
Chromatographic Methods Developed for the Quantification of Quercetin Extracted from Natural Sources: Systematic Review of Published Studies from 2018 to 2022
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Database Search/Search Strategy
2.2. Article Analysis
2.3. Information Collection
2.4. Assessment of the Risk of Bias
2.5. Data Analysis
3. Results
3.1. Quercetin Sources
3.2. Sample Treatment Prior Chromatographic Analysis
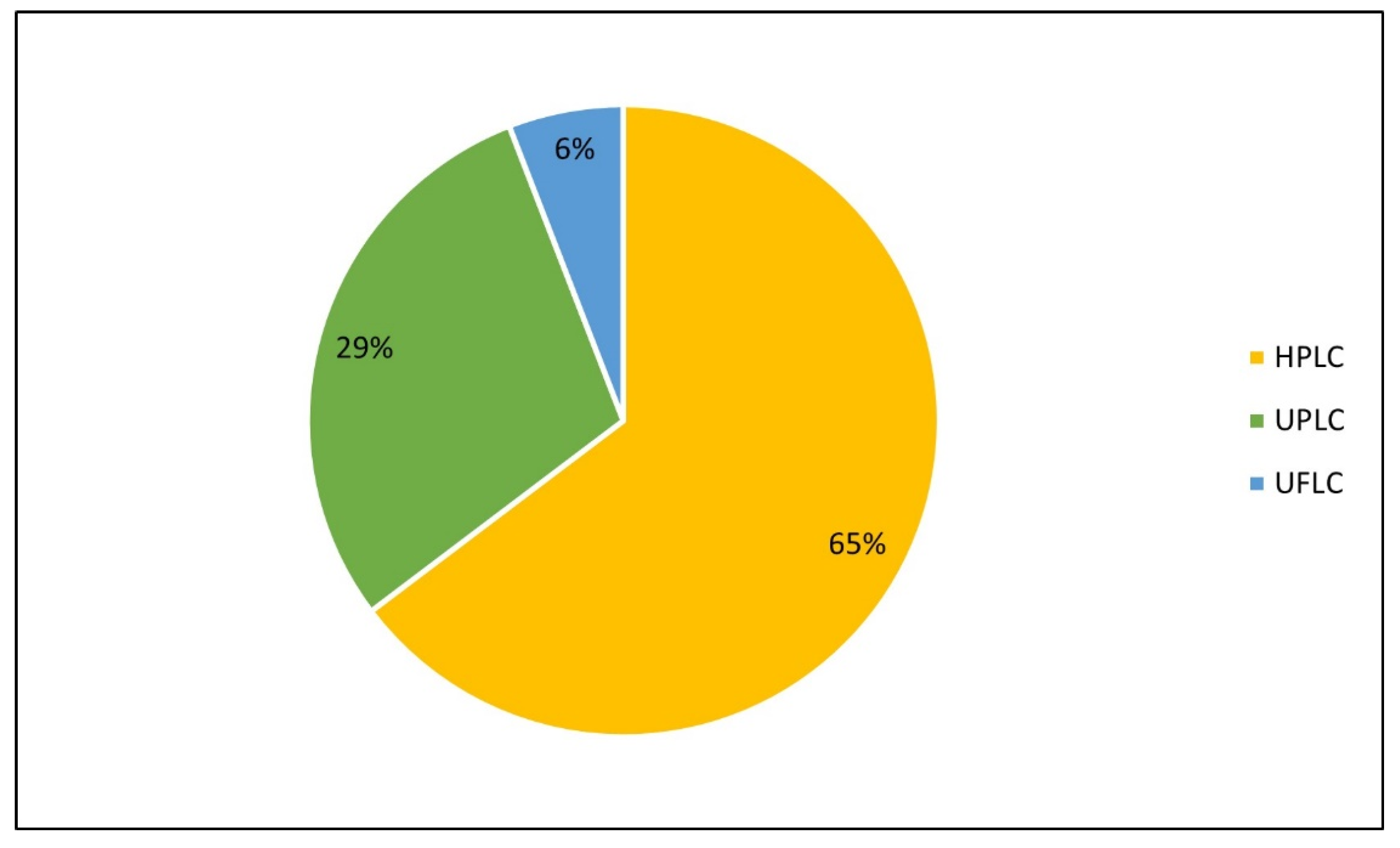
3.3. Chromatographic Conditions
3.4. Validation Parameters
3.5. Bias Assessment
3.6. Assessment of the Methods
3.7. Limitations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- D’Andrea, G. Quercetin: A Flavonol with Multifaceted Therapeutic Applications? Fitoterapia 2015, 106, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Dolatabadi, J.E.N.; Mokhtarzadeh, A.; Ghareghoran, S.M.; Dehghan, G. Synthesis, Characterization and Antioxidant Property of Quercetin-Tb(III) Complex. Adv. Pharm. Bull. 2014, 4, 101–104. [Google Scholar] [CrossRef]
- Corso, M.; Perreau, F.; Mouille, G.; Lepiniec, L. Specialized Phenolic Compounds in Seeds: Structures, Functions, and Regulations. Plant Sci. 2020, 296, 110471. [Google Scholar] [CrossRef] [PubMed]
- Cunha, A.P. da Flavonoides. In Farmacognosia e Fitoquímica; Fundação Calouste Gulbenkian: Lisbon, Portugal, 2010; pp. 238–293. ISBN 972-31-1142-X. [Google Scholar]
- Di Lorenzo, C.; Colombo, F.; Biella, S.; Stockley, C.; Restani, P. Polyphenols and Human Health: The Role of Bioavailability. Nutrients 2021, 13, 273. [Google Scholar] [CrossRef] [PubMed]
- Durazzo, A.; Lucarini, M.; Souto, E.B.; Cicala, C.; Caiazzo, E.; Izzo, A.A.; Novellino, E.; Santini, A. Polyphenols: A Concise Overview on the Chemistry, Occurrence, and Human Health. Phytother. Res. 2019, 33, 2221–2243. [Google Scholar] [CrossRef]
- Es-Safi, N.E.; Ghidouche, S.; Ducrot, P.H. Flavonoids: Hemisynthesis, Reactivity, Characterization and Free Radical Scavenging Activity. Molecules 2007, 12, 2228–2258. [Google Scholar] [CrossRef]
- Singla, R.K.; Dubey, A.K.; Garg, A.; Sharma, R.K.; Fiorino, M.; Ameen, S.M.; Haddad, M.A.; Al-Hiary, M. Natural Polyphenols: Chemical Classification, Definition of Classes, Subcategories, and Structures. J. AOAC Int. 2019, 102, 1397–1400. [Google Scholar] [CrossRef]
- Dias, M.C.; Pinto, D.C.G.A.; Silva, A.M.S. Plant Flavonoids: Chemical Characteristics and Biological Activity. Molecules 2021, 26, 5377. [Google Scholar] [CrossRef]
- Zhao, Z.; Cui, X.; Ma, X.; Wang, Z. Preparation, Characterization, and Evaluation of Antioxidant Activity and Bioavailability of a Self-Nanoemulsifying Drug Delivery System (SNEDDS) for Buckwheat Flavonoids. Acta Biochim. Biophys. Sin. 2020, 52, 1265–1274. [Google Scholar] [CrossRef]
- Beecher, G.R. Overview of Dietary Flavonoids: Nomenclature, Occurence and Intake. J. Nutr. 2003, 133, 3248S–3254S. [Google Scholar] [CrossRef]
- Grande, F.; Parisi, O.I.; Mordocco, R.A.; Rocca, C.; Puoci, F.; Scrivano, L.; Quintieri, A.M.; Cantafio, P.; Ferla, S.; Brancale, A.; et al. Quercetin Derivatives as Novel Antihypertensive Agents: Synthesis and Physiological Characterization. Eur. J. Pharm. Sci. 2016, 82, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yao, J.; Han, C.; Yang, J.; Chaudhry, M.T.; Wang, S.; Liu, H.; Yin, Y. Quercetin, Inflammation and Immunity. Nutrients 2016, 8, 167. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Arif, Y.; Bajguz, A.; Hayat, S. The Role of Quercetin in Plants. Plant Physiol. Biochem. 2021, 166, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Chew, Y.L.; Khor, M.A.; Lim, Y.Y. Choices of Chromatographic Methods as Stability Indicating Assays for Pharmaceutical Products: A Review. Heliyon 2021, 7, e06553. [Google Scholar] [CrossRef]
- Skoog, D.A.; Holler, F.J.; Crouch, S.R. Principles of Instrumental Analysis, 7th ed.; Cengage Learning: Boston, MA, USA, 2018; ISBN 978-1-305-57721-3. [Google Scholar]
- Žuvela, P.; Skoczylas, M.; Jay Liu, J.; Baczek, T.; Kaliszan, R.; Wong, M.W.; Buszewski, B. Column Characterization and Selection Systems in Reversed-Phase High-Performance Liquid Chromatography. Chem. Rev. 2019, 119, 3674–3729. [Google Scholar] [CrossRef]
- Blum, F. High Performance Liquid Chromatography. Br. J. Hosp. Med. 2014, 75, C18–C21. [Google Scholar] [CrossRef]
- International Conference on Harmonization. Validation of Analytical Procedures: Q2(R2); International Conference on Harmonization: Geneva, Switzerland, 2022. [Google Scholar]
- Johnson, R. Assessment of Bias with Emphasis on Method Comparison. Clin. Biochem. Rev. 2008, 29, S37–S42. [Google Scholar]
- Silva, M.C.; dos Anjos, J.P.; Guarieiro, L.L.N.; Machado, B.A.S. A Simple Method for Evaluating the Bioactive Phenolic Compounds’ Presence in Brazilian Craft Beers. Molecules 2021, 26, 4716. [Google Scholar] [CrossRef]
- Whelan, L.C.; Geary, M.; Healy, J. A Novel, Simple Rapid Reverse-Phase HPLC-DAD Analysis, for the Simultaneous Determination of Phenolic Compounds and Abscisic Acid Commonly Found in Foodstuff and Beverages. J. Chromatogr. Sci. 2022, 60, 648–654. [Google Scholar] [CrossRef]
- Du, K.; Li, J.; Guo, X.; Li, Y.; Chang, Y. Quantitative Analysis of Phenolic Acids and Flavonoids in Cuscuta chinensis Lam. by Synchronous Ultrasonic-Assisted Extraction with Response Surface Methodology. J. Anal. Methods Chem. 2018, 2018, 6796720. [Google Scholar] [CrossRef]
- Rajauria, G. Optimization and Validation of Reverse Phase HPLC Method for Qualitative and Quantitative Assessment of Polyphenols in Seaweed. J. Pharm. Biomed. Anal. 2018, 148, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; He, S.; Xiao, N.; Qiao, Y.; Sui, H.; Liang, L.; Chen, J.; Li, W.; Zhang, L. Simultaneous Determination of 15 Flavonoids in Scutellaria barbata-Hedyotis diffusa Herb Pair by HPLC Q-TOF MS. J. AOAC Int. 2019, 102, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, N.; Zhang, H.-F.; Wang, Q.-Q.; Yu, Q.; Wang, F.; Dai, Y.-H.; Wang, D.; Liu, D.-C. Simultaneous Quantitative Analysis of 11 Flavonoid Derivatives with a Single Marker in Persimmon Leaf Extraction and Evaluation of Their Myocardium Protection Activity. J. Nat. Med. 2019, 73, 404–418. [Google Scholar] [CrossRef]
- Srivastava, M.; Singh, M.; Maurya, P.; Srivastava, N.; Gupta, N.; Shanker, K. Simultaneous Quantification of Five Bioactive Phenylethanoid, Iridoid, and Flavonol Glycosides in Duranta erecta L.: Ultra Performance Liquid Chromatography Method Validation and Uncertainty Measurement. J. Pharm. Biomed. Anal. 2019, 174, 711–717. [Google Scholar] [CrossRef]
- Pu, Z.-J.; Yue, S.-J.; Zhou, G.-S.; Yan, H.; Shi, X.-Q.; Zhu, Z.-H.; Huang, S.-L.; Peng, G.-P.; Chen, Y.-Y.; Bai, J.-Q.; et al. The Comprehensive Evaluation of Safflowers in Different Producing Areas by Combined Analysis of Color, Chemical Compounds, and Biological Activity. Molecules 2019, 24, 3381. [Google Scholar] [CrossRef]
- Huang, H.-S.; Yu, H.-S.; Yen, C.-H.; Liaw, E.-T. HPLC-DAD-ESI-MS Analysis for Simultaneous Quantitation of Phenolics in Taiwan Elderberry and Its Anti-Glycation Activity. Molecules 2019, 24, 3861. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hong, Y.; Yang, D.; He, Z.; Lin, X.; Wang, G.; Yu, W. Simultaneous Determination of Phenolic Metabolites in Chinese Citrus and Grape Cultivars. PeerJ 2020, 8, e9083. [Google Scholar] [CrossRef]
- Khan, M.N.; Ul Haq, F.; Rahman, S.; Ali, A.; Musharraf, S.G. Metabolite Distribution and Correlation Studies of Ziziphus jujuba and Ziziphus nummularia Using LC-ESI-MS/MS. J. Pharm. Biomed. Anal. 2020, 178, 112918. [Google Scholar] [CrossRef]
- Jia, Q.; Zhang, S.; Zhang, H.; Yang, X.; Cui, X.; Su, Z.; Hu, P. A Comparative Study on Polyphenolic Composition of Berries from the Tibetan Plateau by UPLC-Q-Orbitrap MS System. Chem. Biodivers. 2020, 17, e2000033. [Google Scholar] [CrossRef]
- Sharma, S.; Joshi, R.; Kumar, D. Quantitative Analysis of Flavonols, Flavonol Glycoside and Homoisoflavonoids in Polygonatum verticillatum Using UHPLC-DAD-QTOF-IMS and Evaluation of Their Antioxidant Potential. Phytochem. Anal. PCA 2020, 31, 333–339. [Google Scholar] [CrossRef]
- Sharma, A.; Katiyar, C.K.; Banerjee, S.; Chanda, J.; Kar, A.; Biswas, S.; Mukherjee, P.K. RP-HPLC and HPTLC Methods for Analysis of Delected Herbs Used as Complexion Promoters in Ayurveda and Unani Systems of Medicine. J. AOAC Int. 2020, 103, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; Gowthamarajan, K.; Priyanka Dwarampudi, L.; Bhaskaran, M.; Kadiyala, M. Analytical Method Development, Validation and Forced Degradation Studies for Rutin, Quercetin, Curcumin, and Piperine by RP-UFLC Method. Drug Dev. Ind. Pharm. 2021, 47, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.; Ali, A.; Khan, M.N.; Rahman, S.; Faizi, S.; Ali, M.S.; Khalifa, S.A.M.; El-Seedi, H.R.; Musharraf, S.G. Rapid Identification of Common Secondary Metabolites of Medicinal Herbs Using High-Performance Liquid Chromatography with Evaporative Light Scattering Detector in Extracts. Metabolites 2021, 11, 489. [Google Scholar] [CrossRef] [PubMed]
- Macêdo, S.K.S.; Almeida, T.S.; Alencar Filho, J.M.T.; Lima, K.S.B.; Libório, R.C.; Costa, M.M.; Rolim Neto, P.J.; Rolim, L.A.; Nunes, X.P. Phytochemical Identification and Quantification of Quercetin in Triplaris gardneriana Wedd. Leaves by HPLC-DAD with Evaluation of Antibacterial Activity. Nat. Prod. Res. 2021, 35, 3083–3088. [Google Scholar] [CrossRef] [PubMed]
- Urbstaite, R.; Raudone, L.; Liaudanskas, M.; Janulis, V. Development, Validation, and Application of the UPLC-DAD Methodology for the Evaluation of the Qualitative and Quantitative Composition of Phenolic Compounds in the Fruit of American Cranberry (Vaccinium macrocarpon Aiton). Molecules 2022, 27, 467. [Google Scholar] [CrossRef] [PubMed]
- Jan, S.; Ahmad, J.; Dar, M.M.; Wani, A.A.; Tahir, I.; Kamili, A.N. Development and Validation of a Reverse Phase HPLC-DAD Method for Separation, Detection & Quantification of Rutin and Quercetin in Buckwheat (Fagopyrum spp.). J. Food Sci. Technol. 2022, 59, 2875–2883. [Google Scholar] [CrossRef]
- Boligon, A.A.; Linde, M. Importance of HPLC in Analysis of Plants Extracts. Austin Chromatogr. 2014, 1, 1–2. [Google Scholar]
- Cos, P.; Vlietinck, A.J.; Berghe, D.V.; Maes, L. Anti-Infective Potential of Natural Products: How to Develop a Stronger in vitro ‘Proof-of-Concept’. J. Ethnopharmacol. 2006, 106, 290–302. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Heinrich, M.; Booker, A. Medicinal Plant Analysis: A Historical and Regional Discussion of Emergent Complex Techniques. Front. Pharmacol. 2020, 10, 1480. [Google Scholar] [CrossRef]
- Kumar, B.R. Application of HPLC and ESI-MS Techniques in the Analysis of Phenolic Acids and Flavonoids from Green Leafy Vegetables (GLVs). J. Pharm. Anal. 2017, 7, 349–364. [Google Scholar] [CrossRef]
- Muyumba, N.W.; Mutombo, S.C.; Sheridan, H.; Nachtergael, A.; Duez, P. Quality Control of Herbal Drugs and Preparations: The Methods of Analysis, Their Relevance and Applications. Talanta Open 2021, 4, 100070. [Google Scholar] [CrossRef]
- Yamagishi, M.; Matsumoto, S.; Nakatsuka, A.; Itamura, H. Identification of Persimmon (Diospyros kaki) Cultivars and Phenetic Relationships between Diospyros Species by More Effective RAPD Analysis. Sci. Hortic. 2005, 105, 283–290. [Google Scholar] [CrossRef]
- Macedo, S.K.S.; Almeida, T.D.S.; Ferraz, C.A.A.; Oliveira, A.P.; Hugo Almeida, A.V.; Almeida, J.R.G.D.S.; Silva, N.D.S.; Nunes, X.P. Identification of Flavonol Glycosides and in Vitro Photoprotective and Antioxidant Activities of Triplaris gardneriana Wedd. J. Med. Plants Res. 2015, 9, 207–215. [Google Scholar] [CrossRef]
- Srivastava, N.; Singh, A.; Kumari, P.; Nishad, J.H.; Gautam, V.S.; Yadav, M.; Bharti, R.; Kumar, D.; Kharwar, R.N. Advances in Extraction Technologies: Isolation and Purification of Bioactive Compounds from Biological Materials. In Natural Bioactive Compounds; Elsevier: Amsterdam, The Netherlands, 2021; pp. 409–433. ISBN 978-0-12-820655-3. [Google Scholar]
- Mukherjee, P.K. Extraction and Other Downstream Procedures for Evaluation of Herbal Drugs. In Quality Control and Evaluation of Herbal Drugs; Elsevier: Amsterdam, The Netherlands, 2019; pp. 195–236. ISBN 978-0-12-813374-3. [Google Scholar]
- Zhang, Q.-W.; Lin, L.-G.; Ye, W.-C. Techniques for Extraction and Isolation of Natural Products: A Comprehensive Review. Chin. Med. 2018, 13, 20. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.M.M. Overview of Sample Preparation and Chromatographic Methods to Analysis Pharmaceutical Active Compounds in Waters Matrices. Separations 2021, 8, 16. [Google Scholar] [CrossRef]
- Nolvachai, Y.; Marriott, P.J. GC for Flavonoids Analysis: Past, Current, and Prospective Trends. J. Sep. Sci. 2013, 36, 20–36. [Google Scholar] [CrossRef]
- Harris, D.C. Quantitative Chemical Analysis, 8th ed.; W.H. Freeman and Company: New York, NY, USA, 2010; ISBN 978-1-4292-1815-3. [Google Scholar]
- Elhamirad, A.H.; Zamanipoor, M.H. Thermal Stability of Some Flavonoids and Phenolic Acids in Sheep Tallow Olein. Eur. J. Lipid Sci. Technol. 2012, 114, 602–606. [Google Scholar] [CrossRef]
- De Souza Dias, F.; Silva, M.F.; David, J.M. Determination of Quercetin, Gallic Acid, Resveratrol, Catechin and Malvidin in Brazilian Wines Elaborated in the Vale Do São Francisco Using Liquid–Liquid Extraction Assisted by Ultrasound and GC-MS. Food Anal. Methods 2013, 6, 963–968. [Google Scholar] [CrossRef]
- Zhang, K.; Zuo, Y. GC-MS Determination of Flavonoids and Phenolic and Benzoic Acids in Human Plasma after Consumption of Cranberry Juice. J. Agric. Food Chem. 2004, 52, 222–227. [Google Scholar] [CrossRef]
- Ang, L.F.; Yam, M.F.; Fung, Y.T.T.; Kiang, P.K.; Darwin, Y. HPLC Method for Simultaneous Quantitative Detection of Quercetin and Curcuminoids in Traditional Chinese Medicines. J. Pharmacopunct. 2014, 17, 36–49. [Google Scholar] [CrossRef]
- Bligh, S.W.A.; Ogegbo, O.; Wang, Z.-T. Flavonoids by HPLC. In Natural Products; Ramawat, K.G., Mérillon, J.-M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 2107–2144. ISBN 978-3-642-22143-9. [Google Scholar]
- Careri, M.; Corradini, C.; Elviri, L.; Nicoletti, I.; Zagnoni, I. Direct HPLC Analysis of Quercetin and Trans-Resveratrol in Red Wine, Grape, and Winemaking Byproducts. J. Agric. Food Chem. 2003, 51, 5226–5231. [Google Scholar] [CrossRef] [PubMed]
- Rajalakshmi, P.V.; Senthil, K.K. Direct HPLC Analysis of Quercetin in Exudates of Abutilon indicum (Linn). Malvaceae. J. Pharm. Sci. Technol. 2009, 1, 80–83. [Google Scholar]
- Rab, R.A.; Zahiruddin, S.; Ibrahim, M.; Husain, F.; Parveen, R.; Khan, W.; Ahmad, F.J.; Khan, A.A.; Ahmad, S. HPTLC and UPLC-MS/MS Methods for Quality Control Analysis of Itrifal Formulations of Unani System of Medicine. J. AOAC Int. 2020, 103, 649–658. [Google Scholar] [CrossRef]
- Bidikar, C.M.; Hurkadale, P.J.; Nandanwadkar, S.M.; Hegde, H.V. A Validated Spectro Densitometric Regulatory Compliant USP-HP-TLC Protocol for Quantification of Polyphenols and Antioxidants from Polyherbal Formulations Containing Terminalia Species. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 2022, 1207, 123379. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.D.; Nahar, L. Applications of High Performance Liquid Chromatography in the Analysis of Herbal Products. In Evidence-Based Validation of Herbal Medicine; Elsevier: Amsterdam, The Netherlands, 2015; pp. 405–425. ISBN 978-0-12-800874-4. [Google Scholar]
- Tátraaljai, D.; Földes, E.; Pukánszky, B. Efficient Melt Stabilization of Polyethylene with Quercetin, a Flavonoid Type Natural Antioxidant. Polym. Degrad. Stab. 2014, 102, 41–48. [Google Scholar] [CrossRef]
- Bird, I.M. High Performance Liquid Chromatography: Principles and Clinical Applications. BMJ 1989, 299, 783–787. [Google Scholar] [CrossRef]
- dos Neto, Á.J.S. Problemas Com o Formato Dos Picos Em Cromatografia Líquida. Sci. Chromatogr. 2010, 2, 71–81. [Google Scholar]
- Issaq, H.J.; Chan, K.C.; Veenstra, T.D. Peptides | Liquid Chromatography. In Encyclopedia of Separation Science; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–9. ISBN 978-0-12-226770-3. [Google Scholar]
- Li, S.; Tian, M.; Row, K.H. Effect of Mobile Phase Additives on the Resolution of Four Bioactive Compounds by RP-HPLC. Int. J. Mol. Sci. 2010, 11, 2229–2240. [Google Scholar] [CrossRef]
- Núñez, O.; Lucci, P. Applications and Uses of Formic Acid in Liquid Chromatography-Mass Spectrometry Analysis. In Advances in Chemical Research; Nova Science Publishers: New York, NY, USA, 2014; Volume 20, pp. 71–86. [Google Scholar]
- Palamareva, M.D. Liquid Chromatography | Overview. In Encyclopedia of Analytical Science; Elsevier: Amsterdam, The Netherlands, 2005; pp. 106–112. ISBN 978-0-12-369397-6. [Google Scholar]
- Ornaf, R.M.; Dong, M.W. Key Concepts of HPLC in Pharmaceutical Analysis. In Separation Science and Technology; Elsevier: Amsterdam, The Netherlands, 2005; Volume 6, pp. 19–45. ISBN 978-0-12-088547-3. [Google Scholar]
- Abdelkawy, K.S.; Balyshev, M.E.; Elbarbry, F. A New Validated HPLC Method for the Determination of Quercetin: Application to Study Pharmacokinetics in Rats. Biomed. Chromatogr. 2017, 31, e3819. [Google Scholar] [CrossRef]
- Asfaram, A.; Arabi, M.; Ostovan, A.; Sadeghi, H.; Ghaedi, M. Simple and Selective Detection of Quercetin in Extracts of Plants and Food Samples by Dispersive-Micro-Solid Phase Extraction Based on Core–Shell Magnetic Molecularly Imprinted Polymers. New J. Chem. 2018, 42, 16144–16153. [Google Scholar] [CrossRef]
- Rafferty, J.L.; Siepmann, J.I.; Schure, M.R. Mobile Phase Effects in Reversed-Phase Liquid Chromatography: A Comparison of Acetonitrile/Water and Methanol/Water Solvents as Studied by Molecular Simulation. J. Chromatogr. A 2011, 1218, 2203–2213. [Google Scholar] [CrossRef] [PubMed]
- Sutton, A.T.; Fraige, K.; Leme, G.M.; Da Silva Bolzani, V.; Hilder, E.F.; Cavalheiro, A.J.; Arrua, R.D.; Funari, C.S. Natural Deep Eutectic Solvents as the Major Mobile Phase Components in High-Performance Liquid Chromatography—Searching for Alternatives to Organic Solvents. Anal. Bioanal. Chem. 2018, 410, 3705–3713. [Google Scholar] [CrossRef] [PubMed]
- Jandera, P. Liquid Chromatography | Normal Phase. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2013; pp. 162–173. ISBN 978-0-12-409547-2. [Google Scholar]
- Poole, C.F. The Column in Liquid Chromatography. In The Essence of Chromatography; Elsevier: Amsterdam, The Netherlands, 2003; pp. 267–429. ISBN 978-0-444-50198-1. [Google Scholar]
- Robards, K.; Haddad, P.R.; Jackson, P.E. High-Performance Liquid Chromatography-Instrumentation and Techniques. In Principles and Practice of Modern Chromatographic Methods; Elsevier: Amsterdam, The Netherlands, 2004; pp. 227–303. ISBN 978-0-08-057178-2. [Google Scholar]
- Murayama, C.; Kimura, Y.; Setou, M. Imaging Mass Spectrometry: Principle and Application. Biophys. Rev. 2009, 1, 131–139. [Google Scholar] [CrossRef] [PubMed]
- De Hoffmann, E.; Stroobant, V. Mass Spectrometry: Principles and Applications, 3rd ed.; J. Wiley: Chichester, UK; Hoboken, NJ, USA, 2007; ISBN 978-0-470-03310-4. [Google Scholar]
- Stoev, G.; Stoyanov, A. Comparison of the Reliability of the Identification with Diode Array Detector and Mass Spectrometry. J. Chromatogr. A 2007, 1150, 302–311. [Google Scholar] [CrossRef]
- Araujo, P. Key Aspects of Analytical Method Validation and Linearity Evaluation. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci. 2009, 877, 2224–2234. [Google Scholar] [CrossRef]
- Moosavi, S.M.; Ghassabian, S. Linearity of Calibration Curves for Analytical Methods: A Review of Criteria for Assessment of Method Reliability. In Calibration and Validation of Analytical Methods-A Sampling of Current Approaches; InTechOpen: London, UK, 2018; pp. 109–128. [Google Scholar]
- Ribani, M.; Grespan Bottoli, C.B.; Collins, C.H.; Fontes Jardim, I.C.S.; Costa Melo, L.F. Validação Em Métodos Cromatográficos e Eletroforéticos. Quim. Nova 2004, 27, 771–780. [Google Scholar] [CrossRef]
- World Health Organization Expert Committee on Specifications for Pharmaceutical Preparations. WHO Technical Report Series 823: Thirty-Second Report; World Health Organization Expert Committee on Specifications for Pharmaceutical Preparations: Geneva, Switzerland, 1992. [Google Scholar]
- Food and Drug Administration. Reviewer Guidance-Validation of Chromatographic Methods; Food and Drug Administration: Silver Spring, MD, USA, 1994.
- Bland, J.M.; Altman, D.G. Statistical Methods for Assessing Agreement between Two Methods of Clinical Measurement. Lancet 1986, 1, 307–310. [Google Scholar] [CrossRef]
- Dimeski, G. Interference Testing. Clin. Biochem. Rev. 2008, 29, S43–S48. [Google Scholar]
- González, A.G.; Herrador, M.Á. A Practical Guide to Analytical Method Validation, Including Measurement Uncertainty and Accuracy Profiles. TrAC Trends Anal. Chem. 2007, 26, 227–238. [Google Scholar] [CrossRef]
- Misra, D.P.; Ravindran, V. An Overview of the Functionalities of PubMed. J. R. Coll. Physicians Edinb. 2022, 52, 8–9. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Analyte | Sample | Source | Sample Preparation and Extraction Procedures | Amount of Quercetin in Real Samples (µg/g) |
|---|---|---|---|---|---|
| Du et al. [23] | Chlorogenic acid; Cryptochlorogenic acid; Neochlorogenic acid; Isochlorogenic acid A; Isochlorogenic acid B; Isochlorogenic acid C; Caffeic acid; Hyperin; Isoquercitrin; Quercetin; Campherol; p-coumaric acid; Isorhamnetin; Rutin; Astragalin; Apigenin; | Cuscuta chinensis Lam. | Undisclosed | Pulverization; Ultra-sonication assisted extraction; Filtration (0.22 µm); | 0.0735 ± 0.0788 |
| Rajauria [24] | Phloroglucinol; Gallic acid; Cyanidin 3-glucoside; Chlorogenic acid; Rutin; Quercetin; | Himanthaliaelongata | Seaweed | Grinding; Percolation; Solid-phase extraction; Filtration (0.22 µm); | 4.2 ± 0.15 |
| Yang et al. [25] | Alpinetin; Apigenin-7-O-β-D-glucopyranoside; Quercetin-3-O-β-D-glucopyranoside; Scutellarein; Apigenin; Wogonoside; Quercetin; Amentoflavone; Wogonin; Chrysin; Luteolin; Rutin; Naringenin; Baicalein; Baicalin; | Scutellaria barbata D. Don and Hedyotis diffusa (Willd.) Roxb. | Dry Grass (Plants) | Reflux extraction (twice); Lyophilization; Solvent resuspension; Liquid–liquid extraction; Filtration (0.22 µm); | 0.02199 ± 0.000618 |
| Zhou et al. [26] | Myricetin-3-O-β-D-galactoside; Myricetin-3-O-glucoside; Quercetin3-O-β-D-galactoside; Quercetin-3-O-β-D-glucoside; Quercetin-3-O-(2″-O-galloyl-β-d-galactoside); Quercetin-3-O(2″-O-galloyl-β-d-glucoside); Kaempferol-3-O-β-D-galactoside; Kaempferol-3-O-β-D-glucoside; Kaempferol-3-O(2″-O-galloyl-β-D-galactoside); Kaempferol-3-O-(2″-O-galloyl-β-D-glucoside); Quercetin; Kaempferol; | Diospyros khaki | Leaves (Plant) | Grinding; Reflux extraction (twice); Defat procedure (twice); Liquid–liquid extraction (twice); Gel Column Chromatography; | 12,700 ± 8000 |
| Srivastava et al. [27] | Acteoside; Isoacteoside; Durantoside-I; Quercetin; Methylapigenin-7-O-D-glucopyranuronate; | Duranta erecta L. | Undisclosed | Pulverization; Ultra-sonication assisted extraction; Filtration (0.22 µm); | 2010 |
| Pu et al. [28] | Hydroxysafflor yellow A; Safflomin C; Anhydrosafflor yellow B; Kaempferol; Kaempferol-3-O-glucoside; Kaempferol-3-O-rutinoside; Kaempferol-3-O-β-sophoroside; 6-hydroxykaempferol; 6-hydroxykaempferol-3-O-β-D-glucoside; 6-hydroxykaempferol-3,6-di-O-β-D-glucoside; 6-hydroxykaempferol-3,6,7-tri-O-β-D-glucoside; Quercetin; Rutin; Luteoloside; Apigenin; Quercetin-3-O-β-D-glucoside; | Carthamus tinctorius L. | Undisclosed | Pulverization; Ultra-sonication assisted extraction; Filtration (0.22 µm); | 65 ± 75 |
| Huang et al. [29] | Chlorogenic acid; Rutin; Isoquercetrin; Nictoflorin; Astragalin; Quercetin; | Sambucus formosana | Stems, leaves, and roots (Plant) | Pulverization; Percolation; Liquid–liquid extraction (twice); | 3500 ± 70 |
| Chen et al. [30] | Gallic acid; Chlorogenic acid; Caffeic acid; Syringic acid; p-coumaric acid; Ferulic acid; Benzoic acid; Salicylic acid; Catechin; Epicatechin; Rutin; Naringin; Hesperidin; Quercetin; Resveratrol; Nobiletin; Tangeritin; | Chinese citrus and grape | Fruit (Plant) | Percolation; Liquid–liquid extraction (twice); Filtration (0.45 µm); | 394,800 ± 527,900 (citrus) 129,700 ± 146,600 (grape) |
| Khan et al. [31] | 6‴-feruloylspinosin; Apigenin; Apigenin-7-O-glucoside; Catechin; Jujuboside A; Jujuboside B; Luteolin; Quercetin; | Ziziphus jujuba and Ziziphus nummularia | Fruits (Plants) | Grinding; Ultra-sonication assisted extraction; Filtration 0.22 µm; | 15.5 ± 12.0 |
| Jia et al. [32] | Phloretin; Gallic acid; Protocatechuat E; Catechin; 2,4-dihydroxybenzoic acid; Chlorogenic acid; Proanthocyanidins-B2; Vanillic acid; O-hydroxybenzene acetic acid; Coffeic acid; Syringate; p-coumaric acid; Proanthocyanidins-A2; Veratronic acid; Ferulic acid; Benzoic acid; Salicylic acid; Naringin; Hesperidin; Rutin; Ellagic acid; Myricetin; Naringenin; Quercetin; Kaempferol; | Berries | Fruit (Plant) | Grinding; Ultra-sonication assisted extraction; Filtration; Lyophilization; Solvent resuspension; Filtration (0.22 µm); | 11.5 ± 15.5 |
| Sharma et al. [33] | Rutin; Quercetin; Kaempherol; 5,7-dihydroxy-3-(2-hydroxy-4-methoxybenzyl)chroman-4-one; 5,7-dihydroxy-3-(2-hydroxy-4-methoxybenzyl)8-methylchroman-4-one; 5,7-dihydroxy-3-(4-methoxybenzyl)8-methylchroman-4-one; | Polygonatum verticillatum | Rhizomes (Plant) | Pulverization; Percolation (fivefold); Liquid–liquid extraction; Filtration (0.25 µm); | 0.0243 ± 0.0044 |
| Sharma et al. [34] | Quercetin; Ferulic acid; Chlorogenic acid; | Myristic fragrans, Hemidesmus indicus, and Inula racemosa | Undisclosed | Maceration; Filtration (11 µm); Lyophilization; Solvent resuspension; Filtration (undisclosed diameter); | 0.0062 |
| Ramaswamy et al. [35] | Curcumin; Piperine; Quercetin; Rutin; | Camellia sinensis L. (1); Glycyrrhiza glabra L. (2); Thymus vulgaris L. (3); Citrus aurantium L. (4); | Leaves (1, 3), rhizomes (2), tuberous roots (2), and rind (4) (Plants) | Ultra-sonication assisted extraction; Filtration 0.22 µm; | C. s: 0.0036 C. a: 0.0011 G. g: 0.00095 T. v: 0.00087 |
| Ali et al. [36] | Rutin; Taxifolin; Quercetin; Apigenin; Kaempferol; Betulinic acid; Oleanolic acid; Betulin; Lupeol; Stigmasterol; β-sitosterol; Ursolic acid; | Caesalpinia pulcherrima (1); Citrus lemon (2); Opuntia dellenii (3); Bauhinia variegata (4); Polyalthia longifolia var. pendula (5); Bombax ceiba (6); Phlox drummondii (7); Olea europea (8); Tagetes patula (9); Melia azedarach (10); | Flower (1, 9, 10), fresh pods (1), seeds (2), cladodes (3), pod (4), root bark (5), wood (6), aerial part (7), leaves (8), and stem bark (6) (Plants) | Ultra-sonication assisted extraction; Filtration 0.22 µm; | C. p (flowers): 234.56 µg/mL C. p (fresh pods): 315.07 µg/mL C. l: < LOQ O. d: < LOQ B. v: < LOQ P. l: 579.51 µg/mL B. c: < LOQ P. d: < LOQ O. e: 94.50 µg/mL T. p: < LOQ |
| Macêdo et al. [37] | Quercetin | Triplaris gardneriana Wedd | Leaves (Plant) | Pulverization; Percolation (threefold); Vacuum Liquid Chromatography; | 9967 ± 1010 |
| Urbstaite et al. [38] | Chlorogenic acid; Myricetin-3-galactoside; Quercetin-3-galactoside; Quercetin-3-glucoside; Quercetin-3-α-Larabinopyranoside; Quercetin-3-α-L-arabinofuranoside; Quercetin-3-rhamnoside; Myricetin; Quercetin; | Vaccinium macrocarpon Aiton | Fruit (Plant) | Pulverization; Ultra-sonication assisted extraction; Filtration (0.22 µm); | 89.76 ± 1.58 |
| Jan et al. [39] | Rutin and Quercetin | Buckwheat (Fagopyrum spp.) | Seeds and Leaves (Plant) | Pulverization; Percolation; Filtration (0.22 µm); | 0.00011 ± 0.00014 |
| Reference | Analytical Method | Validation Parameters | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Chromatographic Method | Detection Method | Chromatographic Run | Mobile Phase | Column | Retention Time (min) | LOD (µg/mL) | LOQ (µg/mL) | Precision (%) | Accuracy (%) | |
| Du et al. [23] | HPLC | ESI-MS | Gradient | acetonitrile + water acidified with 0.05% formic acid | C18 (1.8 μm, 4.6 mm × 150 mm) | 17.25 | 0.03 | 0.1 | Intra-day: 92.2–95.4 Inter-day: 92.1–99.0 | Intra-day: 102.3–110.3 Inter-day: 107.0–115.0 |
| Rajauria [24] | RP-HPLC | DAD-ESI-MS | Gradient | 0.25% aqueous acetic acid and acetonitrile/water (80/20; v/v) containing 0.25% acetic acid | C-18 (5 μm, 4.6 mm × 250 mm) | 37.43 | 0.51 | 1.82 | Retention Time: 98.17 Peak Area: 96.37 | Recovery: 97.2 |
| Yang et al. [25] | HPLC | Q-TOF-MS | Gradient | water containing 0.1% formic acid and acetonitrile containing 0.1% formic acid | C18 (5 μm, 4.6 mm × 150 mm) | 7.07 | 0.003 | 0.0105 | Intra-day: 99.28 Inter-day: 97.65 | Recovery: 96.0–103.0 |
| Zhou et al. [26] | HPLC | DAD-Q-TOF-MS/MS DAD | Gradient Isocratic | acetonitrile and water containing 0.1% formic acid | C18 (5 μm, 2.1 mm × 150 mm, 100 A) | 32.11 | 0.015 | 0.051 | Intra-day: 97.2–99.4 Inter-day: 97.0–99.2 | Recovery: 85.9–106.9 |
| Srivastava et al. [27] | UHPLC | PDA | Gradient | water containing 0.1% formic acid and acetonitrile | C18 (2.5 μm, 2.0 mm × 100 mm) | 6.4 | 0.330 | 1.101 | Intra-day: 99.06 Inter-day: 97.64–98.16 | Recovery: 101.0 |
| Pu et al. [28] | UPLC | QTRAP®-MS2 | Gradient | 0.1% formic acid aqueous solution and acetonitrile | C18 (1.7 μm, 2.1 mm × 100 mm) | 12.56 | 0.007629 | 0.015259 | Intra-day: 96.06 Inter-day: 97.12 | Recovery: 98.67–103.55 |
| Huang et al. [29] | HPLC | DAD-ESI-MS | Gradient | 0.1% formic acid aqueous solution and 0.1% formic acid/acetonitrile | RP-C18 (1.9 μm, 3 mm × 100 mm) | 18.9 | 0.8 | 2.5 | 97.3 | Recovery: 92.7 |
| Chen et al. [30] | HPLC | DAD | Gradient | water with 2% (v/v) acetic acid and acetonitrile | RP-18e (5 μm, 4.0 mm × 250 mm) | 76.52 | 0.13 | 0.39 | Repeatability: 98.03 | Recovery: 94.74 |
| Khan et al. [31] | HPLC | ESI-Q-TOF-MS | Gradient | water with 0.1% formic acid and methanol with 0.1% formic acid | SB-C18 (1.8 μm, 3.0 mm × 50 mm) | 4.9 | 0.00028 | 0.00086 | Intra-day: 96.2–98.3 Inter-day: 97.4–98.5 | Recovery: 98.3–101.4 |
| Jia et al. [32] | UPLC | Q-Orbitrap MS | Gradient | water containing 0.1% formic acid and 0.1% of formic acid in methanol | C18 (2.6 μm 2.1 mm × 150 mm) | 16.31 | 0.00187 | 0.00695 | Intra-day: 98.41 Inter-day: 97.77 | Recovery: 96.2–99.2 |
| Sharma et al. [33] | UHPLC | DAD-Q-TOF-MS | Gradient | water and acetonitrile, containing 0.1% formic acid | C18 (1.8 μm, 2.1 mm × 150 mm) | 5–6 | 0.00004 | 0.00012 | Intra-day: 98.29 Inter-day: 97.74 | Recovery: 93.5 |
| Sharma et al. [34] | RP-HPLC | UV-Vis | Isocratic | Acetonitrile and 0.1 M orthophosphoric acid in water with pH 2.5 in a ratio of 75 + 25 (v/v) | N/A | 7.44 | 1.41 | 6.54 | >98 | Recovery: 94.65–98.14 |
| Ramaswamy et al. [35] | UFLC | PDA | Isocratic | Ammonium acetate buffer (25 mM, pH 3.0) and acetonitrile (20:80, v/v) | C18 (5 μm, 4.6 mm × 250 mm) | 2.8 | 10 | 30 | Intra-day: 98.49–99.01 Inter-day: 98.22–99.31 | Recovery: 98.88 |
| Ali et al. [36] | HPLC | DAD/ESI-MS/MS | Gradient | water plus 0.1% formic acid and acetonitrile with 0.1% formic acid | C18 (1.8 μm, 3 mm × 100 mm) | 8.10 | 19.1 | 57.9 | Intra-day: 92.79–99.5 Inter-day: 98.78–99.47 | Intra-day: 104.59–119.95 Inter-day: 100.91–115.64 |
| Macêdo et al. [37] | HPLC | DAD | Gradient | water containing 0,3% formic acid and methanol | RP C-18 (5 μm, 4.6 mm × 250 mm) | 32.9 | 10.72 | 35.75 | Intra-day: 96.34–99.73 Inter-day: 94.62–98.71 | 94.83–100.84 |
| Urbstaite et al. [38] | UPLC | PDA | Gradient | 0.1% formic acid (v/v) in water and acetonitrile | C18 (1.7 μm, 2.1 mm × 100 mm) | 12.104 | 0.76 | 2.29 | Intra-day: 98.7 Inter-day: 98.24 | Recovery: 97.12–101.19 |
| Jan et al. [39] | HPLC | DAD | Gradient | methanol and methanol:water:acetic acid in the ratio of 100:150:5 | C18 (5 μm, 4.6 mm × 150 mm) | 8.23 | 19.28 | 1.77 | Intra-day: 98.75 Inter-day: 97.27 | Recovery: 96.66–98.63 |
| Bias Assessment Parameter Code | Explanation | Accomplishing the Parameter (n (%)) |
|---|---|---|
| I | Establishment of criteria for acceptable performance | 4 (24%) |
| II | Comparison of test method with reference method using reference material | 17 (100%) |
| III | Presentation of the x–y plot of data with an eye examination | 17 (100%) |
| IV | Consideration of difference plots and statistics of difference | 0 (0%) |
| V | Consideration of regression analysis | 17 (100%) |
| VI | Performance and interpretation of interference test | 2 (12%) |
| VII | Performance and interpretation of linearity test | 13 (76%) |
| VIII | Performance and interpretation of recovery test | 15 (88%) |
| Reference | Bias Assessment Parameters | |||||||
|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | VI | VII | VIII | |
| Du et al. [23] | b | |||||||
| Rajauria [24] | a | |||||||
| Yang et al. [25] | a | b | ||||||
| Zhou et al. [26] | a | b | ||||||
| Srivastava et al. [27] | a | |||||||
| Pu et al. [28] | ||||||||
| Huang et al. [29] | a | b | ||||||
| Chen et al. [30] | a | b | ||||||
| Khan et al. [31] | ||||||||
| Jia et al. [32] | a | b | ||||||
| Sharma et al. [33] | ||||||||
| Sharma et al. [34] | a | b | ||||||
| Ramaswamy et al. [35] | a | b | ||||||
| Ali et al. [36] | a | b | ||||||
| Macêdo et al. [37] | a | |||||||
| Urbstaite et al. [38] | a | b | ||||||
| Jan et al. [39] | b | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carvalho, D.; Pinho, C.; Oliveira, R.; Moreira, F.; Oliveira, A.I. Chromatographic Methods Developed for the Quantification of Quercetin Extracted from Natural Sources: Systematic Review of Published Studies from 2018 to 2022. Molecules 2023, 28, 7714. https://doi.org/10.3390/molecules28237714
Carvalho D, Pinho C, Oliveira R, Moreira F, Oliveira AI. Chromatographic Methods Developed for the Quantification of Quercetin Extracted from Natural Sources: Systematic Review of Published Studies from 2018 to 2022. Molecules. 2023; 28(23):7714. https://doi.org/10.3390/molecules28237714
Chicago/Turabian StyleCarvalho, Daniel, Cláudia Pinho, Rita Oliveira, Fernando Moreira, and Ana Isabel Oliveira. 2023. "Chromatographic Methods Developed for the Quantification of Quercetin Extracted from Natural Sources: Systematic Review of Published Studies from 2018 to 2022" Molecules 28, no. 23: 7714. https://doi.org/10.3390/molecules28237714