
Solid-Phase Synthesis of the Bicyclic Peptide OL-CTOP Containing Two Disulfide Bridges, and an Assessment of Its In Vivo μ-Opioid Receptor Antagonism after Nasal Administration
 and
and
Abstract
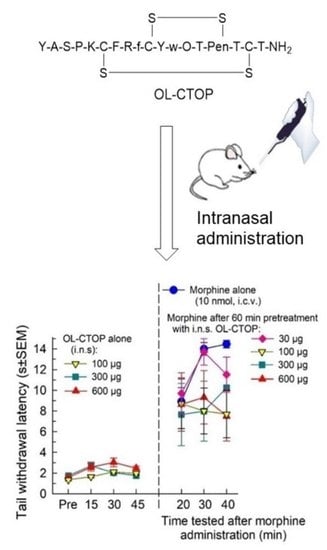
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
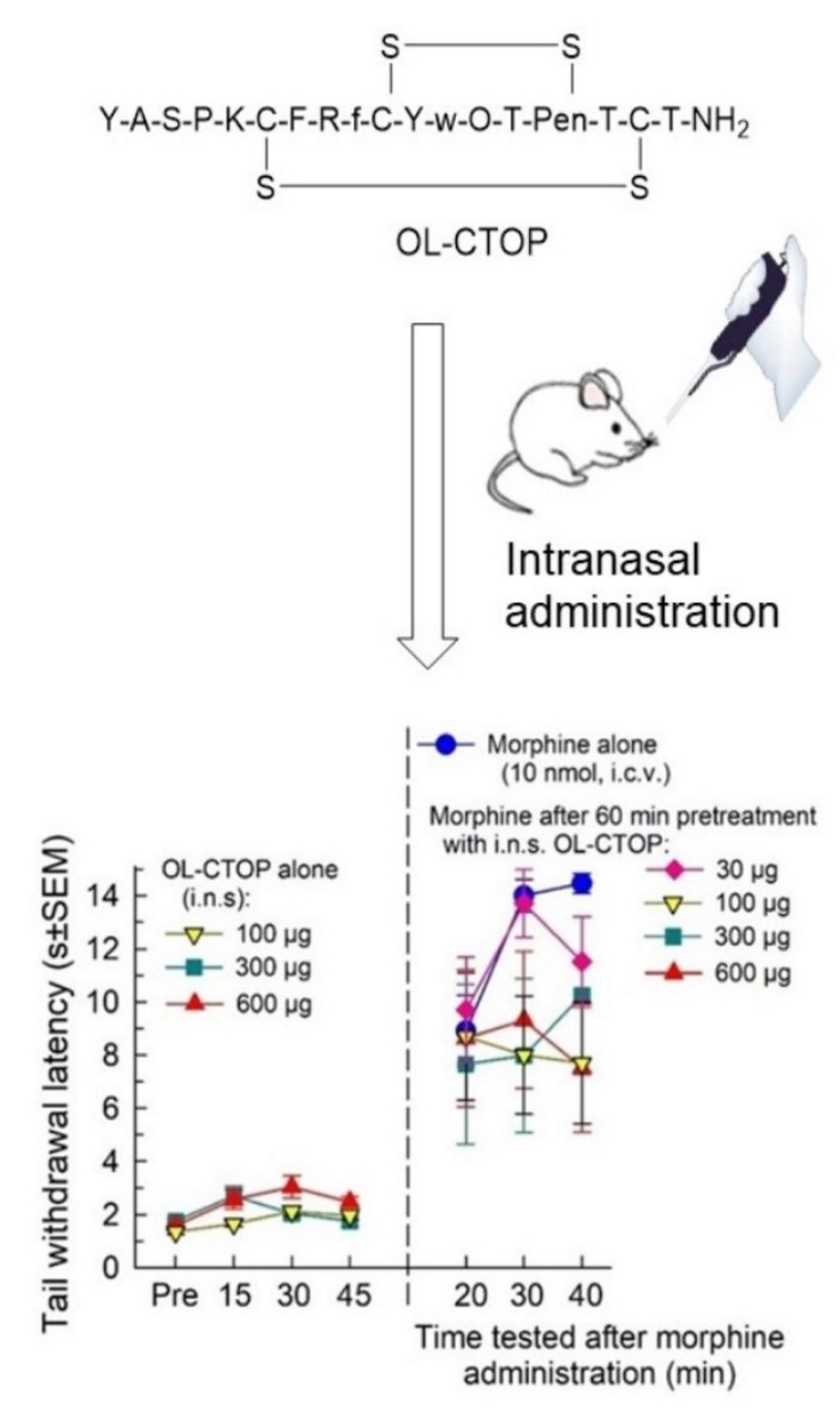
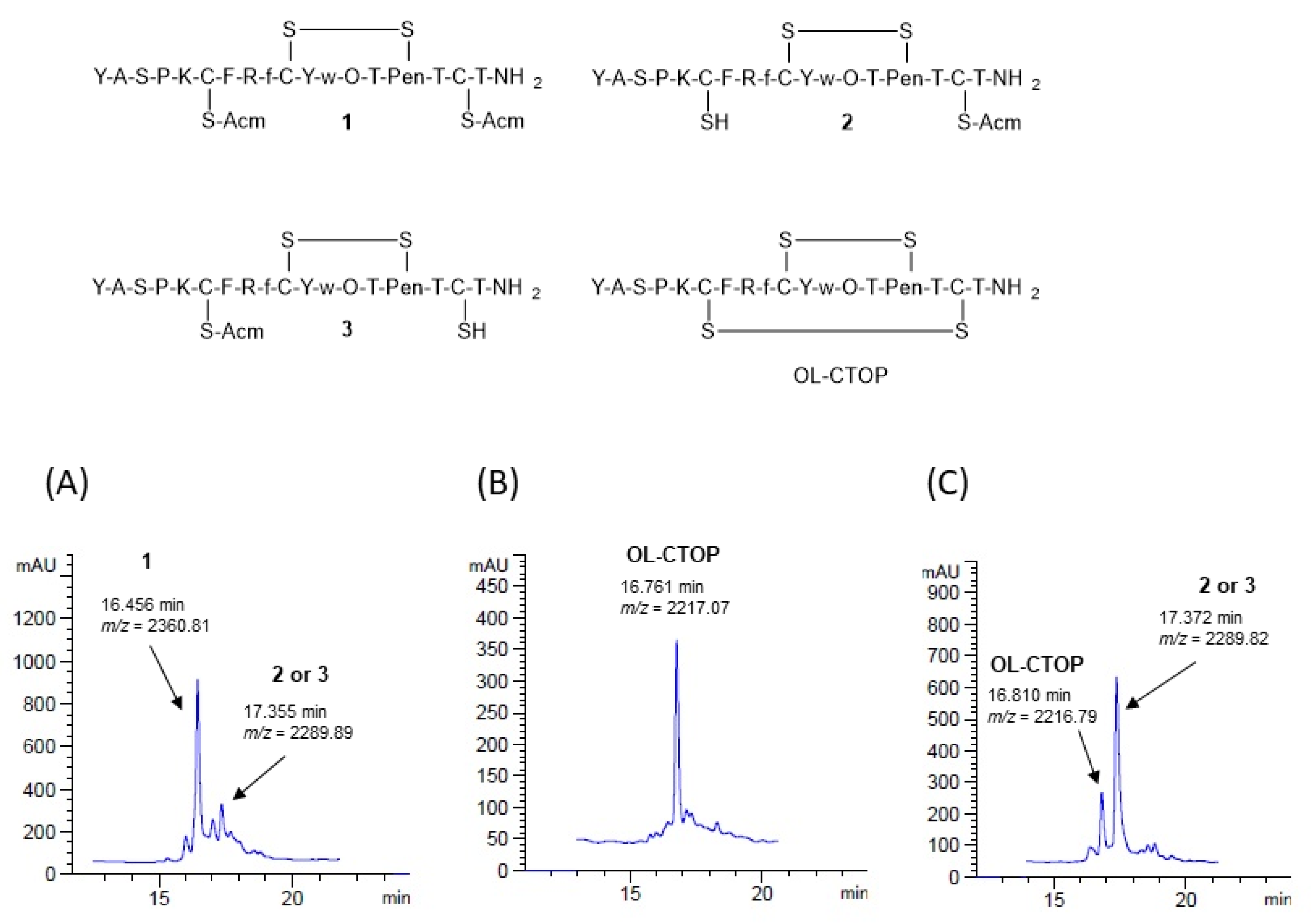
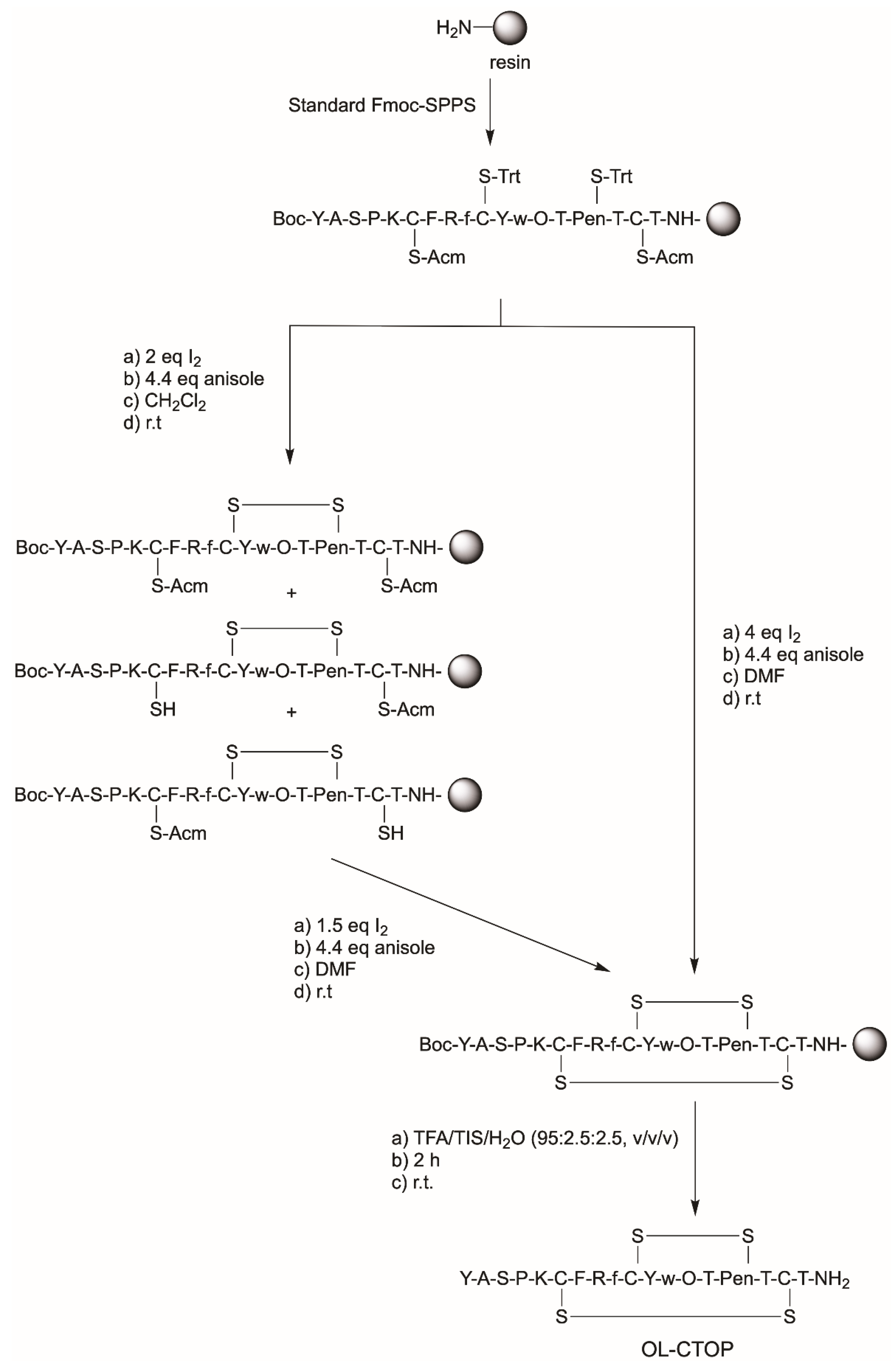
2.1. Synthesis
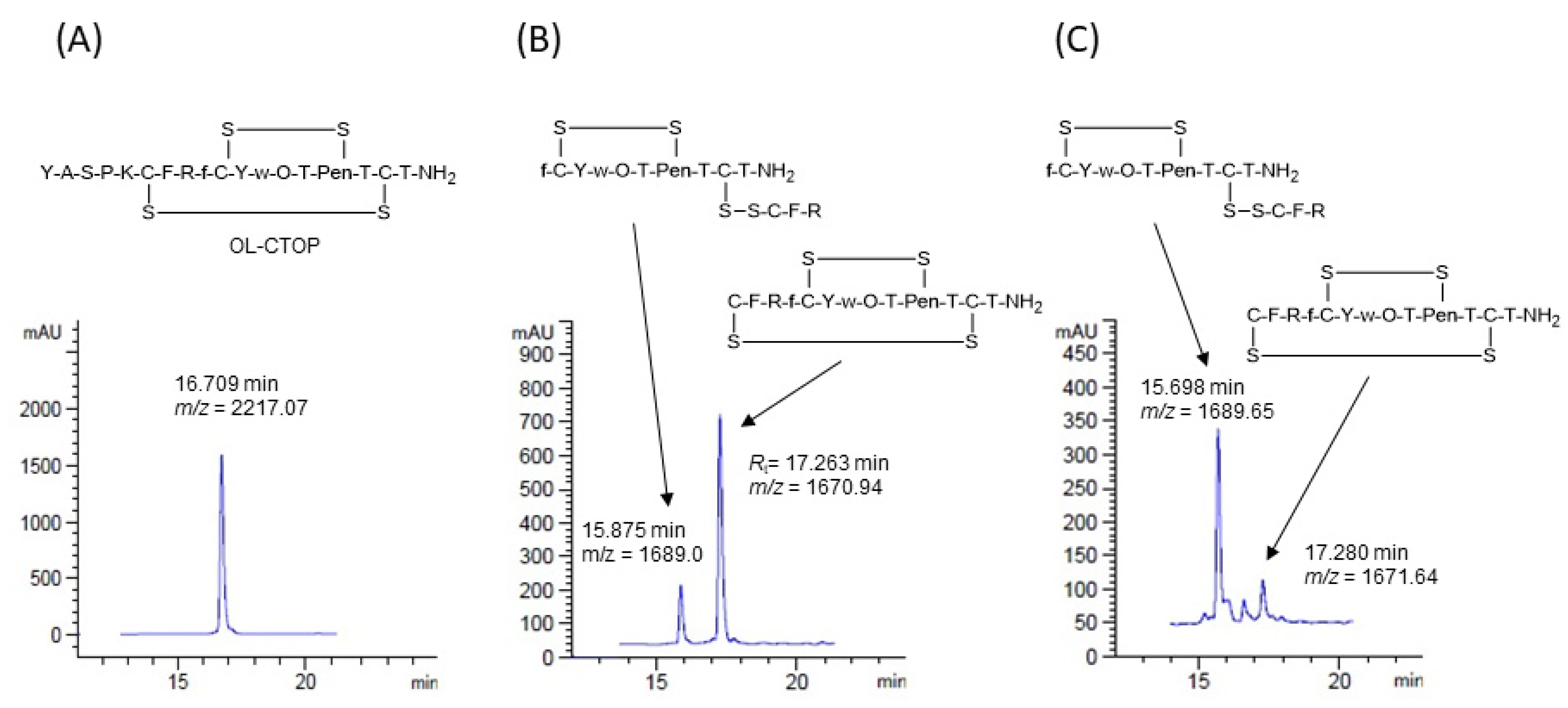
2.2. Proteolytic Stability
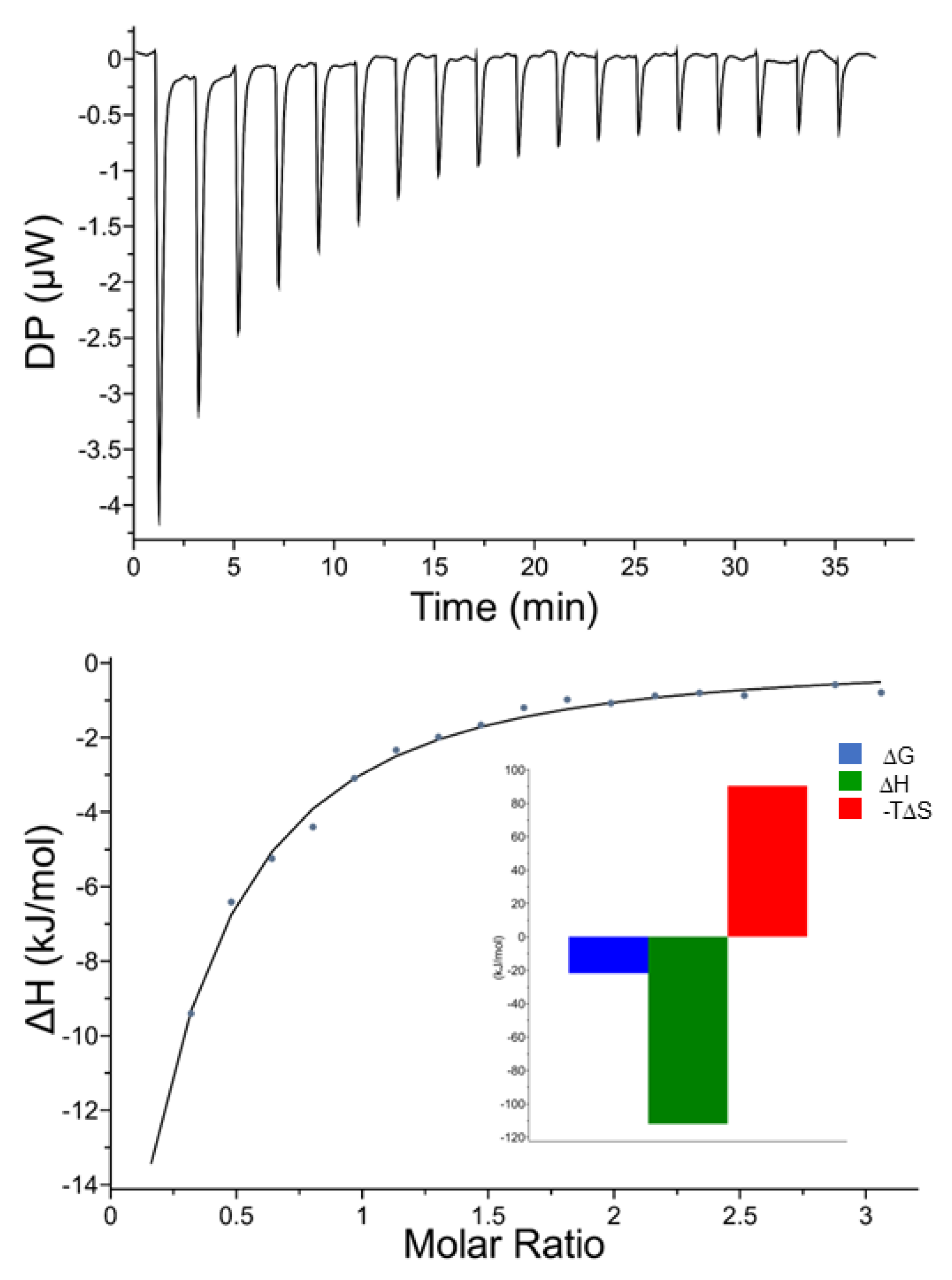
2.3. Carbohydrate-Binding Study Using Isothermal Titration Calorimetry (ITC)
2.4. In Vivo Pharmacological Evaluation
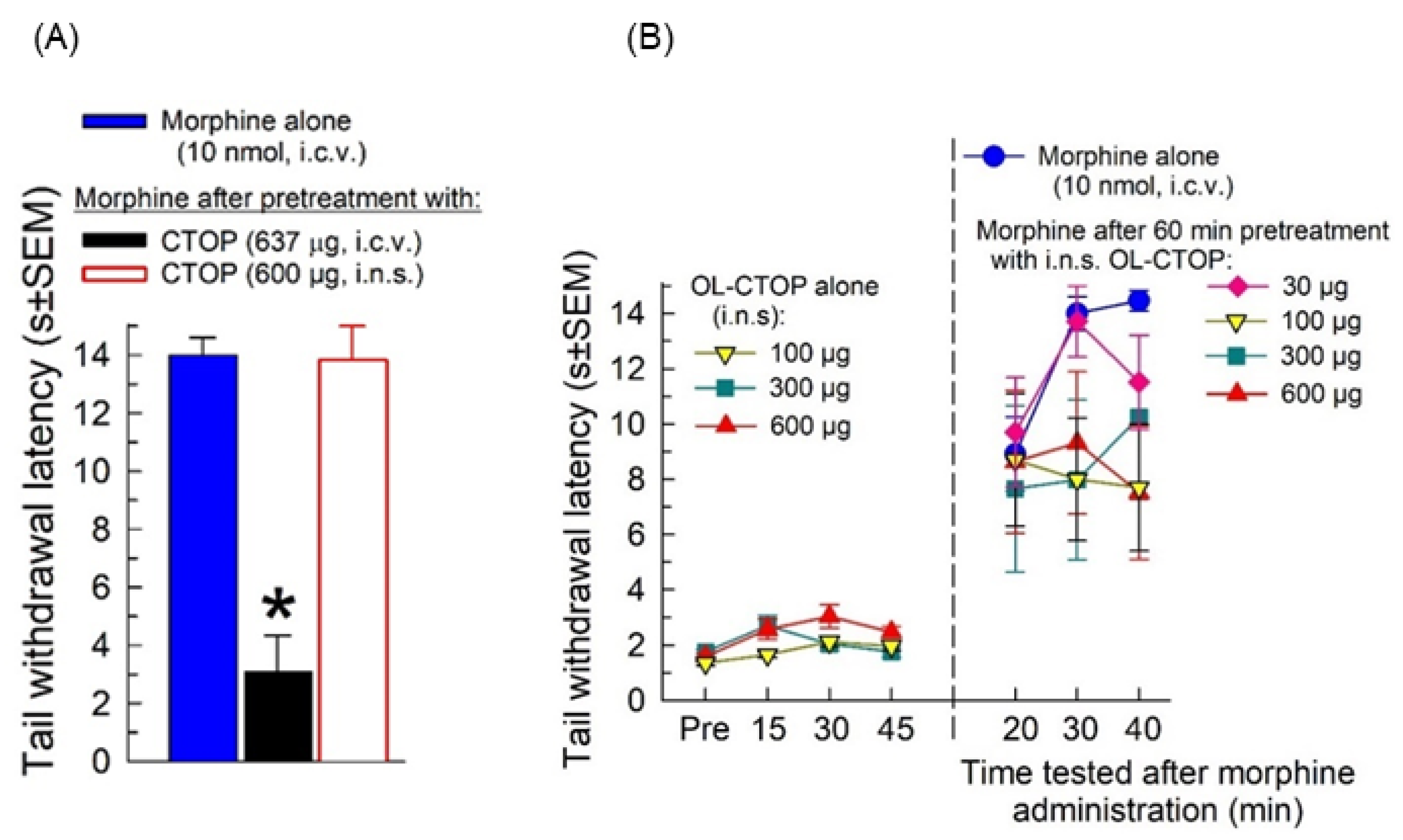
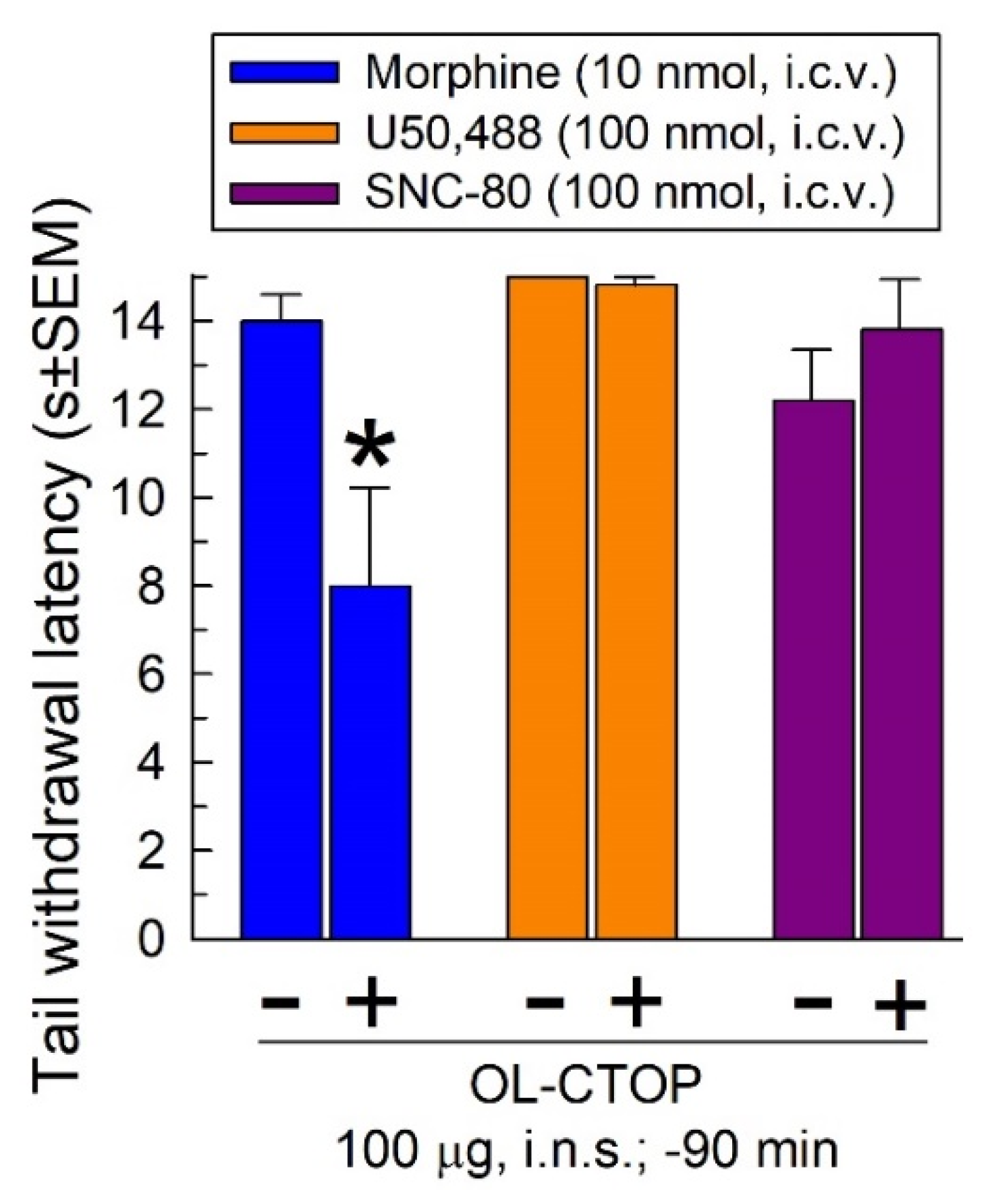
2.4.1. Opioid Receptor Selectivity of OL-CTOP Antagonist Activity
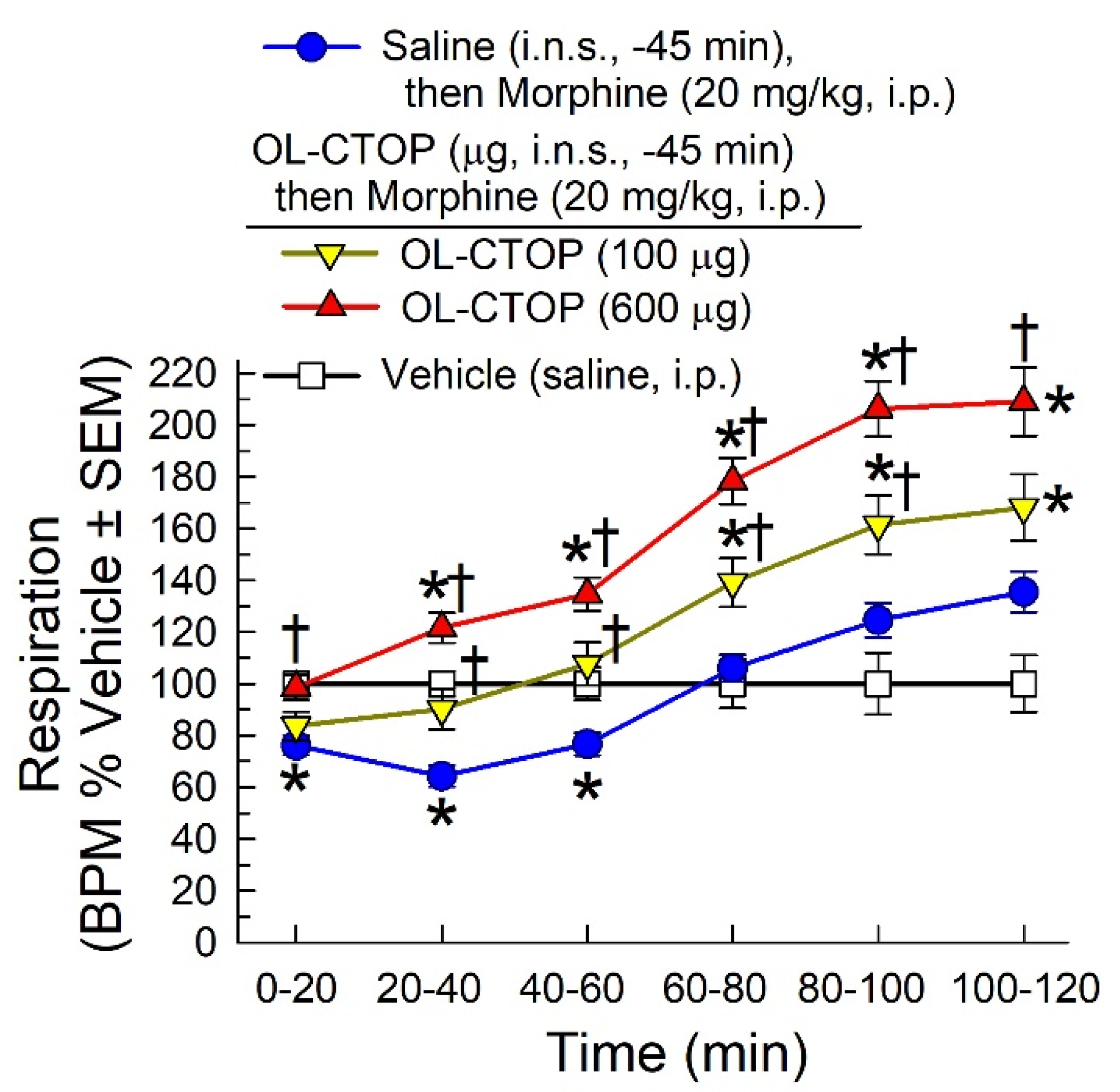
2.4.2. Evaluation of Intranasal OL-CTOP Protection from Morphine-Induced Respiratory Depression
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Peptide Synthesis
4.3. Peptide Stability
4.4. Isothermal Titration Calorimetry (ITC) Experiments
4.5. Behavioral (In Vivo) Pharmacology
4.5.1. Animals
4.5.2. Compound Preparation and Administration
4.5.3. Mouse 55 °C Warm-Water Tail Withdrawal Test
4.5.4. Assessment of Breathing Rate in Mice
4.6. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Mattson, C.L.; Tanz, L.J.; Quinn, K.; Kariisa, M.; Patel, P.; Davis, N.L. Trends and Geographic Patterns in Drug and Synthetic Opioid Overdose Deaths—United States, 2013–2019. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Gomes, T.; Tadrous, M.; Mamdani, M.M.; Paterson, J.M.; Juurlink, D.N. The Burden of Opioid-Related Mortality in the United States. JAMA Netw. Open 2018, 1, e180217. [Google Scholar] [CrossRef]
- Wide-Ranging Online Data for Epidemiologic Research (WONDER). Atlanta, GA: CDC, National Center for Health Statistics. 2021. Available online: http://wonder.cdc.gov (accessed on 12 November 2022).
- Moss, R.B.; Carlo, D.J. Higher doses of naloxone are needed in the synthetic opioid era. Subst. Abus. Treat. Prev. Policy 2019, 14, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, P.J.; Remski, K. Buprenorphine vs methadone treatment: A review of evidence in both developed and developing worlds. J. Neurosci. Rural. Pract. 2012, 03, 45–50. [Google Scholar] [CrossRef]
- Britch, S.C.; Walsh, S.L. Treatment of opioid overdose: Current approaches and recent advances. Psychopharmacology 2022, 239, 2063–2081. [Google Scholar] [CrossRef]
- Hawkins, K.N.; Knapp, R.J.; Lui, G.K.; Gulya, K.; Kazmierski, W.; Wan, Y.P.; Pelton, J.T.; Hruby, V.J.; Yamamura, H. [3H]-[H-D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2] ([3H]CTOP), a potent and highly selective peptide for mu opioid receptors in rat brain. Experiment 1989, 248, 73–80. [Google Scholar]
- Gulya, K.; Kriván, M.; Nyolczas, N.; Sarnyai, Z.; Kovács, G.L. Central effects of the potent and highly selective μ opioid antagonist (CTOP) in mice. Eur. J. Pharmacol. 1988, 150, 355–360. [Google Scholar] [CrossRef]
- Gulya, K.; Lui, G.K.; Pelton, J.T.; Kazmierski, W.; Hruby, V.J.; Yamamura, H. H-D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2: A potent and selective antagonist opioid receptors. NIDA Res. Monogr. 1986, 75, 355–360. [Google Scholar]
- Alam, M.I.; Beg, S.; Samad, A.; Baboota, S.; Kohli, K.; Ali, J.; Ahuja, A.; Akbar, M. Strategy for effective brain drug delivery. Eur. J. Pharm. Sci. 2010, 40, 385–403. [Google Scholar] [CrossRef]
- Hanson, L.R.; Frey, W.H. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008, 9, S5. [Google Scholar] [CrossRef] [Green Version]
- Illum, L. Transport of drugs from the nasal cavity to the central nervous system. Eur. J. Pharm. Sci. 2000, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Mens, W.B.; Witter, A.; Greidanus, T.B.V.W. Penetration of neurohypophyseal hormones from plasma into cerebrospinal fluid (CSF): Half-times of disappearance of these neuropeptides from CSF. Brain Res. 1983, 262, 143–149. [Google Scholar] [CrossRef]
- Robinson, I. Neurohypophysial Peptides in Cerebrospinal Fluid. Prog. Brain Res. 1983, 60, 129–145. [Google Scholar] [CrossRef]
- Burbach, J.; Loeber, J.; Verhoef, J.; De Kloet, E.; Van Ree, J.; De Wied, D. Schizophrenia and degradation of endorphins in cerebrospinal fluid. Lancet 1979, 314, 480–481. [Google Scholar] [CrossRef] [PubMed]
- Burbach, P.H.; De Hoop, M.J.; Schmale, H.; Richter, D.; De Kloet, E.R.; Haaf, J.A.T.; De Wied, D. Differential Responses to Osmotic Stress of Vasopressin-Neurophysin mRNA in Hypothalamic Nuclei. Neuroendocrinology 1984, 39, 582–584. [Google Scholar] [CrossRef] [PubMed]
- Houghten, R.; Swann, R.W.; Li, C.H. beta-Endorphin: Stability, clearance behavior, and entry into the central nervous system after intravenous injection of the tritiated peptide in rats and rabbits. Proc. Natl. Acad. Sci. USA 1980, 77, 4588–4591. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wu, H.; Hong, J.; Xu, X.; Yang, H.; Wu, B.; Wang, Y.; Zhu, J.; Lai, R.; Jiang, X.; et al. Odorranalectin Is a Small Peptide Lectin with Potential for Drug Delivery and Targeting. PLoS ONE 2008, 3, e2381. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, M.C.; Yongye, A.B.; Cudic, M.; Mayorga, K.M.; Liu, E.; Mueller, B.M.; Ainsley, J.; Karabencheva-Christova, T.; Christov, C.Z.; Cudic, M.; et al. Targeting cancer-specific glycans by cyclic peptide lectinomimics. Amino Acids 2017, 49, 1867–1883. [Google Scholar] [CrossRef]
- Singh, Y.; Cudic, P.; Cudic, M. Exploring Glycan Binding Specificity of Odorranalectin by Alanine Scanning Library. Eur. J. Org. Chem. 2022, 28, e202200302. [Google Scholar] [CrossRef]
- Plendl, J.; Sinowatz, F. Glycobiology of the Olfactory System. Cells Tissues Organs 1998, 161, 234–253. [Google Scholar] [CrossRef] [Green Version]
- Baldo, B.A.; Rose, M.A. Mechanisms of opioid-induced respiratory depression. Arch. Toxicol. 2022, 96, 2247–2260. [Google Scholar] [CrossRef]
- Pathan, H.; Williams, J. Basic opioid pharmacology: An update. Br. J. Pain 2012, 6, 11–16. [Google Scholar] [CrossRef]
- Pasternak, G.W.; Pan, Y.-X. Mu Opioids and Their Receptors: Evolution of a Concept. Pharmacol. Rev. 2013, 65, 1257–1317. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.M.; Christie, M.J. Analysis of opioid efficacy, tolerance, addiction and dependence from cell culture to human. Br. J. Pharmacol. 2011, 164, 1322–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Hasani, R.; Bruchas, M.R. Molecular Mechanisms of Opioid Receptor-dependent Signaling and Behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [Green Version]
- Brice-Tutt, A.C.; Wilson, L.L.; Eans, S.O.; Stacy, H.M.; Simons, C.A.; Simpson, G.; Coleman, J.S.; Ferracane, M.J.; Aldrich, J.V.; McLaughlin, J.P. Multifunctional opioid receptor agonism and antagonism by a novel macrocyclic tetrapeptide prevents reinstatement of morphine-seeking behaviour. Br. J. Pharmacol. 2020, 177, 4209–4222. [Google Scholar] [CrossRef] [PubMed]
- Engebretsen, M.; Agner, E.; Sandosham, J.; Fischer, P.M. Unexpected lability of cysteine acetamidomethyl thiol protecting group. Tyrosine ring alkylation and disulfide bond formation upon acidolysis. J. Pept. Res. 2009, 49, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Gabius, H.-J.; Sabesan, S.; Oscarson, S.; Brewer, C.F. Thermodynamic binding studies of bivalent oligosaccharides to galectin-1, galectin-3, and the carbohydrate recognition domain of galectin-3. Glycobiology 2004, 14, 817–825. [Google Scholar] [CrossRef] [Green Version]
- Dam, T.K.; Brewer, C.F. Thermodynamic Studies of Lectin−Carbohydrate Interactions by Isothermal Titration Calorimetry. Chem. Rev. 2002, 102, 387–430. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.S. The cyclization of peptides and depsipeptides. J. Pept. Sci. 2003, 9, 471–501. [Google Scholar] [CrossRef] [PubMed]
- Cary, D.R.; Ohuchi, M.; Reid, P.C.; Masuya, K. Constrained Peptides in Drug Discovery and Development. J. Synth. Org. Chem. Jpn. 2017, 75, 1171–1178. [Google Scholar] [CrossRef] [Green Version]
- White, C.J.; Yudin, A.K. Contemporary strategies for peptide macrocyclization. Nat. Chem. 2011, 3, 509–524. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, S. Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol. 2022, 3, 18–31. [Google Scholar] [CrossRef]
- Sohrabi, C.; Foster, A.; Tavassoli, A. Methods for generating and screening libraries of genetically encoded cyclic peptides in drug discovery. Nat. Rev. Chem. 2020, 4, 90–101. [Google Scholar] [CrossRef]
- Brueckner, A.C.; Deng, Q.; Cleves, A.E.; Lesburg, C.A.; Alvarez, J.C.; Reibarkh, M.Y.; Sherer, E.C.; Jain, A.N. Conformational Strain of Macrocyclic Peptides in Ligand–Receptor Complexes Based on Advanced Refinement of Bound-State Conformers. J. Med. Chem. 2021, 64, 3282–3298. [Google Scholar] [CrossRef] [PubMed]
- Udugamasooriya, D.G.; Spaller, M.R. Conformational constraint in protein ligand design and the inconsistency of binding entropy. Biopolymers 2008, 89, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Joo, S.-H. Cyclic Peptides as Therapeutic Agents and Biochemical Tools. Biomol. Ther. 2012, 20, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Soumana, D.I.; Yilmaz, N.K.; Prachanronarong, K.L.; Aydin, C.; Ali, A.; Schiffer, C.A. Structural and Thermodynamic Effects of Macrocyclization in HCV NS3/4A Inhibitor MK-5172. ACS Chem. Biol. 2016, 11, 900–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hruby, V.J.; Al-Obeidi, F.; Kazmierski, W. Emerging approaches in the molecular design of receptor-selective peptide ligands: Conformational, topographical and dynamic considerations. Biochem. J. 1990, 268, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Lucana, M.C.; Arruga, Y.; Petrachi, E.; Roig, A.; Lucchi, R.; Oller-Salvia, B. Protease-Resistant Peptides for Targeting and Intracellular Delivery of Therapeutics. Pharmaceutics 2021, 13, 2065. [Google Scholar] [CrossRef] [PubMed]
- Zavodszky, M.; Chen, C.-W.; Huang, J.-K.; Zolkiewski, M.; Wen, L.; Krishnamoorthi, R. Disulfide bond effects on protein stability: Designed variants of Cucurbita maxima trypsin inhibitor-V. Protein Sci. 2001, 10, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Postma, T.M.; Albericio, F. Disulfide Formation Strategies in Peptide Synthesis. Eur. J. Org. Chem. 2014, 2014, 3519–3530. [Google Scholar] [CrossRef]
- Imhof, D.; Roy, D.; Albericio, F. Editorial: Chemical Design and Biomedical Applications of Disulfide-rich Peptides: Challenges and Opportunities. Front. Chem. 2020, 8, 586377. [Google Scholar] [CrossRef]
- Kondasinghe, T.D.; Saraha, H.Y.; Jackowski, S.T.; Stockdill, J.L. Raising the bar on-bead: Efficient on-resin synthesis of α-conotoxin LvIA. Tetrahedron Lett. 2019, 60, 23–28. [Google Scholar] [CrossRef]
- Chakraborty, A.; Albericio, F.; de la Torre, B.G. Chemoselective Disulfide Formation by Thiol-Disulfide Interchange in SIT-Protected Cysteinyl Peptides. J. Org. Chem. 2022, 87, 708–712. [Google Scholar] [CrossRef]
- Góngora-Benítez, M.; Tulla-Puche, J.; Albericio, F. Multifaceted Roles of Disulfide Bonds. Peptides as Therapeutics. Chem. Rev. 2014, 114, 901–926. [Google Scholar] [CrossRef]
- McGivern, J.G. Ziconotide: A review of its pharmacology and use in the treatment of pain. Neuropsychiatr. Dis. Treat. 2007, 3, 69–85. [Google Scholar] [CrossRef] [Green Version]
- Fischell, T.; Shah, I.; Khan, S.; Malhotra, S. Eptifibatide: The evidence for its role in the management of acute coronary syndromes. Core Evid. 2010, 4, 49–65. [Google Scholar] [CrossRef] [Green Version]
- Corsetti, M.; Tack, J. Linaclotide: A new drug for the treatment of chronic constipation and irritable bowel syndrome with constipation. United Eur. Gastroenterol. J. 2013, 1, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Bulaj, G. Formation of disulfide bonds in proteins and peptides. Biotechnol. Adv. 2005, 23, 87–92. [Google Scholar] [CrossRef]
- Andreu, D.; Albericio, F.; Sole, N.A.; Munson, M.C.; Ferrer, M.; Barany, G. Formation of Disulfide Bonds in Synthetic Peptides and Proteins; Dunn Pennington, B.M., Ed.; Humane Press: Totowa, NJ, USA, 1994; Volume 35. [Google Scholar]
- Spears, R.J.; McMahon, C.; Chudasama, V. Cysteine protecting groups: Applications in peptide and protein science. Chem. Soc. Rev. 2021, 50, 11098–11155. [Google Scholar] [CrossRef] [PubMed]
- Rovero, P. Homodetic Cyclic Peptides; Steven, A., Kates, F.A., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 2000. [Google Scholar]
- Tam, J.P.; Wu, C.R.; Liu, W.; Zhang, J.W. Disulfide bond formation in peptides by dimethyl sulfoxide—Scope and applications. J. Am. Chem. Soc. 1991, 113, 6657–6662. [Google Scholar] [CrossRef]
- Kamber, B.; Hartmann, A.; Eisler, K.; Riniker, B.; Rink, H.; Sieber, P.; Rittel, W. The Synthesis of Cystine Peptides by Iodine Oxidation of S-Trityl-cysteine and S-Acetamidomethyl-cysteine Peptides. Helvetica Chim. Acta 1980, 63, 899–915. [Google Scholar] [CrossRef]
- Lamthanh, H.; Roumestand, C.; Deprun, C.; Ménez, A. Side reaction during the deprotection of (S-acetamidomethyl)cysteine in a peptide with a high serine and threonine content. Int. J. Pept. Protein Res. 1993, 41, 85–95. [Google Scholar] [CrossRef]
- Lamthanh, H.; Virelizier, H.; Frayssinhes, D. Side reaction of S-to-N acetamidomethyl shift during disulfide bond formation by iodine oxidation of S-acetamidomethyl-cysteine in a glutamine-containing peptide. Pept. Res. 1995, 8, 316–320. [Google Scholar]
- Markaryan, S.A.; Grigoryan, K.R.; Sarkisyan, A.R.; Asatryan, A.M.; Adamyan, T.A. Complexation and reactions of molecular iodine with dimethyl and diethyl sulfoxides. Russ. J. Gen. Chem. 2006, 76, 1801–1803. [Google Scholar] [CrossRef]
- Böttger, R.; Hoffmann, R.; Knappe, D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS ONE 2017, 12, e0178943. [Google Scholar] [CrossRef] [Green Version]
- van der Schier, R.; Roozekrans, M.; van Velzen, M.; Dahan, A.; Niesters, M. Opioid-induced respiratory depression: Reversal by non-opioid drugs. F1000Prime Rep. 2014, 6, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isom, G.E.; Elshowihy, R.M. Naloxone-induced enhancement of carbon dioxide stimulated respiration. Life Sci. 1982, 31, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Badiani, A.; Leone, P.; Stewart, J. Intra-VTA injections of the mu-opioid antagonist CTOP enhance locomotor activity. Brain Res. 1995, 690, 112–116. [Google Scholar] [CrossRef]
- Devine, D.P.; Leone, P.; Wise, R.A. Mesolimbic dopamine neurotransmission is increased by administration of μ-opioid receptor antagonists. Eur. J. Pharmacol. 1993, 243, 55–64. [Google Scholar] [CrossRef]
- Li, C.; Liu, S.; Lu, X.; Tao, F. Role of Descending Dopaminergic Pathways in Pain Modulation. Curr. Neuropharmacol. 2019, 17, 1176–1182. [Google Scholar] [CrossRef]
- Sato, D.; Narita, M.; Hamada, Y.; Mori, T.; Tanaka, K.; Tamura, H.; Yamanaka, A.; Matsui, R.; Watanabe, D.; Suda, Y.; et al. Relief of neuropathic pain by cell-specific manipulation of nucleus accumbens dopamine D1- and D2-receptor-expressing neurons. Mol. Brain 2022, 15, 10. [Google Scholar] [CrossRef]
- Altier, N.; Stewart, J. The role of dopamine in the nucleus accumbens in analgesia. Life Sci. 1999, 65, 2269–2287. [Google Scholar] [CrossRef]
- Aicher, S.A.; Punnoose, A.; Goldberg, A. μ-Opioid Receptors Often Colocalize with the Substance P Receptor (NK1) in the Trigeminal Dorsal Horn. J. Neurosci. 2000, 20, 4345–4354. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, Y.; Asgar, J.; Niu, K.; Lee, J.; Lee, K.; Schneider, M.; Ro, J. Sex differences in μ-opioid receptor expression in trigeminal ganglia under a myositis condition in rats. Eur. J. Pain 2014, 18, 151–161. [Google Scholar] [CrossRef]
- McLaughlin, J.P.; Hill, K.P.; Jiang, Q.; Sebastian, A.; Archer, S.; Bidlack, J.M. Nitrocinnamoyl and chlorocinnamoyl derivatives of dihydrocodeinone: In vivo and in vitro characterization of mu-selective agonist and antagonist activity. Experiment 1999, 289, 304–311. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rayala, R.; Tiller, A.; Majumder, S.A.; Stacy, H.M.; Eans, S.O.; Nedovic, A.; McLaughlin, J.P.; Cudic, P. Solid-Phase Synthesis of the Bicyclic Peptide OL-CTOP Containing Two Disulfide Bridges, and an Assessment of Its In Vivo μ-Opioid Receptor Antagonism after Nasal Administration. Molecules 2023, 28, 1822. https://doi.org/10.3390/molecules28041822
Rayala R, Tiller A, Majumder SA, Stacy HM, Eans SO, Nedovic A, McLaughlin JP, Cudic P. Solid-Phase Synthesis of the Bicyclic Peptide OL-CTOP Containing Two Disulfide Bridges, and an Assessment of Its In Vivo μ-Opioid Receptor Antagonism after Nasal Administration. Molecules. 2023; 28(4):1822. https://doi.org/10.3390/molecules28041822
Chicago/Turabian StyleRayala, Ramanjaneyulu, Annika Tiller, Shahayra A. Majumder, Heather M. Stacy, Shainnel O. Eans, Aleksandra Nedovic, Jay P. McLaughlin, and Predrag Cudic. 2023. "Solid-Phase Synthesis of the Bicyclic Peptide OL-CTOP Containing Two Disulfide Bridges, and an Assessment of Its In Vivo μ-Opioid Receptor Antagonism after Nasal Administration" Molecules 28, no. 4: 1822. https://doi.org/10.3390/molecules28041822