
Combination of Docking-Based and Pharmacophore-Based Virtual Screening Identifies Novel Agonists That Target the Urotensin Receptor
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
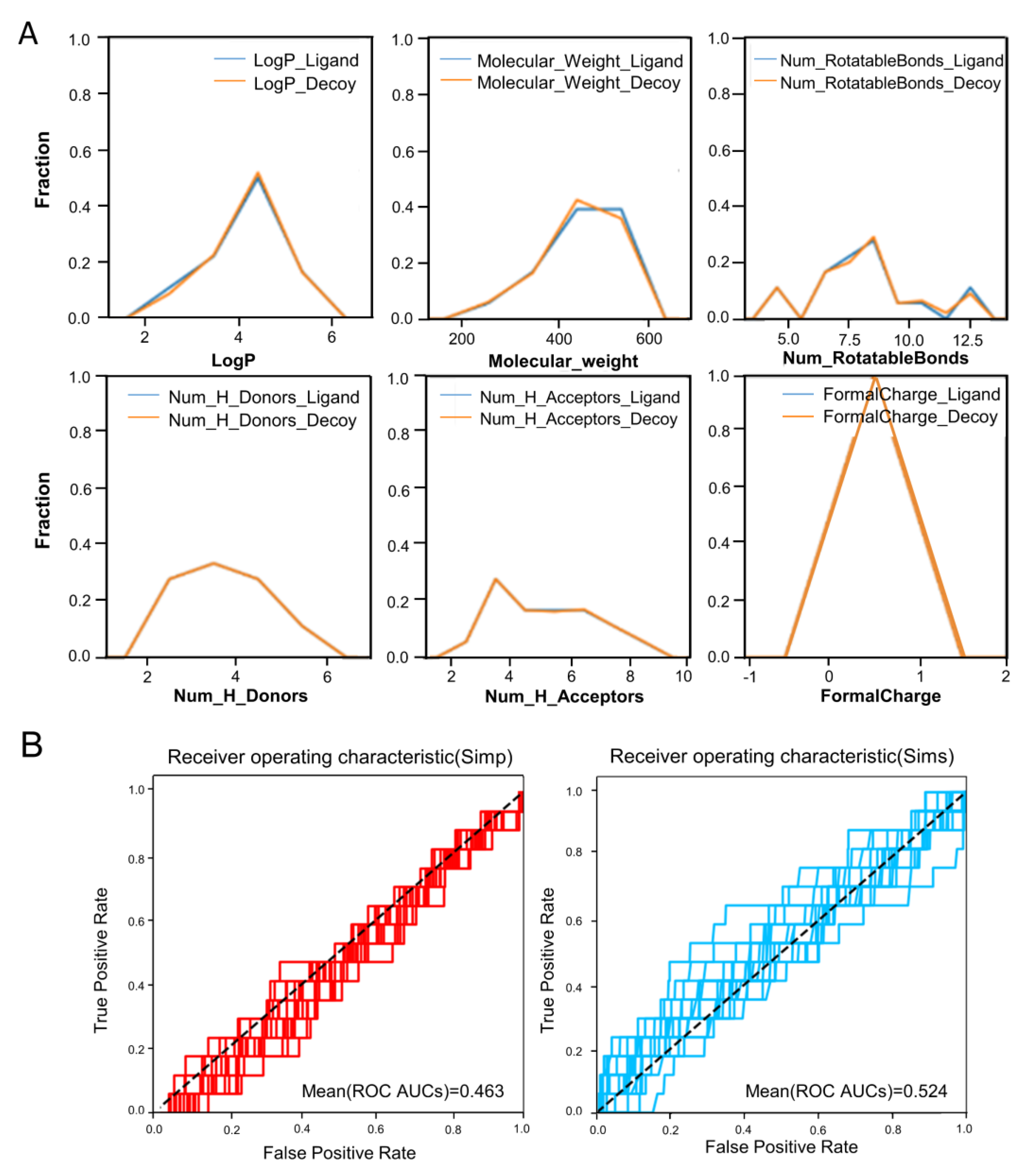
2.1. Construction of a Benchmarking Dataset Targeting UT Receptor and Ligand Enrichment Analysis
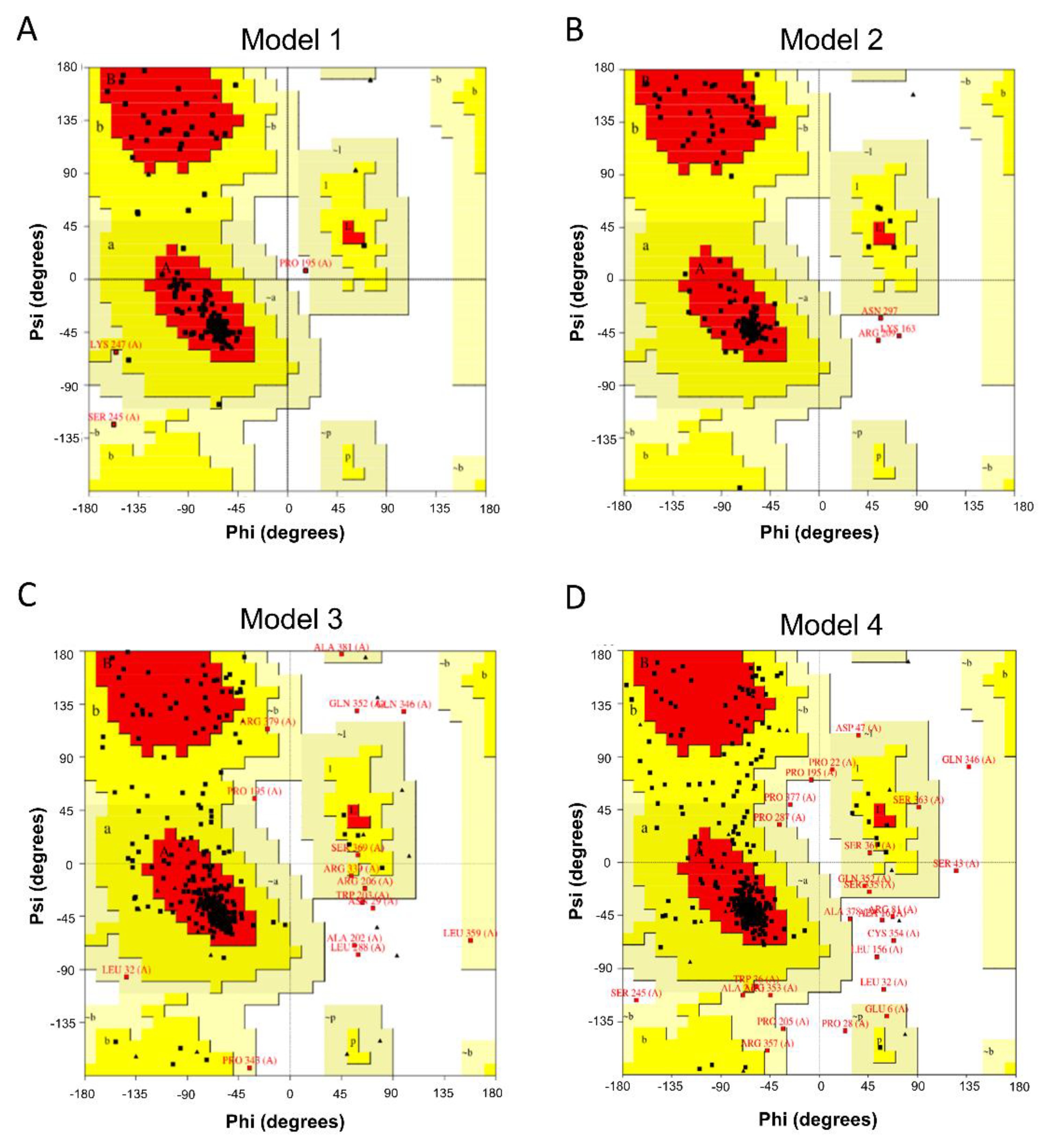
2.2. Homology Modeling of the 3D-Model of the UT Receptor
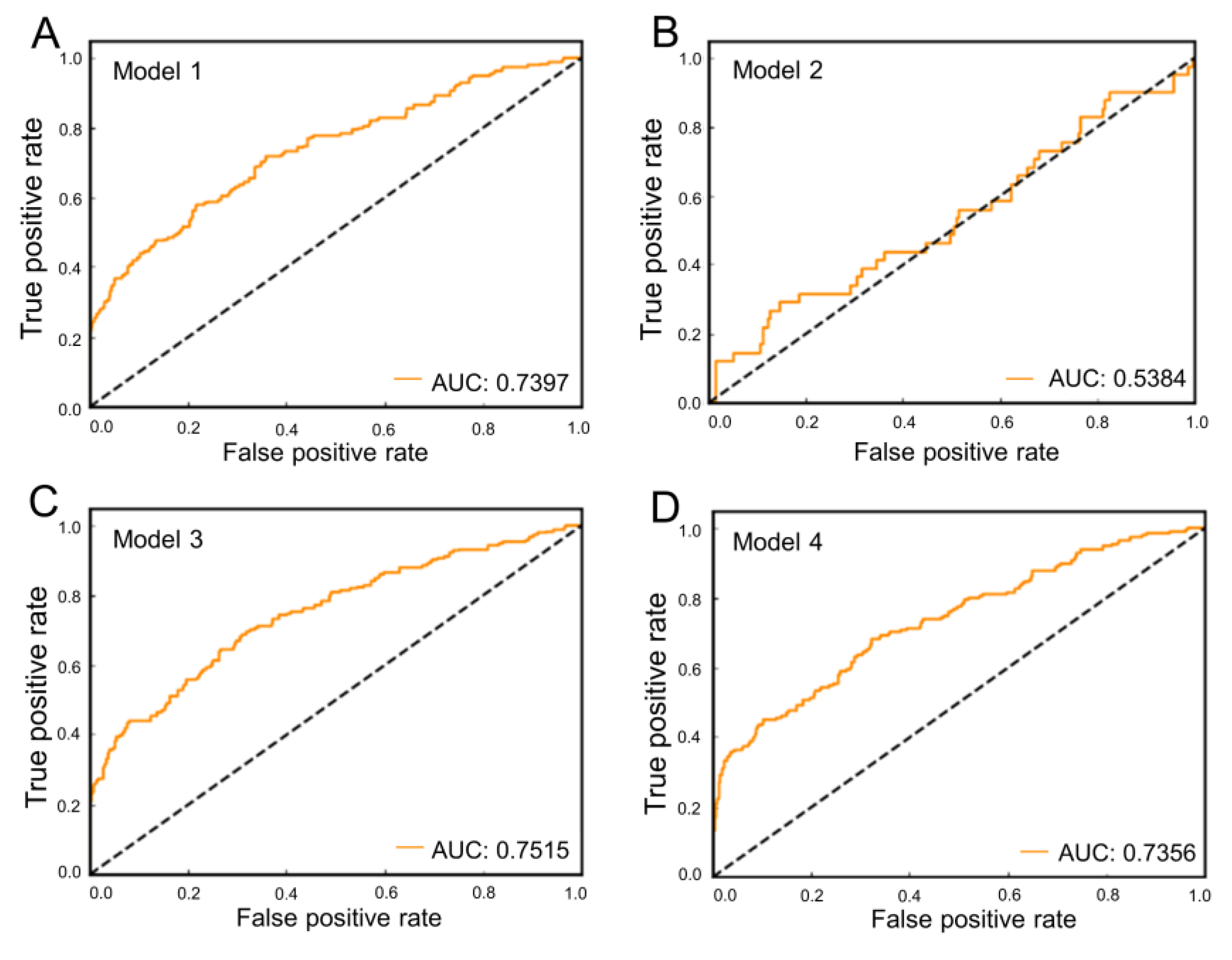
2.3. Validation of Generated Homology Model
2.4. Pharmacophore Generation and Validation
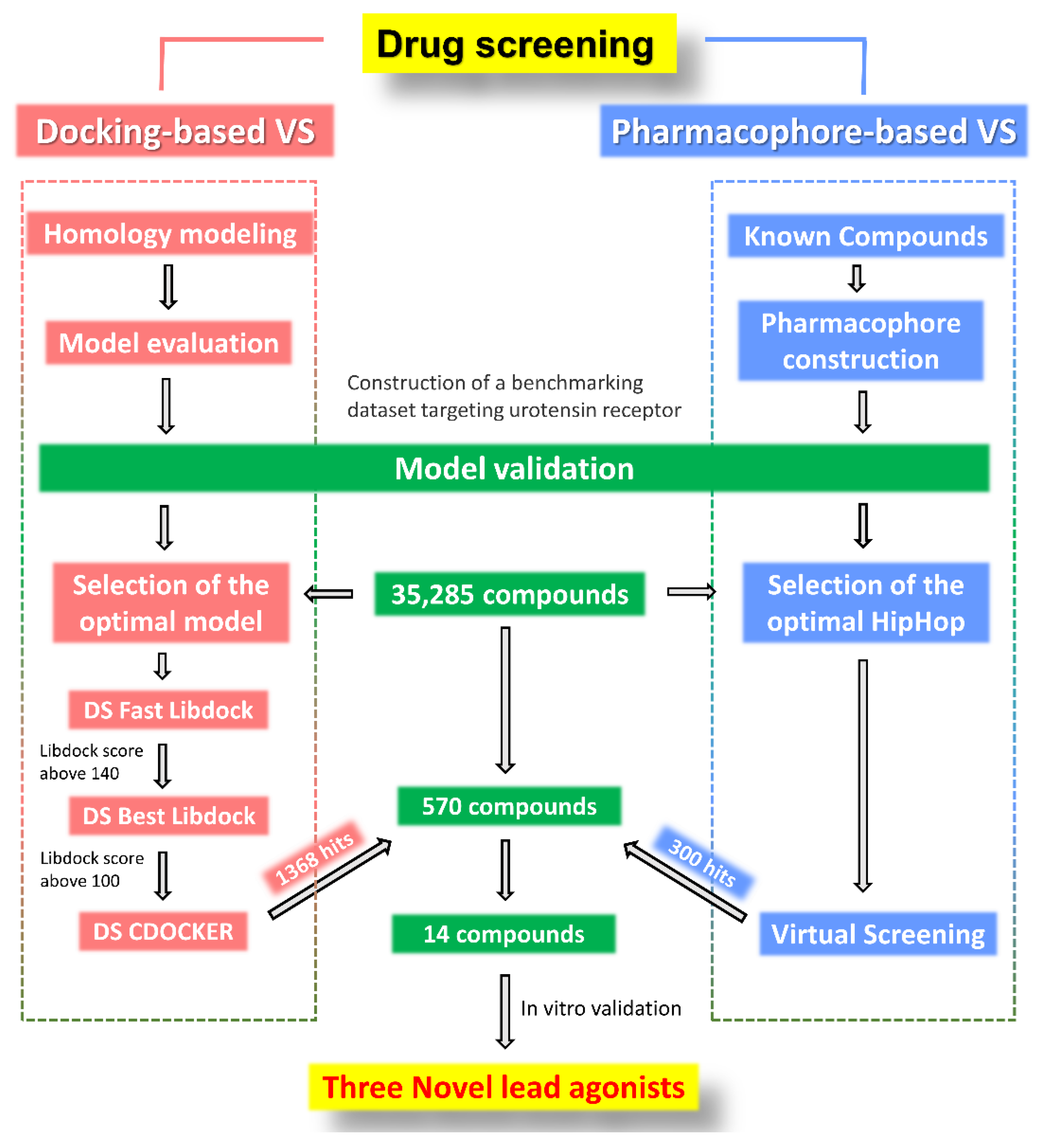
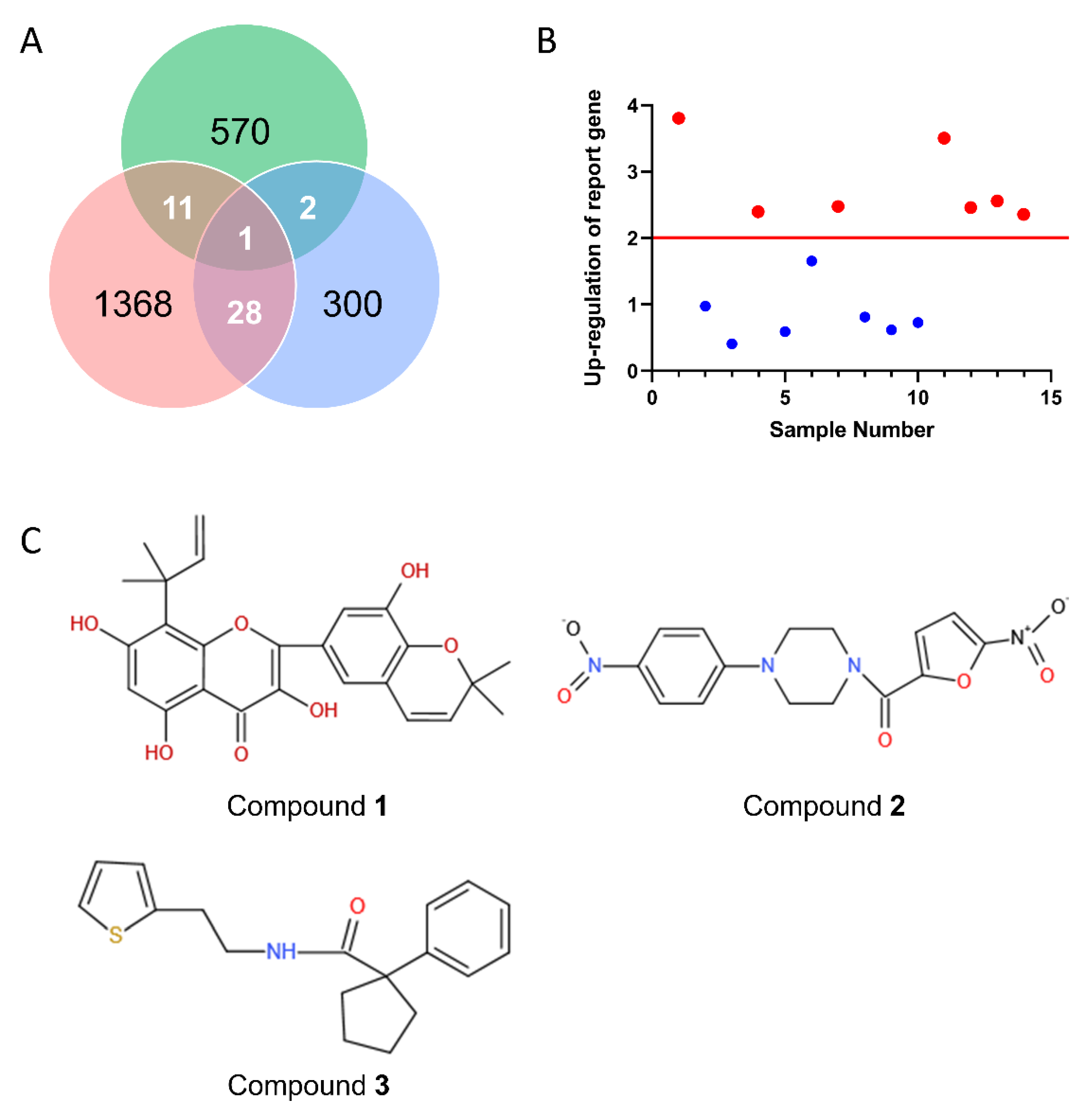
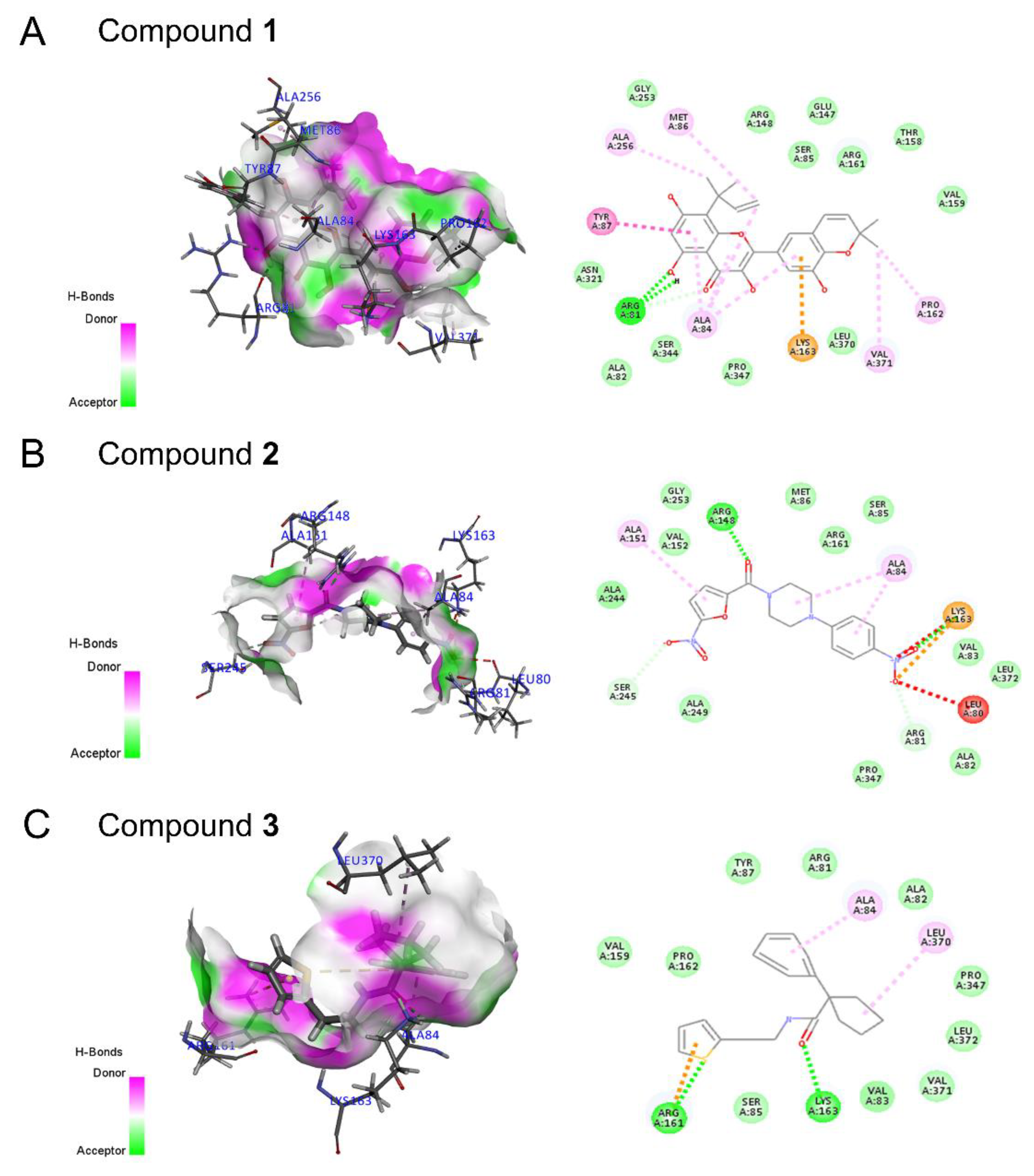
2.5. Drug Screening and Discovery of Novel Lead Compounds
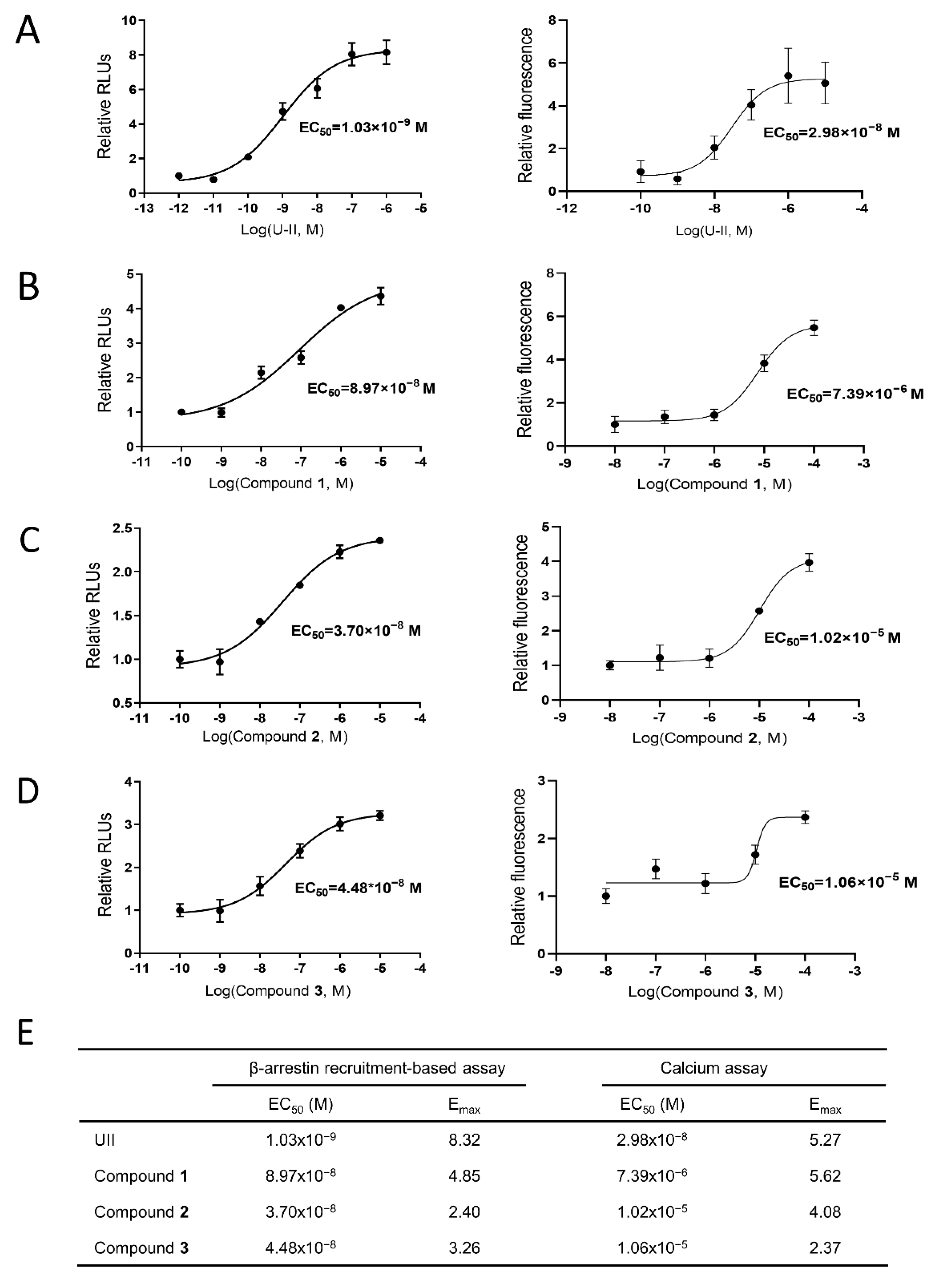
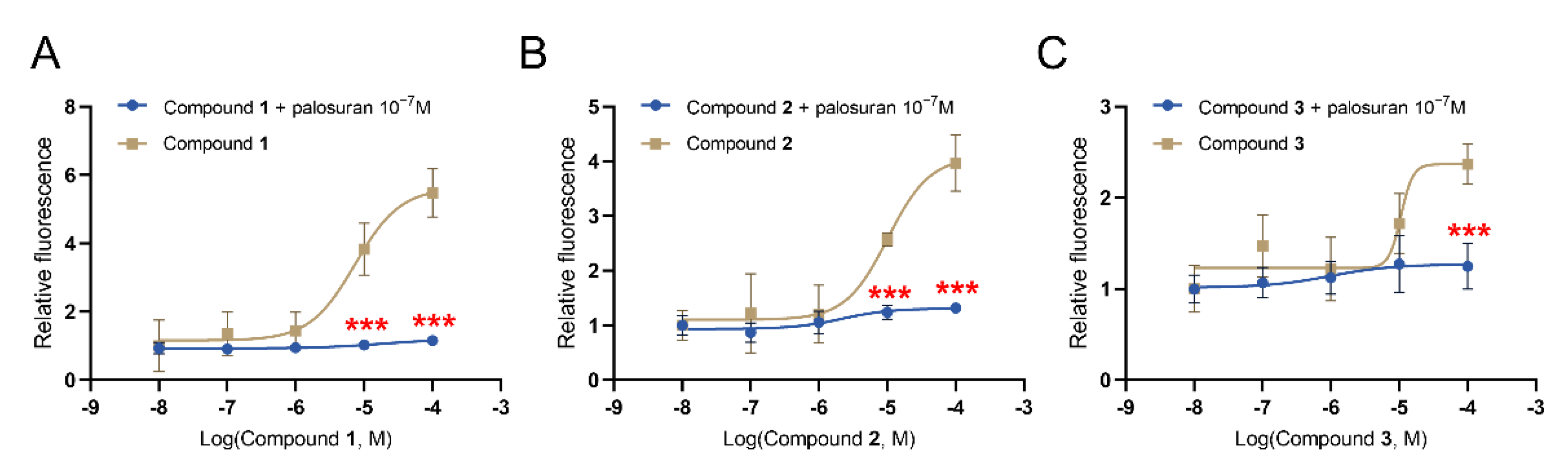
2.6. In Vitro Validation of UT Receptor Agonism
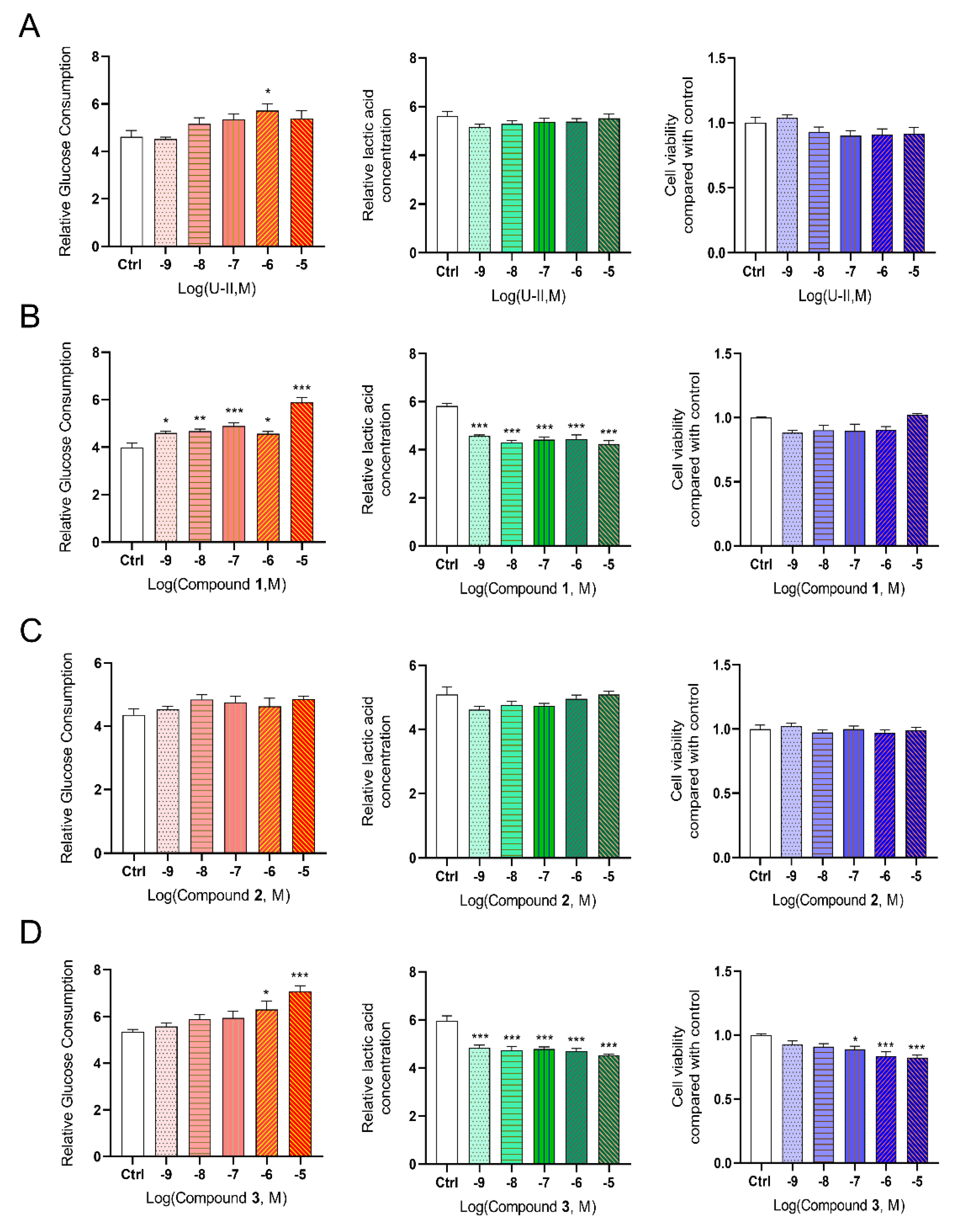
2.7. Effect of Leads on Glucose Consumption and Lactic Acid Release
3. Discussion
4. Materials and Methods
4.1. UT Receptor Docking-Based Virtual Screening
4.1.1. Homology Modeling
4.1.2. Homology Model Evaluation
4.1.3. UT Receptor Docking-Based Virtual Screening
4.2. Pharmacophore-Based Virtual Screening
4.2.1. Pharmacophore Model Generation and Validation
4.2.2. Pharmacophore-Based Virtual Screening
4.3. Benchmarking Dataset Construction
4.3.1. Ligand Preparation
4.3.2. Decoy Generation
4.4. Cell Line Culture
4.5. UT Receptor Activation Experimental Assay
4.6. In Vitro Biology Assay
4.6.1. Cell Viability Detection
4.6.2. Measurements of Glucose Consumption and Lactic Acid Production
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ames, R.S.; Sarau, H.M.; Chambers, J.K.; Willette, R.N.; Aiyar, N.V.; Romanic, A.M.; Louden, C.S.; Foley, J.J.; Sauermelch, C.F.; Coatney, R.W.; et al. Human urotensin-II is a potent vasoconstrictor and agonist for the orphan receptor GPR14. Nature 1999, 401, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Vaudry, H.; Leprince, J.; Chatenet, D.; Fournier, A.; Lambert, D.; Le Mével, J.-C.; Ohlstein, E.H.; Schwertani, A.; Tostivint, H.; Vaudry, D. International Union of Basic and Clinical Pharmacology. XCII. Urotensin II, Urotensin II–Related Peptide, and Their Receptor: From Structure to Function. Pharmacol. Rev. 2015, 67, 214–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, M.; Tagawa, H.; Ueno, M.; Marumoto, S.; Nagayama, T. The urotensin II receptor antagonist DS37001789 ameliorates mortality in pressure-overload mice with heart failure. Heliyon 2020, 6, e03352. [Google Scholar] [CrossRef] [PubMed]
- Avagimyan, A.; Kajaia, A.; Gabunia, L.; Ghambashidze, K.; Sulashvili, N.; Ratiani, L.; Heshmat-Ghahdarijani, K.; Sheibani, M.; Aznauryan, A. Urotensin-II As a Promising Key-Point of Cardiovascular Disturbances Sequel. Curr. Probl. Cardiol. 2021, 47, 101074. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Yu, X.; Xue, X.; Liu, H.; Wang, M.; Li, Y.; Wang, X.; Ding, H. Urotensin II receptor as a potential biomarker for the prognosis of hepatocellular carcinoma patients. Oncol. Lett. 2017, 14, 2749–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozoux, M.L.; Briand, V.; Pelat, M.; Barbe, F.; Schaeffer, P.; Beauverger, P.; Poirier, B.; Guillon, J.M.; Petit, F.; Altenburger, J.M.; et al. Potential therapeutic value of urotensin ii receptor antagonist in chronic kidney disease and associated comorbidities. J. Pharmacol. Exp. Ther. 2020, 374, 24–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Singh, J.; Bala, K.; Singh, J. Association of resistin (rs3745367) and urotensin II (rs228648 and rs2890565) gene polymorphisms with risk of type 2 diabetes mellitus in Indian population. Mol. Biol. Rep. 2020, 47, 9489–9497. [Google Scholar] [CrossRef]
- Bousette, N.; D’Orleans-Juste, P.; Kiss, R.S.; You, Z.; Genest, J.; Al-Ramli, W.; Qureshi, S.T.; Gramolini, A.; Behm, D.; Ohlstein, E.H.; et al. 2009. Urotensin II receptor knockout mice on an ApoE knockout background fed a high-fat diet exhibit an enhanced hyperlipidemic and atherosclerotic phenotype. Circ. Res. 2009, 105, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Yin, L.; Jia, W.H.; Wang, N.Q.; Xu, C.Y.; Hou, B.Y.; Li, N.; Zhang, L.; Qiang, G.F.; Yang, X.Y.; et al. Chronic Urotensin-II Administration Improves Whole-Body Glucose Tolerance in High-Fat Diet-Fed Mice. Front. Endocrinol. 2019, 10, 453. [Google Scholar] [CrossRef] [Green Version]
- Carotenuto, A.; Grieco, P.; Novellino, E.; Rovero, P. Urotensin-II receptor peptide agonists. Med. Res. Rev. 2004, 24, 577–588. [Google Scholar] [CrossRef]
- di Villa Bianca, R.D.E.; Mitidieri, E.; Donnarumma, E.; Fusco, F.; Longo, N.; Rosa, G.D.; Novellino, E.; Grieco, P.; Mirone, V.; Cirino, G.; et al. A new therapeutic approach to erectile dysfunction: Urotensin-II receptor high affinity agonist ligands. Asian J. Androl. 2015, 17, 81–85. [Google Scholar] [PubMed]
- Lehmann, F.; Currier, E.A.; Clemons, B.; Hansen, L.K.; Olsson, R.; Hacksell, U.; Luthman, K. Novel and potent small-molecule urotensin II receptor agonists. Bioorg. Med. Chem. 2009, 17, 4657–4665. [Google Scholar] [CrossRef] [PubMed]
- Maryanoff, B.E.; Kinney, W.A. Urotensin-II receptor modulators as potential drugs. J. Med. Chem. 2010, 53, 2695–2708. [Google Scholar] [CrossRef]
- Lehmann, P.F.; Luthman, I.K.; Kary, M.B.; Andersson, C.M. Urotensin II receptor agents. U.S. Patent 10/458,796, 22 April 2004. [Google Scholar]
- Azam, M.; Sahoo, P.K.; Mohapatra, R.K.; Kumar, M.; Ansari, A.; Moon, I.S.; Chutia, A.; Al-Resayes, S.I.; Biswal, S.K. Structural investigations, Hirsfeld surface analyses, and molecular docking studies of a phenoxo-bridged binuclear Zinc(II) complex. J. Mol. Struct. 2022, 1251, 132039. [Google Scholar] [CrossRef]
- Kansiz, S.; Tolan, A.; Azam, M.; Dege, N.; Alam, M.; Sert, Y.; Al-Resayes, S.I.; Icbudak, H. Acesulfame based Co(II) complex: Synthesis, structural investigations, solvatochromism, Hirshfeld surface analysis and molecular docking studies. Polyhedron Int. J. Inorg. Organomet. Chem. 2022, 218, 115762. [Google Scholar] [CrossRef]
- Kaushik, A.C.; Kumar, S.; Wei, D.Q.; Sahi, S. Structure Based Virtual Screening Studies to Identify Novel Potential Compounds for GPR142 and Their Relative Dynamic Analysis for Study of Type 2 Diabetes. Front. Chem. 2018, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Jockers, R. Structure-Based Virtual Screening Accelerates GPCR Drug Discovery. Trends Pharmacol. Sci. 2020, 41, 382–384. [Google Scholar] [CrossRef]
- Fu, Y.; Sun, Y.-N.; Yi, K.-H.; Li, M.-Q.; Cao, H.-F.; Li, J.-Z.; Ye, F. 3D Pharmacophore-Based Virtual Screening and Docking Approaches toward the Discovery of Novel HPPD Inhibitors. Molecules 2017, 22, 959. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Yin, J.; Yao, J.; Xu, Z.; Tao, Y.; Zhang, H. Pharmacophore-Based Virtual Screening Toward the Discovery of Novel Anti-echinococcal Compounds. Front. Cell Infect. Microbiol. 2020, 10, 118. [Google Scholar] [CrossRef]
- Banerjee, S.; Norman, D.D.; Deng, S.; Fakayode, S.O.; Lee, S.C.; Parrill, A.L.; Li, W.; Miller, D.D.; Tigyi, D.J. Molecular modelling guided design, synthesis and QSAR analysis of new small molecule non-lipid autotaxin inhibitors. Bioorg. Chem. 2020, 103, 104188. [Google Scholar] [CrossRef]
- Coudrat, T.; Simms, J.; Christopoulos, A.; Wootten, D.; Sexton, P.M. Improving virtual screening of G protein-coupled receptors via ligand-directed modeling. PLoS Comput. Biol. 2017, 13, e1005819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Fu, W.; Wang, Z.; Wang, X.; Lei, T.; Zhu, F.; Li, D.; Chang, S.; Xu, L.; Hou, T. Reliability of Docking-Based Virtual Screening for GPCR Ligands with Homology Modeled Structures: A Case Study of the Angiotensin II Type I Receptor. ACS Chem. Neurosci. 2019, 10, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Flohr, S.; Kurz, M.; Kostenis, E.; Brkovich, A.; Fournier, A.; Klabunde, T. Identification of nonpeptidic urotensin II receptor antagonists by virtual screening based on a pharmacophore model derived from structure-activity relationships and nuclear magnetic resonance studies on urotensin II. J. Med. Chem. 2002, 45, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Lescot, E.; Santos, J.S.-D.O.; Colloc’H, N.; Rodrigo, J.; Milazzo-Segalas, I.; Bureau, R.; Rault, S. Three-dimensional model of the human urotensin-II receptor: Docking of human urotensin-II and nonpeptide antagonists in the binding site and comparison with an antagonist pharmacophore model. Proteins Struct. Funct. Bioinform. 2008, 73, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Goddard, W.A., III; Yi, K.Y.; Lee, B.H.; Lim, C.J.; Trzaskowski, B. Predicted ligands for the human urotensin-II G protein-coupled receptor with some experimental validation. ChemMedChem 2014, 9, 1732–1743. [Google Scholar] [PubMed]
- Nassour, H.; Iddir, M.; Chatenet, D. Towards Targeting the Urotensinergic System: Overview and Challenges. Trends Pharmacol. Sci. 2019, 40, 725–734. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Brulé, C.; Perzo, N.; Joubert, J.E.; Sainsily, X.; Leduc, R.; Castel, H.; Prézeau, L. Biased signaling regulates the pleiotropic effects of the urotensin II receptor to modulate its cellular behaviors. FASEB J. 2014, 28, 5148–5162. [Google Scholar] [CrossRef]
- Li, Y.-Y.; Shi, Z.-M.; Yu, X.-Y.; Feng, P.; Wang, X.-J. Urotensin II-induced insulin resistance is mediated by NADPH oxidase-derived reactive oxygen species in HepG2 cells. World J. Gastroenterol. 2016, 22, 5769–5779. [Google Scholar] [CrossRef]
- Kim, M.Y.; Ilyosbek, S.; Lee, B.H.; Yi, K.Y.; Jung, Y.S. A novel urotensin II receptor antagonist, KR-36676, prevents ABCA1 repression via ERK/IL-1β pathway. Eur. J. Pharmacol. 2017, 803, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Kinney, W.A.; Almond, H.R., Jr.; Qi, J.; Smith, C.E.; Santulli, R.J.; de Garavilla, L.; Andrade-Gordon, P.; Cho, D.S.; Everson, A.M.; Feinstein, M.A.; et al. Structure-function analysis of urotensin II and its use in the construction of a ligand-receptor working model. Angew. Chem. Int. Ed. Engl. 2002, 41, 2940–2944. [Google Scholar] [CrossRef] [PubMed]
- Jianyi, Y.; Yang, Z. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar]
- Banegas-Luna, A.J.; Cerón-Carrasco, J.P.; Pérez-Sánchez, H. A review of ligand-based virtual screening web tools and screening algorithms in large molecular databases in the age of big data. Future Med. Chem. 2018, 10, 2641–2658. [Google Scholar] [CrossRef]
- Miles, J.A.; Ross, B.P. Recent advances in virtual screening for cholinesterase inhibitors. ACS Chem. Neurosci. 2020, 12, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Batuwangala, M.; Camarda, V.; McDonald, J.; Marzola, E.; Lambert, D.G.; Ng, L.L.; Calo’, G.; Regoli, D.; Trapella, C.; Guerrini, R.; et al. Structure-activity relationship study on Tyr9 of urotensin-II(4-11): Identification of a partial agonist of the UT receptor. Peptides 2009, 30, 1130–1136. [Google Scholar] [CrossRef]
- Lecointre, C.; Desrues, L.; Joubert, J.E.; Perzo, N.; Guichet, P.O.; Le Joncour, V.; Brulé, C.; Chabbert, M.; Leduc, R.; Prézeau, L.; et al. Signaling switch of the urotensin II vasosactive peptide GPCR: Prototypic chemotaxic mechanism in glioma. Oncogene 2015, 34, 5080–5094. [Google Scholar] [CrossRef]
- Jiang, Z.; Michal, J.J.; Tobey, D.J.; Wang, Z.; MacNeil, M.D.; Magnuson, N.S. Comparative understanding of UTS2 and UTS2R genes for their involvement in type 2 diabetes mellitus. Int. J. Biol. Sci. 2008, 4, 96–102. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Fang, S.; Que, K.; Xu, G.; Zhang, H.; Qi, C.; Yang, N. Urotensin-II gene rs228648 polymorphism associated with the risk of diabetes mellitus. Biosci. Rep. 2018, 38, BSR20181275. [Google Scholar] [CrossRef]
- Tang, C.; Ksiazek, I.; Siccardi, N.; Gapp, B.; Weber, D.; Wirsching, J.; Beck, V.; Reist, M.; Gaudet, L.; Stuber, N.; et al. UTS2B Defines a Novel Enteroendocrine Cell Population and Regulates GLP-1 Secretion Through SSTR5 in Male Mice. Endocrinology 2019, 160, 2849–2860. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2017, 45, D12–D17. [Google Scholar]
- Isberg, V.; Mordalski, S.; Munk, C.; Rataj, K.; Harpsøe, K.; Hauser, A.S.; Vroling, B.; Bojarski, A.J.; Vriend, G.; Gloriam, D.E. GPCRdb: An information system for G protein-coupled receptors. Nucleic Acids Res. 2017, 45, 2936. [Google Scholar] [CrossRef] [PubMed]
- Worth, C.L.; Kreuchwig, F.; Tiemann, J.K.; Kreuchwig, A.; Ritschel, M.; Kleinau, G.; Hildebrand, P.W.; Krause, G. GPCR-SSFE 2.0-a fragment-based molecular modeling web tool for Class A G-protein coupled receptors. Nucleic Acids Res. 2017, 45, W408–W415. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, W.; He, B.; Walker, S.E.; Zhang, H.; Govindarajoo, B.; Virtanen, J.; Xue, Z.; Shen, H.-B.; Zhang, Y. Template-based protein structure prediction in CASP11 and retrospect of I-TASSER in the last decade. Proteins Struct. Funct. Bioinform. 2016, 84 (Suppl. 1), 233–246. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Kenny, P.W. The nature of ligand efficiency. J. Chemin. 2019, 11, 8. [Google Scholar] [CrossRef]
- Xia, J.; Jin, H.; Liu, Z.; Zhang, L.; Wang, X.S. An Unbiased Method To Build Benchmarking Sets for Ligand-Based Virtual Screening and its Application To GPCRs. J. Chem. Inf. Model. 2014, 54, 1433–1450. [Google Scholar] [CrossRef]
- Xia, J.; Li, S.; Ding, Y.; Wu, S.; Wang, X.S. MUBD-DecoyMaker 2.0: A Python GUI Application to Generate Maximal Unbiased Benchmarking Data Sets for Virtual Drug Screening. Mol. Inform. 2020, 39, e1900151. [Google Scholar] [CrossRef]
- Kroeze, W.K.; Sassano, M.F.; Huang, X.-P.; Lansu, K.; McCorvy, J.D.; Giguère, P.M.; Sciaky, N.; Roth, B.L. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 2015, 22, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.Y.; Zhu, X.M.; Chen, B.N.; Qiang, G.F.; Fang, L.H.; Du, G.H. Establishment of GPCR expression cell lines using SNAP-tag technology: A case example of Urotensin II receptor. Life Sci. J. 2013, 10, 2519–2525. [Google Scholar]
- Shi, L.-L.; Jia, W.-H.; Zhang, L.; Xu, C.-Y.; Chen, X.; Yin, L.; Wang, N.-Q.; Fang, L.-H.; Qiang, G.-F.; Yang, X.-Y.; et al. Glucose consumption assay discovers coptisine with beneficial effect on diabetic mice. Eur. J. Pharmacol. 2019, 859, 172523. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Yin, L.; Chen, X.; Shang, J.; Liang, M.; Gao, L.; Qiang, G.; Xia, J.; Du, G.; Yang, X. Combination of Docking-Based and Pharmacophore-Based Virtual Screening Identifies Novel Agonists That Target the Urotensin Receptor. Molecules 2022, 27, 8692. https://doi.org/10.3390/molecules27248692
Li N, Yin L, Chen X, Shang J, Liang M, Gao L, Qiang G, Xia J, Du G, Yang X. Combination of Docking-Based and Pharmacophore-Based Virtual Screening Identifies Novel Agonists That Target the Urotensin Receptor. Molecules. 2022; 27(24):8692. https://doi.org/10.3390/molecules27248692
Chicago/Turabian StyleLi, Na, Lin Yin, Xi Chen, Jiamin Shang, Meidai Liang, Li Gao, Guifen Qiang, Jie Xia, Guanhua Du, and Xiuying Yang. 2022. "Combination of Docking-Based and Pharmacophore-Based Virtual Screening Identifies Novel Agonists That Target the Urotensin Receptor" Molecules 27, no. 24: 8692. https://doi.org/10.3390/molecules27248692