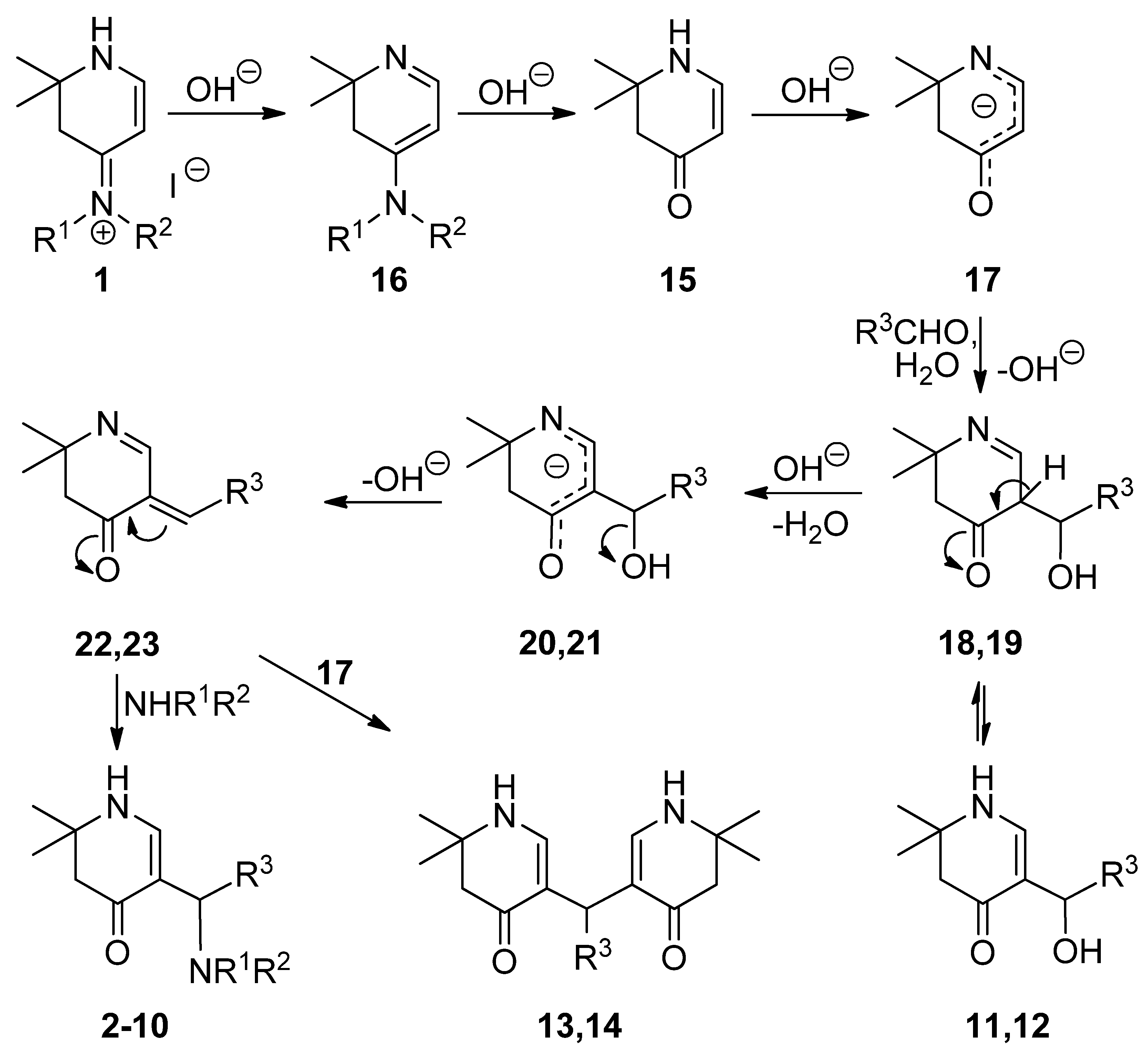
3.2. Syntheses
Compounds
1a–
1d were prepared earlier and their NMR data are in accordance with the reported literature [
1].
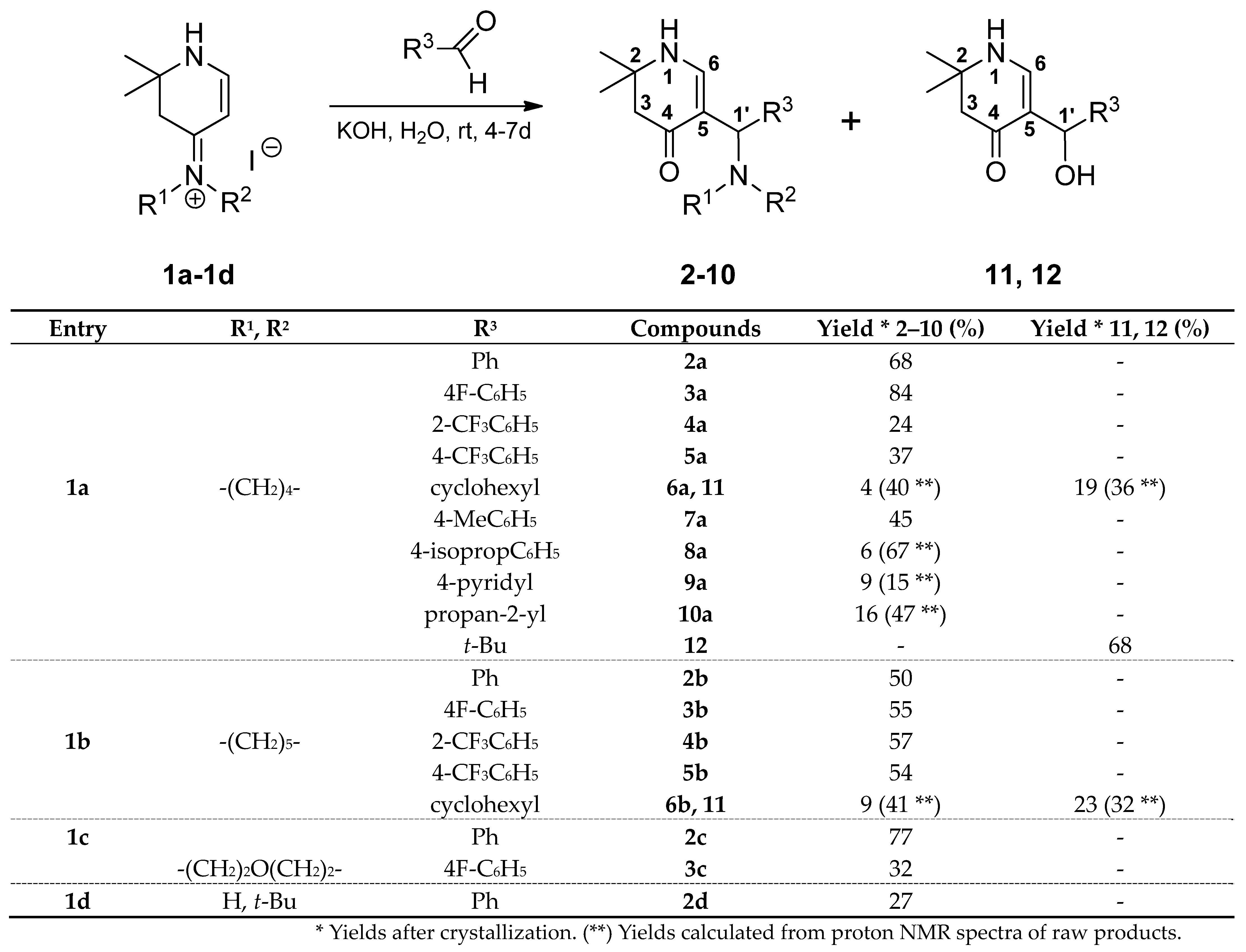
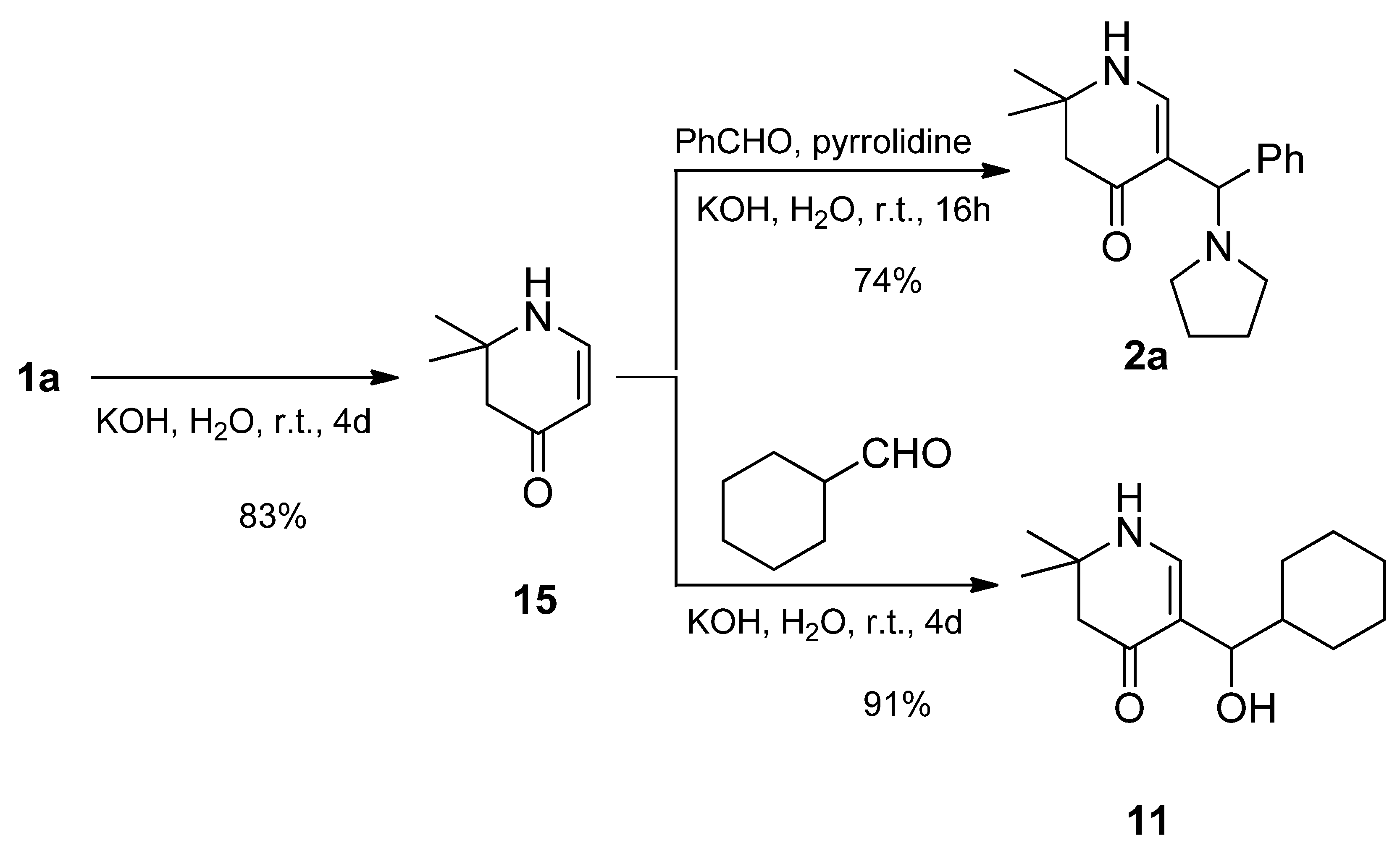
rac-2,2-Dimethyl-5-[(R)-phenyl(pyrrolidin-1-yl)methyl]-2,3-dihydropyridin-4(1H)-one (2a): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (16 mL) and a solution of KOH (1.466 g (26.13 mmol)) in water (16 mL) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5–10 min). Then, benzaldehyde (0.693 g (6.53 mmol)) was added. The reaction mixture was stirred for 4 days at r.t. The separated crystalline solid was filtered with suction, washed with water and acetone, and dried in vacuo yielding 2a (1.261g (68%)) as an orange-yellow solid. For analytical purposes, it was recrystallized from ethyl acetate/cyclohexane resulting in a white solid.
From 15: Compound 15 (0.671 g (5.36 mmol)) was dissolved in a solution of KOH (1.213 g (21.62 mmol)) in distilled water (26 mL). The mixture was sonicated and stirred at r.t. until all was dissolved. To the resulting solution, pyrrolidine (0.409 g (5.75 mmol)) and benzaldehyde (0.582 g (5.48 mmol)) were added. The reaction mixture was stirred for 16 h at r.t. The separated crystalline solid was filtered with suction, washed clean with water, and dried in vacuo resulting in 2a (1.129 g (74%)) as a pale pink solid. For analytical purposes, it was recrystallized from ethyl acetate resulting in colorless needles.
Rf (CH2Cl2:MeOH = 10:1): 0.09; mp: 147–148 °C; IR = 3232, 2967, 2783, 1623, 1568, 1534, 1395, 1231, 700; 1H NMR (CDCl3, 400 MHz) δ = 1.19 (s, 3H, CH3), 1.29 (s, 3H, CH3), 1.75 (br, s, 4H, (CH2)2), 2.37 (s, 2H, 3-H), 2.50 (br, s, 4H, 2NCH2), 4.52 (s, 1H, 1′-H), 4.60 (d, J = 5.5 Hz, 1H, 1-H), 7.14–7.38 (m, 6H, 6-H, ArH); 13C NMR (CDCl3, 100 MHz) δ = 23.49 ((CH2)2), 26.57, 27.57 (2CH3), 49.18 (C-3), 53.17 (2NCH2), 53.80 (C-2), 63.98 (C-1′), 111.43 (C-5), 126.09, 127.14, 128.03 (5ArC), 144.87 (ArCq), 149.23 (C-6), 189.50 (C-4); HRMS (EI): calcd. (C18H24N2O+) [M]+: 284.1889; found: 284.1874.
rac-2,2-Dimethyl-5-[(R)-phenyl(piperidin-1-yl)methyl]-2,3-dihydropyridin-4(1H)-one (2b): Compound 1b (2.091 g (6.53 mmol)) was suspended in water (16 mL) and a solution of KOH (1.470 g (26.20 mmol)) in water (16 mL) was added. The mixture was stirred at r.t. until a solution was formed (ca. 10–20 min). Then, benzaldehyde (0.695 g (6.55 mmol)) was added. The reaction mixture was stirred for 5 days at r.t. The separated crystalline solid was filtered with suction, washed with water and acetone, and dried in vacuo resulting in 2b (0.972 g (50%)) as an orange-yellow solid. For analytical purposes, it was recrystallized from ethyl acetate resulting in a white solid. Rf (CH2Cl2:MeOH = 1:1): 0.86; mp: 158 °C; IR = 2925, 1623, 1571, 1527, 1255, 1232, 700; 1H NMR (DMSO-d6, 400 MHz) δ = 1.09 (s, 3H, CH3), 1.18 (s, 3H, CH3), 1.31–1.51 (m, 6H, (CH2)3), 2.13–2.22 (m, 4H, 3-H, NCH2), 2.28–2.38 (m, 2H, NCH2), 4.37 (s, 1H, 1′-H), 6.95 (d, J = 6.8 Hz, 1H, 6-H), 7.09–7.14 (m, 1H, ArH), 7.20–7.26 (m, 4H, ArH), 7.35 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ 24.62 (CH2), 26.11 (2CH2), 26.19, 26.78 (2CH3), 48.90 (C-3), 52.34 (2NCH2), 53.03 (C-2), 64.17 (C-1′), 106.75 (C-5), 125.98, 127.30, 128.17 (5ArC), 144.62 (ArCq), 149.38 (C-6), 188.49 (C-4); HRMS (HESI): calcd. (C19H27N2O+) [M + H]+: 299.2123; found: 299.2114.
rac-2,2-Dimethyl-5-[(R)-phenyl(morpholin-4-yl)methyl]-2,3-dihydropyridin-4(1H)-one (2c): Compound 1c (2.000 g (6.22 mmol)) was suspended in water (30 mL) and KOH (1.397 g (24.89 mmol)) was added. To the resulting yellow solution benzaldehyde (0.660 g (6.22 mmol)) was added. The reaction mixture was stirred for 4 days at r.t. The separated crystalline solid was filtered with suction, washed with water and acetone, and dried over phosphorus pentoxide in vacuo resulting in 2c (1.436 g (77%)) as an off-white solid. For analytical purposes, it was recrystallized from ethyl acetate resulting in white needles. Rf (CH2Cl2:MeOH = 1:1): 0.89; mp: 165 °C; IR = 3257, 2961, 1623, 1571, 1522, 1395, 1291, 1231, 1181, 1116, 702; 1H NMR (DMSO-d6, 400 MHz) δ 1.09 (s, 3H, CH3), 1.17 (s, 3H, CH3), 2.15–2.25 (m, 4H, 3-H, NCH2), 2.29–2.39 (s, 2H, NCH2), 3.49–3.60 (br, s, 4H, 2OCH2), 4.36 (s, 1H, 1′-H), 7.00 (d, J = 6.8 Hz, 1H, 6-H), 7.13–7.16 (m, 1H, ArH), 7.25–2.26 (m, 4H, ArH), 7.42 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ 26.20, 26.77 (2CH3), 48.82 (C-3), 52.11 (2NCH2), 53.04 (C-2), 64.25 (C-1′), 66.61 (2OCH2), 106.04 (C-5), 126.26, 127.42, 128.33 (5ArC), 143.71 (ArCq), 149.51 (C-6), 188.47 (C-4); HRMS (HESI): calcd. (C14H16ON+) [M + H − C4H9NO]+: 214.1232; found: 214.1223.
rac-5-[(R)-(tert-Butylamino)(phenyl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (2d): Compound 1d (2.013 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.400 g (25 mmol)) was added. Then, benzaldehyde (0.693 g (6.53 mmol)) was added and the reaction mixture was stirred for 6 days at r.t. From the resulting orange resin, the aqueous solution was decanted and the resin was washed with water repeatedly. Then, it was dried overnight in vacuo over phosphorus pentoxide. The dry resin was dissolved in the minimum amount of hot ethyl acetate and left for crystallization at r.t. The solid was sucked off and dried resulting in 2d (0.512 g (27%)) as off-white needles. Rf (MeOH): 0.07; mp: 136 °C; IR = 2964, 1633, 1550, 1477, 1454, 1412, 1367, 1357, 1195, 1174, 947, 875, 828, 727, 713, 697, 678; 1H NMR (DMSO-d6, 400 MHz) δ 1.07 (s, 3H, CH3), 1.10 (s, 3H, CH3), 1.20 (s, 9H 3CH3), 2.31 (d, J = 15.0 Hz, 1H, 3-H), 2.36 (d, J = 15.0 Hz, 1H, 3-H), 5.21 (s, 1H, 1′-H), 5.98 (br, s, 1H, NH*), 6.25 (br, s, 1H, 6-H*), 6.94 (br, s, 1H, 1-H*), 7.12–7.34 (m, 5H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ 26.64, 26.81 (2CH3), 31.66 (3CH3), 41.37 (C-3), 50.97 (C-2), 53.45 (C(CH3)3), 73.61 (C-1′), 109.18 (C-5), 126.17, 126.43, 127.59 (5ArC), 138.93 (C-6), 146.02 (ArCq), 161.70 (C-4); HRMS (HESI): calcd. (C18H27N2O+) [M + H]+: 287.2123; found: 287.2114.
rac-5-[(R)-(4-Fluorophenyl)(pyrrolidin-1-yl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (3a): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (16 mL) and a solution of KOH (1.466 g (26.13 mmol)) in water (16 mL) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5–10 min). Then, 4-fluorobenzaldehyde (0.810 g (6.53 mmol)) was added and the reaction mixture was stirred for 6 days at r.t. The separated crystalline solid was filtered with suction, washed with water, and dried in vacuo over phosphorus pentoxide resulting in 3a (1.658 g (84%)) as a beige solid. It was recrystallized from ethyl acetate resulting in white needles. Rf (CH2Cl2:MeOH = 1:1): 0.29; mp: 169 °C; IR = 3231, 2970, 2783, 1623, 1603, 1568, 1531, 1507, 1395, 1220, 1182; 1H NMR (DMSO-d6, 400 MHz) δ 1.07 (s, 3H, CH3), 1.16 (s, 3H, CH3), 1.62–1.65 (m, 4H, (CH2)2), 2.14 (s, 2H, 3-H), 2.25–2.39 (m, 2H, NCH2), 2.33–2.39 (m, 2H, NCH2), 4.29 (s, 1H, 1′-H), 7.00–7.05 (m, 2H, ArH), 7.10 (d, J = 6.8 Hz, 1H, 6-H), 7.25–7.29 (m, 2H, ArH), 7.36 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ 23.34 ((CH2)2), 26.02, 26.85 (2CH3), 48.92 (C-3), 52.79 (2NCH2), 53.03 (C-2), 63.35 (C-1′), 108.89 (C-5), 114.74 (d, 2J(C,F) = 21.0 Hz, ArC), 128.66 (d, 3J(C,F) = 7.9 Hz, ArC), 141.72 (d, 4J(C,F) = 3.1 Hz, ArCq), 149.08 (C-6), 160.69 (d, 1J(C,F) = 241.3 Hz, ArCq), 187.86 (C-4); HRMS (HESI): calcd. (C18H24FN2O+) [M + H]+: 303.1873; found: 303.1863.
rac-5-[(R)-(4-Fluorophenyl)(piperidin-1-yl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (3b): Compound 1b (2.091 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.470 g (26.20 mmol)) was added. Then, 4-fluorobenzaldehyde (0.810 g (6.53 mmol)) was added and the reaction mixture was stirred for 4 days at r.t. The separated crystalline solid was filtered with suction, washed with water, and dried in vacuo over phosphorus pentoxide yielding 3b as a yellowish solid. It was dissolved in hot ethyl acetate and the insoluble part was removed via filtration. The product crystallized overnight as needles, was sucked off, and washed with ice-cold ethyl acetate resulting in 3b (1.137 g (55%)) as yellowish needles. Rf (CH2Cl2:MeOH = 1:1): 0.30; mp: 183 °C; IR = 3250, 3031, 2965, 2938, 1624, 1571, 1525, 1506, 1393, 1380, 1290, 1255, 1231, 1220, 1180; 1H NMR (DMSO-d6, 400 MHz) δ 1.10 (s, 3H, CH3), 1.18 (s, 3H, CH3), 1.34–1.48 (m, 6H, (CH2)3), 2.14–2.23 (m, 4H, 3-H, NCH2), 2.26–2.36 (m, 2H, NCH2), 4.37 (s, 1H, 1′-H), 6.95 (d, J = 6.8 Hz, 1H, 6-H), 7.05 (t, J = 8.9 Hz, 2H, ArH), 7.25 (dd, J = 8.5, 5.8 Hz, 2H, ArH), 7.38 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 24.56 (CH2), 26.09 (2CH2), 26.15, 26.72 (2CH3), 48.88 (C-3), 52.22 (2NCH2), 53.02 (C-2), 63.60 (C-1′), 106.41 (C-5), 114.78 (d, 2J(C,F) = 21.0 Hz, ArC), 128.96 (d, 3J(C,F) = 7.8 Hz, ArC), 140.55 (d, 4J(C,F) = 3.0 Hz, ArCq), 149.31 (C-6), 160.66 (d, 1J(C,F) = 241.6 Hz, ArCq), 188.51 (C-4); HRMS (HESI): calcd. (C19H26FN2O+) [M + H]+: 317.2029; found: 317.2020.
rac-5-[(R)-4-Fluorophenyl)(morpholin-4-yl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (3c): Compound 1c (0.700 g (2.18 mmol)) was suspended in water (11 mL) and KOH (0.489 g (8.71 mmol)) was added. 4-Fluorobenzaldehyde (0.271 g (2.18 mmol)) was added and the reaction mixture was stirred for 6 days at r.t. The separated resin was washed with water and dried in vacuo. It was triturated with hot ethyl acetate and the insoluble parts were filtered off and a part of the solvent was removed via evaporation. The formed amorphous precipitate was filtered off and the filtrate concentrated in vacuo. The product crystallized and was sucked off, washed with cold ethyl acetate, and dried yielding 3c (0.225 g (32%)). Rf (CH2Cl2:MeOH = 1:1): 0.90; mp: 162 °C; IR = 3244, 2969, 2957, 2842, 2809, 1651, 1621, 1571, 1508, 1394, 1292, 1263, 1249, 1224, 1182, 1118; 1H NMR (DMSO-d6, 400 MHz) δ = 1.09 (s, 3H, CH3), 1.17 (s, 3H, CH3), 2.14–2.23 (m, 4H, 3-H, NCH2), 2.30–2.39 (m, 2H, NCH2), 3.51–3.56 (m, 4H, 2OCH2), 4.35 (s, 1H, 1′-H), 7.00 (d, J = 6.8 Hz, 1H, 6-H), 7.07 (t, J = 8.8 Hz, 2H, ArH), 7.27 (dd, J = 8.5, 5.8 Hz, 2H, ArH), 7.45 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 26.18, 26.73 (2CH3), 48.82 (C-3), 52.02 (2NCH2), 53.05 (C-2), 63.72 (C-1′), 66.60 (2OCH2), 105.78 (C-5), 114.99 (d, 2J(C,F) = 21.0 Hz, ArC), 129.15 (d, 3J(C,F) = 7.7 Hz, ArC), 139.72 (d, 4J(C,F) = 3.0 Hz, ArCq), 149.46 (C-6), 160.81 (d, 1J(C,F) = 241.8 Hz, ArCq), 188.51 (C-4); HRMS (HESI): calcd. (C14H15FNO+) [M + H − C4H9NO]+: 232.1138; found: 232.1129.
rac-2,2-Dimethyl-5-{(R)-(pyrrolidin-1-yl)[(2-trifluoromethyl)phenyl]methyl}-2,3-dihydropyridin-4(1H)-one (4a): Compound 1a (1.000 g (3.27 mmol)) was suspended in water (16 mL) and KOH (0.733 g (13.1 mmol)) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5–10 min). Then, 2-(trifluoromethyl)benzaldehyde (0.569 g (3.27 mmol)) was added and the reaction mixture was stirred for 4 days at r.t. The separated resinous solid was washed with water and dried in vacuo over phosphorus pentoxide. It was dissolved in hot ethyl acetate and cyclohexane was added until the first turbidity appeared. An oil separated which solidified. This amorphous precipitate was filtered off and discarded. The filtrate was evaporated and dissolved in hot ethyl acetate. It was allowed to stand for some days until crystallization took place resulting in 4a (0.282 g (24%)) as white needles. Rf (CH2Cl2:MeOH = 1:1): 0.39; mp: 139 °C; IR = 3281, 2968, 1623, 1606, 1577, 1530, 1392, 1311, 1262, 1158, 1124, 1034, 773; 1H NMR (DMSO-d6, 400 MHz) δ = 1.08 (s, 3H, CH3), 1.14 (s, 3H, CH3), 1.58–1.67 (m, 4H, (CH2)2), 2.12–2.21 (m, 2H, 3-H), 2.28–2.46 (m, 4H, 2NCH2), 4.81 (s, 1H, 1′-H), 6.70 (d, J = 6.9 Hz, 1H, 6-H), 7.25 (d, J = 6.9 Hz, 1H, 1-H), 7.38 (t, J = 7.6 Hz, 1H, ArH), 7.60 (d, J = 7.9 Hz, 1H, ArH), 7.64 (t, J = 7.6 Hz, 1H, ArH), 8.04 (d, J = 7.8 Hz, 1H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ = 23.31 ((CH2)2), 26.33, 26.74 (2CH3), 48.89 (C-3), 52.05 (2NCH2), 52.95 (C-2), 57.93 (C-1′), 107.40 (C-5), 124.46 (q, 1J(C,F) = 274 Hz, CF3), 125.93 (q, 3J(C,F) = 6.1 Hz, ArC), 125.99 (q, 2J(C,F) = 29.5 Hz, ArCq), 126.68 (ArC), 129.67 (ArC), 132.26 (ArC), 143.16 (ArCq), 149.61 (C-6), 187.94 (C-4); HRMS (HESI): calcd. (C19H24F3N2O+) [M + H]+: 353.1841; found: 353.1830.
rac-2,2-Dimethyl-5-{(R)-(piperidin-1-yl)[(2-trifluoromethyl)phenyl]methyl}-2,3-dihydropyridin-4(1H)-one (4b): Compound 1b (2.091 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.400 g (25 mmol) was added. Then, 2-(trifluoromethyl)benzaldehyde (1.137 g (6.53 mmol)) was added and the reaction mixture was stirred for 5 days at r.t. It was poured into a separatory funnel and was extracted 5 times with CH2Cl2. The combined organic phases were dried over sodium sulfate, filtered, and the solvent was evaporated. The orange resinous residue was dissolved in the minimum amount of hot ethyl acetate and left for crystallization over the weekend. The precipitate was sucked off, washed with cold ethyl acetate, and dried at 100 °C in vacuo resulting in 4b (1.370 g (57%)) as a white powder. Rf (CH2Cl2:MeOH = 1:1): 0.83; mp: 168 °C; IR = 2922, 1626, 1575, 1531, 1451, 1386, 1311, 1295, 1266, 1247, 1162, 1153, 1112, 1087, 1058, 1034; 1H NMR (DMSO-d6, 400 MHz) δ = 1.06 (s, 3H, CH3), 1.14 (s, 3H, CH3), 1.27–1.50 (m, 6H, (CH2)3), 2.09–2.28 (m, 4H, 3-H, NCH2), 2.30–2.40 (m, 2H, NCH2), 4.75 (s, 1H, 1′-H), 6.62 (d, J = 6.8 Hz, 1H, 6-H), 7.27 (d, J = 6.9 Hz, 1H, 1-H), 7.36 (t, J = 7.6 Hz, 1H, ArH), 7.60 (d, J = 7.9 Hz, 1H, ArH), 7.63 (t, J = 7.6 Hz, 1H, ArH), 8.05 (d, J = 7.9 Hz, 1H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ = 24.60 (CH2), 26.17 (2CH2), 26.58 (2CH3), 48.81 (C-3), 52.14 (2NCH2), 52.94 (C-2), 59.18 (C-1′), 105.58 (C-5), 124.49 (q, 1J(C,F) = 274 Hz, CF3), 125.98 (q, 3J(C,F) = 6.0 Hz, ArC), 126.65 (ArC), 126.73 (q, 2J(C,F) = 29.4 Hz, ArCq), 129.13 (ArC), 132.26 (ArC), 143.57 (ArCq), 149.81 (C-6), 188.42 (C-4); HRMS (HESI): calcd. (C20H26F3N2O+) [M + H]+: 367.1997; found: 367.1986.
rac-2,2-Dimethyl-5-{(R)-(pyrrolidin-1-yl)[(4-trifluoromethyl)phenyl]methyl}-2,3-dihydropyridin-4(1H)-one (5a): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.400 g (25 mmol)) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5–10 min). Then, 4-(trifluoromethyl)benzaldehyde (1.137 g (6.53 mmol)) was added. The reaction mixture was stirred for 7 days at r.t. It was poured into a separatory funnel and extracted 5 times with CH2Cl2. The combined organic phases were dried over sodium sulfate, filtered, and the solvent was evaporated. The orange resinous residue was dissolved in the minimum amount of hot ethyl acetate and left for crystallization overnight. The solid was sucked off and washed with cold ethyl acetate and dried at 100 °C in vacuo resulting in 5a (0.856 g (37%)) as yellowish needles. Rf (CH2Cl2:MeOH = 1:1): 0.45; mp: 167 °C; IR = 2967, 1568, 1532, 1259, 1391, 1324, 1226, 1151, 1113, 1104, 1064, 1015, 906, 665; 1H NMR (DMSO-d6, 400 MHz) δ = 1.07 (s, 3H, CH3), 1.16 (s, 3H, CH3), 1.62–1.69 (m, 4H, (CH2)2), 2.11–2.19 (m, 2H, 3-H), 2.26–2.33 (m, 2H, NCH2), 2.35–2.41 (m, 2H, NCH2), 4.40 (s, 1H, 1′-H), 7.11 (d, J = 6.8 Hz, 1H, 6-H), 7.45 (d, J = 6.7 Hz, 1H, 1-H), 7.47 (d, J = 8.1 Hz, 2H, ArH), 7.58 (d, J = 8.1 Hz, 2H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ = 23.36 ((CH2)2), 25.98, 26.84 (2CH3), 48.82 (C-3), 52.73 (2NCH2), 53.07 (C-2), 63.78 (C-1′), 108.22 (C-5), 124.61 (q, 1J(C,F) = 272 Hz, CF3), 125.10 (q, 3J(C,F) = 3.8 Hz, ArC), 126.78 (q, 2J(C,F) = 31.5 Hz, ArCq), 127.62 (ArC), 149.36 (C-6), 150.40 (ArCq), 187.82 (C-4); HRMS (HESI): calcd. (C19H24F3N2O+) [M + H]+: 353.1841; found: 353.1830.
rac-2,2-Dimethyl-5-{(R)-(piperidin-1-yl)[(4-trifluoromethyl)phenyl]methyl}-2,3-dihydropyridin-4(1H)-one (5b): Compound 1b (2.091 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.400 g (25 mmol)) was added. Then, 2-(trifluoromethyl)benzaldehyde (1.137 g (6.53 mmol)) was added and the reaction mixture was stirred for 7 days at r.t. It was poured into a separatory funnel and extracted 5 times with CH2Cl2. The combined organic phases were dried over sodium sulfate, filtered, and the solvent was evaporated. The orange resin was dissolved in the minimum amount of hot ethyl acetate and left for crystallization overnight. The solid was sucked off, washed with cold ethyl acetate, and dried at 100 °C in vacuo resulting in 5b (1.304 g (54%)) as white needles. Rf (CH2Cl2:MeOH = 1:1): 0.63; mp: 180 °C; IR = 2933, 1619, 1562, 1516, 1380, 1323, 1292, 1226, 1181, 1156, 1104, 1062, 1035, 1014, 986; 1H NMR (DMSO-d6, 400 MHz) δ = 1.10 (s, 3H, CH3), 1.18 (s, 3H, CH3), 1.31–1.52 (m, 6H, (CH2)3), 2.14–2.24 (m, 4H, 3-H, NCH2), 2.30–2.39 (m, 2H, NCH2), 4.46 (s, 1H, 1′-H), 6.96 (d, J = 6.8 Hz, 1H, 6-H), 7.44–7.48 (m, 3H, 1-H, ArH), 7.59 (d, J = 8.1 Hz, 2H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ = 24.53 (CH2), 26.06 (2CH2), 26.16, 26.69 (2CH3), 48.81 (C-3), 52.25 (2NCH2), 53.09 (C-2), 64.23 (C-1′), 105.73 (C-5), 124.64 (q, 1J(C,F) = 272 Hz, CF3), 125.11 (q, 3J(C,F) = 3.8 Hz, ArC), 126.70 (q, 2J(C,F) = 31.6 Hz, ArCq), 127.91 (ArC), 149.62 (ArCq), 149.67 (C-6), 188.47 (C-4); HRMS (HESI): calcd. (C20H26F3N2O+) [M]+: 367.1997; found: 367.1986.
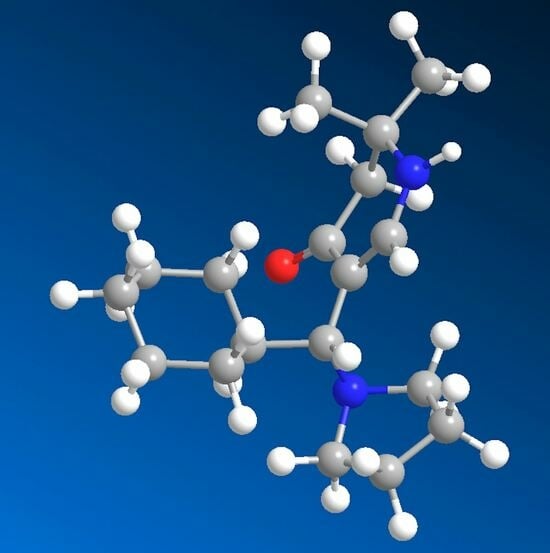
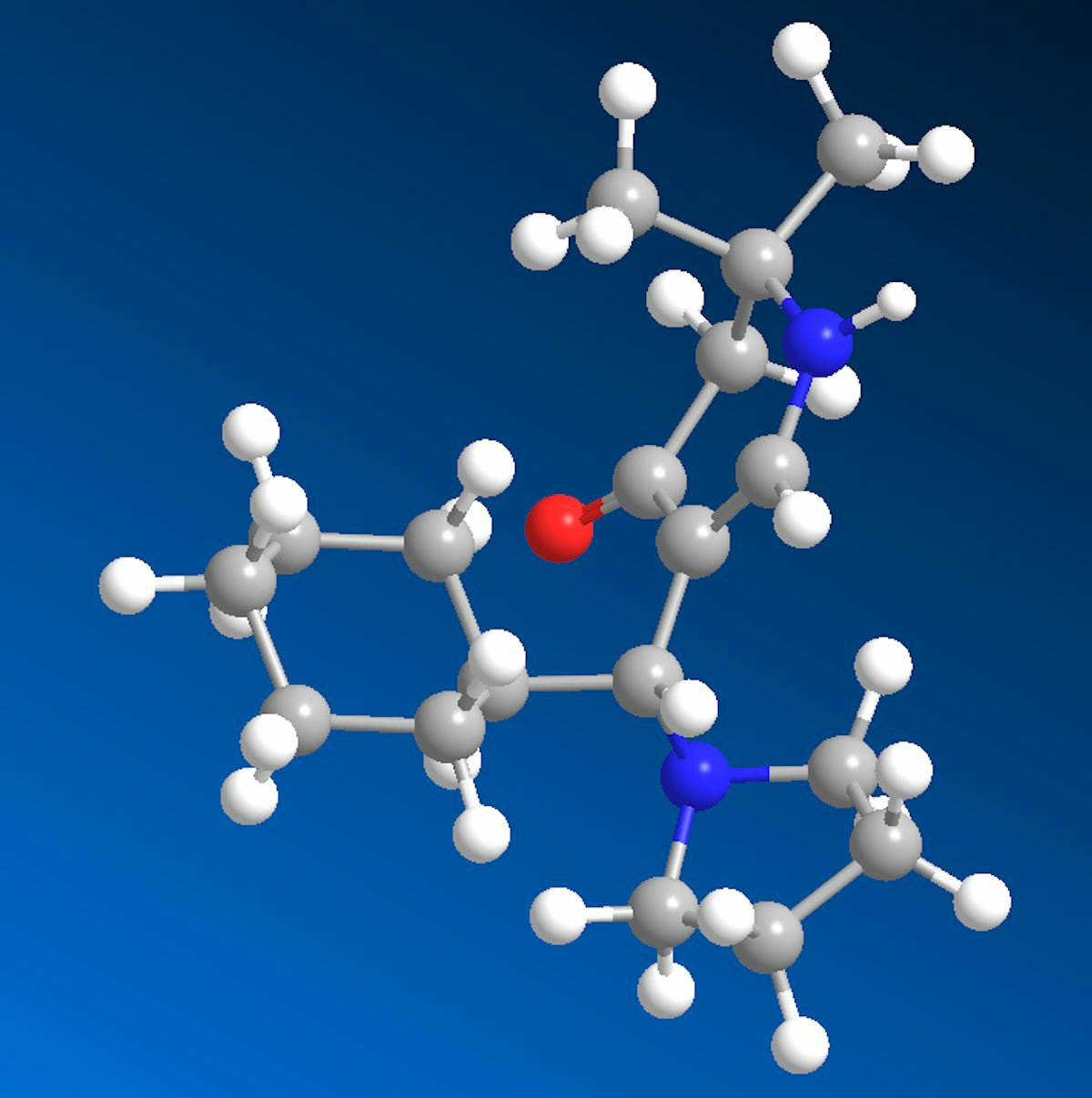
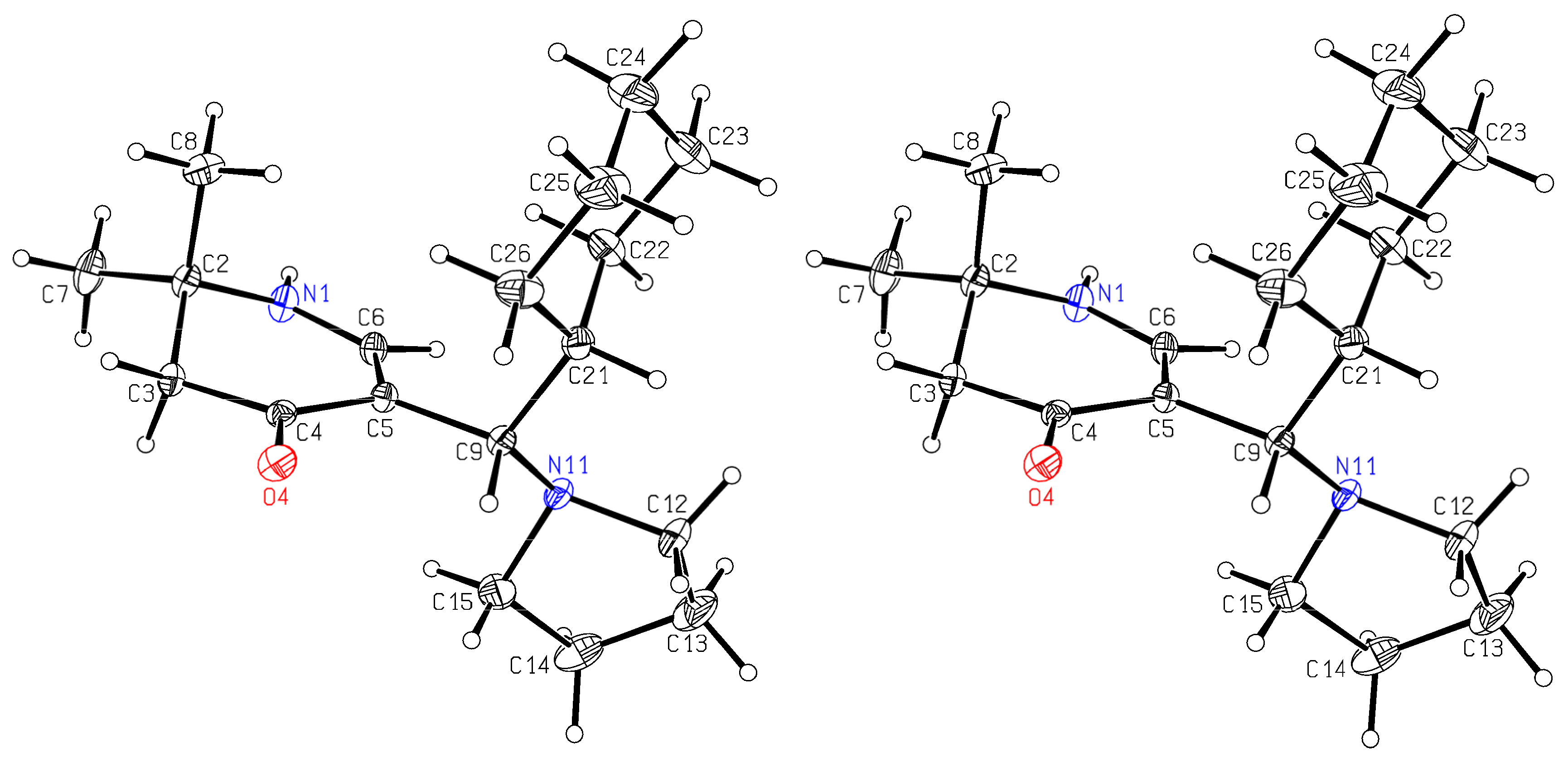
rac-5-[(R)-Cyclohexyl(pyrrolidin-1-yl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (6a) and rac-[(R)-Cyclohexyl(hydroxy)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (13): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.501 g (26.75 mmol)) was added. The mixture was stirred and sonicated at r.t. until most anything was dissolved. Then, cyclohexane carbaldehyde (0.749 g (6.68 mmol)) was added and the reaction mixture was stirred for 7 days at r.t. The separated white precipitate was filtered with suction, washed with water, and dried in vacuo resulting in a white solid. This was treated with dichloromethane and filtered. The filtrate was evaporated and the residue crystallized overnight in hot ethyl acetate in the form of sparkling prisms of 6a (68 mg (4%)). The solid of the dichloromethane filtration was dissolved in hot ethyl acetate. Overnight, compound 11 (0.355 g (19%)) crystallized as silky needles. Compound 6a: Rf (CH2Cl2:MeOH = 1:1): 0.19; mp: 161 °C; IR (KBr) = 3191, 3023, 2962, 2925, 2849, 2776, 1619, 1585, 1561, 1543, 1409, 1293, 1270, 1241, 1210, 1179; 1H NMR (DMSO-d6, 400 MHz) δ = 0.62–0.78 (m, 2H, CH2), 0.95–1.02 (m, 1H, CH2), 1.05–1.20 (m, 7H, CH2, 2CH3), 1.52–1.71 (m, 11H, CH, CH2), 2.18 (br, s, 2H, 3-H), 2.22–2.26 (m, 2H, NCH2), 2.28–2.33 (m, 2H, NCH2), 3.23 (d, J = 6.5 Hz, 1H, 1′-H), 6.95 (d, J = 6.6 Hz, 1H, 6-H), 7.20 (d, J = 6.6 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 22.99 ((CH2)2), 26.03 (CH2), 26.10 (CH3), 26.26 (CH2), 26.55 (CH3), 26.83, 27.79, 31.26 (3CH2), 40.45 (CH), 49.13 (C-3), 50.45 (2NCH2), 52.78 (C-2), 61.81 (C-1′), 103.50 (C-5), 149.44 (C-6), 189.39 (C-4); HRMS (HESI): calcd. (C18H31N2O+) [M + H]+: 291.2436; found: 291.2426.
Crystal Structure Determination of
6a: All the measurements were performed using monochromatized Mo K
α radiation at 100 K: C
18H
30N
2O,
Mr 290.44, orthorhombic, space group P b c a, a = 10.5346(4)Å, b = 11.7374(4)Å, c = 28.1853(11)Å, V = 3485.1(2)Å
3, Z = 8, d
calc = 1.107 g cm
−3, μ = 0.068 mm
−1. A total of 125,061 reflections were collected (Θ
max = 30.0°), from which 5084 were unique (R
int = 0.0642), with 4213 having I > 2σ(I). The structure was solved using direct methods (SHELXS-97) [
29] and refined using full-matrix least-squares techniques against
F2 (SHELXL-2014/6) [
30]. The non-hydrogen atoms were refined with an-isotropic displacement parameters without any constraints. The position of the H atom bonded to N1 was taken from a difference Fourier map, the N–H distance was fixed to 0.88 Å, and this H atom was refined with an individual isotropic displacement parameter without any constraints to the bond angles. The H atom bound to C6 was put at the external bisector of the N–C–C angle at a C–H distance of 0.95 Å and an individual isotropic displacement parameter was refined for it. The H atoms of the tertiary C–H groups were refined with individual isotropic displacement parameters and all X–C–H angles were equal at a C–H distance of 1.00 Å. The H atoms of the CH
2 groups were refined with common isotropic displacement parameters for the H atoms of the same group and idealized geometry with approximately tetrahedral angles and C–H distances of 0.99 Å. The H atoms of the methyl groups were refined with common isotropic displacement parameters for the H atoms of the same group and idealized geometries with tetrahedral angles, enabling rotations around the C–C bonds, and C–H distances of 0.98 Å. For the 211 parameters, final
R indices of R
1 = 0.0431 and wR
2 = 0.1167 (GOF = 1.035) were obtained. The largest peak in a difference Fourier map was 0.417 eÅ
−3. The final atomic parameters, as well as bond lengths and angles, were deposited at the Cambridge Crystallographic Data Centre (CCDC 2193723).
Compound 11 from 15:
Compound 15 (0.412 g (3.29 mmol)) was dissolved in a solution of KOH (0.752 g (13.4 mmol)) in distilled water (15 mL) with sonication. To this solution, cyclohexyl carbaldehyde (0.375 g (3.24 mmol)) was added and the mixture stirred at r.t. After a few minutes a white precipitate was formed. To complete the reaction, the mixture was stirred for a further 4 days at r.t. The white precipitate was sucked off, washed with water, and dried over phosphorous pentaoxide in a desiccator under reduced pressure. Yield: 0.712 g (91%) of pure alcohol 11.
Data of compound 11: Rf (CH2Cl2:MeOH = 1:1): 0.79; mp: 180 °C; IR = 3283, 3221, 3042, 2926, 2849, 1557, 1519, 1409, 1309, 1264, 1244, 1183, 1121; 1H NMR (DMSO-d6, 400 MHz) δ = 0.79–0.95 (m, 2H, CH2), 0.99–1.12 (m, 3H, CH2), 1.15 (s, 3H, CH3), 1.18 (s, 3H, CH3), 1.26–1.35 (m, 1H, CH), 1.42 (br, d, J = 12.9 Hz, 1H, CH2), 1.54–1.67 (m, 3H, CH2), 1.77 (br, d, J = 13.0 Hz, 1H, CH2), 2.11–2.20 (m, 2H, 3-H), 4.07 (dd, J = 6.7, 5.0 Hz, 1H, 1′-H), 4.21 (d, J = 5.0 Hz, 1H, OH), 7.04 (d, J = 6.6 Hz, 1H, 6-H), 7.28 (d, J = 6.7 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 26.02 (CH2), 26.04 (CH3), 26.16 (CH2), 26.56 (CH2), 26.92 (CH3), 28.63, 29.33 (2CH2), 44.23 (CH), 49.17 (C-3), 52.90 (C-2), 70.05 (C-1′), 109.10 (C-5), 148.23 (C-6), 188.81 (C-4); HRMS (HESI): calcd. (C14H22NO2−) [M − H]−: 236.1651; found: 236.1658.
rac-5-[(R)-Cyclohexyl(piperidin-1-yl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (6b): Compound 1b (2.091 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.449 g (25.82 mmol)) was added. The mixture was stirred and sonicated at r.t. until most anything was dissolved. Then, cyclohexane carbaldehyde (0.749 g (6.68 mmol)) was added and the reaction mixture was stirred for 7 days at r.t. The separated off-white solid was filtered with suction, washed with water, and dried in vacuo. It was treated with dichloromethane and the suspension was filtered. The filtrate was evaporated and the residue dissolved in hot ethyl acetate. After the first crystallization, a solid mixture was sucked off. From the filtrate, 6b crystallized as yellowish prisms (0.170 g (9%)). The solid from the treatment with dichloromethane was dissolved in the minimum amount of hot ethyl acetate and left for crystallization overnight. The solid was sucked off and dried yielding 11 (0.355 mg (23%)) as white needles. Compound 6b: Rf (CH2Cl2:MeOH = 1:1): 0.84; mp: 154 °C; IR = 3203, 3026, 2927, 2848, 1616, 1562, 1397, 1383, 1293, 1276, 1259, 1242; 1H NMR (DMSO-d6, 400 MHz) δ = 0.57–0.66 (m, 1H, CH2), 0.74–0.84 (m, 1H, CH2), 1.00–1.16 (m, 3H, CH2), 1.18, 1.19 (2s, 6H, 2CH3), 1.22–1.75 (m, 11H, CH, CH2), 1.92–2.00 (m, 1H, CH2), 2.00–2.09 (m, 2H, NCH2), 2.19 (s, 2H, 3-H), 2.22–2.29 (m, 2H, NCH2), 3.14 (br, s, 1H, 1′-H), 6.90 (d, J = 6.6 Hz, 1H, 6-H), 7.18 (d, J = 6.6 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 24.95 (CH2), 25.84 (CH3), 25.96, 26.03 (2CH2), 26.35 (2CH2), 26.65 (CH3), 26.78, 30.50, 31.33 (3CH2), 36.58 (CH), 49.14 (C-3), 50.24 (2NCH2), 52.77 (C-2), 64.45 (C-1′), 101.84 (C-5), 148.69 (C-6), 190.15 (C-4); HRMS (HESI): calcd. (C14H22NO+) [M + H − C5H11N]+: 220.1701; found: 220.1693.
rac-2,2-Dimethyl-5-[(R)-(4-methylphenyl)(pyrrolidin-1-yl)methyl]-2,3-dihydropyridin-4(1H)-one (7a): Compound 1a (1.000 g (3.27 mmol)) was suspended in water (16 mL) and KOH (0.733 g (13.1 mmol)) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5–10 min). Then, 4-methylbenzaldehyde (0.393 g (3.27 mmol)) was added and the reaction mixture was stirred for 6 days at r.t. The separated beige crystalline solid was filtered with suction, washed with water, and dried in vacuo over phosphorus pentoxide. It was treated with hot ethyl acetate. The insoluble parts were removed via filtration and the filtrate was concentrated in vacuo. Crystallization took place overnight yielding 7a (0.440 g (45%)) as colorless plates. Rf (CH2Cl2:MeOH = 1:1): 0.25; mp: 167 °C; IR = 3240, 2968, 1622, 1571, 1536, 1394, 1240; 1H NMR (DMSO-d6, 400 MHz) δ = 1.07 (s, 3H, CH3), 1.16 (s, 3H, CH3), 1.57–1.67 (m, 4H, (CH2)2), 2.09–2.17 (m, 2H, 3-H), 2.22 (s, 3H, ArCH3), 2.24–2.40 (m, 4H, 2NCH2), 4.27 (s, 1H, 1′-H), 7.02 (d, J = 7.8 Hz, 2H, ArH), 7.08 (d, J = 6.8 Hz, 1H, 6-H), 7.13 (d, J = 7.8 Hz, 2H, ArH), 7.30 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 20.79 (ArCH3), 23.35 ((CH2)2), 26.04, 26.92 (2CH3), 48.98 (C-3), 52.81 (2NCH2), 53.01 (C-2), 63.58 (C-1′), 109.27 (C-5), 126.91, 128.69 (ArC), 134.90, 142.63 (ArCq), 149.13 (C-6), 187.83 (C-4); HRMS (HESI): calcd. (C19H27N2O+) [M + H]+: 299.2123; found: 299.2116.
rac-5-[(R)-(4-Isopropylphenyl)(pyrrolidin-1-yl)methyl]-2,2-dimethyl-2,3-dihydropyridin-4(1H)-one (8a): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (32 mL) and KOH (1.466 g (26.13 mmol)) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5–10 min). Then, 4-isopropylbenzaldehyde (0.968 g (6.53 mmol)) was added and the reaction mixture was stirred for 5 days at r.t. The separated resinous beige product was dried in vacuo over phosphorus pentoxide and then treated with hot ethyl acetate. The insoluble part was filtered off. Cyclohexane was added to the solution and the mixture was allowed to stand overnight. The precipitate was filtered off and discarded, the filtrate was evaporated to dryness, and the residue was recrystallized from ethyl acetate resulting in an almost pure product. Further crystallization from ethyl acetate afforded pure 8a (0.118 g (6%)) as white needles. Rf (CH2Cl2:MeOH = 1:1): 0.91; mp: 152 °C; IR = 3240, 3042, 2963, 1622, 1568, 1461, 1395, 1290, 1239, 1181; 1H NMR (DMSO-d6, 400 MHz) δ = 1.09 (s, 3H, CH3), 1.15–1.16 (m, 9H, CH(CH3)2, CH3), 1.61–1.65 (m, 4H, (CH2)2), 2.14 (s, 2H, 3-H), 2.25–2.37 (m, 4H, 2NCH2), 2.80 (quin, J = 6.9 Hz, 1H, CH(CH3)2), 4.28 (s, 1H, 1′-H), 7.07–7.09 (m, 3H, 6-H, 2ArH), 7.16 (d, J = 8.0 Hz, 2H, 2ArH), 7.31 (d, J = 6.7 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 23.33 ((CH2)2), 24.09, 24.12 (CH(CH3)2), 25.94, 27.10 (2CH3), 33.16 (CH(CH3)2), 48.95 (C-3), 52.86 (2NCH2), 53.02 (C-2), 63.50 (C-1′), 109.23 (C-5), 125.99, 126.90 (4ArC), 142.98, 145.90 (ArCq), 149.23 (C-6), 187.84 (C-4); HRMS (HESI+): calcd. (C21H31N2O+) [M + H]+: 327.2436; found: 327.2426.
rac-2,2-Dimethyl-5-[(R)-pyrrolid-4-yl)(pyrrolidine-1-yl)methyl]-2,3-dihydropyridin-4(1H)-one (9a): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (32 mL) and KOH (1.466 g (26.13 mmol)) was added. The mixture was stirred at r.t. until a solution was formed (ca. 5–10 min). Then, pyridine-4-carbaldehyde (0.700 g (6.53 mmol)) was added and the reaction mixture was stirred for 4 days at r.t. The solution was extracted 5 times with dichloromethane and the combined organic layers were dried over sodium sulfate, filtered, and the solvents evaporated in vacuo resulting in a yellow oil. This was dissolved in hot ethyl acetate and cooled to r.t. Then, cyclohexane was added dropwise until the mixture was opacified permanently. After stirring overnight at r.t., the formed precipitate was sucked off and dissolved in hot ethyl acetate, filtered from insoluble parts, and left for crystallization yielding 9a (0.170 g (9%)) as yellowish needles. Rf (CH2Cl2:MeOH = 1:1): 0.37; mp: 169 °C; IR = 3270, 3026, 2800, 1624, 1594, 1574, 1505, 1386, 1292, 1226, 1180, 1115; 1H NMR (DMSO-d6, 400 MHz) δ = 1.07 (s, 3H, CH3), 1.16 (s, 3H, CH3), 1.64–1.67 (m, 4H, (CH2)2), 2.16 (s, 2H, 3-H), 2.28–2.50 (m, 4H, 2NCH2), 4.33 (s, 1H, 1′-H), 7.10 (d, J = 6.8 Hz, 1H, 6-H), 7.24 (dd, J = 4.5, 1.6 Hz, 2H, ArH), 7.49 (d, J = 6.8 Hz, 1H, 1-H), 8.40 (dd, J = 4.5, 1.6 Hz, 2H, ArH); 13C NMR (DMSO-d6, 100 MHz) δ = 23.34 ((CH2)2), 26.01, 26.76 (2CH3), 48.76 (C-3), 52.56 (2NCH2), 53.08 (C-2), 63.14 (C-1′), 107.57 (C-5), 122.24 (ArC), 149.49, 149.57 (C-6, ArC), 154.17 (ArCq), 187.77 (C-4); HRMS (HESI): calcd. (C17H24N3O+) [M + H]+: 286.1919; found: 286.1909.
rac-2,2-Dimethyl-5-[(R)-2-methyl-1-(pyrrolidin-1-yl)propyl]-2,3-dihydropyridin-4(1H)-one (10a): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (30 mL) and KOH (1.473 g (26.25 mmol)) was added. To the mixture, isobutyric aldehyde (475 mg (6.59 mmol)) was added and it was stirred and sonicated at r.t. until nearly all was dissolved. The reaction mixture was stirred for 7 days at r.t. The raw product contained small amounts of Morita–Baylis–Hillman product, which was not isolated. The separated red oil was exhaustively extracted with dichloromethane, and then the combined organic layers were dried over sodium sulfate, filtered, and the solvent was evaporated in vacuo resulting in a red oil. This was dissolved in the minimum amount of hot dichloromethane. The product crystallized in the form of yellowish needles overnight, and then was sucked off and dried. Yield: 0.255 g (16%). Rf (CH2Cl2:MeOH = 1:1): 0.14; mp: 148 °C; IR = 2955, 2771, 1617, 1584, 1562, 1537, 1456, 1408, 1382, 1364, 1268, 1239, 1211, 1181, 1125, 1110, 667; 1H NMR (DMSO-d6, 400 MHz) δ = 0.67 (d, J = 6.6 Hz, 3H, CH(CH3)2), 0.73 (d, J = 6.6 Hz, 3H, CH(CH3)2), 1.17 (s, 3H, CH3), 1.18 (s, 3H, CH3), 1.52–1.64 (m, 4H, 2CH2), 1.94 (dsept, J = 6.6 Hz, 1H, CH(CH3)2), 2.14–2.23 (m, 2H, 3-H), 2.22–2.36 (m, 4H, 2NCH2), 3.18 (d, J = 6.3 Hz, 1H, 1‘-H), 6.98 (d, J = 6.7 Hz, 1H, 6-H), 7.24 (d, J = 6.7 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 17.21 (CH(CH3)2), 20.83 (CH(CH3)2), 23.05 (2CH2), 25.90, 26.72 (2CH3), 30.12 (CH(CH3)2), 49.12 (C-3), 50.84 (2NCH2), 52.75 (C-2), 62.71 (C-1‘), 103.16 (C-5), 149.56 (C-6), 189.34 (C-4); HRMS (HESI): calcd. (C15H27N2O+) [M + H]+: 251.2118; found: 251.2114.
rac-2,2-Dimethyl-5-[(R)-(1-Hydroxy-2,2-dimethylpropyl)-2,3-dihydropyridin-4(1H)-one (12): Compound 1a (2.413 g (7.88 mmol)) was suspended in water (32 mL) and KOH (1.496 g (26.66 mmol)) was added. Then, pivalaldehyde (0.678 g (7.88 mmol)) was added to the mixture. The reaction mixture was stirred for 4 days at r.t. The formed white fluffy solid was sucked off and dried overnight in a desiccator over phosphorous pentaoxide. Yield: 1.129 g (68%) Rf (CH2Cl2:MeOH = 1:1): 0.80; mp: 168 °C; IR = 3249, 2950, 1526, 1402, 1389, 1366, 1258, 1239, 1211, 1177, 1000; 1H NMR (DMSO-d6, 400 MHz) δ = 0.75 (s, 9H, 3CH3), 1.16 (s, 3H, CH3), 1.20 (s, 3H, CH3), 2.12 (d, J = 16.0 Hz, 1H, 3-H), 2.19 (d, J = 16.0 Hz, 1H, 3-H), 4.15 (d, J = 4.6 Hz, 1H, 1′-H), 4.43 (d, J = 4.8 Hz, 1H, OH), 7.07 (d, J = 6.6 Hz, 1H, 6-H), 7.39 (d, J = 6.8 Hz, 1H, 1-H); 13C NMR (DMSO-d6, 100 MHz) δ = 25.45 (CH3), 26.13 (3CH3), 27.23 (CH3), 36.55 (C(CH3)3), 49.11 (C-3), 52.62 (C-2), 72.25 (C-1′), 107.94 (C-5), 149.17 (C-6), 188.70 (C-4); HRMS (DIP EI): calcd. (C12H21NO2) [M+]: 211.1567; found: 211.1573.
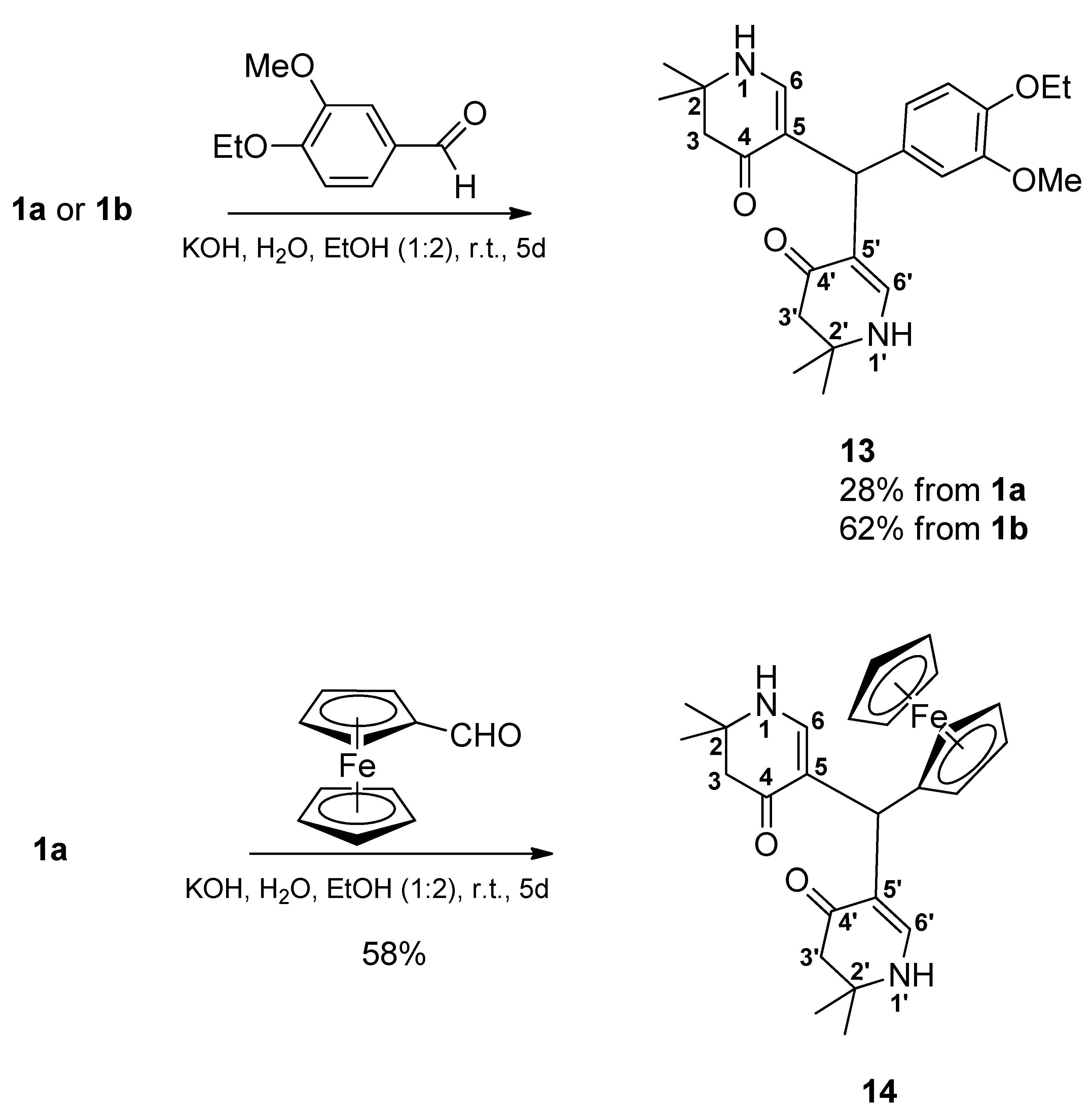
5,5’-[(4-Ethoxy-3-methoxyphenyl)methylene]bis(2,2-dimethyl-2,3-dihydropyridin-4(1H)-one) (13): From 1a: Compound 1a (2.000 g (6.53 mmol)) was suspended in water (15 mL) and ethanol (30 mL). Then, a solution of KOH (1.488 mg (26.52 mmol)) in a mixture of water (15 mL) and ethanol (30 mL) was added. Finally, 4-ethoxy-3-methoxybenzaldehyde (1.177 g (6.53 mmol)) was added and the reaction mixture was stirred for 5 days. Most of the ethanol was evaporated in vacuo. The remaining solution was diluted with water, transferred into a separatory funnel, and then extracted 4 times with dichloromethane. The combined organic layers were dried over sodium sulfate, filtered, and the solvent was evaporated yielding a yellow oil. This was dissolved in a minimum amount of hot ethyl acetate. Then, cyclohexane was added dropwise until the mixture was opacified permanently. The formed beige precipitate was sucked off, washed with a cold mixture of cyclohexane and ethyl acetate, and dried at 100 °C at reduced pressure resulting in 11 (0.375 g (28%)). For analytical purposes, it was recrystallized from ethyl acetate giving the product an off-white precipitate.
From 1b: Compound 1b (2.091 g (6.53 mmol)) was suspended in water (15 mL) and ethanol (30 mL). Then, a solution of KOH (1.579 g (28.14 mmol)) in a mixture of water (15 mL) and ethanol (30 mL) was added. Finally, 4-ethoxy-3-methoxybenzaldehyde (1.177 g (6.53 mmol)) was added and the reaction mixture was stirred for 5 days. Most of the ethanol was evaporated in vacuo. The remaining solution was diluted with water, transferred into a separatory funnel, and then extracted 4 times with dichloromethane. The combined organic layers were dried over sodium sulfate, filtered, and the solvent was evaporated resulting in a yellow oil. This oil was dissolved in a minimum amount of hot ethyl acetate. Cyclohexane was added dropwise until the mixture was opacified permanently. The formed beige precipitate was sucked off, washed with a cold mixture of cyclohexane and ethyl acetate, and dried at 100 °C at reduced pressure resulting in 13 (0.830 g (62%)). For analytical purposes, it was recrystallized from ethyl acetate resulting in the product as an off-white precipitate. Rf (CH2Cl2:MeOH = 1:1): 0.80; mp: 125 °C; IR = 2968, 1578, 1512, 1378, 1294, 1241, 1182, 1135; 1H NMR (DMSO-d6, 400 MHz) δ = 1.17 (s, 6H, 2CH3), 1.18 (s, 6H, 2CH3), 1.28 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.14–2.22 (m, 4H, 3-H, 3′-H), 3.65 (s, 3H, OCH3), 3.92 (q, J = 7.0 Hz, 2H, OCH2CH3), 4.84 (s, 1H, CH), 6.50 (dd, J = 8.5, 2.0 Hz, 1H, ArH), 6.55–6.62 (m, 3H, 6-H, 6′-H, ArH), 6.78 (d, J = 8.2 Hz, 1H, ArH), 6.99 (d, J = 6.7 Hz, 2H, 1-H, 1′-H); 13C NMR (DMSO-d6, 100 MHz) δ = 15.08 (OCH2CH3), 26.36, 26.80 (4CH3), 37.99 (CH), 49.35 (C-3, C-3′), 53.12 (C-2, C-2′), 55.42 (OCH3), 63.90 (OCH2), 109.22 (C-5, C-5′), 112.38, 112.89, 119.87 (ArC), 138.27, 145.83, 148.65 (ArCq), 148.81 (C-6, C-6′), 188.74 (C-4, C-4′); HRMS (HESI): calcd. (C24H33N2O4+) [M + H]+: 413.2440; found: 413.2433.
5,5’-(Ferrocenylmethylene)bis(2,2-dimethyl-2,3-dihydropyridin-4(1H)-one) (14): Compound 1a (2.000 g (6.53 mmol)) was suspended in water (15 mL) and ethanol (30 mL). Then, a solution of KOH (1.435 g (25.57 mmol)) in a mixture of water (15 mL) and ethanol (30 mL) was added. Finally, ferrocene carbaldehyde (1.398 g (6.53 mmol)) was added. The reaction mixture was stirred for 5 days. The formed precipitate was sucked off and dried in vacuo over phosphorus pentoxide. It was treated with hot ethyl acetate and filtered. The filtrate was discarded. The precipitate was suspended in a mixture of dichloromethane and ethanol (4:1) and filtered off. Then, the filtrate was evaporated and the residue was dissolved in a hot mixture of ethyl acetate and ethanol (1:1). The mixture was filtered and the filtrate was left for crystallization overnight. The formed solid was sucked off and dried at 150 °C in vacuo resulting in 14 (0.850 g (58%)) as a yellow solid. Rf (CH2Cl2:MeOH = 1:1): 0.83; mp: 262 °C (decomp.); IR = 3441, 3288, 2965, 1620, 1569, 1528, 1382, 1297, 1243, 1177; 1H NMR (DMSO-d6, 400 MHz) δ = 1.14 (s, 6H, 2CH3), 1.17 (s, 6H, 2CH3), 2.09–2.18 (m, 4H, 3-H, 3′-H), 3.85 (br, s, 2H, ArH), 4.01 (br, s, 2H, ArH), 4.07 (s, 5H, ArH), 4.63 (s, 1H, CH), 6.80 (d, J = 6.7 Hz, 2H, 6-H, 6′-H), 6.92 (d, J = 6.7 Hz, 2H, 1-H, 1′-H); 13C NMR (DMSO-d6, 100 MHz) δ = 26.32, 26.48 (4CH3), 32.76 (CH), 49.49 (C-3, C-3′), 53.01 (C-2, C-2′), 66.50 (2ArC), 68.01 (2ArC), 68.41 (5ArC), 94.48 (ArCq), 110.75 (C-5, C-5′), 148.78 (C-6, C-6′), 188.07 (C-4, C-4′); HRMS (HESI): calcd. (C25H31FeN2O2+) [M + H]+: 447.1735; found: 447.1721.
2,2-Dimethyl-2,3-dihydropyridin-4(1H)-one (
15): Compound
1b (2.50 g (7.81 mmol)) was suspended in 37.5 mL water and KOH (2.127g (37.91 mmol)) was added. The mixture was stirred at r.t. for 4 days. The solution was transferred into a separatory funnel and extracted 10 times with dichloromethane. The combined organic phases were dried over sodium sulfate, filtered, and the solvent evaporated. The remaining oil crystallized spontaneously resulting in
15 as an off-white solid. Yield: 812 mg (6.49 mmol; 83%). The melting point (93°) corresponds well with that reported (93–94°) by Gusev [
9] for this compound.
1H NMR (DMSO-d
6, 400 MHz)
δ = 1.17 (s, 6H, 2CH
3), 2.16 (br, s, 2H, 3-H), 4.62 (dd,
J = 7.1, 1.2 Hz, 1H, 5-H), 7.13 (dd,
J = 7.0, 6.9 Hz, 1H, 6-H), 7.41 (br, s, 1H, 1-H);
13C NMR (DMSO-d
6, 100 MHz)
δ = 26.36 (2CH
3), 49.25 (C-3), 53.01 (C-2), 95.00 (C-5), 150.22 (C-6), 190.60 (C-4).

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}