
Virtual Screening, Synthesis, and Biological Evaluation of Some Carbohydrazide Derivatives as Potential DPP-IV Inhibitors
 , , , , ,
, , , , ,  and
and
Abstract
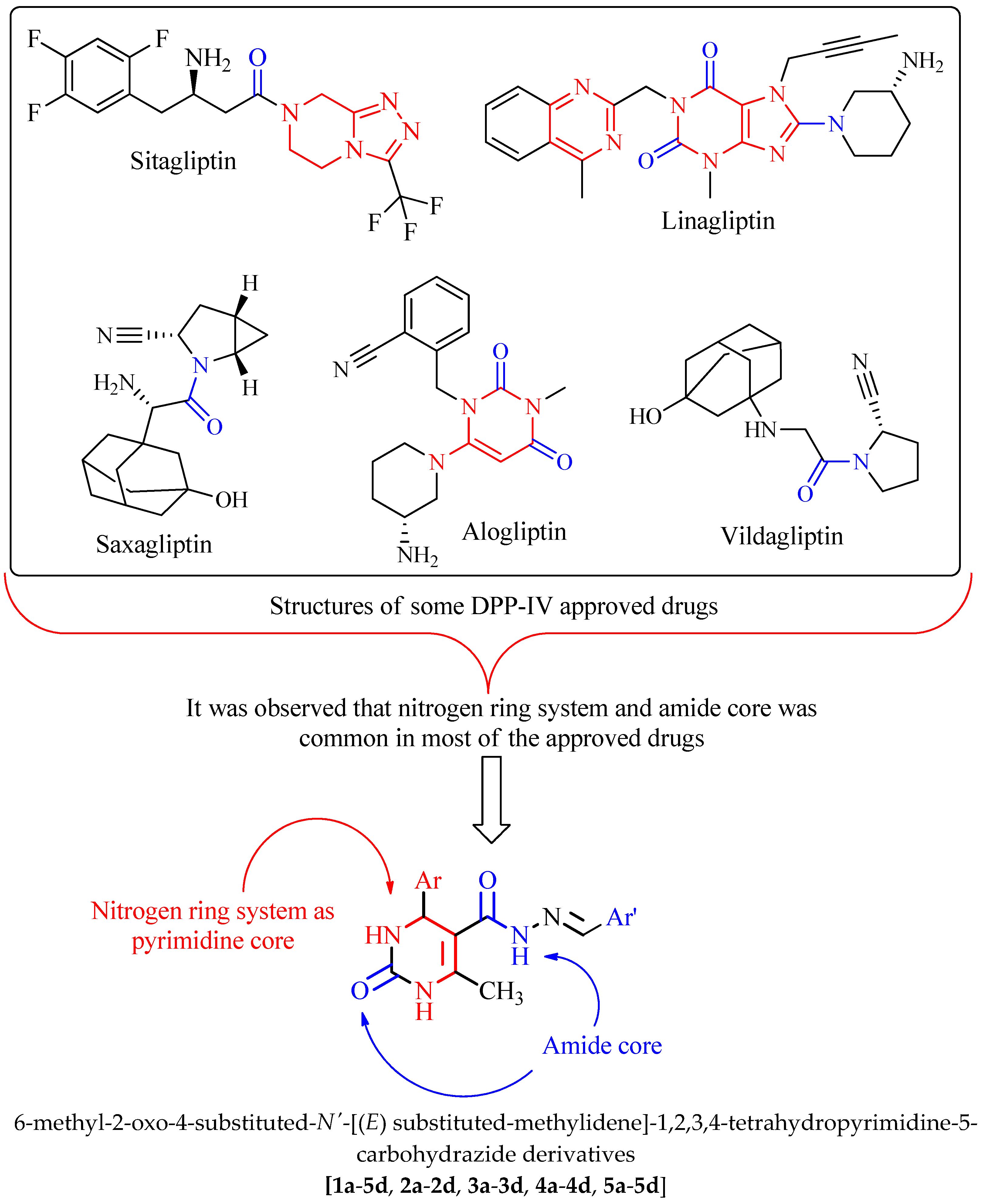
:1. Introduction
2. Results and Discussion
2.1. Virtual Screening of the Designed Derivatives
2.1.1. In Silico ADMET Analysis
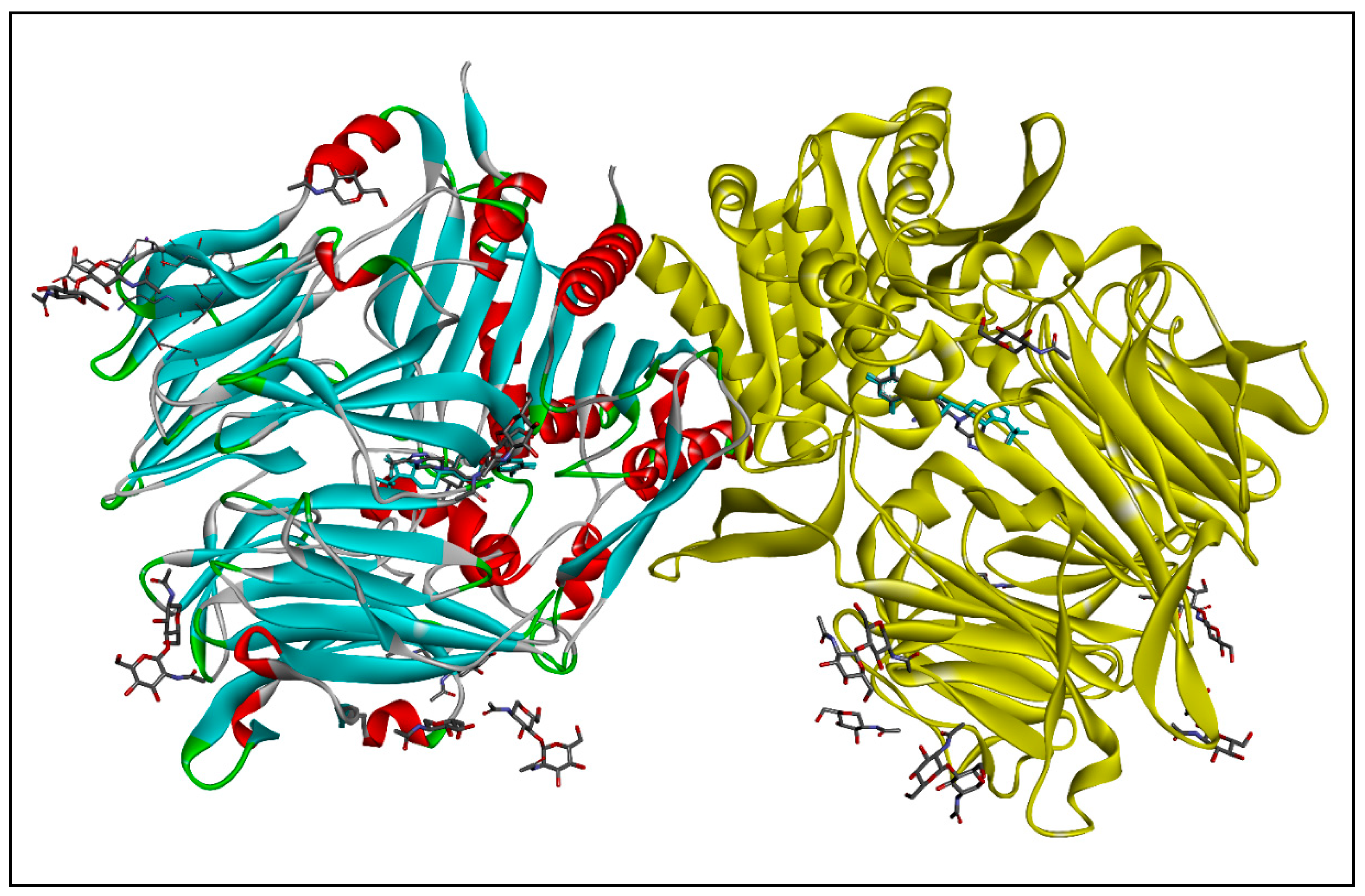
2.1.2. Molecular Docking
2.2. In vitro DPP-IV Enzyme Assay
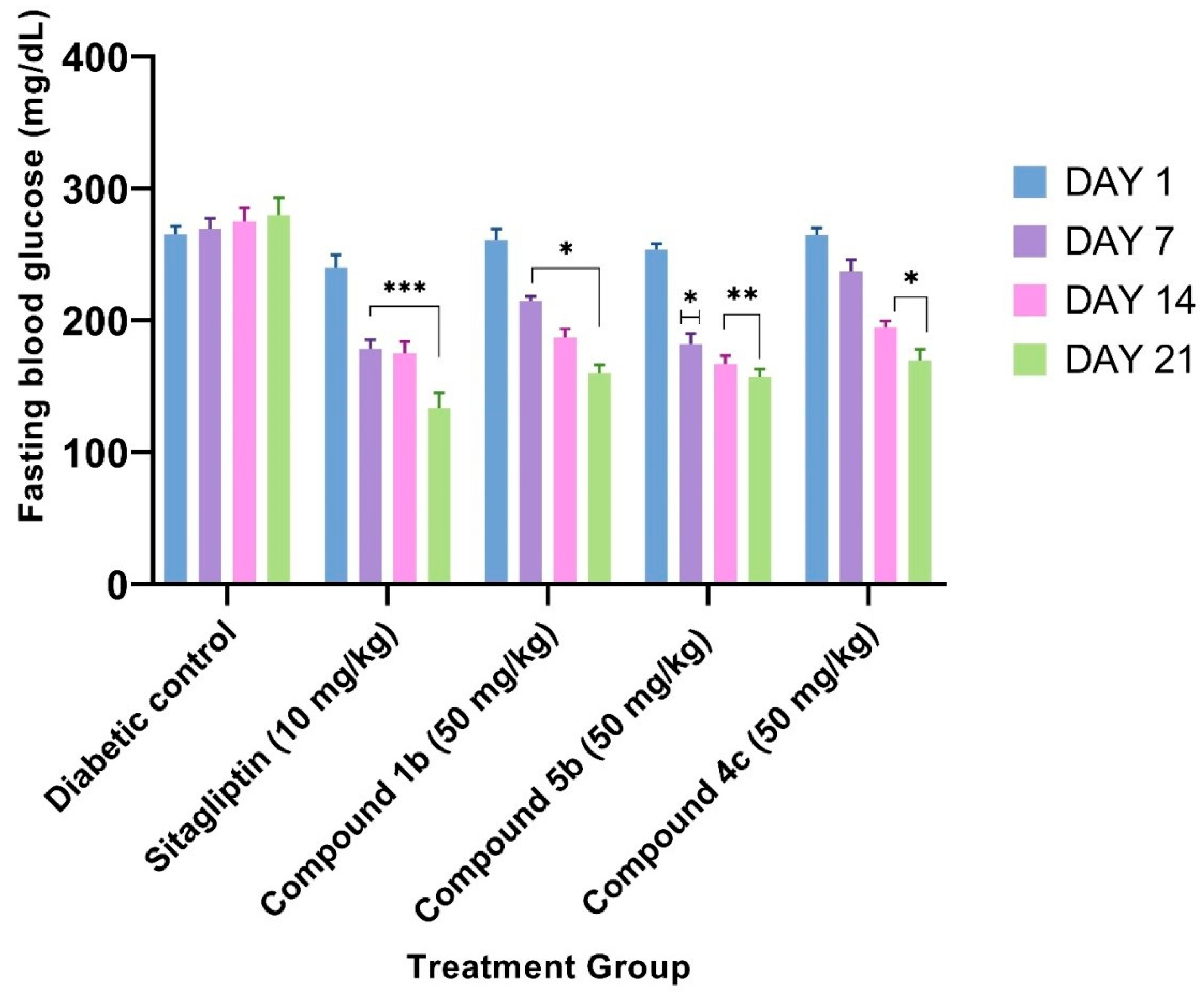
2.3. In Vivo Antidiabetic Activity
3. Materials and Methods
3.1. Virtual Screening of the Designed Derivatives
3.1.1. In Silico ADMET Analysis
3.1.2. Molecular Docking
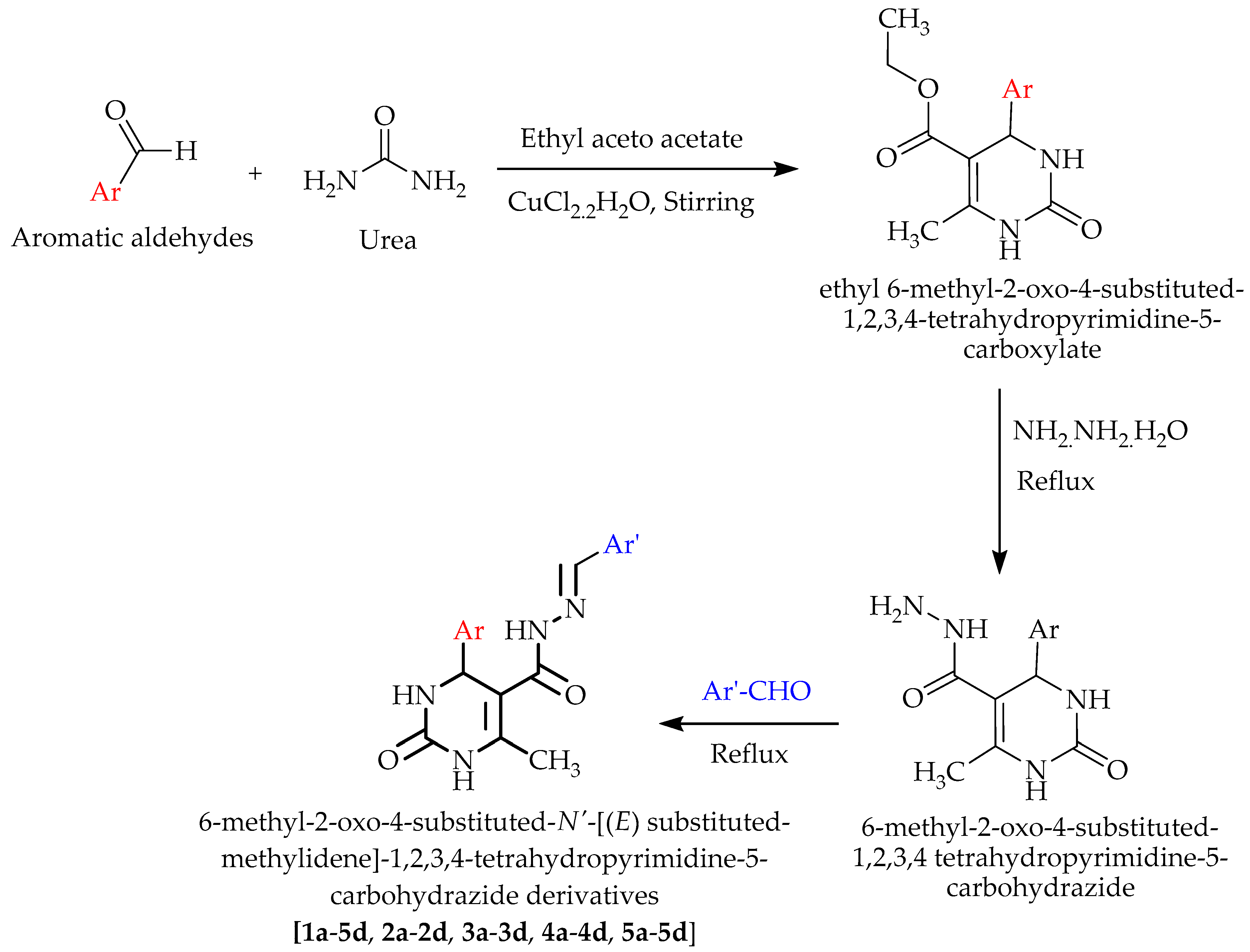
3.2. Chemistry
3.2.1. Synthesis of ethyl 6-Methyl-2-oxo-4- -1,2,3,4-tetrahydropyrimidine-5-carboxylate
3.2.2. Synthesis of 6-Methyl-2-oxo-4-substituted-1,2,3,4 Tetrahydropyrimidine-5-carbohydrazide
3.2.3. Synthesis of 6-Methyl-2-oxo-4-substituted-N’-((E) Substituted-methylidene)-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide Derivatives
- Characterization data(E)-N-Benzylidene-6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (1a).
- (E)-N-(2-Hydroxybenzylidene)-6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (1b).
- (E)-N-(2-Methoxybenzylidene)-6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (1c).
- (E)-N-(4-Methoxybenzylidene)-6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (1d).
- (E)-N-Benzylidene-4-(2-chlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidin-5-carbohydrazide (2a).
- (E)-4-(2-Chlorophenyl)-N-(2-hydroxybenzylidene)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (2b).
- (E)-4-(2-Chlorophenyl)-N-(2-methoxybenzylidene)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (2c).
- (E)-4-(2-Chlorophenyl)-N-(4-methoxybenzylidene)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (2d).
- (E)-N-Benzylidene-4-(2,4-dichlorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (3a).
- (E)--4-(2,4-Dichlorophenyl)-N-(2-hydroxybenzyldene)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (3b).
- (E)-N-(2,4-Diclorobenzylidene)-4-(2-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (3c).
- (E)-N-(2,4-Diclorobenzylidene)-4-(4-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (3d).
- (E)-N-Benzylidene-4-(2-fluorophenyl)-6-methyl-2-oxo-4-phenyl-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (4a).
- (E)--4-(2-Fluorophenyl)-N-(2-hydroxybenzyldene)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (4b).
- (E)--4-(2-Fluorophenyl)-N-(2-methoxybenzyldene)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (4c).
- (E)--4-(2-Fluorophenyl)-N-(4-methoxybenzyldene)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (4d).
- (E)-N-Benzylidene-4-(2,4-difluorophenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (5a).
- (E)--4-(2,4-Difluorophenyl)-N-(2-hydroxybenzyldene)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (5b).
- (E)-N-(2,4-Difluorobenzylidene)-4-(2-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (5c).
- (E)-N-(2,4-Difluorobenzylidene)-4-(4-methoxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carbohydrazide (5d).
3.3. In Vitro DPP-IV Enzymatic Assay
3.4. In Vivo Antidiabetic Activity
3.4.1. Animals and Ethical Approvals
3.4.2. Acute Toxicity Studies
3.4.3. STZ-Induced T2DM Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hawash, M.; Jaradat, N.; Shekfeh, S.; Abualhasan, M.; Eid, A.M.; Issa, L. Molecular Docking, Chemo-Informatic Properties, Alpha-Amylase, and Lipase Inhibition Studies of Benzodioxol Derivatives. BMC Chem. 2021, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Jaradat, N.; Khasati, A.; Hawi, M.; Hawash, M.; Shekfeh, S.; Qneibi, M.; Eid, A.M.; Arar, M.; Qaoud, M.T. Antidiabetic, Antioxidant, and Anti-Obesity Effects of Phenylthio-Ethyl Benzoate Derivatives, and Molecular Docking Study Regarding α-Amylase Enzyme. Sci. Rep. 2022, 12, 3108. [Google Scholar] [CrossRef] [PubMed]
- Arulmozhi, D.K.; Portha, B. GLP-1 Based Therapy for Type 2 Diabetes. Eur. J. Pharm. Sci. 2006, 28, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Deacon, C.F. A Review of Dipeptidyl Peptidase-4 Inhibitors. Hot Topics from Randomized Controlled Trials. Diabetes Obes. Metab. 2018, 20, 34–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallwitz, B. Clinical Use of DPP-4 Inhibitors. Front. Endocrinol. 2019, 10, 389. [Google Scholar] [CrossRef] [Green Version]
- Kirby, M.; Yu, D.M.T.; O’Connor, S.P.; Gorrell, M.D. Inhibitor Selectivity in the Clinical Application of Dipeptidyl Peptidase-4 Inhibition. Clin. Sci. 2010, 118, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Singla, R.; Jaitak, V. In Silico Study of Flavonoids as DPP-4 and α-Glucosidase Inhibitors. Lett. Drug Des. Discov. 2017, 14, 634–642. [Google Scholar] [CrossRef]
- Bittle, P.A. Use of Dipeptidyl Peptidase-4 Inhibitors in Patients with Type 2 Diabetes & Chronic & Kidney Disease. Nurse Pract. 2017, 42, 31–37. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Bandeira-Echtler, E.; Bergerhoff, K.; Lerch, C.L. Dipeptidyl Peptidase-4 (DPP-4) Inhibitors for Type 2 Diabetes Mellitus. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef]
- Barnett, A. DPP-4 Inhibitors and Their Potential Role in the Management of Type 2 Diabetes. Int. J. Clin. Pract. 2006, 60, 1454–1470. [Google Scholar] [CrossRef]
- Sesti, G.; Avogaro, A.; Belcastro, S.; Bonora, B.M.; Croci, M.; Daniele, G.; Dauriz, M.; Dotta, F.; Formichi, C.; Frontoni, S.; et al. Ten Years of Experience with DPP-4 Inhibitors for the Treatment of Type 2 Diabetes Mellitus. Acta Diabetol. 2019, 56, 605–617. [Google Scholar] [CrossRef]
- Akhtar, W.; Khan, M.F.; Verma, G.; Shaquiquzzaman, M.; Rizvi, M.A.; Mehdi, S.H.; Akhter, M.; Alam, M.M. Therapeutic Evolution of Benzimidazole Derivatives in the Last Quinquennial Period. Eur. J. Med. Chem. 2017, 126, 705–753. [Google Scholar] [CrossRef]
- Kamal, A.; Shaik, A.B.; Jain, N.; Kishor, C.; Nagabhushana, A.; Supriya, B.; Bharath Kumar, G.; Chourasiya, S.S.; Suresh, Y.; Mishra, R.K.; et al. Design and Synthesis of Pyrazole-Oxindole Conjugates Targeting Tubulin Polymerization as New Anticancer Agents. Eur. J. Med. Chem. 2015, 92, 501–513. [Google Scholar] [CrossRef]
- Barret, R. Lipinski’s Rule of Five. In Therapeutical Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; pp. 97–100. [Google Scholar] [CrossRef]
- Khan, S.; Kale, M.; Siddiqui, F.; Nema, N. Novel Pyrimidine-Benzimidazole Hybrids with Antibacterial and Antifungal Properties and Potential Inhibition of SARS-CoV-2 Main Protease and Spike Glycoprotein. Digit. Chin. Med. 2021, 4, 102–119. [Google Scholar] [CrossRef]
- Krzywinski, M.; Altman, N. Points of Significance: Significance, P Values and t-Tests. Nat. Methods 2013, 10, 1041–1042. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.; Singh, S.; Sethi, S.; Roy, S.; Mittra, S.; Rayasam, G.; Bansal, V.; Sattigeri, J.; Ray, A. Nature of Action of Sitagliptin, the Dipeptidyl Peptidase-IV Inhibitor in Diabetic Animals. Indian J. Pharmacol. 2010, 42, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Eswaramoorthy, R.; Hailekiros, H.; Kedir, F.; Endale, M. In Silico Molecular Docking, Dft Analysis and Admet Studies of Carbazole Alkaloid and Coumarins from Roots of Clausena Anisata: A Potent Inhibitor for Quorum Sensing. Adv. Appl. Bioinform. Chem. 2021, 14, 13–24. [Google Scholar] [CrossRef] [PubMed]
- James, J.P.; Mathew, J.B.; Bhat, K.I.; Kumar, P.; Aiswarya, T.C. Design And Synthesis Of Novel Pyrimidine Analogs As Anti-Tubercular Agents Targeting Thymidine Kinase Domain. J. Microbiol. Biotechnol. Food Sci. 2021, 11, 1–4. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Chavan, P.A.; Jadhav, S.B. Synthesis, Characterization and Screening of Some Novel 2-Methyl-N’- [(Z)-Substituted-Phenyl Ethylidene] Imidazo [1, 2-a] Pyridine-3-Carbohy Drazide Derivatives as DPP-IV Inhibitors for the Treatment of Type 2 Diabetes Mellitus. Lett. Drug Des. Discov. 2021, 19, 160–174. [Google Scholar] [CrossRef]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Accelrys Software, Inc. Discovery Studio Modeling Environment, Release 3.1; Accelrys Software, Inc.: San Diego, CA, USA, 2012; pp. 98–104. [Google Scholar]
- Khan, S.L.; Siddiui, F.A. Beta-Sitosterol: As Immunostimulant, Antioxidant and Inhibitor of SARS-CoV-2 Spike Glycoprotein. Arch. Pharmacol. Ther. 2020, 2, 12–16. [Google Scholar] [CrossRef]
- Siddiqui, F.A.; Khan, S.L.; Marathe, R.P.; Nema, N.V. Design, Synthesis, and In Silico Studies of Novel N-(2-Aminophenyl)-2,3- Diphenylquinoxaline-6-Sulfonamide Derivatives Targeting Receptor-Binding Domain (RBD) of SARS-CoV-2 Spike Glycoprotein and Their Evaluation as Antimicrobial and Antimalarial Agents. Lett. Drug Des. Discov. 2021, 18, 915–931. [Google Scholar] [CrossRef]
- Chaudhari, R.N.; Khan, S.L.; Chaudhary, R.S.; Jain, S.P.; Siddiqui, F.A. Β-Sitosterol: Isolation from Muntingia Calabura Linn Bark Extract, Structural Elucidation And Molecular Docking Studies As Potential Inhibitor of SARS-CoV-2 Mpro (COVID-19). Asian J. Pharm. Clin. Res. 2020, 13, 204–209. [Google Scholar] [CrossRef]
- Khan, S.L.; Siddiqui, F.A.; Shaikh, M.S.; Nema, N.V.; Shaikh, A.A. Discovery of Potential Inhibitors of the Receptor-Binding Domain (RBD) of Pandemic Disease-Causing SARS-CoV-2 Spike Glycoprotein from Triphala through Molecular Docking. Curr. Chin. Chem. 2021, 1, e220321192390. [Google Scholar] [CrossRef]
- Khan, S.L.; Sonwane, G.M.; Siddiqui, F.A.; Jain, S.P.; Kale, M.A.; Borkar, V.S. Discovery of Naturally Occurring Flavonoids as Human Cytochrome P450 (CYP3A4) Inhibitors with the Aid of Computational Chemistry. Indo Glob. J. Pharm. Sci. 2020, 10, 58–69. [Google Scholar] [CrossRef]
- Shntaif, A.H.; Khan, S.; Tapadiya, G.; Chettupalli, A.; Saboo, S.; Shaikh, M.S.; Siddiqui, F.; Amara, R.R. Rational Drug Design, Synthesis, and Biological Evaluation of Novel N-(2-Arylaminophenyl)-2,3-Diphenylquinoxaline-6-Sulfonamides as Potential Antimalarial, Antifungal, and Antibacterial Agents. Digit. Chin. Med. 2021, 4, 290–304. [Google Scholar] [CrossRef]
- Bhoi, M.N.; Borad, M.A.; Pithawala, E.A.; Modi, S.; Patel, H.D. Synthesis of N’-(7-Chloroquinolin-4-Yl)-6-Methyl-2-Oxo-4-Phenyl-1,2,3,4-Tetrahydropyrimidine-5-Carbohydrazide Derivatives as Potent Antibacterial Agents. Int. Lett. Chem. Phys. Astron. 2015, 56, 82–90. [Google Scholar] [CrossRef]
- Kumar, R.; Mittal, A.; Ramachandran, U. Design and Synthesis of 6-Methyl-2-Oxo-1,2,3,4-Tetrahydro-Pyrimidine-5-Carboxylic Acid Derivatives as PPARγ Activators. Bioorg. Med. Chem. Lett. 2007, 17, 4613–4618. [Google Scholar] [CrossRef]
- Barreira da Silva, R.; Ingersoll, M.; Albert, M. Measurement of Dipeptidylpeptidase Activity in Vitro and in Vivo. Bio-Protocol 2017, 7, e2184. [Google Scholar] [CrossRef]
- Wilhelm, K.P.; Maibach, H.I. OECD Guidelines for Testing of Chemicals. In Dermatotoxicology, 8th ed.; CRC Press: Boca Raton, FL, USA, 2012; pp. 497–499. ISBN 9781841848570. [Google Scholar] [CrossRef]
- Cassano, V.; Leo, A.; Tallarico, M.; Nesci, V.; Cimellaro, A.; Fiorentino, T.V.; Citraro, R.; Hribal, M.L.; De Sarro, G.; Perticone, F.; et al. Metabolic and Cognitive Effects of Ranolazine in Type 2 Diabetes Mellitus: Data from an in Vivo Model. Nutrients 2020, 12, 382. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, T.; Kanemoto, N.; Ohbuchi, Y.; Okano, M.; Fukui, H.; Sudo, T. Characterization of STZ-Induced Type 2 Diabetes in Zucker Fatty Rats. Exp. Anim. 2008, 57, 335–345. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
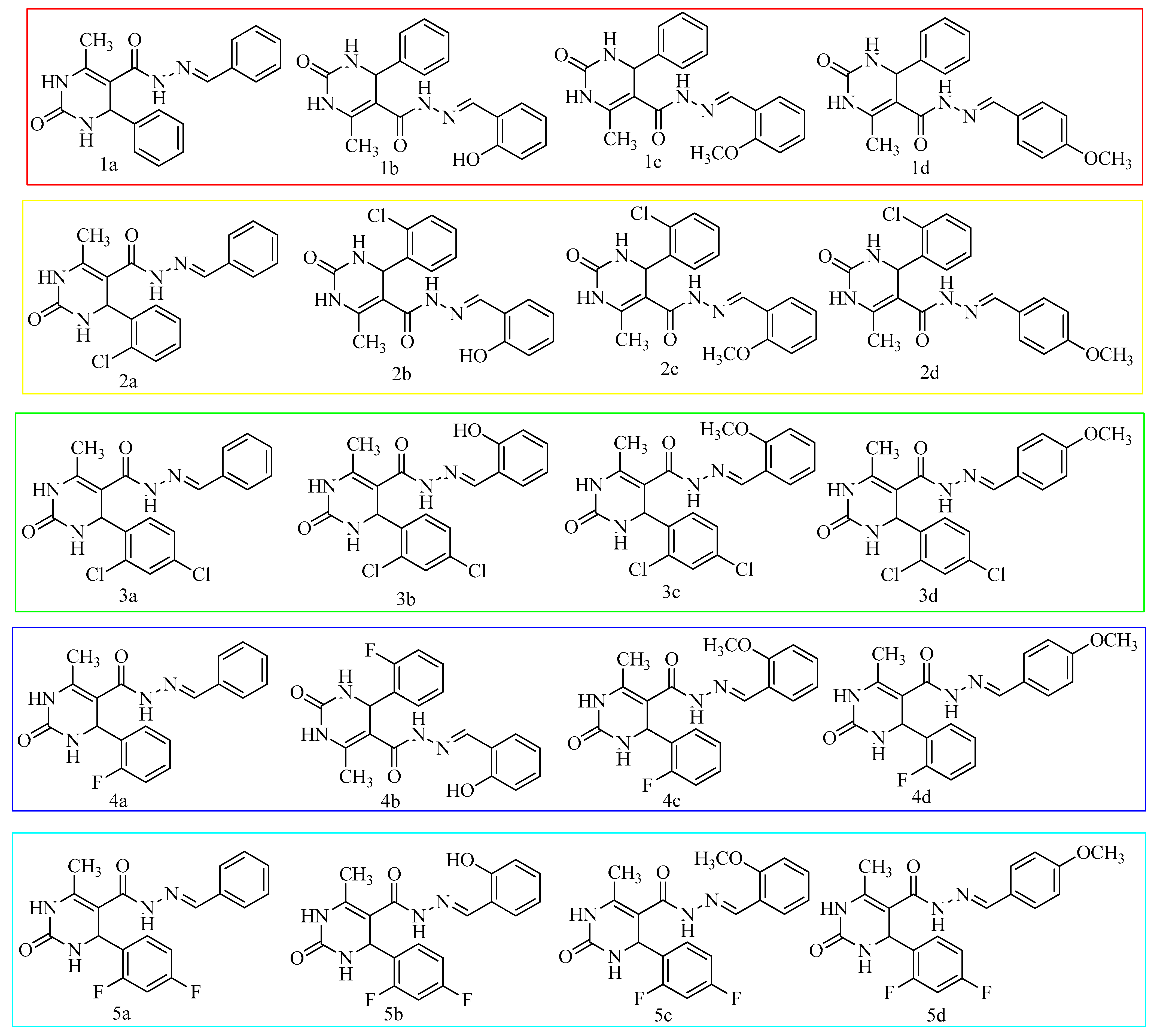
| Codes | Ar | Ar’ | Codes | Ar | Ar’ |
|---|---|---|---|---|---|
| 1a | Phenyl | Phenyl | 3c | 2,4-dichlorophenyl | 2-methoxyphenyl |
| 1b | Phenyl | 2-hydroxyphenyl | 3d | 2,4-dichlorophenyl | 4-methoxyphenyl |
| 1c | Phenyl | 2-methoxyphenyl | 4a | 2-fluorophenyl | Phenyl |
| 1d | Phenyl | 4-methoxyphenyl | 4b | 2-fluorophenyl | 2-hydroxyphenyl |
| 2a | 2-chlorophenyl | Phenyl | 4c | 2-fluorophenyl | 2-methoxyphenyl |
| 2b | 2-chlorophenyl | 2-hydroxyphenyl | 4d | 2-fluorophenyl | 4-methoxyphenyl |
| 2c | 2-chlorophenyl | 2-methoxyphenyl | 5a | 2,4-difluorophenyl | Phenyl |
| 2d | 2-chlorophenyl | 4-methoxyphenyl | 5b | 2,4-difluorophenyl | 2-hydroxyphenyl |
| 3a | 2,4-dichlorophenyl | Phenyl | 5c | 2,4-difluorophenyl | 2-methoxyphenyl |
| 3b | 2,4-dichlorophenyl | 2-hydroxyphenyl | 5d | 2,4-difluorophenyl | 4-methoxyphenyl |
| Compound Codes | Lipinski Rule of Five | Veber’s Rule | |||||
|---|---|---|---|---|---|---|---|
| Log P (<5) | Mol. Wt. (<500 Da) | HBA (<10) | HBD (<5)s | Violations | Total Polar Surface Area (Å2) (<140 Å2) | No. of Rotatable Bonds (<10) | |
| NL | 3.29 | 419.37 | 10 | 01 | 0 | 59.97 | 03 |
| 1a | 1.97 | 334.37 | 3 | 3 | 0 | 82.59 | 5 |
| 1b | 1.61 | 350.37 | 4 | 4 | 0 | 102.82 | 5 |
| 1c | 2.1 | 364.4 | 4 | 3 | 0 | 91.82 | 6 |
| 1d | 2.07 | 364.4 | 4 | 3 | 0 | 91.82 | 6 |
| 2a | 2.44 | 368.82 | 3 | 3 | 0 | 82.59 | 5 |
| 2b | 2.06 | 384.82 | 4 | 4 | 0 | 102.82 | 5 |
| 2c | 2.61 | 398.84 | 4 | 3 | 0 | 91.82 | 6 |
| 2d | 2.59 | 398.84 | 4 | 3 | 0 | 91.82 | 6 |
| 3a | 2.98 | 403.26 | 3 | 3 | 0 | 82.59 | 5 |
| 3b | 2.58 | 419.26 | 4 | 4 | 0 | 102.82 | 5 |
| 3c | 3.16 | 433.29 | 4 | 3 | 0 | 91.82 | 6 |
| 3d | 3.12 | 433.29 | 4 | 3 | 0 | 91.82 | 6 |
| 4a | 2.24 | 352.36 | 4 | 3 | 0 | 82.59 | 5 |
| 4b | 1.86 | 368.36 | 5 | 4 | 0 | 102.82 | 5 |
| 4c | 2.41 | 382.39 | 5 | 3 | 0 | 91.82 | 6 |
| 4d | 2.38 | 382.39 | 5 | 3 | 0 | 91.82 | 6 |
| 5a | 2.56 | 370.35 | 5 | 3 | 0 | 82.59 | 5 |
| 5b | 2.19 | 386.35 | 6 | 4 | 0 | 102.82 | 5 |
| 5c | 2.73 | 400.38 | 6 | 3 | 0 | 91.82 | 6 |
| 5d | 2.7 | 400.38 | 6 | 3 | 0 | 91.82 | 6 |
| Compound Codes | Pharmacokinetics | Drug-Likeness | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GI abs. | BBB pen. | P-gp sub. | CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 | Log Kp (Skin Permeation, cm/s) | Ghose | Egan | Muegge | Bioavailability Score | |
| Inhibitors | |||||||||||||
| NL | H | Y | Y | N | N | N | Y | N | –7.43 | Y | Y | Y | 0.55 |
| 1a | H | N | Y | N | N | N | N | N | –6.87 | 0 | 0 | 0 | 0.55 |
| 1b | H | N | Y | N | N | N | N | N | –7.22 | 0 | 0 | 0 | 0.55 |
| 1c | H | N | Y | N | N | Y | N | N | –7.07 | 0 | 0 | 0 | 0.55 |
| 1d | H | N | Y | N | N | Y | N | N | –7.07 | 0 | 0 | 0 | 0.55 |
| 2a | H | N | Y | N | N | Y | N | N | –6.63 | 0 | 0 | 0 | 0.55 |
| 2b | H | N | Y | N | N | N | N | N | –6.99 | 0 | 0 | 0 | 0.55 |
| 2c | H | N | Y | N | N | Y | N | Y | –6.84 | 0 | 0 | 0 | 0.55 |
| 2d | H | N | Y | N | N | Y | N | Y | –6.84 | 0 | 0 | 0 | 0.55 |
| 3a | H | N | Y | Y | Y | Y | N | Y | –6.4 | 0 | 0 | 0 | 0.55 |
| 3b | H | N | Y | Y | N | N | N | Y | –6.75 | 0 | 0 | 0 | 0.55 |
| 3c | H | N | Y | Y | Y | Y | N | Y | –6.6 | 0 | 0 | 0 | 0.55 |
| 3d | H | N | Y | Y | Y | Y | N | Y | –6.6 | 0 | 0 | 0 | 0.55 |
| 4a | H | N | Y | N | N | N | N | N | –6.91 | 0 | 0 | 0 | 0.55 |
| 4b | H | N | Y | N | N | N | N | N | –7.26 | 0 | 0 | 0 | 0.55 |
| 4c | H | N | Y | N | N | Y | N | Y | –7.11 | 0 | 0 | 0 | 0.55 |
| 4d | H | N | Y | N | N | Y | N | Y | –7.11 | 0 | 0 | 0 | 0.55 |
| 5a | H | N | Y | N | N | N | N | N | -6.95 | 0 | 0 | 0 | 0.55 |
| 5b | H | N | Y | N | N | N | N | N | –7.29 | 0 | 0 | 0 | 0.55 |
| 5c | H | N | Y | N | N | Y | N | Y | –7.15 | 0 | 0 | 0 | 0.55 |
| 5d | H | N | Y | N | N | Y | N | Y | –7.15 | 0 | 0 | 0 | 0.55 |
| Compound Codes | Parameters | |||||||
|---|---|---|---|---|---|---|---|---|
| LD50 (mg/kg) | Toxicity Class | Prediction Accuracy (%) | Hepatotoxicity (Probability) | Carcinogenicity (Probability) | Immunotoxicity (Probability) | Mutagenicity (Probability) | Cytotoxicity (Probability) | |
| NL | 800 | 4 | 23 | I (0.60) | A (0.50) | A (0.80) | I (0.65) | I (0.71) |
| 1a | 711 | 4 | 54.26 | A (0.64) | A (0.71) | I (0.98) | I (0.60) | I (0.72) |
| 1b | 1644 | 4 | 54.26 | A (0.64) | A (0.66) | I (0.65) | I (0.62) | I (0.78) |
| 1c | 1880 | 4 | 54.26 | A (0.61) | A (0.60) | A (0.55) | I (0.59) | I (0.80) |
| 1d | 1880 | 4 | 54.26 | A (0.62) | A (0.60) | I (0.89) | I (0.59) | I (0.77) |
| 2a | 1000 | 4 | 54.26 | A (0.59) | A (0.52) | I (0.96) | I (0.69) | I (0.76) |
| 2b | 1000 | 4 | 23 | A (0.62) | A (0.52) | A (0.69) | I (0.70) | I (0.79) |
| 2c | 1000 | 4 | 23 | A (0.61) | I (0.53) | A (0.84) | I (0.68) | I (0.77) |
| 2d | 1190 | 4 | 100 | A (0.69) | I (0.62) | A (0.96) | I (0.97) | I (0.93) |
| 3a | 1000 | 4 | 54.26 | A (0.59) | A (0.52) | I (0.93) | I (0.69) | I (0.76) |
| 3b | 1190 | 4 | 100 | A (0.69) | I (0.62) | A (0.96) | I (0.97) | I (0.93) |
| 3c | 1000 | 4 | 23 | A (0.61) | I (0.53) | A (0.91) | I (0.68) | I (0.77) |
| 3d | 1000 | 4 | 23 | A (0.61) | I (0.53) | A (0.50) | I (0.68) | I (0.77) |
| 4a | 1000 | 4 | 23 | A (0.60) | A (0.57) | I (0.97) | I (0.68) | I (0.77) |
| 4b | 1644 | 4 | 23 | A (0.64) | A (0.55) | A (0.59) | I (0.67) | I (0.79) |
| 4c | 711 | 4 | 23 | A (0.63) | I (0.51) | A (0.78) | I (0.66) | I (0.78) |
| 4d | 1880 | 4 | 23 | A (0.63) | I (0.51) | I (0.76) | I (0.66) | I (0.78) |
| 5a | 1000 | 4 | 23 | A (0.60) | A (0.57) | I (0.60) | I (0.68) | I (0.77) |
| 5b | 1644 | 4 | 23 | A (0.64) | A (0.55) | A (0.97) | I (0.67) | I (0.79) |
| 5c | 711 | 4 | 23 | A (0.63) | I (0.51) | A (0.98) | I (0.66) | I (0.78) |
| 5d | 1880 | 4 | 23 | A (0.63) | I (0.51) | A (0.91) | I (0.66) | I (0.78) |
| Active Amino Acid Residues | Atom from Ligand | Bond Length (Å) | Bond Type | Bond Category | Ligand Energy | Docking Scores |
|---|---|---|---|---|---|---|
| (kcal/mol) | ||||||
| Native Ligand | ||||||
| TYR662 | N-H | 1.66907 | Hydrogen Bond | Conventional Hydrogen Bond | 447.3 | −9.1 |
| ARG125 | Pi-Orbitals | 4.39768 | Electrostatic | Pi-Cation | ||
| ARG358 | 3.52293 | |||||
| ARG358 | 5.41244 | Hydrophobic | Pi-Alkyl | |||
| PHE357 | Aromatic Carbon | 3.79334 | ||||
| 1b | ||||||
| GLU378 | N | 4.2858 | Electrostatic | Attractive Charge | 299.89 | −9.4 |
| GLU378 | N | 4.09602 | ||||
| SER349 | H | 2.37918 | Hydrogen Bond | Conventional Hydrogen Bond | ||
| SER376 | H | 2.55398 | ||||
| SER376 | NH | 2.862 | ||||
| THR351 | O | 1.98084 | ||||
| THR351 | O | 1.67832 | ||||
| GLU378 | C | 3.06114 | Carbon Hydrogen Bond | |||
| THR350 | Pi-Orbitals | 3.1604 | Pi-Donor Hydrogen Bond | |||
| SER376 | 3.06845 | |||||
| PHE396 | 4.9139 | Hydrophobic | Pi-Pi T-shaped | |||
| CYS394 | Alkyl | 5.29122 | Pi-Alkyl | |||
| VAL354 | 5.28734 | |||||
| 2b | ||||||
| ARG560 | NH | 2.90874 | Hydrogen Bond | Conventional Hydrogen Bond | 343.49 | −9.3 |
| ASN562 | H | 2.81998 | ||||
| LYS512 | Pi-Orbitals | 3.51422 | Hydrophobic | Pi-Sigma | ||
| ILE529 | 3.43887 | |||||
| ARG560 | 3.75323 | |||||
| PHE559 | 5.21502 | Pi-Pi T-shaped | ||||
| PRO475 | Cl | 5.41082 | Alkyl | |||
| LYS512 | Cl | 4.09612 | ||||
| ALA564 | Pi-Orbitals | 5.11889 | Pi-Alkyl | |||
| PRO475 | 4.72305 | |||||
| PHE559 | 5.12368 | |||||
| 3b | ||||||
| GLU205 | N | 5.12407 | Electrostatic | Attractive Charge | 340.69 | −9.3 |
| GLU205 | N | 5.23339 | ||||
| HIS740 | H | 2.57758 | Hydrogen Bond | Conventional Hydrogen Bond | ||
| PHE357 | Pi-Orbitals | 3.79816 | Hydrophobic | Pi-Pi Stacked | ||
| TYR662 | 4.0191 | |||||
| TYR547 | 4.68336 | |||||
| TYR666 | 4.89163 | Pi-Pi T-shaped | ||||
| SER630; TYR631 | 4.99633 | Amide-Pi Stacked | ||||
| LYS554 | Cl | 4.43461 | Alkyl | |||
| VAL656 | Pi-Orbitals | 5.41552 | Pi-Alkyl | |||
| TYR547 | Cl | 4.69875 | ||||
| TYR547 | Cl | 4.67797 | ||||
| 4a | ||||||
| PHE559 | F | 3.12032 | Hydrogen Bond;Halogen | Carbon Hydrogen Bond;Halogen (Fluorine) | 316.76 | −9.5 |
| VAL558 | F | 3.51875 | Halogen | Halogen (Fluorine) | ||
| LEU514 | Pi-Orbitals | 4.68451 | Hydrophobic | Pi-Alkyl | ||
| PRO510 | 5.49372 | |||||
| LYS512 | 4.13088 | |||||
| ILE529 | 4.76619 | |||||
| 5b | ||||||
| ILE102 | H | 2.81295 | Hydrogen Bond | Conventional Hydrogen Bond | 316.62 | −9.6 |
| HIS100 | H | 1.85943 | ||||
| ILE102 | O | 2.2207 | ||||
| ASN74 | F | 3.14132 | Halogen | Halogen (Fluorine) | ||
| LYS71 | Pi-Orbitals | 4.97015 | Electrostatic | Pi-Cation | ||
| TYR105 | 5.06176 | Hydrophobic | Pi-Pi T-shaped | |||
| ILE76 | 5.43513 | Pi-Alkyl | ||||
| ILE102 | 5.10056 | |||||
| 3D-Docking Poses | 2D-Docking Poses |
|---|---|
| Native Ligand | |
 |  |
| Compound 1b | |
 |  |
| Compound 2b | |
 |  |
| Compound 3b | |
 |  |
| Compound 4a | |
 |  |
| Compound 5b | |
 |  |
| Compound Code | % Inhibition at 50 µM | IC50 (µM) | Compound Code | % Inhibition at 50 µM | IC50 (µM) |
|---|---|---|---|---|---|
| 1a | 40.74 ± 0.38 | 58.95 ± 0.0012 | 3c | 61.00 ± 0.08 | 33.73 ± 0.0024 |
| 1b | 42.69 ± 0.28 | 54.73 ± 0.0025 | 3d | 63.25 ± 0.05 | 32.38 ± 0.0021 |
| 1c | 56.47 ± 0.37 | 39.72 ± 0.0027 | 4a | 64.78 ± 0.33 | 30.47 ± 0.0025 |
| 1d | 57.74 ± 0.38 | 38.26 ± 0.0050 | 4b | 63.80 ± 0.16 | 31.75 ± 0.0027 |
| 2a | 61.25 ± 0.16 | 32.41 ± 0.0033 | 4c | 60.40 ± 0.08 | 34.94 ± 0.0016 |
| 2b | 65.50 ± 0.08 | 31.93 ± 0.0029 | 4d | 57.74 ± 0.38 | 38.62 ± 0.0025 |
| 2c | 63.25 ± 0.05 | 33.97 ± 0.0025 | 5a | 44.20 ± 0.28 | 65.31 ± 0.0021 |
| 2d | 64.78 ±0.33 | 31.72 ± 0.0021 | 5b | 70.70 ± 0.37 | 28.13 ± 0.0029 |
| 3a | 61.25 ± 0.16 | 33.91 ± 0.0029 | 5c | 58.54 ± 0.27 | 37.63 ± 0.0026 |
| 3b | 61.00 ± 0.08 | 33.79 ± 0.0021 | 5d | 62.28 ± 0.29 | 34.13 ± 0.0027 |
| Sitagliptin | 101.7 ± 0.09 | 0.018 ± 0.0012 | --- | ||
| Treatment Group | Fasting Blood Glucose (mg/dL) ± SEM | |||
|---|---|---|---|---|
| On Day 1 | On Day 7 | On Day 14 | On Day 21 | |
| Diabetic control | 265.33 ± 6.27 | 269.66 ± 7.96 | 275.33 ± 1 0.04 | 280.00 ± 13.29 |
| Sitagliptin (10 mg/kg) | 240.16 ± 9.88 | 178.66 ± 6.92 *** | 175.16 ± 8.61 *** | 133.50 ± 11.80 *** |
| Compound 1b (50 mg/kg) | 261.00 ± 8.44 | 215.03 ± 3.29 * | 187.05 ± 6.48 * | 160.12 ± 6.18 * |
| Compound 5b (50 mg/kg) | 253.83 ± 4.49 | 182.00 ± 8.11 * | 167.16 ± 6.23 ** | 157.33 ± 5.75 ** |
| Compound 4c (50 mg/kg) | 264.66 ± 5.70 | 237.16 ± 9.01 | 194.83 ± 4.81 * | 169.66 ± 8.53 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jadhav, P.B.; Jadhav, S.B.; Zehravi, M.; Mubarak, M.S.; Islam, F.; Jeandet, P.; Khan, S.L.; Hossain, N.; Rashid, S.; Ming, L.C.; et al. Virtual Screening, Synthesis, and Biological Evaluation of Some Carbohydrazide Derivatives as Potential DPP-IV Inhibitors. Molecules 2023, 28, 149. https://doi.org/10.3390/molecules28010149
Jadhav PB, Jadhav SB, Zehravi M, Mubarak MS, Islam F, Jeandet P, Khan SL, Hossain N, Rashid S, Ming LC, et al. Virtual Screening, Synthesis, and Biological Evaluation of Some Carbohydrazide Derivatives as Potential DPP-IV Inhibitors. Molecules. 2023; 28(1):149. https://doi.org/10.3390/molecules28010149
Chicago/Turabian StyleJadhav, Prerana B., Shailaja B. Jadhav, Mehrukh Zehravi, Mohammad S. Mubarak, Fahadul Islam, Philippe Jeandet, Sharuk L. Khan, Nazmul Hossain, Salma Rashid, Long Chiau Ming, and et al. 2023. "Virtual Screening, Synthesis, and Biological Evaluation of Some Carbohydrazide Derivatives as Potential DPP-IV Inhibitors" Molecules 28, no. 1: 149. https://doi.org/10.3390/molecules28010149