Then, 14 was prepared by a Pd-catalyzed direct biaryl coupling methodology [23,24,25]. If the reaction was carried out at a lower temperature for a long time, debrominated byproducts appeared. The selection of ligands was also particularly important, because different ligands had different stability, which will greatly affect the yield. In terms of the choice of solvent, we chose 1,4-dioxane. Compared with DMA, DMF or DMSO, our advantage was that we can choose to directly steam the solvent after the reaction was complete, avoiding the product loss and tedious operation steps brought by extraction. We tested many reaction conditions and determined the most suitable reaction conditions (Pd(OAc)2 (0.2 equiv.), Cs2CO3 (3 equiv.), (t-Bu)2PMeHBF4 (0.4 equiv.), 1,4-dioxane, 101 °C, (t-Bu)2PMeHBF4 = di-tert-butylmethylphosphine tetrafluor). It was worth noting that the reaction conditions were harsh and required very strict vacuum and nitrogen protection. The synthesis of 15 was achieved by deprotecting the benzyl (Bn) group of 14 by using Pd/C under a hydrogen atmosphere. Finally, the synthesis of laurolitsine (16) was achieved by deprotecting the Boc group of 15 by using anhydrous ZnBr2 [18]. Spectral data, including 1H NMR, 13C NMR, were collected for both the natural and synthetic sample (16) and found to be in good agreement (Table 1) [4]. All these compounds were well characterized by using 1H NMR, 13C NMR, and high-resolution (HR) ESI-MS, as showed in Supplementary Materials (Scheme 4).
3. Materials and Methods
3.1. General Experimental Details
All chemicals were purchased from commercial sources. Silica gel (200–300 mesh, Qingdao Marine Chemistry Co. Ltd., Qingdao, Tsingtao, China) was used for column chromatography. Reactions were monitored by thin-layer chromatography (TLC). Silica gel plates (Qingdao Marine Chemistry Co. Ltd., GF254, 0.20–0.25 mm) were used for the TLC analyses, which were visualized under model ZF-20D ultraviolet analyzing equipment (Shanghai Baoshan Gucun Photoelectricity Instrument, Shanghai, China) at 254 nm. 1H and 13C NMR experiments were performed on a JNM-ECZ400S NMR Spectrometer (Nippon Electric Company Limited, Tokyo, Japan) at 400 MHz for 1H NMR and 100 MHz for 13C NMR. Chemical shifts (δ) are given in parts per million (ppm), and coupling constants are given in Hz. The multiplicity of 1H NMR signals is reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, dd = doublet of doublets. The HRMS (ESI) spectroscopic data were obtained from an Agilent 1290II Mass Spectrometer (Agilent Technologies Inc., Santa Clara, CA, USA).
3.2. Synthesis and Characterization of the Compounds
3.2.1. 3-(Benzyloxy)-4-methoxybenzaldehyde (2)
A solution of 3-hydroxy-4-methoxybenzaldehyde (1; 1.52 g, 10.0 mmol, 1 equiv.), BnCl (1.52 g, 12.0 mmol, 1.2 equiv.), K2CO3 (4.15 g, 30.0 mmol, 3 equiv.), and KI (4.15 mg, 0.025 mmol, 0.025 equiv.) in anhydrous CH3CN (10 mL) was stirred at 82 °C for 2 h. The mixture was then cooled to 20–30 °C, after which the precipitate was filtered off. The solvent was removed under reduced pressure, and the obtained residue was dissolved in EtOAc (20 mL) and washed sequentially with Sad. NaHCO3 solution (2 × 10 mL), water (2 × 10 mL), and brine (2 × 10 mL). The organic layer was dried (anhydrous NaSO4) and concentrated under reduced pressure. The resultant crude products were purified by recrystallization (hexane), which furnished 3-(benzyloxy)-4-methoxybenzaldehyde (2) (83% yield).
3-(Benzoxy)-4-methoxybenzaldehyde (2): white solid; Rf (Hexane/EtOAc 80:20) = 0.62; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 90:10); 1H-NMR (400 MHz, CDCl3): δ = 9.80 (s, 1H), 7.46–7.44 (m, 4H), 7.38–7.34 (m, 2H), 7.32 (m, 1H), 7.00 (d, J = 8.8 Hz, 1H), 5.17 (s, 2H), 3.94 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ = 190.97, 155.15, 148.79, 136.38, 130.07, 128.75 (2C), 128.23, 127.59 (2C), 127.01, 111.44, 110.88, 70.93, 56.27. HRMS (ESI) calculated for C15H15O3+ [M + H]+ 243.1016, found 243.1013.
3.2.2. 2-(Benzyloxy)-1-methoxy-4-((E)-2-nitrovinyl)benzene (3)
A solution of 3-(benzyloxy)-4-methoxybenzaldehyde (2; 2.42 g, 10.0 mmol, 1 equiv.), NH4OAc (1.00 g, 13.0 mmol, 1.3 equiv.), and CH3NO2 (2.44 g, 40.0 mmol, 4 equiv.) in anhydrous HOAc (10 mL) was stirred at 118 °C for 4 h. The mixture was then cooled to 20–30 °C, after which the precipitate was filtered off. The precipitate was washed with water to neutral, which furnished 2-(benzyloxy)-1-methoxy-4-((E)-2-nitrovinyl)benzene (3) (89% yield).
2-(Benzoxy)-1-methoxy-4-((E)-2-nitrovinyl)benzene (3): yellow solid; Rf (Hexane/EtOAc 80:20) = 0.66; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 90:10) 1H-NMR (400 MHz, CDCl3): δ = 7.89 (d, J = 13.6 Hz, 1H), 7.42–7.30 (m, 6H), 7.15–7.13 (m, 1H), 7.01 (s, 1H), 6.91–6.89 (m, 1H), 5.14 (s, 2H), 3.91 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ = 153.56, 148.68, 139.42, 136.37, 135.20, 128.83 (2C), 128.32, 127.43 (2C), 125.02, 122.74, 113.22, 111.83, 71.25, 56.20. HRMS (ESI) calculated for C16H16NO4+ [M + H]+ 286.1074, found 286.1072.
3.2.3. 2-(3-(Benzyloxy)-4-methoxyphenyl)ethanamine (4)
A flask containing anhydrous THF (15 mL) was cooled to 0 °C under N2, after which LiAlH4 (1.52 g, 40.0 mmol, 4 equiv.) was added cautiously. A solution of 2-(benzyloxy)-1-methoxy-4-((E)-2-nitrovinyl)benzene (3; 2.85 g, 10.0 mmol, 1 equiv.) in anhydrous THF (30 mL) was added dropwise at 35 °C. The temperature of the system was controlled at 35 °C under N2 for 4 h. Upon completion, the reaction mixture was cooled to 0 °C and slowly quenched with water (6 mL). After 15 min, 15% (w/w) aq. NaOH (3 mL) was added. The resultant mixture was stirred for 30 min at 20–30 °C, and the mixture was filtered through a Celite pad with anhydrous MgSO4. The filtrate was concentrated, and the residue was purified as follows: methanol (15 mL) was added to the residue and allowed to dissolve completely by stirring. Anhydrous oxalic acid (1.05 g) was also added at 45 °C. Once completely dissolved, ethyl acetate (50 mL) was introduced into the mixture, and a significant amount of white solid was precipitated. The reaction was stirred for 2 h before cooling it down to 20–30 °C, after which we filtered the solution and air dried the obtained filter cake. Next, we adjusted the pH value to approximately 9–10 by adding a solution containing 30% (w/w) aq. NaOH (40 mL) while continuously stirring magnetically for 2 h. The solid was filtered, and then we beat the filter cake with water (50 mL) for 2 h before filtering it once more. Finally, the resulting filter cake was dried by air, which furnished the 2-(3-(benzyloxy)-4-methoxyphenyl)ethanamine (4) (50% yield).
2-(3-(Benzoxy)-4-methoxyphenyl)ethanamine (4): white solid; Rf (MeOH/CH2Cl2 90:10, 0.02% NH3·H2O) = 0.56; purification by flash column chromatography (deactivated silica gel, MeOH/CH2Cl2 95:5) 1H-NMR (400 MHz, CDCl3): δ = 7.39–7.37 (m, 2H), 7.30–7.26 (m, 2H), 7.23–7.20 (m, 1H), 7.96 (d, J = 8.0 Hz, 1H), 6.70–6.66 (m, 2H), 5.06 (s, 2H), 3.76 (s, 3H); 13C-NMR (100 MHz, CDCl3): δ = 148.30, 148.09, 137.28, 132.27, 128.58 (2C), 127.90, 127.48 (2C), 121.56, 115.14, 112.10, 71.07, 56.10, 43.47, 39.16. HRMS (ESI) calculated for C16H20NO2+ [M + H]+ 258.1489, found 258.1489.
3.2.4. Ethyl 2-(3-Hydroxy-4-methoxyphenyl)acetate (6)
A solution of 2-(3-hydroxy-4-methoxyphenyl)acetic acid (5; 1.82 g, 10.0 mmol, 1 equiv.) and H2SO4 (1.83 g, 1 mL, 18.7 mmol, 1.87 equiv.) in anhydrous EtOH (10 mL) was stirred at 78 °C for 4 h. The mixture was then cooled to 20–30 °C. The solvent was removed under reduced pressure, and the obtained residue was dissolved in EtOAc (10 mL) and washed sequentially with Sad. NaHCO3 solution (2 × 2 mL), water (2 × 5 mL), and brine (2 × 5 mL). The organic layer was dried (anhydrous NaSO4) and concentrated under reduced pressure, which furnished ethyl 2-(3-hydroxy-4-methoxyphenyl)acetate (6) (90% yield).
Ethyl 2-(3-hydroxy-4-methoxyphenyl)acetate (6): colorless oil; Rf (Hexane/EtOAc 35:65) = 0.66; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 60:40) 1H-NMR (400 MHz, CDCl3): δ = 6.78–6.74 (m, 1H), 6.61–6.57 (m, 2H), 6.49 (br, 1H), 4.01–3.95 (m, 2H), 3.59–3.55 (m, 3H), 3.39–3.35 (m, 2H), 1.10–1.02 (m, 3H); 13C-NMR (100 MHz, CDCl3): δ = 172.10, 146.14, 145.82, 127.17, 120.70, 115.88, 111.06, 60.81, 55.71, 40.58, 13.99. HRMS (ESI) data were calculated for C11H14O4Na+ [M + Na]+ 233.0784, found 233.0788.
3.2.5. Ethyl 2-(3-(Benzyloxy)-4-methoxyphenyl)acetate (7)
A solution of ethyl 2-(3-hydroxy-4-methoxyphenyl)acetate (6; 2.1 g, 10.0 mmol, 1 equiv.), BnCl (1.52 g, 12.0 mmol, 1.2 equiv.), K2CO3 (4.15 g, 30.0 mmol, 3 equiv.), and KI (4.15 mg, 0.025 mmol, 0.025 equiv.) in anhydrous CH3CN (10 mL) was stirred at 82 °C for 3 h. The mixture was then cooled to 20–30 °C, after which the precipitate was filtered off. The solvent was removed under reduced pressure, and the obtained residue was dissolved in EtOAc (20 mL) and washed sequentially with Sad. NaHCO3 solution (2 × 10 mL), water (2 × 10 mL), and brine (2 × 10 mL). The organic layer was dried (anhydrous NaSO4) and concentrated under reduced pressure. The resultant crude products were purified by recrystallization (hexane), which furnished ethyl 2-(3-(benzyloxy)-4-methoxyphenyl)acetate (7) (82% yield).
2-(3-(Benzoxy)-4-methoxyphenyl)acetate (7): white solid; Rf (Hexane/EtOAc 35:65) = 0.66; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 60:40) 1H-NMR (400 MHz, CDCl3): δ = 7.45–7.27 (m, 5H), 6.87–6.83 (m, 3H), 5.13 (s, 1H), 4.11 (q, 2H), 3.85 (s, 3H), 3.49 (s, 2H), 1.21 (t, 3H); 13C-NMR (100 MHz, CDCl3): δ = 171.88, 148.93, 148.24, 137.20, 128.62 (2C), 127.93, 127.47 (2C), 126.68, 122.13, 115.18, 111.95, 71.09, 60.88, 56.13, 40.99, 14.28. HRMS (ESI) calculated for C18H21O4+ [M + H]+ 301.1434, found 301.1436.
3.2.6. 2-(3-(Benzyloxy)-4-methoxyphenyl)acetic Acid (8)
A solution of ethyl 2-(3-(benzyloxy)-4-methoxyphenyl)acetate (7; 3.00 g, 10.0 mmol, 1 equiv.) and 20% (w/w) aq. NaOH (40 mL) in anhydrous EtOH (15 mL) was stirred at 78 °C for 3 h. The mixture was then cooled to 20–30 °C. The solvent was removed under reduced pressure, and the obtained residue was dissolved in EtOAc (10 mL) and water (10 mL). Following this, the 10% (w/w) aq. HCl was added to adjust the pH to 2–3. The precipitate was filtered off. The precipitate was washed with water to neutral, which furnished 2-(3-(benzyloxy)-4-methoxyphenyl)acetic acid (8) (93% yield).
2-(3-(Benzoxy)-4-methoxyphenyl)acetic acid (8): white solid; Rf (Hexane/EtOAc 80:20) = 0.53; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 50:50) 1H-NMR (400 MHz, CDCl3): δ = 7.43–7.27 (m, 5H), 6.84–6.83 (m, 3H), 5.11 (s, 2H), 3.85 (s, 3H), 3.52 (s, 2H); 13C-NMR (100 MHz, CDCl3): δ = 177.96, 149.13, 148.27, 137.04, 128.63 (2C), 127.98, 127.57 (2C), 125.75, 122.29, 115.34, 111.96, 71.15, 56.12, 40.60. HRMS (ESI) calculated for C16H16O4Na+ [M + Na]+ 295.0941, found 295.0944.
3.2.7. 2-(5-(Benzyloxy)-2-bromo-4-methoxyphenyl)acetic Acid (9)
Bromine (11.2 mmol, 1.12 equiv.) was cautiously added to a solution of 2-(3-(benzyloxy)-4-methoxyphenyl)acetic acid (8; 2.72 g, 10.0 mmol, 1 equiv.) and anhydrous sodium acetate (1.36 g, 34.0 mmol, 3.4 equiv.) in acetic acid (15 mL), and the mixture was stirred for 1 h at 20–30 °C. The precipitate was filtered off and washed with water to neutral pH, which furnished 2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetic acid (9) (61% yield).
2-(5-(Benzoxy)-2-bromo-4-methoxyphenyl)acetic acid (9): white solid; Rf (Hexane/EtOAc 65:35) = 0.46; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 85:15) 1H-NMR (400 MHz, CDCl3): δ = 8.02 (br, 1H), 7.41–7.29 (m, 5H), 7.05 (s, 1H), 6.82 (s, 1H), 5.09 (s, 2H), 3.84 (s, 3H), 3.70 (s, 2H); 13C-NMR (100 MHz, CDCl3): δ = 176.87, 149.80, 147.63, 136.56, 128.71 (2C), 128.18, 127.56 (2C), 125.25, 116.74, 116.04, 115.80, 71.39, 56.32, 40.85. HRMS (ESI) calculated for C16H15BrO4Na+ [M + Na]+ 373.0046, found 373.0050.
3.2.8. N-(3-(Benzoxy)-4-methoxyphenethyl)-2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetamide (10)
Next, 1-hydroxybenzotriazole (2.03 g, 15.0 mmol, 1.5 equiv.) was added to a solution of 2-(3-(benzyloxy)-4-methoxyphenyl)ethanamine (4; 2.57 g, 10.0 mmol, 1 equiv.) and 2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetic acid (9; 3.69 g, 10.5 mmol, 1.05 equiv.) in dry DMF (50 mL). The reaction mixture was cooled to 0 °C, after which 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride (2.3 g, 12.0 mmol, 1.2 equiv.) was added. The reaction mixture was warmed slowly to 20–30 °C and then stirred for 4 h before being quenched by the addition of NaHCO3 (25 mL, sat., aq.) and extracted with EtOAc (3 × 25 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated to afford N-(3-(benzyloxy)-4-methoxyphenethyl)-2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetamide (10) (74% yield).
N-(3-(benzyloxy)-4-methoxyphenethyl)-2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetamide (10): white solid; Rf (Hexane/EtOAc 65:35) = 0.62; purification by flash column chromatography (deactivated silica gel, Hexane/EtOAc 85:15) 1H-NMR (400 MHz, CDCl3): δ = 7.43–7.28 (m, 10H), 6.99 (s, 1H), 6.78 (s, 1H), 6.73 (d, J = 8.1 Hz, 1H), 6.66 (d, J = 2.0 Hz, 1H), 6.58 (dd, J = 8.1, 2.0 Hz, 1H), 5.33 (t, J = 5.8 Hz, 1H), 5.08 (s, 2H), 5.06 (s, 2H), 3.82 (s, 3H), 3.81 (s, 3H), 3.49 (s, 2H), 3.37 (q, J = 7.0 Hz, 2H), 2.60 (t, J = 6.9 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 169.83, 149.73, 148.46, 148.30, 147.81, 137.15, 136.42, 131.05, 128.70 (2C), 128.62 (2C), 128.19, 127.96, 127.54 (2C), 127.47 (2C), 126.49, 121.39, 116.43, 116.11, 115.48, 114.69, 111.97, 71.17, 71.10, 56.29, 56.10, 43.64, 40.72, 34.94, 29.78. HRMS (ESI) data were calculated for C32H33BrNO5+ [M + H]+ 590.1537, found 590.1538.
3.2.9. 1-(5-(Benzoxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline (11)
A solution of N-(3-(benzyloxy)-4-methoxyphenethyl)-2-(5-(benzyloxy)-2-bromo-4-methoxyphenyl)acetamide (10; 5.91 g, 10.0 mmol, 1 equiv.) and POCl3 (1.84 g, 12.0 mmol, 1.2 equiv.) in anhydrous CH3CN (10 mL) was stirred at 82 °C for 4 h. The mixture was then cooled to 20–30 °C and quenched slowly with water (6 mL). The solvent was removed under reduced pressure. The resultant crude products were purified by column chromatography on silica gel (CH2Cl2), which furnished 1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline (11) (71% yield).
1-(5-(Benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline (11): White solid; Rf (CH2Cl2/MeOH 95:5) = 0.52; purification by flash column chromatography (deactivated silica gel, CH2Cl2) 1H-NMR (400 MHz, CDCl3): δ = 7.42–7.18 (m, 10H), 7.03 (s, 1H), 6.90 (s, 1H), 6.79 (s, 1H), 6.62 (s, 1H), 5.15 (s, 2H), 5.00 (s, 2H), 4.04 (s, 2H), 3.82 (s, 3H), 3.79 (s, 3H), 3.59 (t, J = 7.5 Hz, 2H), 2.43 (t, J = 7.6 Hz, 2H); 13C-NMR (100 MHz, CDCl3): δ = 165.65, 150.04, 148.98, 147.99, 147.70, 136.71, 136.68, 131.43, 129.43, 128.76 (2C), 128.54 (2C), 128.13, 127.88, 127.28 (2C), 127.16 (2C), 121.61, 115.78, 114.87, 114.69, 112.31, 109.75, 70.97, 70.85, 56.38, 56.27, 47.21, 42.05, 25.68. HRMS (ESI) calculated for C32H31BrNO4+ [M + H]+ 572.1431, found 572.1432.
3.2.10. 1-(5-(Benzoxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-1,2,3,4-tetrahydro-7-methoxyisoquinoline (12)
NaBH4 (0.57 g, 15.0 mmol, 1.5 equiv.) was added slowly to a stirred ice-cooled solution of 1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline (11; 5.72 g, 10.0 mmol, 1.0 equiv.) in EtOH (30 mL) at 0 °C. The mixture was stirred at 0 °C for 1 h and then at 20–30 °C for 2 h. The mixture was cooled to 0 °C and diluted with water (10 mL), after which the precipitate was filtered. The precipitate was washed with water to neutral, which furnished 1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-1,2,3,4-tetrahydro-7-methoxyisoquinoline (12) (72% yield).
1-(5-(Benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-1,2,3,4-tetrahydro-7-methoxyisoquinoline (12): white solid; Rf (CH2Cl2/MeOH 90:10) = 0.55; purification by flash column chromatography (deactivated silica gel, CH2Cl2) 1H-NMR (400 MHz, DMSO-d6): δ = 9.16 (br, 1H), 7.41–7.32 (m, 10H), 7.28 (s, 1H), 7.18 (s, 1H), 6.88 (s, 1H), 6.36 (s, 1H), 5.07 (d, J = 11.6 Hz, 1H), 5.03 (s, 2H), 4.98 (d, J = 11.6 Hz, 1H), 4.53 (t, J = 7.4 Hz, 2H), 3.75 (s, 3H), 3.53 (s, 3H), 3.41–3.33 (m, 2H), 3.20–3.15 (m, 2H), 3.02–2.95 (m, 1H), 2.87–2.79 (m, 1H); 13C-NMR (100 MHz, DMSO-d6): δ = 149.59, 147.90, 147.80, 137.47, 137.13, 130.19, 129.00 (2C), 128.96 (2C), 128.58 (2C), 128.43, 128.31 (2C), 127.74, 125.02, 117.68, 116.44, 115.43, 113.93, 110.55, 70.74, 70.34, 56.57, 55.88, 54.35, 39.19, 29.61, 29.12, 27.08, 25.37. HRMS (ESI) data were calculated for C32H33BrNO4+ [M + H]+ 574.1588, found 574.1587.
3.2.11. Tert-Butyl 1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline-2(1H)-carboxylate (13)
Triethylamine (1.32 g, 13.0 mmol, 1.3 equiv.) and Boc2O (2.62 g, 12.0 mmol, 1.2 equiv.) were added to a stirred solution of 1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-1,2,3,4-tetrahydro-7-methoxyisoquinoline (12; 5.75 g, 10.0 mmol, 1 equiv.) in CH2Cl2 (15 mL) at 20–30 °C, and the resulting mixture was stirred for 2 h and washed sequentially with water (2 × 10 mL) and brine (2 × 10 mL). The organic layer was dried (anhydrous NaSO4) and concentrated under reduced pressure, which furnished tert-butyl1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline-2(1H)-carboxylate (13) (80% yield).
Tert-butyl1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline-2(1H)-carboxylate (13): white solid; Rf (CH2Cl2/MeOH 97:3) = 0.52; purification by flash column chromatography (deactivated silica gel, CH2Cl2) 1H-NMR (400 MHz, CDCl3): δ = 7.44–7.27 (m, 10H), 7.06 (s, 1H), 6.72 (s, 1H), 6.63 (s, 1H), 6.55 (s, 1H), 5.24 (dd, J = 10.1, 3.8 Hz, 1H), 5.11–5.05 (m, 4H), 4.27–4.21 (m, 1H), 3.86 (s, 3H), 3.84 (s, 3H), 3.18–3.13 (m, 2H), 2.85–2.79 (m, 2H), 2.57–2.52 (m, 1H), 1.20 (s, 9H); 13C-NMR (400 MHz, CDCl3): δ = 154.17, 149.84, 146.58, 143.65, 130.39, 130.28, 127.43, 123.82, 123.01, 115.68, 114.98, 112.55, 79.48, 67.53, 59.61, 56.34, 56.10, 30.02, 28.63 (3C), 28.22, 25.64. HRMS (ESI) calculated for C37H41BrNO6+ [M + H]+ 674.2112, found 674.2114.
3.2.12. Tert-Butyl 1-(5-(Benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline-2(1H)-carboxylate (14)
Pd(OAc)2 (4.49 g, 2.0 mmol, 0.2 equiv.), the ligand di-tert-butyl(methyl)phosphonium tetrafluoroborate (9.89 g, 4.0 mmol, 0.4 equiv.), and Cs2CO3 (9.77 g, 3.0 mmol, 3 equiv.) were added to a solution of tert-butyl1-(5-(benzyloxy)-2-bromo-4-methoxybenzyl)-6-(benzyloxy)-3,4-dihydro-7-methoxyisoquinoline-2(1H)-carboxylate (13; 6.75 g, 10.0 mmol, 1 equiv.) in 1,4-dioxane (40 mL) by purging with nitrogen for 8 h at 101 °C. After cooling to 20–30 °C, the solvent was removed under reduced pressure. The precipitate was extracted with CH2Cl2 (3 × 50 mL). The combined organic layer was dried (anhydrous Na2SO4), and the precipitate was loaded onto a deactivated silica gel column (200–300 mesh) and eluted with hexane/EtOAc (90:10, v/v) to afford 6H-dibenzo[de,g]quinoline-6-carboxylic acid, 4,5,6a,7-tetrahydro-1,9,10-trimethoxy-2-(phenylmethoxy)-,1,1-dimethylethyl ester (14) (53%).
6H-Dibenzo[de,g]quinoline-6-carboxylic acid, 4,5,6a,7-Tetrahydro-1,9,10-trimethoxy-2-(phenylmethoxy)-, 1,1-dimethylethyl ester (14): white solid; Rf (Hexanes/EtOAc 80:20) = 0.64; purification by flash column chromatography (deactivated silica gel, Hexanes/EtOAc 94:6) 1H-NMR (400 MHz, CDCl3): δ = 8.17 (s, 1H), 7.47–7.27 (m, 10H), 6.77 (s, 1H), 6.67 (s, 1H), 5.19–5.08 (m, 4H), 4.63–4.60 (m, 1H), 4.38–4.35 (m, 1H), 3.91 (s, 3H), 3.70 (s, 3H), 2.92–2.68 (m, 5H), 2.59 (d, J = 14.7 Hz, 1H), 1.43 (s, 9H); 13C-NMR (400 MHz, CDCl3): δ = 154.28, 149.23, 148.14, 147.81, 147.19, 137.18, 136.96, 130.35, 129.53, 128.75 (2C), 128.66 (2C), 128.05, 127.96, 127.37 (2C), 127.33 (2C), 126.71, 117.20, 116.02, 115.71, 114.15, 110.71, 79.47, 71.68, 71.13, 56.55, 56.27, 54.33, 42.12, 36.48, 28.53, 28.17 (3C). HRMS (ESI) calculated for C37H39NO6Na+ [M + Na]+ 616.2670, found 616.2667.
3.2.13. Tert-Butyl 6H-Dibenzo[de,g]quinoline-6-carboxylic acid,4,5,6a,7-tetrahydro-9-hydroxy-1,2,10-trimethoxy-,1,1-dimethylethyl Ester (15)
Palladium 10% on carbon (wetted with ca. 55% water) (Pd/C) (1.18 g) and HOAc (0.5 mL) were added to a solution of 6H-dibenzo[de,g]quinoline-6-carboxylic acid, 4,5,6a,7-tetrahydro-1,9,10-trimethoxy-2-(phenylmethoxy)-, and 1,1-dimethylethyl ester (14; 5.94 g, 10.0 mmol, 1 equiv.) in THF (40 mL) by purging with hydrogen for 8 h at 20–30 °C. The precipitate was filtered off. The solvent was removed under reduced pressure, and the obtained residue was dissolved in EtOAc (40 mL) and washed sequentially with water (2 × 20 mL) and brine (2 × 20 mL). The organic layer was dried (anhydrous NaSO4) and concentrated under reduced pressure, which furnished 6H-dibenzo[de,g]quinoline-6-carboxylic acid and 4,5,6a,7-tetrahydro-9-hydroxy-1,2,10-trimethoxy-, 1,1-dimethylethyl ester (15) (82% yield).
6H-Dibenzo[de,g]quinoline-6-carboxylic acid,4,5,6a,7-tetrahydro-9-hydroxy-1,2,10-trimethoxy-,1,1-dimethylethyl ester (15): white solid; Rf (Hexanes/EtOAc 66:34) = 0.46; purification by flash column chromatography (deactivated silica gel, Hexanes/EtOAc 80:20) 1H-NMR (400 MHz, DMSO-d6): δ = 9.18 (s, 1H), 9.16 (s, 1H), 7.89 (s, 1H), 6.65 (s, 1H), 6.54 (s, 1H), 4.38–4.33 (m, 1H), 4.15 (d, J = 12.7 Hz, 1H), 3.75 (s, 3H), 3.51 (s, 3H), 2.66–2.49 (m, 5H), 1.37 (s, 9H); 13C-NMR (100 MHz, DMSO-d6): δ = 154.17, 149.84, 146.58, 143.65, 130.39, 130.28, 127.43, 123.82, 123.01, 115.68, 114.98, 112.55, 79.48, 67.53, 59.61, 56.34, 56.10, 30.02, 28.63 (3C), 28.22, 25.64. HRMS (ESI) calculated for C23H27NO6Na+ [M + H]+ 436.1731, found 436.1729.
3.2.14. 5,6,6a,7-Tetrahydro-1,10-dimethoxy-4H-dibenzo[de,g]quinoline-2,9-diol (16)
ZnBr2 (9.01 g, 40.0 mmol, 4 equiv.) was added to a solution of 6H-dibenzo[de,g]quinoline-6-carboxylic acid, 4,5,6a,7-tetrahydro-9-hydroxy-1,2,10-trimethoxy-, 1,1-dimethylethyl ester (15; 4.13 g, 10.0 mmol, 1 equiv.) in dry CH2Cl2 (50 mL) under a nitrogen atmosphere, and the mixture was stirred at 20–30 °C for 48 h. The mixture was then quenched with a solution of saturated NaHCO3 (50 mL) and extracted with CH2Cl2 (3 × 250 mL). The combined organic layer was dried (anhydrous Na2SO4) and concentrated under reduced pressure to afford the resultant crude products. Then, the resultant crude products were purified by column chromatography on silica gel (CH2Cl2/MeOH 90:10), which furnished 5,6,6a,7-Tetrahydro-1,10-dimethoxy-4H-dibenzo[de,g]quinoline-2,9-diol (16) (49% yield).
5,6,6a,7-Tetrahydro-1,10-dimethoxy-4H-dibenzo[de,g]quinoline-2,9-diol (16): brown solid; Rf (CH2Cl2/MeOH 80:20) = 0.45; purification by flash column chromatography (deactivated silica gel, CH2Cl2/MeOH 90:10) 1H-NMR (400 MHz, DMSO-d6): δ = 10.21 (br, 1H), 9.48 (s, 1H), 9.37 (s, 1H), 7.85 (s, 1H), 6.73 (s, 1H), 6.62 (s, 1H), 4.09–4.06 (m, 1H), 3.73 (s, 3H), 3.55 (s, 3H), 3.46–3.44 (m, 1H), 3.13–3.02 (m, 2H), 2.89–2.84 (m, 1H), 2.81–2.73 (m, 2H); 13C-NMR (100 MHz, DMSO-d6): δ = 151.23, 147.01, 146.88, 143.85, 127.11, 127.07, 126.78, 122.73, 120.16, 115.75, 114.94, 112.54, 59.87, 56.16, 52.42, 40.61, 32.82, 25.14. HRMS (ESI) calculated for C18H20NO4+ [M + H]+ 314.1387, found 314.1384.
4. Conclusions
The total synthesis of laurolitsine, an alkaloid extracted from Litsea glutinosa bark, was achieved in 14 steps with a 2.3% yield (this was calculated using 3-hydroxy-4-methoxybenzaldehyde as the starting material) starting from 3-hydroxy-4-methoxybenzaldehyde (1) and 2-(3-hydroxy-4-methoxyphenyl)acetic acid (5), and the longest linear sequence consisted of 11 steps. In this study, many experimental steps were optimized, and an innovative postprocessing method for salting 2-(3-(benzyloxy)-4-methoxyphenyl)ethanamine (4) with oxalic acid was proposed.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules29030745/s1. Detailed procedures for the synthesis of 16 and 1H- and 13C-NMR charts of all the compounds.
Author Contributions
Conceptualization, M.C. and N.C.; methodology, M.C.; software, Y.W.; validation, Y.Z.; formal analysis, C.Z.; investigation, N.C.; resources, X.Z.; data curation, M.C.; writing—original draft preparation, M.C.; writing—review and editing, N.C.; visualization, X.Z.; supervision, X.Z.; project administration, X.Z.; funding acquisition, X.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was founded by the Key Research and Development Program of Hainan Province (No. ZDYF2022SHFZ037) and the National Natural Science Foundation of China (No. 82060778).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, Y.; Wang, R.; Yang, Y.; Ma, N.; Zhou, Z.; Tan, Y.; Dong, L.; Li, Y.; Lu, W.; Wu, C.; et al. Laurolitsine ameliorates type 2 diabetes by regulating the hepatic LKB1-AMPK pathway and gut microbiota. Phytomedicine 2022, 106, 154423. [Google Scholar]
- Zhang, X.; Jin, Y.; Wu, Y.; Zhang, C.; Jin, D.; Zheng, Q.; Li, Y. Anti-hyperglycemic and anti-hyperlipidemia effects of the alkaloid-rich extract from barks of Litsea glutinosa in ob/ob mice. Sci. Rep. 2018, 8, 12646. [Google Scholar] [CrossRef] [PubMed]
- Nakasato, T.; Nomura, S. Studies on the Alkaloids of Lauraceous Plants. III: Alkaloids isolated from the Leaves of Neolitsea sericea (BLUME) KOIDZ. Yakugaku Zasshi J. Pharm. Soc. Jpn. 1959, 79, 1267–1272. [Google Scholar] [CrossRef]
- Sun, C.; Li, J.; Wang, D.; Yu, J.; Wang, X.; Huang, L. Preparative separation of alkaloids from Litsea cubeba using combined applications of pH-zone-refining and high-speed counter-current chromatography. RSC Adv. 2015, 5, 75831–75837. [Google Scholar] [CrossRef]
- Chen, K.S.; Ko, F.N.; Teng, C.M.; Wu, Y.C. Antiplatelet and vasorelaxing actions of some aporphinoids. Planta Med. 1996, 62, 133–136. [Google Scholar] [CrossRef]
- Chia, Y.C.; Chen, K.S.; Chang, Y.L.; Teng, C.M.; Wu, Y.C. Antiplatelet actions of aporphinoids from Formosan plants. Bioorganic Med. Chem. Lett. 1999, 9, 3295–3300. [Google Scholar] [CrossRef] [PubMed]
- Munoz, V.; Sauvain, M.; Mollinedo, P.; Callapa, J.; Rojas, I.; Gimenez, A.; Valentin, A.; Mallie, M. Antimalarial activity and cytotoxicity of (-)-roemrefidine isolated from the stem bark of Sparattanthelium amazonum. Planta Med. 1999, 65, 448–449. [Google Scholar] [CrossRef]
- Corsini, G.U.; Del Zompo, M.; Gessa, G.L.; Mangoni, A. Therapeutic efficacy of apomorphine combined with an extracerebral inhibitor of dopamine receptors in Parkinson’s disease. Lancet 1979, 1, 954–956. [Google Scholar] [CrossRef]
- Guinaudeau, H.; Lebœuf, M.; Cavé, A. Aporphinoid Alkaloids, V. J. Nat. Prod. 1994, 57, 1033–1135. [Google Scholar] [CrossRef]
- Guinaudeau, H.; Leboeuf, M.; Cavé, A. Aporphinoid Alkaloids, III. J. Nat. Prod. 1983, 46, 761–835. [Google Scholar] [CrossRef]
- Xin, A.; Liu, J.; Di, D. Research progress on aporphine alkaloids (in Chinese). Res. Prog. Aporphine Alkaloids 2018, 49, 712–724. [Google Scholar]
- Lafrance, M.; Gorelsky, S.I.; Fagnou, K. High-yielding palladium-catalyzed intramolecular alkane arylation: Reaction development and mechanistic studies. J. Am. Chem. Soc. 2007, 129, 14570–14571. [Google Scholar] [CrossRef]
- Gao, K.; Wu, J. Synthesis of functionalized 1,2-dihydroisoquinolines via multicomponent one-pot reaction of 2-alkynylbenzaldehyde, amine, zinc, and allylic bromide or benzyl bromide. J. Org. Chem. 2007, 72, 8611–8613. [Google Scholar] [CrossRef]
- Chaudhary, S.; Pecic, S.; Legendre, O.; Navarro, H.A.; Harding, W.W. (+/−)-Nantenine analogs as antagonists at human 5-HT(2A) receptors: C1 and flexible congeners. Bioorganic Med. Chem. Lett. 2009, 19, 2530–2532. [Google Scholar] [CrossRef]
- Pieper, P.; Mchugh, E.; Amaral, M.; Tempone, A.G.; Anderson, E.A. Enantioselective synthesis and anti-parasitic properties of aporphine natural products. Tetrahedron 2019, 76, 130814. [Google Scholar] [CrossRef]
- Nishimoto, S.; Nakahashi, H.; Toyota, M. Development of Pd(OAc)2-catalyzed tandem oxidation of C N, C C, and C(sp3)–H bonds: Concise synthesis of 1-aroylisoquinoline, oxoaporphine, and 8-oxyprotoberberine alkaloids. Tetrahedron Lett. 2020, 61, 152599. [Google Scholar] [CrossRef]
- Zhang, Y.-N.; Zhong, X.-G.; Zheng, Z.-P.; Hu, X.-D.; Zuo, J.-P.; Hu, L.-H. Discovery and synthesis of new immunosuppressive alkaloids from the stem of Fissistigma oldhamii (Hemsl.) Merr. Bioorganic Med. Chem. 2007, 15, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Jaiswal, P.K.; Kumar, S.; Mathur, M.; Swami, A.K.; Yadav, D.K.; Chaudhary, S. Discovery of Aporphine Analogues as Potential Antiplatelet and Antioxidant Agents: Design, Synthesis, Structure-Activity Relationships, Biological Evaluations, and in silico Molecular Docking Studies. ChemMedChem 2018, 13, 1817–1832. [Google Scholar] [CrossRef] [PubMed]
- Colligs, V.; Hansen, S.P.; Imbri, D.; Seo, E.J.; Kadioglu, O.; Efferth, T.; Opatz, T. Synthesis and biological evaluation of a D-ring-contracted analogue of lamellarin D. Bioorg Med. Chem. 2017, 25, 6137–6148. [Google Scholar] [CrossRef] [PubMed]
- Leese, M.P.; Jourdan, F.L.; Major, M.R.; Dohle, W.; Hamel, E.; Ferrandis, E.; Fiore, A.; Kasprzyk, P.G.; Potter, B.V. Tetrahydroisoquinolinone-based steroidomimetic and chimeric microtubule disruptors. ChemMedChem 2014, 9, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Li, H.; Sun, F.; Bao, C.; Bao, X.; Chen, G. The synthesis of precursor of FP- (+) DTBZ. Synth. Commun. 2019, 49, 3218–3225. [Google Scholar] [CrossRef]
- Wang, Y.C.; Georghiou, P.E. First enantioselective total synthesis of (-)-tejedine. Org. Lett. 2002, 4, 2675–2678. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Harding, W.W. Synthesis of C-Homoaporphines via Microwave-Assisted Direct Arylation. Tetrahedron 2011, 67, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Pecic, S.; Legendre, O.; Harding, W.W. Microwave-Assisted Direct Biaryl Coupling: First Application to the Synthesis of Aporphines. Tetrahedron Lett. 2009, 50, 2437–2439. [Google Scholar] [CrossRef]
- Hellal, M.; Singh, S.; Cuny, G.D. Monoligated Pd(0)-catalyzed intramolecular ortho- and para-arylation of phenols for the synthesis of aporphine alkaloids. Synthesis of (−)-lirinine. Tetrahedron 2012, 68, 1674–1681. [Google Scholar] [CrossRef]
Figure 1.
The structure of laurolitsine.

Scheme 1.
Retrosynthetic analysis of laurolitsine.

Scheme 2.
Conditions: (a) K2CO3, BnCl, CH3CN, 82 °C, 83%; (b) NH4OAc, CH3NO2, HOAc, 118 °C, 89%; (c) LiAlH4, THF, 30 °C, 50%.
Scheme 2.
Conditions: (a) K2CO3, BnCl, CH3CN, 82 °C, 83%; (b) NH4OAc, CH3NO2, HOAc, 118 °C, 89%; (c) LiAlH4, THF, 30 °C, 50%.

Scheme 3.
Conditions: (d) EtOH, H2SO4, 78 °C, 90%; (e) K2CO3, BnCl, CH3CN, 82 °C, 82%; (f) NaOH, EtOH, 78 °C, 93%; (g) Br2, NaOAc, HOAc, 25 °C, 61%.
Scheme 3.
Conditions: (d) EtOH, H2SO4, 78 °C, 90%; (e) K2CO3, BnCl, CH3CN, 82 °C, 82%; (f) NaOH, EtOH, 78 °C, 93%; (g) Br2, NaOAc, HOAc, 25 °C, 61%.

Scheme 4.
Conditions: (h) DMF, HOBt, EDC, 74%; (i) POCl3, CH3CN, 82 °C, 71%; (j) NaBH4, EtOH, 25 °C, 72%; (k) (Boc)2O, Et3N, CH2Cl2, 25 °C, 80%; (l) Cs2CO3, Pd(OAc)2, (t-Bu)2PMeHBF4, 1,4-dioxane, 100 °C, 53%; (m) Pd/C, THF, HOAc, 25 °C, 82%; (n) ZnBr2, CH2Cl2, 25 °C, 49%.
Scheme 4.
Conditions: (h) DMF, HOBt, EDC, 74%; (i) POCl3, CH3CN, 82 °C, 71%; (j) NaBH4, EtOH, 25 °C, 72%; (k) (Boc)2O, Et3N, CH2Cl2, 25 °C, 80%; (l) Cs2CO3, Pd(OAc)2, (t-Bu)2PMeHBF4, 1,4-dioxane, 100 °C, 53%; (m) Pd/C, THF, HOAc, 25 °C, 82%; (n) ZnBr2, CH2Cl2, 25 °C, 49%.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of the 13C NMR data of 16 with literature data [4].
Table 1.
Comparison of the 13C NMR data of 16 with literature data [4].
| No. | Natural (δ1) | Sample 16 (δ2) | Δ = δ1 − δ2 |
|---|---|---|---|
| 1 | 143.1 | 143.9 | −0.8 |
| 1a | 127.1 | 126.8 | 0.3 |
| 1b | 126.3 | 122.7 | 3.6 |
| 2 | 149.6 | 151.2 | −1.6 |
| 3 | 115.1 | 114.9 | 0.2 |
| 3a | 129.7 | 127.1 | 2.6 |
| 4 | 28.9 | 25.1 | 3.8 |
| 5 | 43.1 | 40.6 | 2.5 |
| 6a | 53.9 | 52.4 | 1.5 |
| 7 | 36.5 | 32.8 | 3.7 |
| 7a | 130.1 | 127.1 | 3.0 |
| 8 | 115.6 | 115.8 | −0.2 |
| 9 | 146.5 | 147.0 | −0.5 |
| 10 | 146.3 | 146.9 | −0.6 |
| 11 | 112.6 | 112.5 | 0.1 |
| 11a | 123.5 | 120.2 | 3.3 |
| 1-OCH3 | 59.7 | 59.9 | −0.2 |
| 10-OCH3 | 56.2 | 56.2 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cao, M.; Wang, Y.; Zhang, Y.; Zhang, C.; Chen, N.; Zhang, X. Total Synthesis of the Racemate of Laurolitsine. Molecules 2024, 29, 745. https://doi.org/10.3390/molecules29030745
AMA Style
Cao M, Wang Y, Zhang Y, Zhang C, Chen N, Zhang X. Total Synthesis of the Racemate of Laurolitsine. Molecules. 2024; 29(3):745. https://doi.org/10.3390/molecules29030745
Chicago/Turabian StyleCao, Mingyu, Yiming Wang, Yong Zhang, Caiyun Zhang, Niangen Chen, and Xiaopo Zhang. 2024. "Total Synthesis of the Racemate of Laurolitsine" Molecules 29, no. 3: 745. https://doi.org/10.3390/molecules29030745