

Ligand−Structure Effects on N−Heterocyclic Carbene Rhenium Photo− and Electrocatalysts of CO2 Reduction
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results and Discussion
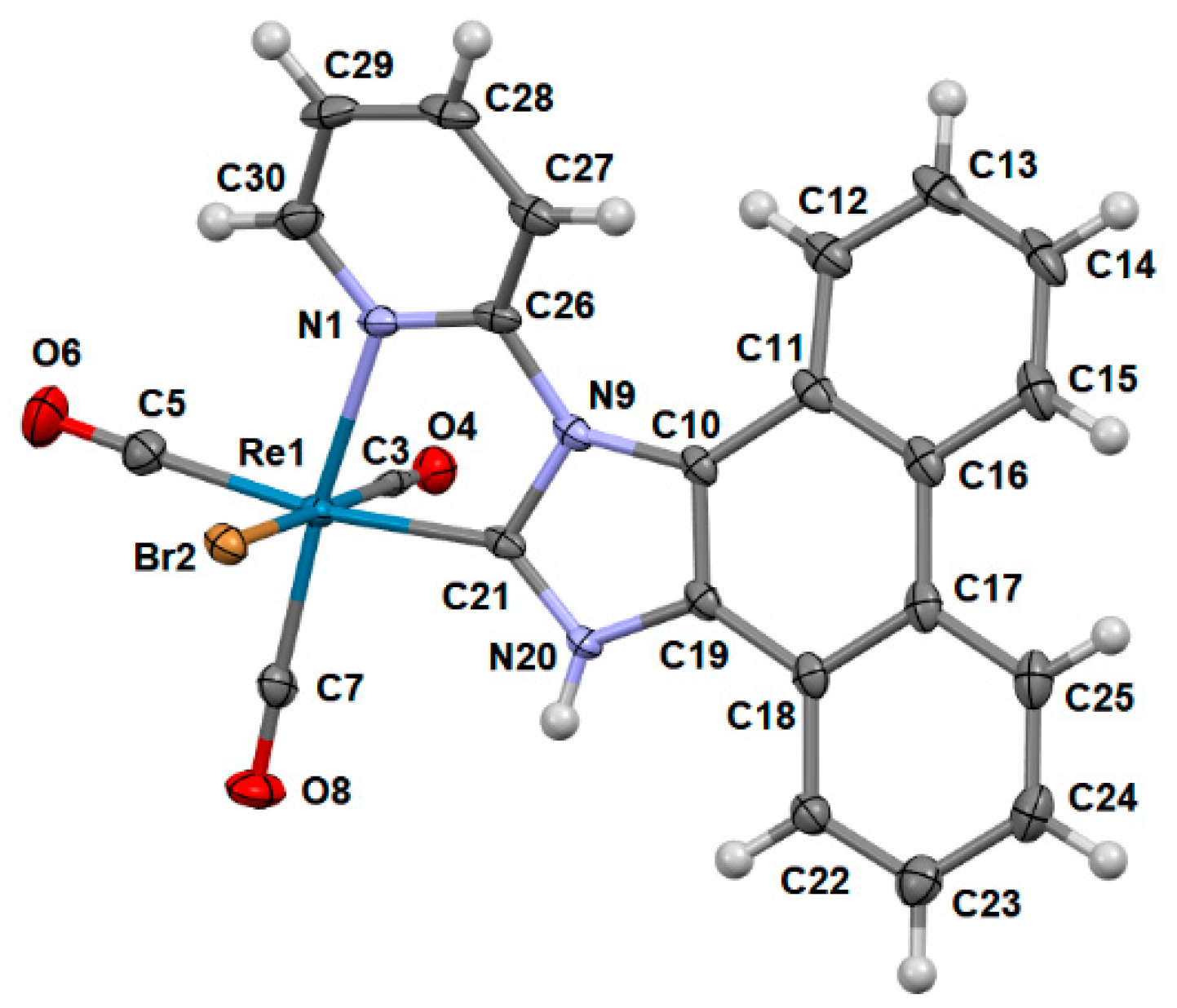
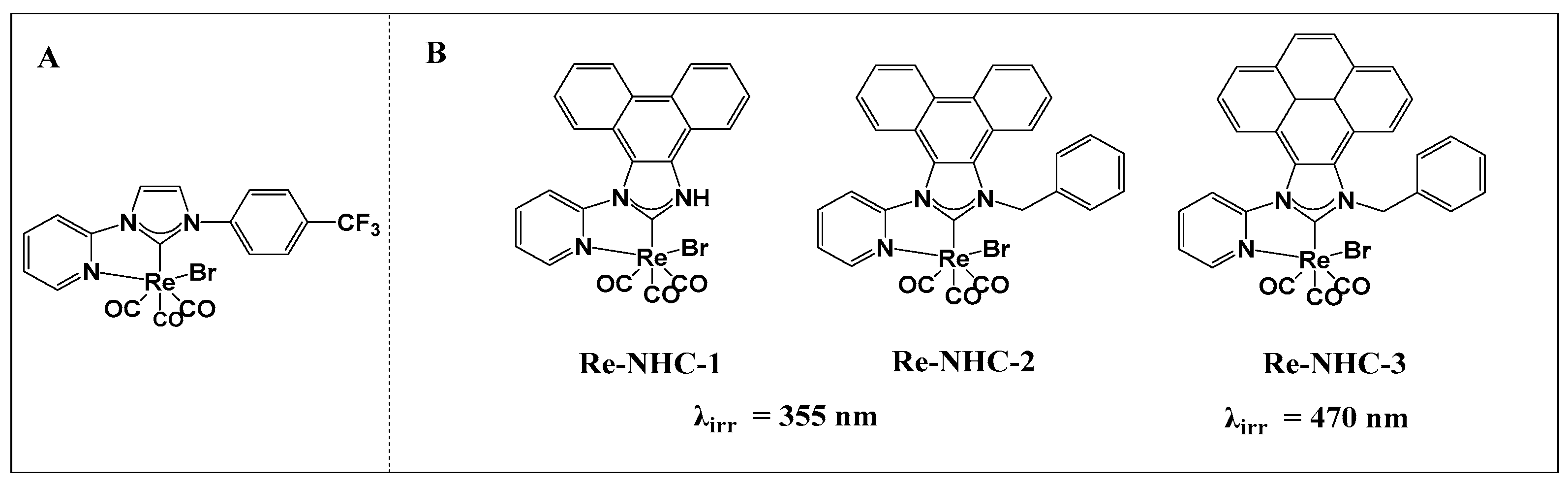
2.1. Synthesis and Structural Characterization
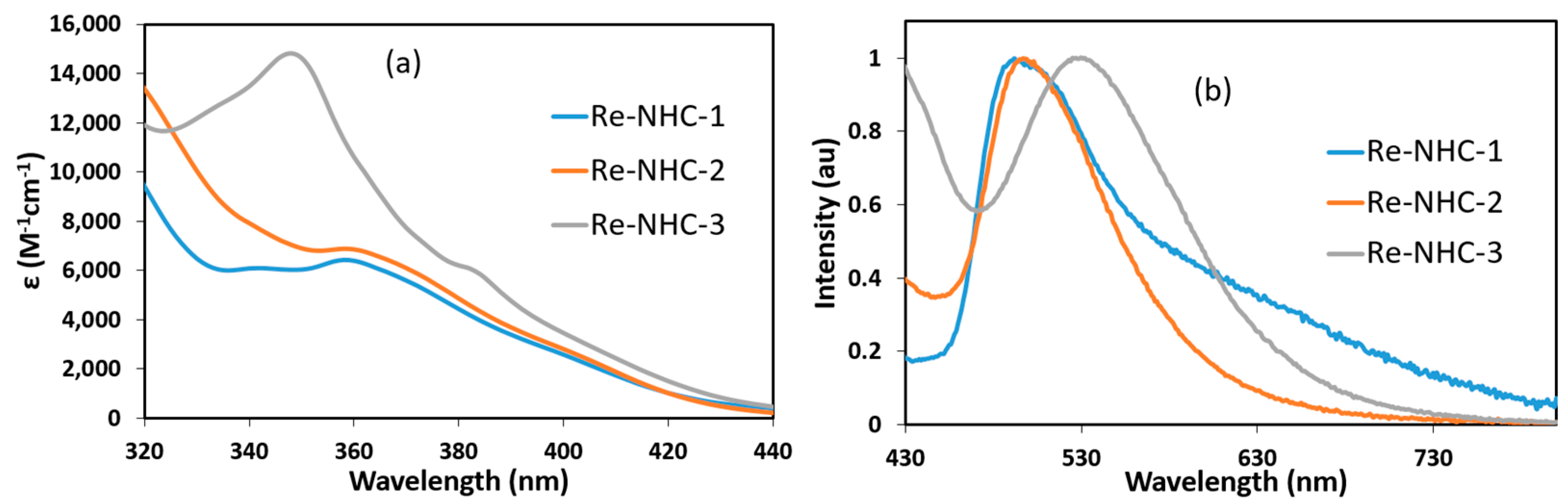
2.2. Photophysical Investigations
2.2.1. Electronic Absorption Spectroscopy
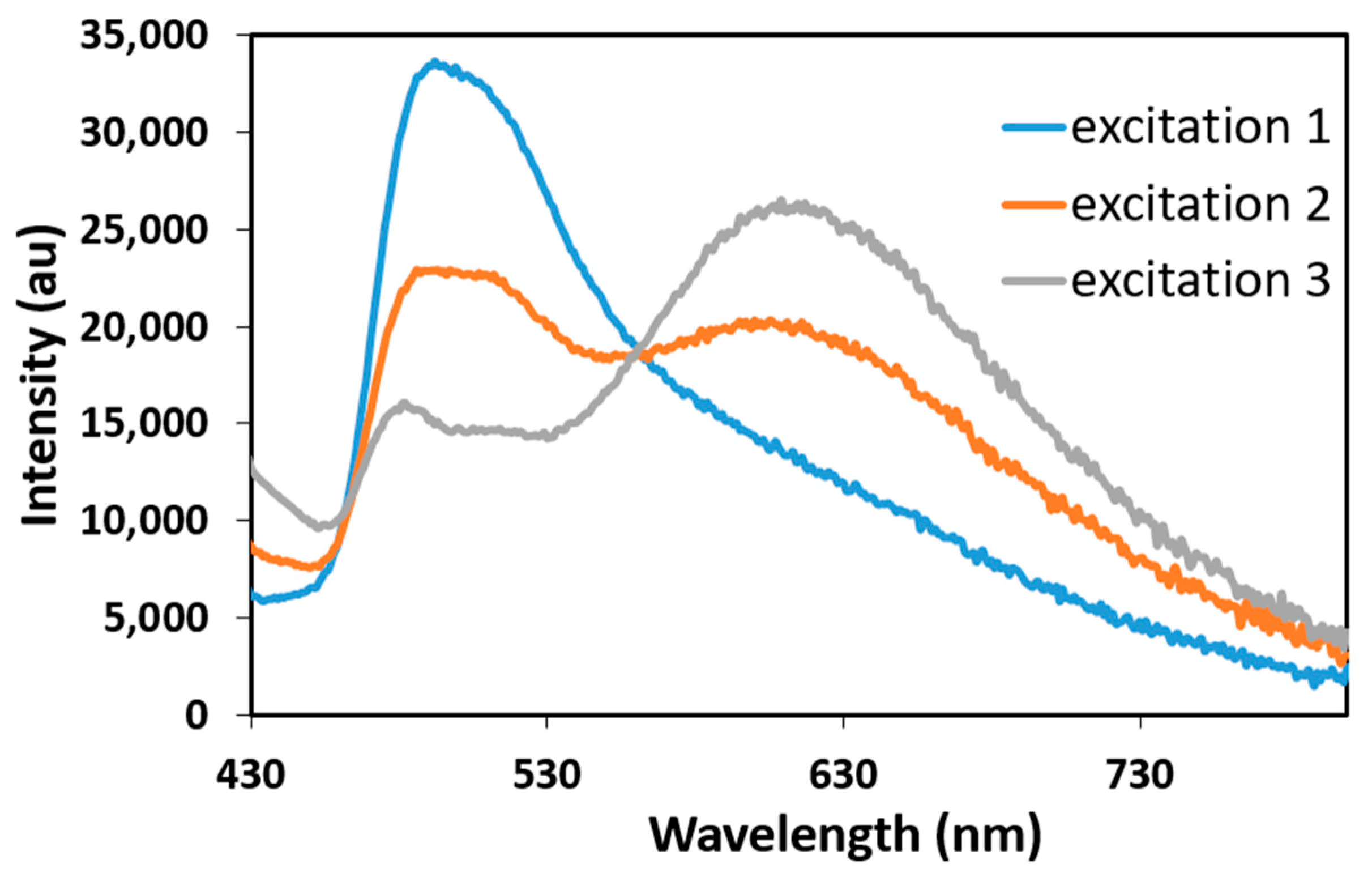
2.2.2. Luminescence Spectroscopy
2.2.3. Photostability
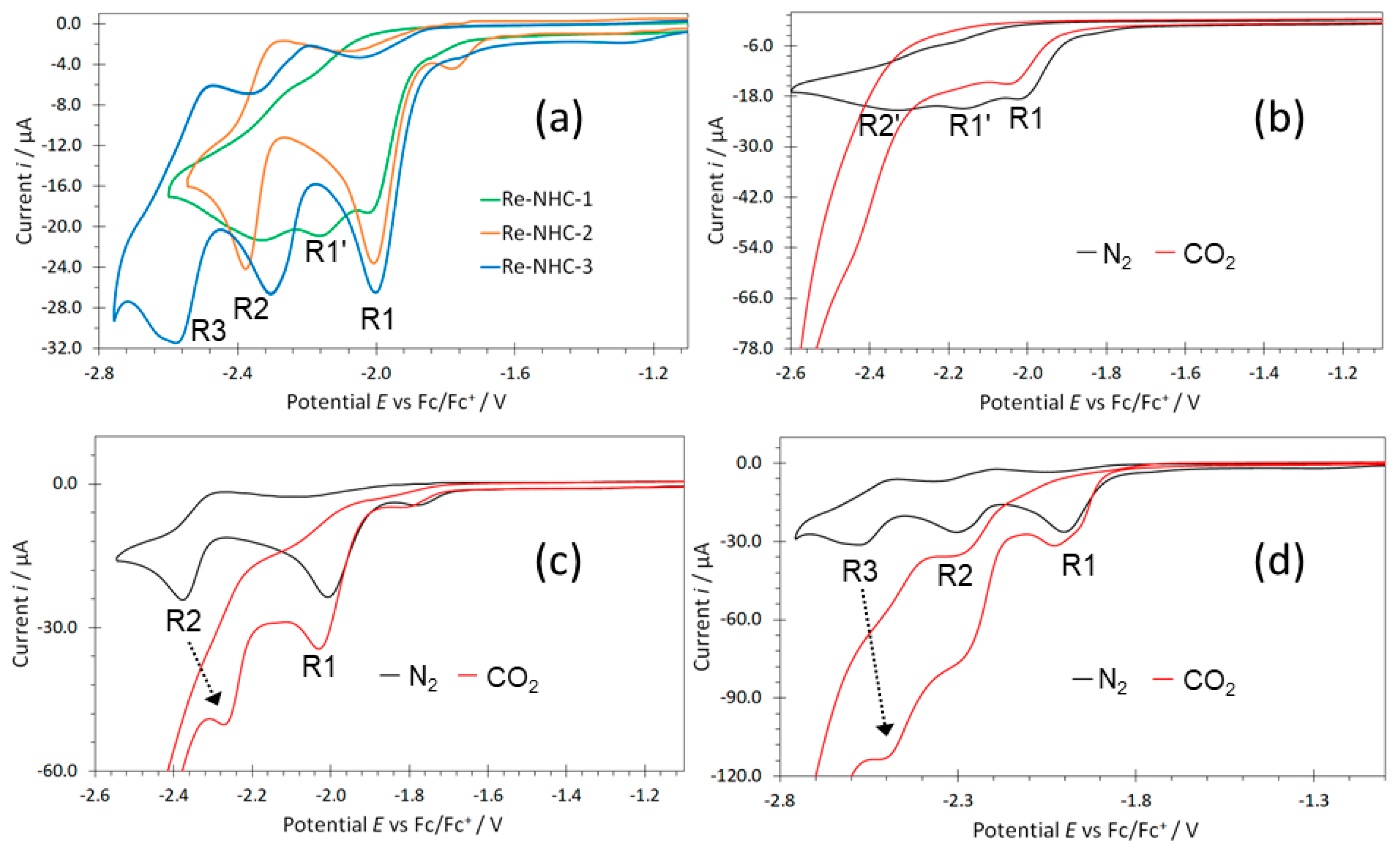
2.3. Redox Behaviour
2.3.1. Electrochemical Reduction and Electrocatalysis
2.3.2. Electrochemical Oxidation
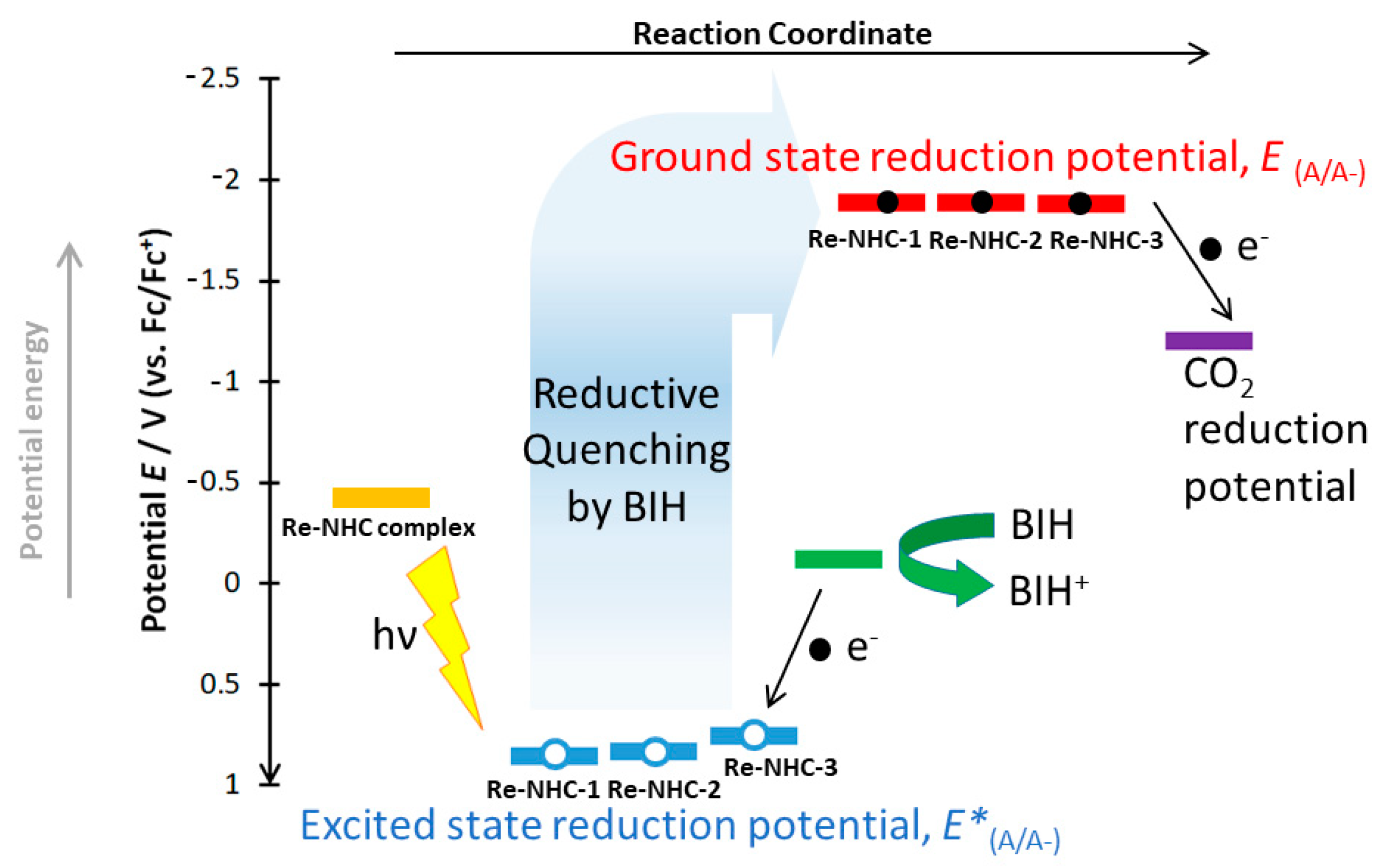
2.4. Photocatalytic CO2 Reduction
3. Materials and Methods
3.1. Syntheses
3.1.1. 1−(Pyridin−2−yl)−1H−phenanthro [9,10−d]imidazole (NHC−1)

3.1.2. 1−Benzyl−3−(pyridine−2−yl)−1H−3λ4−phenanthro[9,10−d]imidazolium bromide (NHC−2)

3.1.3. 1−(Pyridin−2−yl)−3a1, 5a1 −dihydro−9H−11λ4 −pyreno[4,5−d]imidazole

3.1.4. (9−Benzyl−11−(pyridin−2−yl)−3a1, 5a1 −dihydro−9H−11λ4 −pyreno[4,5−d]imidazolium bromide (NHC−3)

3.1.5. Re−NHC−1
3.1.6. Re−NHC−2
3.1.7. Re−NHC−3
3.2. X-ray Crystallography
3.3. Photophysical Studies
3.4. Cyclic Voltammetry
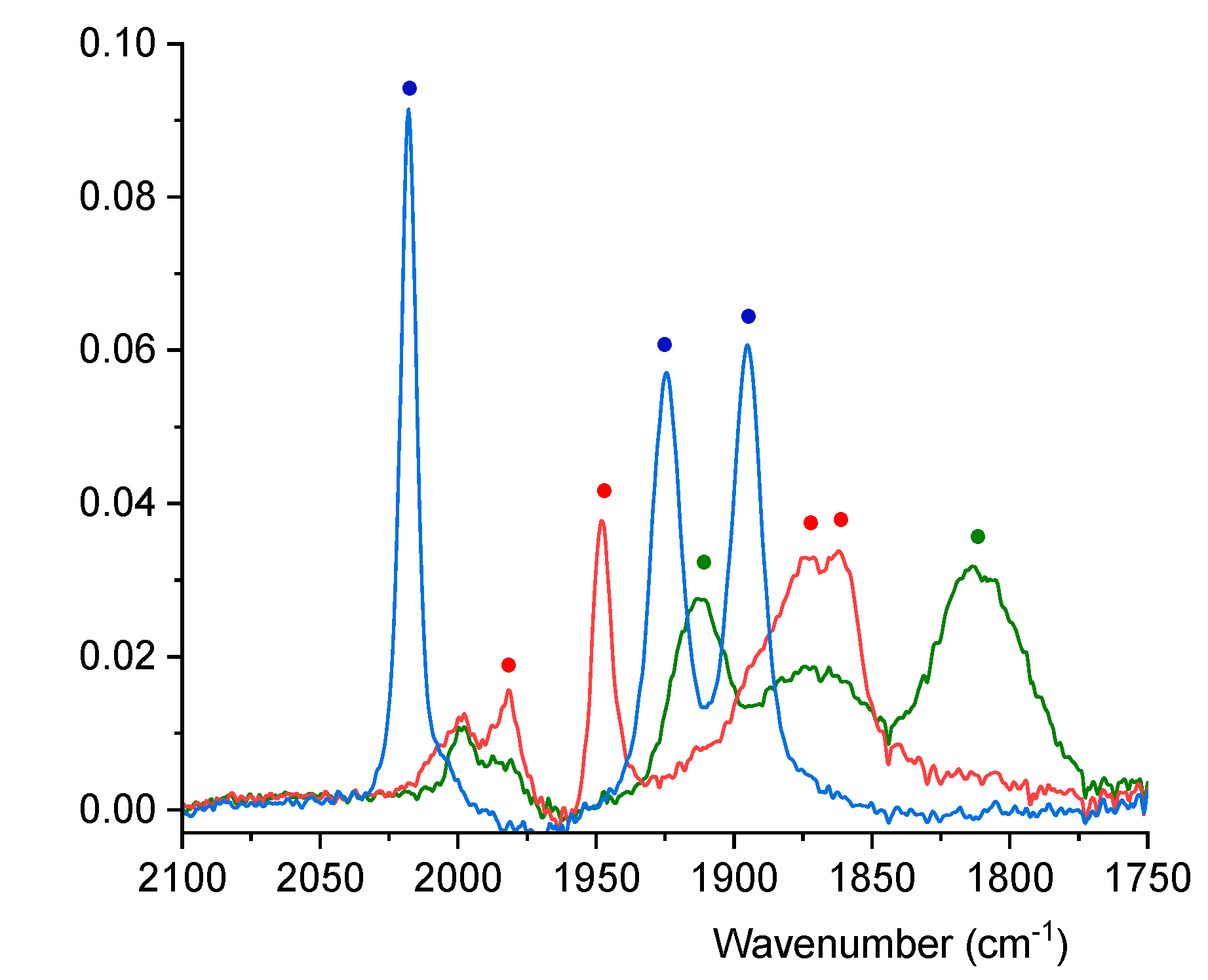
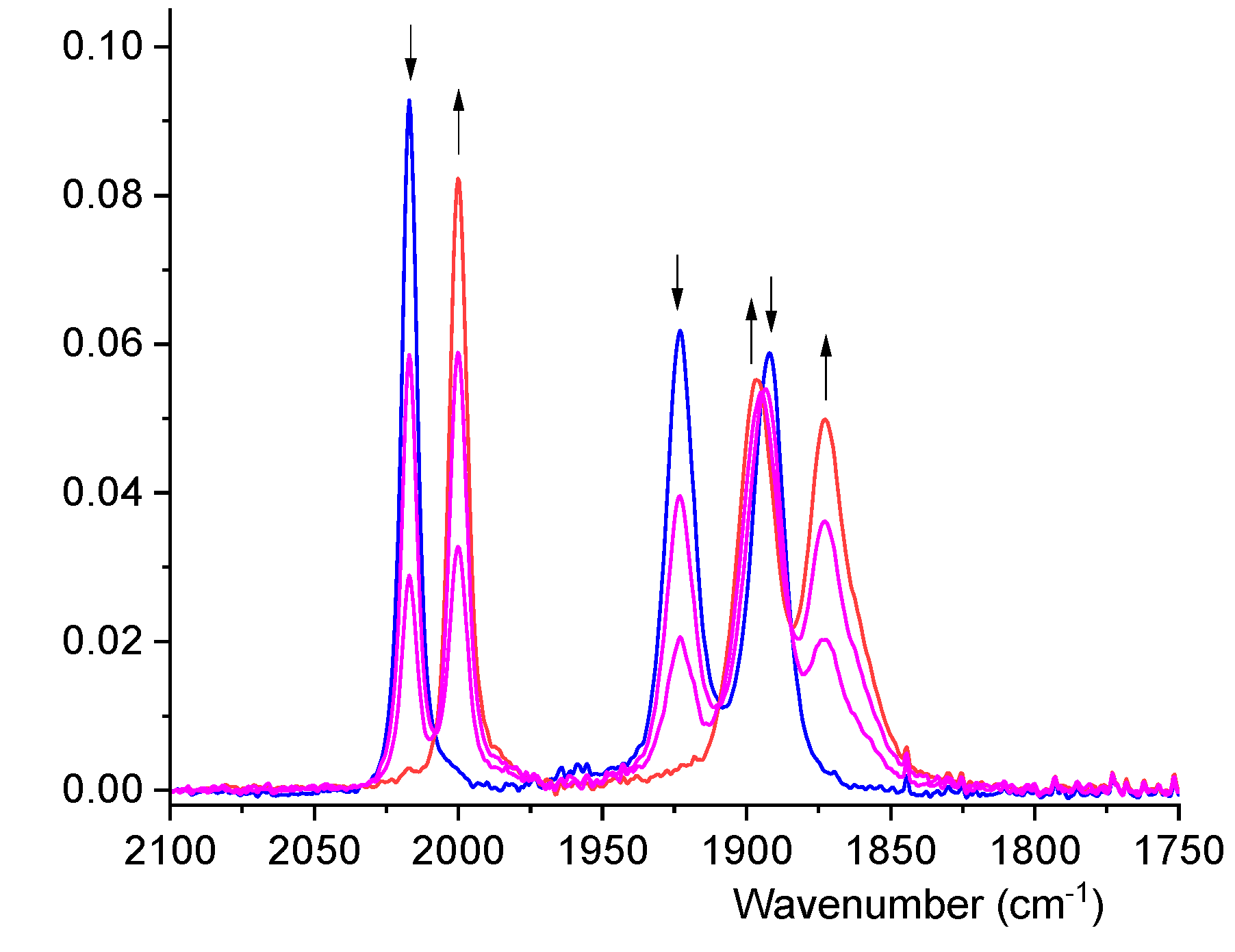
3.5. IR Spectroelectrochemistry
3.6. Photocatalytic CO2 Reduction
3.7. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Gasser, T.; Guivarch, C.; Tachiiri, K.; Jones, C.D.; Ciais, P. Negative Emissions Physically Needed to Keep Global Warming below 2 °C. Nat. Commun. 2015, 6, 7958. [Google Scholar] [CrossRef] [PubMed]
- Ochedi, F.O.; Liu, D.; Yu, J.; Hussain, A.; Liu, Y. Photocatalytic, Electrocatalytic and Photoelectrocatalytic Conversion of Carbon Dioxide: A Review. Environ. Chem. Lett. 2021, 19, 941–967. [Google Scholar] [CrossRef]
- Majumdar, A.; Deutch, J. Research Opportunities for CO2 Utilization and Negative Emissions at the Gigatonne Scale. Joule 2018, 2, 805–809. [Google Scholar] [CrossRef]
- Bushuyev, O.S.; De Luna, P.; Dinh, C.T.; Tao, L.; Saur, G.; van de Lagemaat, J.; Kelley, S.O.; Sargent, E.H. What Should We Make with CO2 and How Can We Make It? Joule 2018, 2, 825–832. [Google Scholar] [CrossRef]
- Tu, W.; Zhou, Y.; Zou, Z. Photocatalytic Conversion of CO2 into Renewable Hydrocarbon Fuels: State-of-the-Art Accomplishment, Challenges, and Prospects. Adv. Mater. 2014, 26, 4607–4626. [Google Scholar] [CrossRef] [PubMed]
- Robert, M. Running the Clock: CO2 Catalysis in the Age of Anthropocene. ACS Energy Lett. 2016, 1, 281–282. [Google Scholar] [CrossRef]
- Francke, R.; Schille, B.; Roemelt, M. Homogeneously Catalyzed Electroreduction of Carbon Dioxide—Methods, Mechanisms, and Catalysts. Chem. Rev. 2018, 118, 4631–4701. [Google Scholar] [CrossRef]
- Chang, X.; Wang, T.; Gong, J. CO2 Photo-Reduction: Insights into CO2 Activation and Reaction on Surfaces of Photocatalysts. Energy Environ. Sci. 2016, 9, 2177–2196. [Google Scholar] [CrossRef]
- Zada, A.; Ali, N.; Subhan, F.; Anwar, N.; Ali Shah, M.I.; Ateeq, M.; Hussain, Z.; Zaman, K.; Khan, M. Suitable Energy Platform Significantly Improves Charge Separation of G-C3N4 for CO2 Reduction and Pollutant Oxidation under Visible-Light. Prog. Nat. Sci. Mater. Int. 2019, 29, 138–144. [Google Scholar] [CrossRef]
- Huckaba, A.J.; Sharpe, E.A.; Delcamp, J.H. Photocatalytic Reduction of CO2 with Re-Pyridyl-NHCs. Inorg. Chem. 2016, 55, 682–690. [Google Scholar] [CrossRef]
- Wang, Y.; He, D.; Chen, H.; Wang, D. Catalysts in Electro-, Photo- and Photoelectrocatalytic CO2 Reduction Reactions. J. Photochem. Photobiol. C Photochem. Rev. 2019, 40, 117–149. [Google Scholar] [CrossRef]
- Yaashikaa, P.R.; Senthil Kumar, P.; Varjani, S.J.; Saravanan, A. A Review on Photochemical, Biochemical and Electrochemical Transformation of CO2 into Value-Added Products. J. CO2 Util. 2019, 33, 131–147. [Google Scholar] [CrossRef]
- Herron, J.A.; Kim, J.; Upadhye, A.A.; Huber, G.W.; Maravelias, C.T. A General Framework for the Assessment of Solar Fuel Technologies. Energy Environ. Sci. 2014, 8, 126–157. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Ishitani, O.; Ishida, H. Reaction Mechanisms of Catalytic Photochemical CO2 Reduction Using Re(I) and Ru(II) Complexes. Coord. Chem. Rev. 2018, 373, 333–356. [Google Scholar] [CrossRef]
- Kumaravel, V.; Bartlett, J.; Pillai, S.C. Photoelectrochemical Conversion of Carbon Dioxide (CO2) into Fuels and Value-Added Products. ACS Energy Lett. 2020, 5, 486–519. [Google Scholar] [CrossRef]
- White, J.L.; Baruch, M.F.; Pander, J.E.; Hu, Y.; Fortmeyer, I.C.; Park, J.E.; Zhang, T.; Liao, K.; Gu, J.; Yan, Y.; et al. Light-Driven Heterogeneous Reduction of Carbon Dioxide: Photocatalysts and Photoelectrodes. Chem. Rev. 2015, 115, 12888–12935. [Google Scholar] [CrossRef]
- Zhao, G.; Huang, X.; Wang, X.; Wang, X. Progress in Catalyst Exploration for Heterogeneous CO2 Reduction and Utilization: A Critical Review. J. Mater. Chem. A 2017, 5, 21625–21649. [Google Scholar] [CrossRef]
- Raziq, F.; Khan, K.; Ali, S.; Ali, S.; Xu, H.; Ali, I.; Zada, A.; Muhammad Ismail, P.; Ali, A.; Khan, H.; et al. Accelerating CO2 Reduction on Novel Double Perovskite Oxide with Sulfur, Carbon Incorporation: Synergistic Electronic and Chemical Engineering. J. Chem. Eng. 2022, 446, 137161. [Google Scholar] [CrossRef]
- Luo, Y.-H.; Dong, L.-Z.; Liu, J.; Li, S.-L.; Lan, Y.-Q. From Molecular Metal Complex to Metal-Organic Framework: The CO2 Reduction Photocatalysts with Clear and Tunable Structure. Coord. Chem. Rev. 2019, 390, 86–126. [Google Scholar] [CrossRef]
- Frayne, L.; Das, N.; Paul, A.; Amirjalayer, S.; Buma, W.J.; Woutersen, S.; Long, C.; Vos, J.G.; Pryce, M.T. Photo- and Electrochemical Properties of a CO2 Reducing Ruthenium–Rhenium Quaterpyridine-Based Catalyst. ChemPhotoChem 2018, 2, 323–331. [Google Scholar] [CrossRef]
- Grills, D.C.; Ertem, M.Z.; McKinnon, M.; Ngo, K.T.; Rochford, J. Mechanistic Aspects of CO2 Reduction Catalysis with Manganese-Based Molecular Catalysts. Coord. Chem. Rev. 2018, 374, 173–217. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Efficient Photochemical Reduction of CO2 to CO by Visible Light Irradiation of Systems Containing Re(Bipy)(CO)3X or Ru(Bipy)32+–Co2+ Combinations as Homogeneous Catalysts. J. Chem. Soc. Chem. Commun. 1983, 536–538. [Google Scholar] [CrossRef]
- Frantz, S.; Fiedler, J.; Hartenbach, I.; Schleid, T.; Kaim, W. A Complete Series of Tricarbonylhalidorhenium(I) Complexes (Abpy)Re(CO)3(Hal), Hal=F, Cl, Br, I., Abpy=2,2′-Azobispyridine: Structures, Spectroelectrochemistry and EPR of Reduced Forms. J. Organomet. Chem. 2004, 689, 3031–3039. [Google Scholar] [CrossRef]
- Friaes, S.; Realista, S.; Mourao, H.; Royo, B. N-Heterocyclic and Mesoionic Carbenes of Manganese and Rhenium in Catalysis. Eur. J. Inorg. Chem. 2022, 2022, e202100884. [Google Scholar] [CrossRef]
- Liyanage, N.P.; Dulaney, H.A.; Huckaba, A.J.; Jurss, J.W.; Delcamp, J.H. Electrocatalytic Reduction of CO2 to CO With Re-Pyridyl-NHCs: Proton Source Influence on Rates and Product Selectivities. Inorg. Chem. 2016, 55, 6085–6094. [Google Scholar] [CrossRef] [PubMed]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An Overview of N-Heterocyclic Carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, M.C.; Hahn, F.E. CHAPTER 1 Introduction to N-Heterocyclic Carbenes: Synthesis and Stereoelectronic Parameters. In N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools; Royal Society of Chemistry: London, UK, 2016; pp. 1–45. [Google Scholar] [CrossRef]
- Shirley, H.; Sexton, T.M.; Liyanage, N.P.; Palmer, C.Z.; McNamara, L.E.; Hammer, N.I.; Tschumper, G.S.; Delcamp, J.H. Effect of “X” Ligands on the Photocatalytic Reduction of CO2 to CO with Re(PyridylNHC-CF3)(CO)3X Complexes. Eur. J. Inorg. Chem. 2020, 2020, 1844–1851. [Google Scholar] [CrossRef]
- Shirley, H.; Sexton, T.M.; Liyanage, N.P.; Perkins, M.A.; Autry, S.A.; McNamara, L.E.; Hammer, N.I.; Parkin, S.R.; Tschumper, G.S.; Delcamp, J.H. Probing the Effects of Electron Deficient Aryl Substituents and a π-System Extended NHC Ring on the Photocatalytic CO2 Reduction Reaction with Re-PyNHC-Aryl Complexes**. ChemPhotoChem 2021, 5, 353–361. [Google Scholar] [CrossRef]
- Maurin, A.; Ng, C.-O.; Chen, L.; Lau, T.-C.; Robert, M.; Ko, C.-C. Photochemical and Electrochemical Catalytic Reduction of CO 2 with NHC-Containing Dicarbonyl Rhenium(i) Bipyridine Complexes. Dalton Trans. 2016, 45, 14524–14529. [Google Scholar] [CrossRef]
- Feng, Y.; Ng, C.-O.; Tong, K.-M.; Cheng, S.-C.; Chan, L.-L.; Ko, C.-C. Study of Re(I) Carbene Complexes for Photocatalytic Reduction of Carbon Dioxide. Energy Fuels 2021, 35, 19170–19177. [Google Scholar] [CrossRef]
- Carpenter, C.A.; Brogdon, P.; McNamara, L.E.; Tschumper, G.S.; Hammer, N.I.; Delcamp, J.H. A Robust Pyridyl-NHC-Ligated Rhenium Photocatalyst for CO2 Reduction in the Presence of Water and Oxygen. Inorganics 2018, 6, 22. [Google Scholar] [CrossRef]
- Stanton, C.J.; Machan, C.W.; Vandezande, J.E.; Jin, T.; Majetich, G.F.; Schaefer, H.F.; Kubiak, C.P.; Li, G.; Agarwal, J. Re(I) NHC Complexes for Electrocatalytic Conversion of CO2. Inorg. Chem. 2016, 55, 3136–3144. [Google Scholar] [CrossRef]
- Suntrup, L.; Stein, F.; Klein, J.; Wilting, A.; Parlane, F.G.L.; Brown, C.M.; Fiedler, J.; Berlinguette, C.P.; Siewert, I.; Sarkar, B. Rhenium Complexes of Pyridyl-Mesoionic Carbenes: Photochemical Properties and Electrocatalytic CO2 Reduction. Inorg. Chem. 2020, 59, 4215–4227. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Berdichevsky, E.K.; Warren, J.J. Electrocatalytic CO2 Reduction Using Rhenium(I) Complexes with Modified 2-(2′-Pyridyl)Imidazole Ligands. Inorg. Chim. Acta 2017, 460, 63–68. [Google Scholar] [CrossRef]
- Vaughan, J.G.; Reid, B.L.; Ramchandani, S.; Wright, P.J.; Muzzioli, S.; Skelton, B.W.; Raiteri, P.; Brown, D.H.; Stagni, S.; Massi, M. The Photochemistry of Rhenium(I) Tricarbonyl N-Heterocyclic Carbene Complexes. Dalton Trans. 2013, 42, 14100–14114. [Google Scholar] [CrossRef]
- Casson, L.A.; Muzzioli, S.; Raiteri, P.; Skelton, B.W.; Stagni, S.; Massi, M.; Brown, D.H. N-Heterocyclic Carbenes as Π*-Acceptors in Luminescent Re(I) Triscarbonyl Complexes. Dalton Trans. 2011, 40, 11960–11967. [Google Scholar] [CrossRef]
- Li, X.-W.; Li, H.-Y.; Wang, G.-F.; Chen, F.; Li, Y.-Z.; Chen, X.-T.; Zheng, Y.-X.; Xue, Z.-L. Blue-Green Luminescent Rhenium(I) Tricarbonyl Complexes with Pyridine-Functionalized N-Heterocyclic Carbene Ligands. Organometallics 2012, 31, 3829–3835. [Google Scholar] [CrossRef]
- Luengo, A.; Fernández-Moreira, V.; Marzo, I.; Gimeno, M.C. Bioactive Heterobimetallic Re(I)/Au(I) Complexes Containing Bidentate N-Heterocyclic Carbenes. Organometallics 2018, 37, 3993–4001. [Google Scholar] [CrossRef]
- Nicholls, T.P.; Burt, L.K.; Simpson, P.V.; Massi, M.; Bissember, A.C. Tricarbonyl Rhenium(I) Tetrazolato and N-Heterocyclic Carbene Complexes: Versatile Visible-Light-Mediated Photoredox Catalysts. Dalton Trans. 2019, 48, 12749–12754. [Google Scholar] [CrossRef]
- Siegmund, D.; Lorenz, N.; Gothe, Y.; Spies, C.; Geissler, B.; Prochnow, P.; Nuernberger, P.; Bandow, J.E.; Metzler-Nolte, N. Benzannulated Re(I)–NHC Complexes: Synthesis, Photophysical Properties and Antimicrobial Activity. Dalton Trans. 2017, 46, 15269–15279. [Google Scholar] [CrossRef]
- Xue, W.-M.; Chan, M.C.-W.; Su, Z.-M.; Cheung, K.-K.; Liu, S.-T.; Che, C.-M. Spectroscopic and Excited-State Properties of Luminescent Rhenium(I) N-Heterocyclic Carbene Complexes Containing Aromatic Diimine Ligands. Organometallics 1998, 17, 1622–1630. [Google Scholar] [CrossRef]
- Sacksteder, L.; Zipp, A.P.; Brown, E.A.; Streich, J.; Demas, J.N.; DeGraff, B.A. Luminescence Studies of Pyridine.Alpha.-Diimine Rhenium(I) Tricarbonyl Complexes. Inorg. Chem. 1990, 29, 4335–4340. [Google Scholar] [CrossRef]
- Zipp, A.P.; Sacksteder, L.; Streich, J.; Cook, A.; Demas, J.N.; DeGraff, B.A. Luminescence of Rhenium(I) Complexes with Highly Sterically Hindered.Alpha.-Diimine Ligands. Inorg. Chem. 1993, 32, 5629–5632. [Google Scholar] [CrossRef]
- Koike, K.; Okoshi, N.; Hori, H.; Takeuchi, K.; Ishitani, O.; Tsubaki, H.; Clark, I.P.; George, M.W.; Johnson, F.P.A.; Turner, J.J. Mechanism of the Photochemical Ligand Substitution Reactions of fac-[Re(Bpy)(CO)3(PR3)]+ Complexes and the Properties of Their Triplet Ligand-Field Excited States. J. Am. Chem. Soc. 2002, 124, 11448–11455. [Google Scholar] [CrossRef] [PubMed]
- Worl, L.A.; Duesing, R.; Chen, P.; Ciana, L.D.; Meyer, T.J. Photophysical Properties of Polypyridyl Carbonyl Complexes of Rhenium(I). J. Chem. Soc. Dalton Trans. 1991, 849–858. [Google Scholar] [CrossRef]
- Johnson, F.P.A.; George, M.W.; Hartl, F.; Turner, J.J. Electrocatalytic Reduction of CO2 Using the Complexes [Re(Bpy)(CO)3L]n (n = +1, L = P(OEt)3, CH3CN; n = 0, L = Cl−, Otf−; Bpy = 2,2‘-Bipyridine; Otf− = CF3SO3) as Catalyst Precursors: Infrared Spectroelectrochemical Investigation. Organometallics 1996, 15, 3374–3387. [Google Scholar] [CrossRef]
- Machan, C.W.; Chabolla, S.A.; Kubiak, C.P. Reductive Disproportionation of Carbon Dioxide by an Alkyl-Functionalized Pyridine Monoimine Re(I) Fac-Tricarbonyl Electrocatalyst. Organometallics 2015, 34, 4678–4683. [Google Scholar] [CrossRef]
- Abramov, P.A.; Dmitriev, A.A.; Kholin, K.V.; Gritsan, N.P.; Kadirov, M.K.; Gushchin, A.L.; Sokolov, M.N. Mechanistic Study of the [(Dpp-Bian)Re(CO)3Br] Electrochemical Reduction Using in Situ EPR Spectroscopy and Computational Chemistry. Electrochim. Acta 2018, 270, 526–534. [Google Scholar] [CrossRef]
- Bonfiglio, A.; Magra, K.; Cebrián, C.; Polo, F.; Gros, P.C.; Mercandelli, P.; Mauro, M. Red-Emitting Neutral Rhenium(I) Complexes Bearing a Pyridyl Pyridoannelated N-Heterocyclic Carbene. Dalton Trans. 2020, 49, 3102–3111. [Google Scholar] [CrossRef]
- Suntrup, L.; Klenk, S.; Klein, J.; Sobottka, S.; Sarkar, B. Gauging Donor/Acceptor Properties and Redox Stability of Chelating Click-Derived Triazoles and Triazolylidenes: A Case Study with Rhenium(I) Complexes. Inorg. Chem. 2017, 56, 5771–5783. [Google Scholar] [CrossRef]
- Zhao, Y.; Xue, D.; Qi, H.; Zhang, C. Twisted Configuration Pyrene Derivative: Exhibiting Pure Blue Monomer Photoluminescence and Electrogenerated Chemiluminescence Emissions in Non-Aqueous Media. RSC Adv. 2017, 7, 22882–22891. [Google Scholar] [CrossRef]
- Heinze, J. Cathodic Reactions of Hydrocarbons, Olefins and Aromatic Compounds. In Encyclopedia of Electrochemistry, Vol. 8, Organic Electrochemistry; Bard, A.J., Straatmann, M., Schäfer, H.J., Eds.; Wiley VCH: Weinheim, Germany, 2004; Chapter 4; pp. 93–124. [Google Scholar]
- Taylor, J.O.; Neri, G.; Banerji, L.; Cowan, A.J.; Hartl, F. Strong Impact of Intramolecular Hydrogen Bonding on the Cathodic Path of [Re(3,3′-Dihydroxy-2,2′-Bipyridine)(CO)3Cl] and Catalytic Reduction of Carbon Dioxide. Inorg. Chem. 2020, 59, 5564–5578. [Google Scholar] [CrossRef] [PubMed]
- Asbai, Z.; Bonfiglio, A.; Mercandelli, P.; Polo, F.; Mauro, M. Cationic Rhenium(I) Complexes Bearing a π-Accepting Pyridoannulated N-Heterocyclic Carbene Ligand: Synthesis, Photophysical, Electrochemical and Theoretical Investigation. Polyhedron 2021, 197, 115025. [Google Scholar] [CrossRef]
- Paolucci, F.; Marcaccio, M.; Paradisi, C.; Roffia, S.; Bignozzi, C.A.; Amatore, C. Dynamics of the Electrochemical Behavior of Diimine Tricarbonyl Rhenium(I) Complexes in Strictly Aprotic Media. J. Phys. Chem. B 1998, 102, 4759–4769. [Google Scholar] [CrossRef]
- Cerpentier, F.J.R.; Karlsson, J.; Lalrempuia, R.; Brandon, M.P.; Sazanovich, I.V.; Greetham, G.M.; Gibson, E.A.; Pryce, M.T. Ruthenium Assemblies for CO2 Reduction and H2 Generation: Time Resolved Infrared Spectroscopy, Spectroelectrochemistry and a Photocatalysis Study in Solution and on NiO. Front. Chem. 2021, 9, 795877. [Google Scholar] [CrossRef]
- O’Neill, J.S.; Kearney, L.; Brandon, M.P.; Pryce, M.T. Design Components of Porphyrin-Based Photocatalytic Hydrogen Evolution Systems: A Review. Coord. Chem. Rev. 2022, 467, 214599. [Google Scholar] [CrossRef]
- Nganga, J.K.; Samanamu, C.R.; Tanski, J.M.; Pacheco, C.; Saucedo, C.; Batista, V.S.; Grice, K.A.; Ertem, M.Z.; Angeles-Boza, A.M. Electrochemical Reduction of CO2 Catalyzed by Re(Pyridine-Oxazoline)(CO)3Cl Complexes. Inorg. Chem. 2017, 56, 3214–3226. [Google Scholar] [CrossRef]
- Stratakes, B.M.; Dempsey, J.L.; Miller, A.J.M. Determining the Overpotential of Electrochemical Fuel Synthesis Mediated by Molecular Catalysts: Recommended Practices, Standard Reduction Potentials, and Challenges. ChemElectroChem 2021, 8, 4161–4180. [Google Scholar] [CrossRef]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90–94. [Google Scholar] [CrossRef]
- Jo, M.; Choi, S.; Jo, J.H.; Kim, S.-Y.; Kim, P.S.; Kim, C.H.; Son, H.-J.; Pac, C.; Kang, S.O. Utility of Squaraine Dyes for Dye-Sensitized Photocatalysis on Water or Carbon Dioxide Reduction. ACS Omega 2019, 4, 14272–14283. [Google Scholar] [CrossRef]
- Cardona, C.M.; Li, W.; Kaifer, A.E.; Stockdale, D.; Bazan, G.C. Electrochemical Considerations for Determining Absolute Frontier Orbital Energy Levels of Conjugated Polymers for Solar Cell Applications. Adv. Mater. 2011, 23, 2367–2371. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Koike, K.; Inoue, H.; Ishitani, O. Development of an Efficient Photocatalytic System for CO2 Reduction Using Rhenium(I) Complexes Based on Mechanistic Studies. J. Am. Chem. Soc. 2008, 130, 2023–2031. [Google Scholar] [CrossRef] [PubMed]
- Rigaku, O.D. CrysAlisPRO; Oxford Difraction, Rigaku Corporation: Oxford, UK, 2019. [Google Scholar]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A Computer Program for the Solution of Crystal Structures by Charge Flipping in Arbitrary Dimensions. J. Appl. Cryst. 2007, 40, 786–790. [Google Scholar] [CrossRef]
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS Version 12: Software for Guided Crystal Structure Analysis. J. Appl. Cryst. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Krejčik, M.; Daněk, M.; Hartl, F. Simple Construction of an Infrared Optically Transparent Thin-Layer Electrochemical Cell: Applications to the Redox Reactions of Ferrocene, Mn2(CO)10 and Mn(CO)3(3,5-Di-t-Butyl-Catecholate)−. J. Electroanal. Chem. Interfacial Electrochem. 1991, 317, 179–187. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phy. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-Adjustedab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theoret. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Sundermann, A. Correlation Consistent Valence Basis Sets for Use with the Stuttgart–Dresden–Bonn Relativistic Effective Core Potentials: The Atoms Ga–Kr and In–Xe. J. Chem. Phys. 2001, 114, 3408–3420. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | λabs (nm) a,b | λem (nm) b 298 K | λem (nm) c 77 K | τ1 (ns) d | τ2 (ns) d | TON f |
|---|---|---|---|---|---|---|
| Re−NHC−1 | 250, 358, ca. 400 sh | 490 | 472, 503 | 5.6 (5.02%) e | 83.7 (94.98%) e | 11 g |
| Re−NHC−2 | 252, 358, ca. 400 sh | 496 | 477, 508 | 11.6 (8.60%) e | 940.9 (91.40%) e | 10 g |
| Re−NHC−3 | 244, 295, 328 sh, 348, 384 sh, 400 sh | 527 | 580, 642 | 6.3 (20.61%) e | 25.8 (79.39%) e | 26 h |
| Complex | R1 (V) | R1′ (V) | R2 (V) | R3 (V) | O1 (V) | E(MLCT) (eV) | E*(A/A−) (V) |
|---|---|---|---|---|---|---|---|
| Re−NHC−1 | −2.03 (Ep,c) −1.89 (Eo) | −2.17 (Ep,c) | −2.33 (Ep,c) | 0.80 (Ep,a) 0.73 (Eo) | 2.74 | 0.85 | |
| Re−NHC−2 | −2.01 (Ep,c) −1.89 (Eo) | −2.27 (E1/2) | 0.87 (Ep,a) 0.80 (Eo) | 2.72 | 0.83 | ||
| Re−NHC−3 | −2.00 (Ep,c) −1.88 (Eo) | −2.19 (E1/2) | −2.49 (E1/2) | 0.87 (Ep,a) 0.79 (Eo) | 2.63 | 0.75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kearney, L.; Brandon, M.P.; Coleman, A.; Chippindale, A.M.; Hartl, F.; Lalrempuia, R.; Pižl, M.; Pryce, M.T. Ligand−Structure Effects on N−Heterocyclic Carbene Rhenium Photo− and Electrocatalysts of CO2 Reduction. Molecules 2023, 28, 4149. https://doi.org/10.3390/molecules28104149
Kearney L, Brandon MP, Coleman A, Chippindale AM, Hartl F, Lalrempuia R, Pižl M, Pryce MT. Ligand−Structure Effects on N−Heterocyclic Carbene Rhenium Photo− and Electrocatalysts of CO2 Reduction. Molecules. 2023; 28(10):4149. https://doi.org/10.3390/molecules28104149
Chicago/Turabian StyleKearney, Lauren, Michael P. Brandon, Andrew Coleman, Ann M. Chippindale, František Hartl, Ralte Lalrempuia, Martin Pižl, and Mary T. Pryce. 2023. "Ligand−Structure Effects on N−Heterocyclic Carbene Rhenium Photo− and Electrocatalysts of CO2 Reduction" Molecules 28, no. 10: 4149. https://doi.org/10.3390/molecules28104149