
Targeting Replication Stress Response Pathways to Enhance Genotoxic Chemo- and Radiotherapy

Department of Environmental and Radiological Health Sciences, Colorado State University, Fort Collins, CO 80523, USA
Molecules 2022, 27(15), 4736; https://doi.org/10.3390/molecules27154736
Submission received: 1 July 2022
/
Revised: 20 July 2022
/
Accepted: 22 July 2022
/
Published: 25 July 2022
(This article belongs to the Special Issue Research and Development of DNA Repair Inhibitors)
Abstract
:Proliferating cells regularly experience replication stress caused by spontaneous DNA damage that results from endogenous reactive oxygen species (ROS), DNA sequences that can assume secondary and tertiary structures, and collisions between opposing transcription and replication machineries. Cancer cells face additional replication stress, including oncogenic stress that results from the dysregulation of fork progression and origin firing, and from DNA damage induced by radiotherapy and most cancer chemotherapeutic agents. Cells respond to such stress by activating a complex network of sensor, signaling and effector pathways that protect genome integrity. These responses include slowing or stopping active replication forks, protecting stalled replication forks from collapse, preventing late origin replication firing, stimulating DNA repair pathways that promote the repair and restart of stalled or collapsed replication forks, and activating dormant origins to rescue adjacent stressed forks. Currently, most cancer patients are treated with genotoxic chemotherapeutics and/or ionizing radiation, and cancer cells can gain resistance to the resulting replication stress by activating pro-survival replication stress pathways. Thus, there has been substantial effort to develop small molecule inhibitors of key replication stress proteins to enhance tumor cell killing by these agents. Replication stress targets include ATR, the master kinase that regulates both normal replication and replication stress responses; the downstream signaling kinase Chk1; nucleases that process stressed replication forks (MUS81, EEPD1, Metnase); the homologous recombination catalyst RAD51; and other factors including ATM, DNA-PKcs, and PARP1. This review provides an overview of replication stress response pathways and discusses recent pre-clinical studies and clinical trials aimed at improving cancer therapy by targeting replication stress response factors.
1. Introduction
Cancer is treated with one or more modalities comprising surgery, genotoxic (DNA damaging) chemo- and radiotherapy, and, increasingly, targeted therapeutics that block oncogenic pathways or enhance immunologic responses in “personalized” therapy. Each treatment modality has advantages and limitations. Surgery can be curative, but it is only effective against local disease, it is invasive, which causes pain and poses risks of infection, and many tumors are unresectable. Genotoxic chemo- and radiotherapy can effectively kill rapidly dividing tumor cells, but some tumor cells are resistant because they divide slowly or are quiescent, such as cancer stem cells and dormant cells [1,2,3]. General genotoxic agents are also limited because they also damage normal tissues, which is dose-limiting. Targeted therapies block oncogenic (cell growth) pathways or enhance immune responses and are therefore cancer-specific, with generally milder side effects than non-targeted therapeutics.
The complexity and built-in redundancy of oncogenic pathways allows resistance to targeted agents to develop by mutation of target genes or by activation of alternative oncogenic cell growth pathways, limiting their long-term effectiveness. For example, the tyrosine kinase inhibitors (TKIs) gefitinib and erlotinib are first-line therapeutics for EGFR-mutant non-small cell lung cancer, but resistance usually develops within two years [4]. This resistance can be overcome by second- and third generation TKIs, but these too soon succumb to resistance [4]. Resistance to agents that target oncogenic/immunologic pathways is driven by genome instability and resultant tumor heterogeneity, two key hallmarks of cancer [5]. In fact, it has been calculated that high mutation rates typically observed in tumors essentially guarantee that several resistance mutations exist before therapy begins [6,7]. In addition to extant resistance mutations, new resistance mutations can arise by several mechanisms, including induction by genotoxic therapeutics, upregulated mutagenesis in response to therapeutic stress (‘adaptive mutability’), and when tumors modulate their microenvironment [8,9,10,11]. Therapeutic resistance is a widespread problem, limiting the long-term efficacy of both targeted and non-targeted therapeutics. Quoting a recent review:
“…regardless of the mechanism of administered therapies (either targeted agents, chemotherapy, or immunotherapy), resistance is a near-universal occurrence.”[5]
Similar to surgery, radiotherapy is generally limited to treatment of local disease. However, recent advances in our understanding of the abscopal effect, wherein radiation-ablated tumor cells stimulate an immune attack on tumor cells outside the radiation field, has suggested combining radiotherapy with the new generation of immune checkpoint inhibitors to enhance the abscopal effect [12,13,14]. Given a sufficient dose, there is no absolute tumor resistance to radiotherapy [15]. However, as with genotoxic chemotherapeutics, normal tissue tolerance limits radiotherapy doses and many tumors show natural radio-resistance that reflects, for example, slow growing cancer stem cells, dormant cells, hypoxia, enhanced DNA repair, and accelerated tumor cell repopulation [15]. Despite these limitations, genotoxic chemo- and radiotherapy remain critical weapons in the cancer fight.
The effectiveness of genotoxic agents, whether chemical or physical, reflects the sensitivity of tumor cells to DNA damage, and, in particular, DNA double-strand breaks (DSBs), a type of double-strand damage. Along with inter-strand crosslinks induced by bifunctional reactive chemicals such as cyclophosphamide, melphalan, mitomycin C, cisplatin, and psoralen compounds [16], DSBs are among the most cytotoxic DNA lesions, accounting for the widespread use of DSB-inducing agents in cancer therapy [17,18,19,20]. Unlike single-strand lesions, DSBs and inter-strand crosslinks lack an undamaged repair template in the complementary strand in duplex DNA. With double-strand damage, cells are forced to engage a homologous repair template elsewhere in the genome (homologous chromosome, repetitive elements, or sister chromatids in S and G2 phases) to achieve accurate repair, or repair proceeds without a template by error-prone pathways.
Ionizing radiation and most genotoxic chemotherapeutics induce DSBs either directly, or indirectly when single-strand DNA lesions block replicative polymerases, causing replication stress that can lead to replication fork collapse to DSBs [21]. Most DSBs are repaired by canonical non-homologous end-joining (cNHEJ) and homologous recombination (HR). cNHEJ is the dominant DSB repair pathway throughout the cell cycle, while HR is largely restricted to S and G2 phases when sister chromatids are available to serve as homologous repair templates [22]. cNHEJ is fast and efficient, but it is error-prone: repair junctions typically show loss or gain of a few nucleotides [23]. Because HR uses a homologous sequence as a repair template, it is generally accurate. However, any sequence differences (e.g., single-nucleotide polymorphisms) are copied from the donor (undamaged) duplex to the recipient (repaired) duplex, a phenomenon termed gene conversion [24]. In addition, crossovers during HR can cause rearrangements including deletions, inversions, translocations, and large-scale loss of heterozygosity, depending on the linkage and configuration of the interacting homologous sequences [25]. cNHEJ and HR are backed up by alternative NHEJ and single-strand annealing, two intrinsically error-prone pathways that gain importance in cells with defects in cNHEJ and HR factors [26,27,28]. These DSB repair pathways are the effector modules of the complex network of DNA damage sensing, signaling, and repair processes termed the DNA damage response (DDR). The DDR plays a critical role in maintaining genome stability. Key components of the DDR are often dysregulated in tumor cells, as such defects can increase mutation rates and block apoptosis, allowing cells with excessive genomic damage to survive. Ongoing genome instability allows tumor subpopulations to test genetic adaptations that might promote cell growth in the face of intrinsic and extrinsic stressors. Such adaptations can further dysregulate cell growth; promote survival in hostile microenvironments (i.e., low oxygen and nutrient levels, attack by the immune system); gain cell migratory functions that drive invasion and metastasis; develop resistance to intrinsic oncogenic stress reflecting dysregulated DNA replication [29,30]; and develop resistance to extrinsic stress from genotoxic therapeutics [15,31].
Genotoxic chemotherapeutics do not induce DSBs directly, but instead create single-strand DNA lesions (adducts, oxidized bases) that generate DSBs indirectly by blocking DNA replication [20,32]. Although ionizing radiation can induce DSBs directly, most DNA lesions are single-stranded, comprising more than 100 different types including single-strand breaks [33], and these also cause replication stress that results in replication-associated DSBs [34]. Thus, all genotoxic cancer therapeutics induce replication stress, and tumor cells survive these treatments by activating parts of the DDR that manage replication stress. This review focuses on replication stress response factors that have emerged as potential targets to augment the efficacy of genotoxic chemo- and radiotherapy.
2. Key Features of the Cellular DNA Damage Response
Cellular responses to DNA damage can be described in three phases: (i) lesion recognition (sensing), (ii) signaling through phosphorylation cascades and other signal amplification systems, and (iii) activation of checkpoint responses that arrest the cell cycle; promote DNA repair, promote repair and restart stalled or collapsed replication forks; and trigger programmed cell death by apoptosis, autophagy, or other death pathways when damage is excessive [35,36,37]. The DDR is regulated by a trio of kinases in the phosphatidyl inositol 3’ kinase-related kinase (PIKK) family, DNA-PKcs (DNA dependent protein kinase catalytic subunit), ATM (ataxia telangiectasia mutated), and ATR (ATM and RAD3-related). These are very large proteins (300 to 470 kDa) with common structural elements, including FRAP-ATM-TRRAP (FAT) and FAT C-terminal (FATC) domains that flank and regulate kinase domains, and HEAT repeats that mediate protein–protein interactions [35]. These PIKKs are quite promiscuous: ATM and ATR phosphorylate at least 900 targets in >700 proteins, typically at consensus SQ or TQ motifs [38], although non-consensus targets have been identified [39]. In response to DSBs and replication stress, activated PIKKs are themselves targets, via auto- and trans-phosphorylation.
There is significant crosstalk among the PIKKs, as certain autophosphorylation targets may be phosphorylated by other PIKKs under specific conditions [40,41,42]. Replication protein A (RPA) has several residues targeted by multiple PIKKs, demonstrating further crosstalk [43,44]. Despite this crosstalk, the primary roles of DNA-PKcs and ATM are to promote the repair of two-ended (frank) DSBs by cNHEJ and HR, respectively (Figure 1A). The primary roles of ATR are to regulate replication, respond to replication stress by protecting stalled replication forks by preventing replisome dissociation and fork collapse to DSBs [45,46], and to promote HR-mediated repair and restart of collapsed forks [47,48].
PIKK kinases are activated in response to DNA damage by complex mechanisms. Phosphorylation of ATM was suggested to contribute to activation of its kinase through a feed-forward mechanism [49,50,51], although later studies in cells and mice questioned this conclusion [52,53,54,55]. Phosphorylation of DNA-PKcs is an important step in DSB repair by cNHEJ [39,56,57]. ATR is activated/phosphorylated in response to single-stranded DNA (ssDNA) coated with the trimeric RPA complex [58,59], a process mediated by the ATR interacting protein ATRIP [60]. ssDNA forms naturally and is bound by RPA at replication forks when the MCM helicase unwinds DNA ahead of the fork. Constitutive, low-level ATR activation by physiologic levels of ssDNA-RPA regulates replication origin firing to achieve a relatively stable number of active replication forks during the S phase [61]. When a replisome encounters a blocking lesion, the MCM helicase can decouple from the leading-strand polymerase, producing long segments of ssDNA-RPA, and this causes full activation of ATR (Figure 1B). Recent evidence indicates that ATR and ATRIP form a tetrameric complex with two ATR and two ATRIP subunits, and that ATR activation depends on either TopBP1 (in response to DNA damage) or ETAA1 (regulating ATR during normal DNA replication and mitosis) [62,63,64].
3. Sources of DNA Replication Stress
Cells experience replication stress continuously from spontaneous DNA damage, difficult-to-replicate DNA sequences, and collisions between replication and transcription machineries. Nearly all DNA lesions block replicative polymerases, causing fork stalling, including single-strand breaks, DSBs, pyrimidine dimers, adducted and oxidized bases, and broken rings. DNA lesions cause localized replication stress, but genome-wide stress can be induced by the depletion of nucleotide pools with hydroxyurea, and by aphidicolin, a DNA polymerase inhibitor [32,65,66]. Spontaneous DNA damage results from normal DNA lability, ribonucleotide misincorporation, and reactive oxygen species (ROS), including hydroxyl radicals (OH⦁) and superoxide anions (O2⦁−) generated during mitochondrial oxidative metabolism [67,68]. ROS causes oxidative DNA damage, abasic sites, single-strand breaks, and other lesions [69] that have been linked to mutagenesis, cellular senescence, redox imbalance, and aging [70]. For the most part, highly transcribed genes are arranged within chromosomal replication domains to minimize collisions between replication and transcription, but chromosomal rearrangements, including translocations and inversions, can upset this order and increase replication stress. Highly transcribed ribosomal RNA genes, telomeres, and fragile sites are also sources of replication stress [71,72,73,74,75]. Certain DNA sequences are difficult to replicate, and these are additional endogenous sources of replication stress, including G-rich sequences in which stable R-loops [76,77,78], and G-quadruplex structures can form [71,79,80,81,82]. Replication stress is also caused by induced DNA lesions from incidental exposures to exogenous genotoxic chemicals in air, water, and food; from ultraviolet light (primarily from sun exposure) which causes damage directly and through ROS generation; from intentional exposures to genotoxic chemotherapeutics and ionizing radiation during cancer therapy; and by topoisomerase I inhibitors that induce replication stress by causing DNA overwinding ahead of the replication fork, driven by the MCM helicase [83,84,85].
4. Repair and Restart of Stressed Replication Forks
Several DNA damage tolerance pathways exist that allow lesions to be bypassed and replication to continue, leaving the lesions to be repaired at a later time. One tolerance mechanism is direct lesion bypass by less structurally constrained translesion DNA synthesis (TLS) polymerases. These include members of the DNA polymerase Y-family, Rev1, Pol η, Pol ι and Pol κ; Pol θ in the A-family, Pol ζ in the B-family, and PRIMPOL [86,87]. Reflecting their flexibility in catalyzing synthesis on damaged DNA templates, TLS polymerases are error-prone, and therefore represent important drivers of mutagenesis and carcinogenesis [88,89,90,91]. Pol β with a primary role in base excision repair, is another error-prone polymerase that has been linked to cancer predisposition and is thus accurately described as a tumor suppressor [92,93,94]. Other lesion tolerance pathways are repriming by PRIMPOL (which results in under-replicated single-strand gaps [95]), HR-mediated template switching, and passive rescue of a stalled fork by an adjacent fork [21,96].
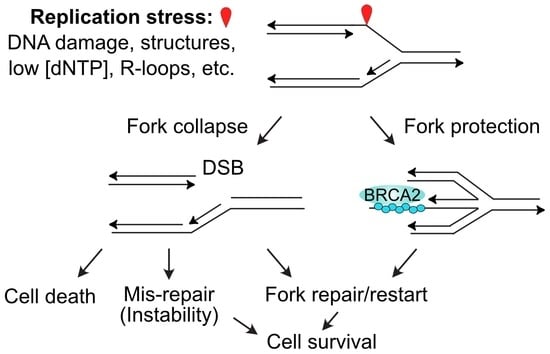
Stalled replication forks may also be restarted by at least three mechanisms that involve fork regression or fork cleavage. A blocked fork may regress, creating a 4-strand branched structure that resembles a Holliday junction, and this allows the blocked strand to anneal to the nascent strand of the sister chromatid, and prime synthesis using the nascent strand as a template (Figure 2A). If this synthesis extends far enough, the regressed fork can reverse in a reaction catalyzed by RECQ1 [83], reestablishing a functional replication fork, and bypassing the blocking lesion (Figure 2A). Regression of a stalled fork also produces a structure with a one-ended DSB that is prone to attack by nucleases. Cells protect these ends with two key HR proteins, RAD51 and BRCA2 (Figure 2B), along with a host of other factors including RIF1, FANCA, FANCC, FANCD2, FANCG, ABRO1, VHL, RADX, and BOD1L [97,98,99]. There are at least two distinct fork regression mechanisms, one promoted by HLTF, SMARCAL1 and ZRANB3, and another by FBH1; 53BP1 is a fork protection factor for FBH1-remodeled forks [97]. The RAD51 filament in the protected regressed fork can catalyze strand invasion to reestablish a functional replication fork (Figure 2B). A third class of fork restart pathway involves cleavage of the stalled fork by branched structure-selective nucleases MUS81 (with its EME2 cofactor) or EEPD1 [100,101,102,103,104] (Figure 2C). Cleavage of a stalled fork creates a one-ended DSB that poses a significant risk of large-scale genome rearrangement if cNHEJ mediates rejoining with another DSB elsewhere in the genome (i.e., at another broken fork or one of the ends at a frank, two-ended DSB). To avoid genome destabilization by cNHEJ-mediated mis-repair of the one-ended DSBs at cleaved forks, the EEPD1 pathway includes an important safeguard. EEPD1 interacts with and promotes EXO1-mediated end-resection of the one-ended DSB to inhibit cNHEJ and promote accurate fork repair and restart by HR [21,102]. An analogous system to promote the resection of one-ended DSBs induced by MUS81-EME2 has yet to be described. MUS81 is a 3′ nuclease first identified in Saccharomyces cerevisiae [105] and this forces invasion of the one-ended DSB into the discontinuous lagging strand duplex. In contrast, the 5′ nuclease EEPD1 evolved more recently in chordates/vertebrates, and this mode of cleavage allows invasion by the resected end into the continuous, leading strand duplex. The EEPD1 cleavage mechanism may speed fork restart and thereby reduce the opportunity for stressed forks to assume toxic structures, promoting both genome stability and cell survival under stress [43].
5. Targeting Replication Stress Signaling and Fork Repair/Restart Pathways in Cancer Therapy
The importance of replication stress responses in cells exposed to genotoxic chemo- or radiotherapy has prompted considerable research focused on how tumor cells might be selectively killed by combined treatments with genotoxins and agents targeting DDR and replication stress factors. TLS polymerases and other DNA damage tolerance pathways contribute to cancer chemoresistance, hence key factors in these pathways have been explored as potential targets to augment genotoxic therapies. These targets are not discussed further here as several excellent treatises have been published on this topic [87,106,107,108,109,110,111,112,113,114,115]. Table 1 summarizes the replication stress factors targeted in cancer therapy, discussed below.
5.1. Targeting Downstream DNA Damage Checkpoint Factors (Chk1, Chk2, Wee1)
Many DDR targets along the DNA damage sensing-signaling-repair continuum have been explored as cancer therapeutic targets. In the 1990s and early 2000s, there was considerable excitement about combining genotoxic chemo- or radiotherapy with inhibitors of checkpoint effector kinases. The general idea was that by inhibiting effector checkpoint kinases Chk1, Chk2, or Wee1, cells experiencing genotoxic stress would fail to arrest, and continued cell cycle progression in the face of significant DNA damage would lead to cell death by mitotic catastrophe or other mechanisms. This prompted the early development of Chk1 inhibitors (Chk1i) including UCN-01. Despite success in preclinical studies, in a Phase I trial, UCN-01 had a long half-life but limited bioavailability due to high avidity to plasma α1-acid glycoprotein, and serious side effects were observed when doses were increased to exceed the plasma binding capacity [116]. By 2013, at least 12 additional Chk1i were developed, but like UCN-01, many were cross-inhibitory with other targets (e.g., Chk2, CDK1, VEGFR2, PIM1) and only one, LY2603618, combined with the antifolate antineoplastic drug Pemetredex, reached a Phase I/II trial [117]. The severe side effects of UCN-01 may reflect the rather broad impact that Chk1 has on cellular functions, including replication initiation, replication fork stabilization, cell cycle progression, DNA repair, and apoptosis (Figure 1B). Alternatively, the disappointing results with early Chk1i may reflect their lack of specificity. More potent/specific Chk1i have been developed and several have been tested in recent Phase I or Phase II trials in mono- or combination therapies. The Chk1i GDC-0575 (±gemcitabine) had manageable side effects that were primarily hematologic, but GDC-0575 showed limited antitumor activity against advanced solid tumors [118]. A phase I trial with the Chk1i MK-8776 (SCH 900776), also in mono- or combination therapy with gemcitabine against advanced solid tumors, had adverse effects on cardiac function and caused abdominal pain, despite minimal clinical benefit (NCT00779584). In a Phase II trial, MK-8776 tested as an adjunct to AraC (Cytarabine) treatment for relapsed acute myeloid leukemia also showed adverse effects on cardiac function and other organ toxicities but no clinical benefit beyond that of AraC alone (NCT01870596). LY2606368 (Prexasertib) is a Chk1i that causes mitotic catastrophe (also termed replication catastrophe) in vitro and showed antitumor activity in preclinical animal studies [164], yet in several Phase I/II trials, LY2606368 has shown limited clinical benefit with occasional serious side effects [120,121,122,123]. Wee1 is a checkpoint kinase in the ATR/Chk1/Wee1 pathway that negatively regulates the G2/M transition, among other functions [165]. The Wee1i MK1775 (AZD1775, Adavosertib) showed a significant increase in progression free survival in a Phase II trial of ovarian cancer treated with Paclitaxel and carboplatin ± MK1775 [124].
5.2. Targeting the Upstream DNA Damage Checkpoint Kinase ATR and Its Activation Partner TopBP1
Given the propensity of serious side effects and limited clinical benefits to date for Chk1i, it may seem counterintuitive that targeting ATR, which acts upstream of Chk1, would have fewer adverse effects on normal tissues. Nonetheless, potent and relatively specific ATRi have been developed and several have been tested in mono- or combination therapy in Phase I/II trials. The ATRi BAY1895344 has shown initially promising results in a Phase I trial against advanced solid tumors, with ~20% of patients showing partial responses and nearly 40% showing stable disease. Although adverse effects with BAY1895344 were common, including hematologic problems, fatigue, and nausea, these were manageable [125]. The ATRi AZD6738 (Ceralasertib) was well tolerated in combination with carboplatin in a Phase I trial that also showed moderate clinical benefit [126]. Interestingly, in both of these ATRi trials, patients who responded had DDR defects, including loss of ATM. These results should motivate further studies with ATRi and other DNA damage checkpoint inhibitors in patients with DDR defects to further develop personalized therapies.
TopBP1 has many binding partners and functions in a variety of cellular processes [166], including itself, topoisomerase IIβ, p53, and RAD51 [167], and it plays a critical role in ATR signaling to Chk1 (Figure 1A) [168]. TopBP1 expression is frequently increased in osteosarcoma and other sarcomas, and this correlates with poor prognosis [169]. Interestingly, low or moderate levels of TopBP1 correlate with normal ATR/Chk1 activation during stress, but TopBP1 overexpression has an inhibitory effect on ATR/Chk1 activation [170], which, in theory, would promote the resistance of tumor cells to DNA damage by suppressing apoptosis. Because TopBP1 functions depend on protein–protein interactions, specific inhibitors that block these interactions are of interest. The cell viability dye calcein acetoxymethyl ester (calcein AM) was shown to interfere with TopBP1 oligomerization and p53 binding, resulting in the reactivation of apoptosis and interference with mutant/oncogenic p53 [127]. Calcein AM showed antitumor activity against breast cancer xenografts [127] and it increased the antitumor activity of the topoisomerase inhibitor doxorubicin against lung tumor xenografts [128]. To date no TopBP1 inhibitors have advanced to clinical trials.
5.3. Targeting Replication Stress Nucleases (CtIP, MUS81, EEPD1, Metnase)
Several nucleases play key roles in the replication stress response [43]. CtIP is a nuclease and key factor in the early steps of DNA end resection at DSBs, functioning with BRCA1 in both end resection and the protection of stressed replication forks [171,172,173]. It thus plays an important role in creating the ssDNA-RPA substrate required for ATR activation (Figure 1B). A recent study demonstrated that a peptide mimic that blocks CtIP tetramerization is a specific CtIPi that impairs DSB repair, interferes with stressed fork protection, sensitizes cells to DNA damage and PARP1i, and is cytotoxic to BRCA1-defective cells [129]. CtIP is also a target of a lncRNA (lnc15.2)-encoded micropeptide termed PACMP. PACMP stabilizes CtIP by blocking its ubiquitination, and it also promotes poly(ADP)ribosylation of substrates by PARP1 [130]. siRNA knockdown of lnc15.2 causes hypersensitivity to PARP1i, ATRi, CDK4/6i, the TopoI inhibitor camptothecin, the TopoII inhibitor epirubicin, and radiation, and it has antitumor activity against breast tumor xenografts [130]. Together, these results warrant studies to further test the antitumor effects of CtIPi in preclinical and clinical trials.
Several nucleases that process stressed replication forks are being explored as therapeutic targets. MUS81 cleaves stalled replication forks to initiate HR-mediated fork restart [100], and it has shown promise as a target in preclinical studies [131,174]. EEPD1 also cleaves stalled replication forks [103,104] and given that MUS81 and EEPD1 cleave forks with different polarity, combining MUS81i and EEPD1i may be potent approach to augment the cytotoxic effects of replication stress. Metnase is a nuclease that promotes replication fork restart, but it does not cleave stalled forks [103]. Metnase nuclease can be inhibited by the frequently administered antibiotic Ciprofloxacin [132], suggesting a safe and potentially effective means to augment genotoxic cancer therapy, perhaps in combination with drugs targeting other DDR factors.
5.4. Targeting DSB Repair Proteins (RAD51, PARP1)
RAD51 plays a central role in HR, and HR plays a critical role in fork protection and the accurate repair and restart of collapsed replication forks (Figure 2B,C). RAD51 is frequently overexpressed in cancer cells [175], thus RAD51 has emerged as a potentially useful target in cancer therapy. RI-1 is a RAD51i that inhibits RAD51-RAD51 interactions that are important for RAD51-ssDNA nucleoprotein filament formation, the key complex that catalyzes HR [176]. RI-1 has been shown by two groups to radiosensitize glioblastoma cells [133,134] and it enhances the chemosensitivity of glioblastoma cells to genotoxic damage by alkylating agents [135]. The RAD51i B02 interferes with RAD51-mediated strand invasion [177], and B02 sensitizes glioblastoma cells to alkylating agents [135], and it sensitizes multiple myeloma cells to doxorubicin [136]. Tumors with HR defects, such as BRCA1- and BRCA2-defective breast cancer are sensitive to PARP1i because PARP1 promotes the repair of single-strand lesions that block replication forks and therefore require HR to repair/restart these forks [178,179]. The original B02 compound has been modified into a B02-isomer with greater potency, and the HR defect induced by B02-isomer shows strong synergistic sensitization of tumor cells with PARP1i [180].
PARP1 adds poly(ADP)ribose (PAR) groups to numerous target proteins. PARylation of repair factors is seen in response to DNA damage processed by base excision repair, nucleotide excision repair, single-strand break repair, and DSB repair by cNHEJ, alternative NHEJ, and HR [181]. PARP1i induces replication stress by at least two mechanisms: PARP1i increases the load of unrepaired endogenous DNA lesions (analogous to increased lesion load by exogenous genotoxins), and it blocks replication directly by trapping PARP1 on DNA [182]. In addition, recent studies have revealed new roles for PARP1 in chromatin remodeling. DNA repair occurs within the chromatin environment, so it is no surprise that proteins that modify or remodel chromatin influence DNA repair [183,184,185,186,187,188,189,190] and replication stress responses [191,192]. PARP1 has emerged has an important factor in chromatin modeling, and PARylation of both repair factors and chromatin promote the recruitment of repair factors to damaged sites [181,193].
Normal cells cope with PARP1i-induced replication stress by marshaling HR to repair and restart collapsed replication forks, but in cancer cells with HR defects, PARP1i are synthetically lethal [194,195]. First exploited in cancer cells with mutant BRCA1 or BRCA2, PARP1i are potentially valuable against tumors with defects in other HR proteins due to inherited germline mutations or sporadic somatic mutations. The PARP1i olaparib was the first targeted therapeutic approved to treat BRCA-defective ovarian cancer, and PARP1i are being explored to treat BRCA-mutant breast, ovarian, prostate, and pancreatic cancers [196,197,198,199]. In 2020 the FDA approved two PARP1i, olaparib and rucaparib to treat metastatic castration-resistant prostate cancer, and it approved olaparib to treat eligible patients with pancreatic cancer. Because PARP1 plays many roles in DNA repair, replication stress responses, and chromatin remodeling, additional uses for PARP1i in cancer monotherapy, combination therapy, and maintenance therapy continue to be explored (Table 1) [157,158,159,160,161,162,163].
5.5. Exploiting Synthetic Lethality of TKIs Targeting Activated Oncogenes and ATM Inhibitors
When cancer cells activate oncogenes that promote growth, an important consequence is oncogenic stress, also known as oncogenic replicative stress [29]. Oncogenic stress reflects dysregulated replication initiation and progression, including mis-timed origin firing. Many drugs targeting activated oncogenes have been developed and brought to clinical practice, including drugs that target activated HER2/ERBB2, ALK, KRAS, BRAF, or EGFR [200,201,202,203,204,205]. It was recently shown that blocking oncogenic pathways with targeted therapeutics (TKIs) causes moderate stress which results in sublethal DSBs induced by caspase-activated DNase (CAD; also known as DFF40 and DFFB) [156]. When fully activated during apoptosis, CAD is responsible for digesting the genome into ~180 bp “ladders” by cleaving linker DNA between nucleosomes [206]. Although cells survive these TKI/CAD-induced DSBs, their repair depends on ATM, thus ATMi kills cells treated with various TKIs targeting activated oncogenes [156]. This effect appears to be general, as ATMi kills tumor cells treated with cognate TKIs targeting different activated oncogenes (EGFR, ALK, KRAS, and BRAF) in different tumor types (lung, pancreatic, melanoma, and acute myeloid leukemia). Interestingly, TKI + ATMi is cytotoxic even in cells that have gained resistance to the cognate TKI, suggesting ATMi may prove beneficial to patients with TKI-resistant tumors [156]. This TKI + ATMi effect represents a novel approach to exploit replication stress (oncogenic stress) and DDR inhibition to selectively target cancer cells.
6. Perspectives
The DDR has emerged as a rich source of targets that may be exploited to augment the cytotoxic effects of genotoxic chemo- and radiotherapy that cause replication stress. The value of DDR targeting became abundantly clear with the discovery in 2005 that PARP1i selectively kills tumor cells with HR defects [194,195], and this has spurred research seeking additional tumor-specific vulnerabilities related to the DDR, including those based on the addiction of tumor cells to DNA repair [207]. Because DDR functions are important in normal cells, cross-toxicity presents a barrier to effective eradication of tumor cells. The unacceptable side effects caused by early Chk1 inhibitors highlight these challenges. Nonetheless, moving the target up- or downstream along DDR signaling and repair pathways has shown success in preclinical studies, and, as discussed above, several DDR inhibitors are in clinical trials. Each DDR factor presents unique targeting opportunities, from kinase inhibitors and nuclease inhibitors to inhibitors of protein–protein interactions, as evidenced by the RAD51i RI-1. Interactions between PIKKs and other proteins are mediated by their HEAT repeats, so blocking these interactions may prove effective in inhibiting the DDR. It took nearly a decade from the discovery of PARP1i effects on BRCA-defective cancer cells to FDA approval of olaparib in combination therapy of suspected BRCA-deficient/defective ovarian cancer. It took 15 years for olaparib to be approved by the FDA as an ovarian cancer monotherapy. Efforts focused on inhibitors of PARP1, and other replication stress factors, are growing research fields. Given the central role of replication stress in genotoxic cancer therapy, the complexity of the DDR, and the challenges presented by tumor heterogeneity and therapeutic resistance, opportunities to advance replication stress targeting in basic, preclinical, and clinical arenas will continue far into the future.
Funding
Research in the Nickoloff lab is supported by American Lung Association grant LCD-686552 and a grant from the Colorado State University College of Veterinary Medicine and Biomedical Sciences College Research Council.
Acknowledgments
Helpful discussions with S. Bailey, M. Weil, L. Argueso, C. Wiese, T. Kato, N. Sharma, L. Taylor, and R. Hromas are greatly appreciated.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
Calcein AM, calcein acetoxymethyl ester; CAD, caspase-activated deoxyribonuclease; cNHEJ, canonical non-homologous end-joining; DDR, DNA damage response; DSB, DNA double-strand break; FAT, FRAP-ATM-TRRAP domain; FATC, FAT C-terminal domain; HEAT, protein–protein interaction domain named for Huntingtin, EF3, PP2A, and TOR1; HR, homologous recombination; PAR, poly(ADP)ribose; PIKK, phosphatidyl inositol 3’ kinase-related kinase; ROS, reactive oxygen species; ssDNA, single-stranded DNA; TKI, tyrosine kinase inhibitor; TLS, translesion DNA synthesis.
References
- Fattore, L.; Mancini, R.; Ciliberto, G. Cancer stem cells and the slow cycling phenotype: How to cut the gordian knot driving resistance to therapy in melanoma. Cancers 2020, 12, 3368. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Dong, Y.; Kumar, R.; Jeter, C.; Tang, D.G. Slow-cycling (dormant) cancer cells in therapy resistance, cancer relapse and metastasis. Semin. Cancer Biol. 2021, 78, 90–103. [Google Scholar] [CrossRef]
- Tang, H.; Shrager, J.B. CRISPR /Cas-mediated genome editing to treat EGFR -mutant lung cancer: A personalized molecular surgical therapy. EMBO Mol. Med. 2016, 8, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Loeb, L.A. Human cancers express a mutator phenotype: Hypothesis, origin, and consequences. Cancer Res. 2016, 76, 2057–2059. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, M.W.; Loeb, L.A.; Salk, J.J. The influence of subclonal resistance mutations on targeted cancer therapy. Nat. Rev. Clin. Oncol. 2016, 13, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef]
- Gerlinger, M. Targeted drugs ramp up cancer mutability. Science 2019, 366, 1452–1453. [Google Scholar] [CrossRef]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magrì, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Fitzgerald, D.M.; Hastings, P.; Rosenberg, S.M. Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance. Annu. Rev. Cancer Biol. 2017, 1, 119–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daguenet, E.; Louati, S.; Wozny, A.-S.; Vial, N.; Gras, M.; Guy, J.-B.; Vallard, A.; Rodriguez-Lafrasse, C.; Magné, N. Radiation-induced bystander and abscopal effects: Important lessons from preclinical models. Br. J. Cancer 2020, 123, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Reynders, K.; Illidge, T.; Siva, S.; Chang, J.Y.; De Ruysscher, D. The abscopal effect of local radiotherapy: Using immunotherapy to make a rare event clinically relevant. Cancer Treat. Rev. 2015, 41, 503–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Dong, Y.; Kong, L.; Shi, F.; Zhu, H.; Yu, J. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Willers, H.; Azzoli, C.G.; Santivasi, W.; Xia, F. Basic mechanisms of therapeutic resistance to radiation and chemotherapy in lung cancer. Cancer J. 2013, 19, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Nickoloff, J.A.; Sharma, N.; Allen, C.P.; Taylor, L.; Allen, S.J.; Jaiswal, A.S.; Hromas, R. Roles of homologous recombination in response to ionizing radiation-induced DNA damage. Int. J. Radiat. Biol. 2021, 1–12. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Sharma, N.; Taylor, L. Clustered dna double-strand breaks: Biological effects and relevance to cancer radiotherapy. Genes 2020, 11, 99. [Google Scholar] [CrossRef] [Green Version]
- Trenner, A.; Sartori, A.A. Harnessing DNA double-strand break repair for cancer treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef]
- Hsieh, H.-J.; Peng, G. Cellular responses to replication stress: Implications in cancer biology and therapy. DNA Repair 2017, 49, 9–20. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Sharma, N.; Taylor, L.; Allen, S.J.; Hromas, R. The safe path at the fork: Ensuring replication-associated DNA double-strand breaks are repaired by homologous recombination. Front. Genet. 2021, 12, 748033. [Google Scholar] [CrossRef] [PubMed]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [Green Version]
- Nickoloff, J.A. Recombination: Mechanisms and roles in tumorigenesis. In Encyclopedia of Cancer, 2nd ed.; Bertino, J.R., Ed.; Elsevier Science (USA): San Diego, CA, USA, 2002; Volume 4, pp. 49–59. [Google Scholar]
- Tutt, A.; Bertwistle, D.; Valentine, J.; Gabriel, A.; Swift, S.; Ross, G.; Griffin, C.; Thacker, J.; Ashworth, A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001, 20, 4704–4716. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Hills, S.A.; Diffley, J.F. DNA Replication and Oncogene-Induced Replicative Stress. Curr. Biol. 2014, 24, R435–R444. [Google Scholar] [CrossRef] [Green Version]
- Primo, L.M.F.; Teixeira, L.K. DNA replication stress: Oncogenes in the spotlight. Genet. Mol. Biol. 2020, 43, e20190138. [Google Scholar] [CrossRef]
- Li, L.-Y.; Guan, Y.-D.; Chen, X.-S.; Yang, J.-M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11, 629266. [Google Scholar] [CrossRef] [PubMed]
- Vesela, E.; Chroma, K.; Turi, Z.; Mistrik, M. Common Chemical Inductors of Replication Stress: Focus on Cell-Based Studies. Biomolecules 2017, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Ward, J. Complexity of Damage Produced by Ionizing Radiation. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Groth, P.; Orta, M.L.; Elvers, I.; Majumder, M.M.; Lagerqvist, A.; Helleday, T. Homologous recombination repairs secondary replication induced DNA double-strand breaks after ionizing radiation. Nucleic Acids Res. 2012, 40, 6585–6594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA Damage Response and Autophagy: A Meaningful Partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-H.; Tseng, W.H.-S.; Chi, J.-T. The Intersection of DNA Damage Response and Ferroptosis—A Rationale for Combination Therapeutics. Biology 2020, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Jette, N.; Lees-Miller, S.P. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 2014, 117, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.P.C.; Uematsu, N.; Kobayashi, J.; Lerenthal, Y.; Krempler, A.; Yajima, H.; Löbrich, M.; Shiloh, Y.; Chen, D.J. Ataxia Telangiectasia Mutated (ATM) Is Essential for DNA-PKcs Phosphorylations at the Thr-2609 Cluster upon DNA Double Strand Break. J. Biol. Chem. 2007, 282, 6582–6587. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Crowe, J.L.; Liu, X.; Nakajima, S.; Wang, Y.; Li, C.; Lee, B.J.; Dubois, R.L.; Liu, C.; Yu, X.; et al. Differential Phosphorylation of DNA-PKcs Regulates the Interplay between End-Processing and End-Ligation during Nonhomologous End-Joining. Mol. Cell 2015, 58, 172–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yajima, H.; Lee, K.-J.; Chen, B.P.C. ATR-dependent DNA-PKcs phosphorylation in response to UV-induced replication stress. Mol. Cell. Biol. 2006, 26, 7520–7528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickoloff, J.A.; Sharma, N.; Taylor, L.; Allen, S.J.; Hromas, R. Nucleases and Co-Factors in DNA Replication Stress Responses. DNA 2022, 2, 68–85. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2014, 25, 9–23. [Google Scholar] [CrossRef] [Green Version]
- Rickman, K.; Smogorzewska, A. Advances in understanding DNA processing and protection at stalled replication forks. J. Cell Biol. 2019, 218, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickman, K.A.; Noonan, R.J.; Lach, F.; Sridhar, S.; Wang, A.; Abhyankar, A.; Huang, A.; Kelly, M.; Auerbach, A.D.; Smogorzewska, A. Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links. Genes Dev. 2020, 34, 832–846. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Zou, L. ATR: A master conductor of cellular responses to DNA replication stress. Trends Biochem. Sci. 2011, 36, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.M.; Zhang, X. Roles of ATM and ATR in DNA double strand breaks and replication stress. Prog. Biophys. Mol. Biol. 2021, 163, 109–119. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Kozlov, S.V.; Graham, M.E.; Peng, C.; Chen, P.; Robinson, P.J.; Lavin, M.F. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 2006, 25, 3504–3514. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.V.; Graham, M.; Jakob, B.; Tobias, F.; Kijas, A.W.; Tanuji, M.; Chen, P.; Robinson, P.J.; Taucher-Scholz, G.; Suzuki, K.; et al. Autophosphorylation and ATM Activation: Additional sites add to the complexity. J. Biol. Chem. 2011, 286, 9107–9119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupré, A.; Boyer-Chatenet, L.; Gautier, J. Two-step activation of ATM by DNA and the Mre11–Rad50–Nbs1 complex. Nat. Struct. Mol. Biol. 2006, 13, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Paull, T.T. ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.A.; Pellegrini, M.; Lee, J.-H.; Paull, T.T.; Feigenbaum, L.; Nussenzweig, A. Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. J. Cell Biol. 2008, 183, 777–783. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, M.; Celeste, A.; Difilippantonio, S.; Guo, R.; Wang, W.; Feigenbaum, L.; Nussenzweig, A. Autophosphorylation at serine 1987 is dispensable for murine Atm activation in vivo. Nature 2006, 443, 222–225. [Google Scholar] [CrossRef]
- Chen, B.P.C.; Chan, D.W.; Kobayashi, J.; Burma, S.; Asaithamby, A.; Morotomi-Yano, K.; Botvinick, E.; Qin, J.; Chen, D.J. Cell Cycle Dependence of DNA-dependent Protein Kinase Phosphorylation in Response to DNA Double Strand Breaks. J. Biol. Chem. 2005, 280, 14709–14715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.W.; Chen, B.P.-C.; Prithivirajsingh, S.; Kurimasa, A.; Story, M.D.; Qin, J.; Chen, D.J. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002, 16, 2333–2338. [Google Scholar] [CrossRef] [Green Version]
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.X.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Räschle, M.; Mailand, N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207. [Google Scholar] [CrossRef] [Green Version]
- Nam, E.A.; Cortez, D. ATR signalling: More than meeting at the fork. Biochem. J. 2011, 436, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldivar, J.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thada, V.; Cortez, D. ATR activation is regulated by dimerization of ATR activating proteins. J. Biol. Chem. 2021, 296, 100455. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C.; et al. An intrinsic S/G 2 checkpoint enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bass, T.E.; Cortez, D. Quantitative phosphoproteomics reveals mitotic function of the ATR activator ETAA1. J. Cell Biol. 2019, 218, 1235–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, X.; Chastain, M.; Chai, W. Genome-wide mapping and profiling of γH2AX binding hotspots in response to different replication stress inducers. BMC Genom. 2019, 20, 579. [Google Scholar] [CrossRef] [Green Version]
- Mazouzi, A.; Stukalov, A.; Müller, A.C.; Chen, D.; Wiedner, M.; Prochazkova, J.; Chiang, S.-C.; Schuster, M.; Breitwieser, F.P.; Pichlmair, A.; et al. A Comprehensive Analysis of the Dynamic Response to Aphidicolin-Mediated Replication Stress Uncovers Targets for ATM and ATMIN. Cell Rep. 2016, 15, 893–908. [Google Scholar] [CrossRef] [Green Version]
- Sarmiento-Salinas, F.L.; Perez-Gonzalez, A.; Acosta-Casique, A.; Ix-Ballote, A.; Diaz, A.; Treviño, S.; Rosas-Murrieta, N.H.; Millán-Perez-Peña, L.; Maycotte, P. Reactive oxygen species: Role in carcinogenesis, cancer cell signaling and tumor progression. Life Sci. 2021, 284, 119942. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, D.E.; Lindahl, T. Repair and Genetic Consequences of Endogenous DNA Base Damage in Mammalian Cells. Annu. Rev. Genet. 2004, 38, 445–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, A.R.; Yousefzadeh, M.J.; Rozgaja, T.A.; Wang, J.; Li, X.; Tilstra, J.S.; Feldman, C.H.; Gregg, S.Q.; Johnson, C.H.; Skoda, E.M.; et al. Spontaneous DNA damage to the nuclear genome promotes senescence, redox imbalance and aging. Redox Biol. 2018, 17, 259–273. [Google Scholar] [CrossRef]
- Billard, P.; Poncet, D.A. Replication Stress at Telomeric and Mitochondrial DNA: Common Origins and Consequences on Ageing. Int. J. Mol. Sci. 2019, 20, 4959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef]
- Gómez-González, B.; Aguilera, A. Transcription-mediated replication hindrance: A major driver of genome instability. Genes Dev. 2019, 33, 1008–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamperl, S.; Cimprich, K.A. Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell 2016, 167, 1455–1467. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Muse, T.; Aguilera, A. Transcription–replication conflicts: How they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 2016, 17, 553–563. [Google Scholar] [CrossRef]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170, 774–786.e19. [Google Scholar] [CrossRef] [Green Version]
- Freudenreich, C.H. R-loops: Targets for nuclease cleavage and repeat instability. Curr. Genet. 2018, 64, 789–794. [Google Scholar] [CrossRef]
- Poggi, L.; Richard, G.-F. Alternative DNA Structures In Vivo: Molecular Evidence and Remaining Questions. Microbiol. Mol. Biol. Rev. 2021, 85, e00110-20. [Google Scholar] [CrossRef]
- Spiegel, J.; Adhikari, S.; Balasubramanian, S. The Structure and Function of DNA G-Quadruplexes. Trends Chem. 2020, 2, 123–136. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, S.; Freudenreich, C.H. The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosom. Cancer 2018, 58, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Gadaleta, M.C.; Noguchi, E. Regulation of DNA Replication through Natural Impediments in the Eukaryotic Genome. Genes 2017, 8, 98. [Google Scholar] [CrossRef] [Green Version]
- Berti, M.; Chaudhuri, A.R.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Emanuelli, M.; Sartini, D.; Molinelli, E.; Campagna, R.; Pozzi, V.; Salvolini, E.; Simonetti, O.; Campanati, A.; Offidani, A. The Double-Edged Sword of Oxidative Stress in Skin Damage and Melanoma: From Physiopathology to Therapeutical Approaches. Antioxidants 2022, 11, 612. [Google Scholar] [CrossRef]
- Yang, W. An Overview of Y-Family DNA Polymerases and a Case Study of Human DNA Polymerase η. Biochemistry 2014, 53, 2793–2803. [Google Scholar] [CrossRef]
- Ler, A.A.L.; Carty, M.P. DNA Damage Tolerance Pathways in Human Cells: A Potential Therapeutic Target. Front. Oncol. 2021, 11, 822500. [Google Scholar] [CrossRef]
- Goodman, M.F.; Woodgate, R. Translesion DNA Polymerases. Cold Spring Harb. Perspect. Biol. 2013, 5, a010363. [Google Scholar] [CrossRef]
- Ma, X.; Tang, T.; Guo, C. Regulation of translesion DNA synthesis in mammalian cells. Environ. Mol. Mutagen. 2020, 61, 680–692. [Google Scholar] [CrossRef]
- Sale, J.E. Translesion DNA Synthesis and Mutagenesis in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012708. [Google Scholar] [CrossRef]
- Vaisman, A.; Woodgate, R. Translesion DNA polymerases in eukaryotes: What makes them tick? Crit. Rev. Biochem. Mol. Biol. 2017, 52, 274–303. [Google Scholar] [CrossRef] [Green Version]
- Wallace, S.S.; Murphy, D.L.; Sweasy, J.B. Base excision repair and cancer. Cancer Lett. 2012, 327, 73–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donigan, K.A.; Sun, K.-W.; Nemec, A.A.; Murphy, D.L.; Cong, X.; Northrup, V.; Zelterman, D.; Sweasy, J.B. Human POLB Gene Is Mutated in High Percentage of Colorectal Tumors. J. Biol. Chem. 2012, 287, 23830–23839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makridakis, N.M.; Reichardt, J.K.V. Translesion DNA Polymerases and Cancer. Front. Genet. 2012, 3, 174. [Google Scholar] [CrossRef] [Green Version]
- Quinet, A.; Tirman, S.; Cybulla, E.; Meroni, A.; Vindigni, A. To skip or not to skip: Choosing repriming to tolerate DNA damage. Mol. Cell 2021, 81, 649–658. [Google Scholar] [CrossRef]
- Conti, B.A.; Smogorzewska, A. Mechanisms of direct replication restart at stressed replisomes. DNA Repair 2020, 95, 102947. [Google Scholar] [CrossRef]
- Liu, W.; Krishnamoorthy, A.; Zhao, R.; Cortez, D. Two replication fork remodeling pathways generate nuclease substrates for distinct fork protection factors. Sci. Adv. 2020, 6, eabc3598. [Google Scholar] [CrossRef]
- Mukherjee, C.; Tripathi, V.; Manolika, E.M.; Heijink, A.M.; Ricci, G.; Merzouk, S.; De Boer, H.R.; Demmers, J.; Van Vugt, M.A.T.M.; Chaudhuri, A.R. RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 2019, 10, 3287. [Google Scholar] [CrossRef]
- Bhat, K.P.; Krishnamoorthy, A.; Dungrawala, H.; Garcin, E.B.; Modesti, M.; Cortez, D. RADX Modulates RAD51 Activity to Control Replication Fork Protection. Cell Rep. 2018, 24, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Pepe, A.; West, S.C. MUS81-EME2 Promotes Replication Fork Restart. Cell Rep. 2014, 7, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Pepe, A.; West, S.C. Substrate specificity of the MUS81-EME2 structure selective endonuclease. Nucleic Acids Res. 2013, 42, 3833–3845. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-S.; Nickoloff, J.A.; Wu, Y.; Williamson, E.A.; Sidhu, G.S.; Reinert, B.L.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Kong, K.; et al. Endonuclease EEPD1 Is a Gatekeeper for Repair of Stressed Replication Forks. J. Biol. Chem. 2017, 292, 2795–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Speed, M.C.; Allen, C.P.; Maranon, D.G.; Williamson, E.; Singh, S.; Hromas, R.; Nickoloff, J.A. Distinct roles of structure-specific endonucleases EEPD1 and Metnase in replication stress responses. NAR Cancer 2020, 2, zcaa008. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lee, S.-H.; Williamson, E.A.; Reinert, B.L.; Cho, J.H.; Xia, F.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Brantley, A.; et al. EEPD1 Rescues Stressed Replication Forks and Maintains Genome Stability by Promoting End Resection and Homologous Recombination Repair. PLoS Genet. 2015, 11, e1005675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Interthal, H.; Heyer, W.-D. MUS81 encodes a novel Helix-hairpin-Helix protein involved in the response to UV- and methylation-induced DNA damage in Saccharomyces cerevisiae. Mol. Gen. Genet. 2000, 263, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Sweasy, J.B. DNA polymerase κ: Friend or foe? Sci. Signal. 2020, 13, eabb2934. [Google Scholar] [CrossRef]
- Temprine, K.; Campbell, N.R.; Huang, R.; Langdon, E.M.; Simon-Vermot, T.; Mehta, K.; Clapp, A.; Chipman, M.; White, R.M. Regulation of the error-prone DNA polymerase Polκ by oncogenic signaling and its contribution to drug resistance. Sci. Signal. 2020, 13, eaau1453. [Google Scholar] [CrossRef]
- Nayak, S.; Calvo, J.A.; Cong, K.; Peng, M.; Berthiaume, E.; Jackson, J.; Zaino, A.M.; Vindigni, A.; Hadden, M.K.; Cantor, S.B. Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci. Adv. 2020, 6, eaaz7808. [Google Scholar] [CrossRef]
- Nayak, S.; Calvo, J.A.; Cantor, S.B. Targeting translesion synthesis (TLS) to expose replication gaps, a unique cancer vulnerability. Expert Opin. Ther. Targets 2021, 25, 27–36. [Google Scholar] [CrossRef]
- Tonzi, P.; Huang, T.T. Role of Y-family translesion DNA polymerases in replication stress: Implications for new cancer therapeutic targets. DNA Repair 2019, 78, 20–26. [Google Scholar] [CrossRef]
- Saha, P.; Mandal, T.; Talukdar, A.D.; Kumar, D.; Kumar, S.; Tripathi, P.P.; Wang, Q.; Srivastava, A.K. DNA polymerase eta: A potential pharmacological target for cancer therapy. J. Cell. Physiol. 2020, 236, 4106–4120. [Google Scholar] [CrossRef]
- Wilson, D.M.; Duncton, M.A.J.; Chang, C.; Luo, C.L.; Georgiadis, T.M.; Pellicena, P.; Deacon, A.M.; Gao, Y.; Das, D. Early Drug Discovery and Development of Novel Cancer Therapeutics Targeting DNA Polymerase Eta (POLH). Front. Oncol. 2021, 11, 778925. [Google Scholar] [CrossRef]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase θ in Cancer Therapy. Trends Cancer 2020, 7, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszek, J.L.; Chatterjee, N.; Najeeb, J.; Ramos, A.; Lee, M.; Bian, K.; Xue, J.Y.; Fenton, B.A.; Park, H.; Li, D.; et al. A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell 2019, 178, 152–159.e11. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, K.; Chatterjee, N.; Hemann, M.T.; Walker, G.C. Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy? PLoS Genet. 2017, 13, e1006842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuse, E.; Tanii, H.; Kurata, N.; Kobayashi, H.; Shimada, Y.; Tamura, T.; Sasaki, Y.; Tanigawara, Y.; Lush, R.D.; Headlee, D.; et al. Unpredicted clinical pharmacology of UCN-01 caused by specific binding to human alpha1-acid glycoprotein. Cancer Res. 1998, 58, 3248–3253. [Google Scholar]
- Thompson, R.; Eastman, A. The cancer therapeutic potential of Chk1 inhibitors: How mechanistic studies impact on clinical trial design. Br. J. Clin. Pharmacol. 2013, 76, 358–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiano, A.; Infante, J.; Shapiro, G.; Moore, K.; LoRusso, P.; Hamilton, E.; Cousin, S.; Toulmonde, M.; Postel-Vinay, S.; Tolaney, S.; et al. Phase I study of the checkpoint kinase 1 inhibitor GDC-0575 in combination with gemcitabine in patients with refractory solid tumors. Ann. Oncol. 2018, 29, 1304–1311. [Google Scholar] [CrossRef]
- Webster, J.A.; Tibes, R.; Morris, L.; Blackford, A.L.; Litzow, M.; Patnaik, M.; Rosner, G.L.; Gojo, I.; Kinders, R.; Wang, L.; et al. Randomized phase II trial of cytosine arabinoside with and without the CHK1 inhibitor MK-8776 in relapsed and refractory acute myeloid leukemia. Leuk. Res. 2017, 61, 108–116. [Google Scholar] [CrossRef]
- Byers, L.A.; Navarro, A.; Schaefer, E.; Johnson, M.; Özgüroğlu, M.; Han, J.-Y.; Bondarenko, I.; Cicin, I.; Dragnev, K.H.; Abel, A.; et al. A Phase II Trial of Prexasertib (LY2606368) in Patients with Extensive-Stage Small-Cell Lung Cancer. Clin. Lung Cancer 2021, 22, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Cash, T.; Fox, E.; Liu, X.; Minard, C.G.; Reid, J.M.; Scheck, A.C.; Weigel, B.J.; Wetmore, C. A phase 1 study of prexasertib (LY2606368), a CHK1/2 inhibitor, in pediatric patients with recurrent or refractory solid tumors, including CNS tumors: A report from the Children’s Oncology Group Pediatric Early Phase Clinical Trials Network (ADVL1515). Pediatr. Blood Cancer 2021, 68, e29065. [Google Scholar] [CrossRef] [PubMed]
- Gatti-Mays, M.E.; Karzai, F.H.; Soltani, S.N.; Zimmer, A.; Green, J.E.; Lee, M.-J.; Trepel, J.B.; Yuno, A.; Lipkowitz, S.; Nair, J.; et al. A Phase II Single Arm Pilot Study of the CHK1 Inhibitor Prexasertib (LY2606368) in BRCA Wild-Type, Advanced Triple-Negative Breast Cancer. Oncologist 2020, 25, 1013–e1824. [Google Scholar] [CrossRef]
- Hong, D.; Infante, J.; Janku, F.; Jones, S.; Nguyen, L.M.; Burris, H.A.; Naing, A.; Bauer, T.M.; Piha-Paul, S.; Johnson, F.M.; et al. Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients with Advanced Cancer. J. Clin. Oncol. 2016, 34, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Estevez-Diz, M.D.P.; Grischke, E.-M.; Hall, M.; Marmé, F.; Provencher, D.M.; Uyar, D.S.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A Biomarker-enriched, Randomized Phase II Trial of Adavosertib (AZD1775) Plus Paclitaxel and Carboplatin for Women with Platinum-sensitive TP53-mutant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, G.B.C.V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-Human Trial of the Oral Ataxia Telangiectasia and RAD3-Related (ATR) Inhibitor BAY 1895344 in Patients with Advanced Solid Tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Krebs, M.G.; Postel-Vinay, S.; El-Khouiery, A.; Soria, J.-C.; Lopez, J.; Berges, A.; Cheung, S.Y.A.; Irurzun-Arana, I.; Goldwin, A.; et al. Ceralasertib (AZD6738), an Oral ATR Kinase Inhibitor, in Combination with Carboplatin in Patients with Advanced Solid Tumors: A Phase I Study. Clin. Cancer Res. 2021, 27, 5213–5224. [Google Scholar] [CrossRef]
- Chowdhury, P.; Lin, G.E.; Liu, K.; Song, Y.; Lin, F.-T.; Lin, W.-C. Targeting TopBP1 at a convergent point of multiple oncogenic pathways for cancer therapy. Nat. Commun. 2014, 5, 5476. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Liu, R.; Xie, S.; Zheng, X.; Mao, J.; Cai, Y.; Chen, W. Calcein-acetoxymethy ester enhances the antitumor effects of doxorubicin in nonsmall cell lung cancer by regulating the TopBP1/p53RR pathway. Anti-Cancer Drugs 2017, 28, 861–868. [Google Scholar] [CrossRef]
- Kuster, A.; Mozaffari, N.L.; Wilkinson, O.J.; Wojtaszek, J.L.; Zurfluh, C.; Przetocka, S.; Zyla, D.; von Aesch, C.; Dillingham, M.S.; Williams, R.S.; et al. A stapled peptide mimetic of the CtIP tetramerization motif interferes with double-strand break repair and replication fork protection. Sci. Adv. 2021, 7, eabc6381. [Google Scholar] [CrossRef]
- Zhang, C.; Zhou, B.; Gu, F.; Liu, H.; Wu, H.; Yao, F.; Zheng, H.; Fu, H.; Chong, W.; Cai, S.; et al. Micropeptide PACMP inhibition elicits synthetic lethal effects by decreasing CtIP and poly(ADP-ribosyl)ation. Mol. Cell 2022, 82, 1297–1312.e8. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, P.; Li, C.; Liu, W.; Shen, Q.; Yang, L.; Xie, G.; Bai, J.; Li, R.; Tao, K.; et al. MUS81 Inhibition Enhances the Anticancer Efficacy of Talazoparib by Impairing ATR/CHK1 Signaling Pathway in Gastric Cancer. Front. Oncol. 2022, 12, 844135. [Google Scholar] [CrossRef]
- Williamson, E.A.; Damiani, L.; Leitao, A.; Hu, C.; Hathaway, H.; Oprea, T.; Sklar, L.; Shaheen, M.; Bauman, J.; Wang, W.; et al. Targeting the Transposase Domain of the DNA Repair Component Metnase to Enhance Chemotherapy. Cancer Res. 2012, 72, 6200–6208. [Google Scholar] [CrossRef] [Green Version]
- Balbous, A.; Cortes, U.; Guilloteau, K.; Rivet, P.; Pinel, B.; Duchesne, M.; Godet, J.; Boissonnade, O.; Wager, M.; Bensadoun, R.J.; et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC Cancer 2016, 16, 604. [Google Scholar] [CrossRef] [Green Version]
- King, H.O.; Brend, T.; Payne, H.L.; Wright, A.; Ward, T.A.; Patel, K.; Egnuni, T.; Stead, L.F.; Patel, A.; Wurdak, H.; et al. RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem Cell Rep. 2017, 8, 125–139. [Google Scholar] [CrossRef] [Green Version]
- Berte, N.; Piée-Staffa, A.; Piecha, N.; Wang, M.; Borgmann, K.; Kaina, B.; Nikolova, T. Targeting Homologous Recombination by Pharmacological Inhibitors Enhances the Killing Response of Glioblastoma Cells Treated with Alkylating Drugs. Mol. Cancer Ther. 2016, 15, 2665–2678. [Google Scholar] [CrossRef] [Green Version]
- Alagpulinsa, D.A.; Ayyadevara, S.; Reis, R.J.S. A Small-Molecule Inhibitor of RAD51 Reduces Homologous Recombination and Sensitizes Multiple Myeloma Cells to Doxorubicin. Front. Oncol. 2014, 4, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghafoor, A.; Mian, I.; Wagner, C.; Mallory, Y.; Agra, M.G.; Morrow, B.; Wei, J.S.; Khan, J.; Thomas, A.; Sengupta, M.; et al. Phase 2 Study of Olaparib in Malignant Mesothelioma and Correlation of Efficacy with Germline or Somatic Mutations in BAP1 Gene. JTO Clin. Res. Rep. 2021, 2, 100231. [Google Scholar] [CrossRef] [PubMed]
- Poveda, A.M.; Davidson, R.; Blakeley, C.; Milner, A. Olaparib maintenance monotherapy in platinum-sensitive, relapsed ovarian cancer without germline BRCA mutations: OPINION Phase IIIb study design. Future Oncol. 2019, 15, 3651–3663. [Google Scholar] [CrossRef]
- Gelmon, K.A.; Fasching, P.A.; Couch, F.J.; Balmaña, J.; Delaloge, S.; Labidi-Galy, I.; Bennett, J.; McCutcheon, S.; Walker, G.; O’Shaughnessy, J.; et al. Clinical effectiveness of olaparib monotherapy in germline BRCA-mutated, HER2-negative metastatic breast cancer in a real-world setting: Phase IIIb LUCY interim analysis. Eur. J. Cancer 2021, 152, 68–77. [Google Scholar] [CrossRef]
- Cadoo, K.; Simpkins, F.; Mathews, C.; Liu, Y.L.; Provencher, D.; McCormick, C.; ElNaggar, A.C.; Altman, A.D.; Gilbert, L.; Black, D.; et al. Olaparib treatment for platinum-sensitive relapsed ovarian cancer by BRCA mutation and homologous recombination deficiency status: Phase II LIGHT study primary analysis. Gynecol. Oncol. 2022. [Google Scholar] [CrossRef]
- Fasching, P.; Link, T.; Hauke, J.; Seither, F.; Jackisch, C.; Klare, P.; Schmatloch, S.; Hanusch, C.; Huober, J.; Stefek, A.; et al. Neoadjuvant paclitaxel/olaparib in comparison to paclitaxel/carboplatinum in patients with HER2-negative breast cancer and homologous recombination deficiency (GeparOLA study). Ann. Oncol. 2020, 32, 49–57. [Google Scholar] [CrossRef]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.-A.; Park, Y.H.; Delord, J.-P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): An open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Arrieta, O.; Giménez-Capitán, A.; Aldeguer, E.; Drozdowskyj, A.; Chaib, I.; Reguart, N.; Garcia-Campelo, R.; Chen, J.-H.; Molina-Vila, M.A.; et al. BRCA1 Expression and Outcome in Patients With EGFR-Mutant NSCLC Treated with Gefitinib Alone or in Combination with Olaparib. JTO Clin. Res. Rep. 2021, 2, 100113. [Google Scholar] [CrossRef] [PubMed]
- Penson, R.T.; Valencia, R.V.; Cibula, D.; Colombo, N.; Leath, C.A., 3rd; Bidziński, M.; Kim, J.-W.; Nam, J.H.; Madry, R.; Hernández, C.; et al. Olaparib Versus Nonplatinum Chemotherapy in Patients with Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J. Clin. Oncol. 2020, 38, 1164–1174. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men with Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients with Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; LoRusso, P.; Patel, M.R.; Domchek, S.M.; Balmaña, J.; Drew, Y.; Chen, L.-M.; et al. A Phase I–II Study of the Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef] [Green Version]
- Molin, G.Z.D.; Omatsu, K.; Sood, A.K.; Coleman, R.L. Rucaparib in ovarian cancer: An update on safety, efficacy and place in therapy. Ther. Adv. Med. Oncol. 2018, 10, 1758835918778483. [Google Scholar] [CrossRef]
- Okamoto, A.; Kondo, E.; Nakamura, T.; Yanagida, S.; Hamanishi, J.; Harano, K.; Hasegawa, K.; Hirasawa, T.; Hori, K.; Komiyama, S.; et al. Phase 2 single-arm study on the efficacy and safety of niraparib in Japanese patients with heavily pretreated, homologous recombination-deficient ovarian cancer. J. Gynecol. Oncol. 2021, 32, e16. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Thara, E.; Awad, M.M.; Dowlati, A.; Haque, B.; Stinchcombe, T.E.; Dy, G.K.; Spigel, D.R.; Lu, S.; Ms, N.I.S.; et al. JASPER: Phase 2 trial of first-line niraparib plus pembrolizumab in patients with advanced non–small cell lung cancer. Cancer 2021, 128, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined with Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.R.; Lundqvist, E.; Birrer, M.J.; Christensen, R.D.; Nyvang, G.-B.; Malander, S.; Anttila, M.; Werner, T.L.; Lund, B.; Lindahl, G.; et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): A randomised, phase 2, superiority trial. Lancet Oncol. 2019, 20, 1409–1419. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Ali, M.; Lu, M.; Ang, H.X.; Soderquist, R.S.; Eyler, C.E.; Hutchinson, H.M.; Glass, C.; Bassil, C.F.; Lopez, O.M.; Kerr, D.L.; et al. Small-molecule targeted therapies induce dependence on DNA double-strand break repair in residual tumor cells. Sci. Transl. Med. 2022, 14, eabc7480. [Google Scholar] [CrossRef]
- Luo, L.; Keyomarsi, K. PARP inhibitors as single agents and in combination therapy: The most promising treatment strategies in clinical trials for BRCA-mutant ovarian and triple-negative breast cancers. Expert Opin. Investig. Drugs 2022, 31, 607–631. [Google Scholar] [CrossRef]
- Barayan, R.; Ran, X.; Lok, B.H. PARP inhibitors for small cell lung cancer and their potential for integration into current treatment approaches. J. Thorac. Dis. 2020, 12, 6240–6252. [Google Scholar] [CrossRef]
- Rao, A.; Antonarakis, E.S. The growing role of rucaparib in contemporary treatment of metastatic prostate cancer: A review of efficacy and guidance for side effect management. Expert Rev. Anticancer Ther. 2022, 22, 671–679. [Google Scholar] [CrossRef]
- Xia, M.; Guo, Z.; Hu, Z. The Role of PARP Inhibitors in the Treatment of Prostate Cancer: Recent Advances in Clinical Trials. Biomolecules 2021, 11, 722. [Google Scholar] [CrossRef]
- Kamel, D.; Gray, C.; Walia, J.S.; Kumar, V. PARP Inhibitor Drugs in the Treatment of Breast, Ovarian, Prostate and Pancreatic Cancers: An Update of Clinical Trials. Curr. Drug Targets 2018, 19, 21–37. [Google Scholar] [CrossRef]
- Buege, M.; Mahajan, P. Clinical Trials of Poly(ADP-Ribose) Polymerase Inhibitors for Cancer Therapy: A Review. Rev. Recent Clin. Trials 2015, 10, 326–339. [Google Scholar] [CrossRef] [PubMed]
- McCann, K.E.; Hurvitz, S.A. Advances in the use of PARP inhibitor therapy for breast cancer. Drugs Context 2018, 7, 212540. [Google Scholar] [CrossRef] [PubMed]
- King, C.; Diaz, H.B.; McNeely, S.; Barnard, D.; Dempsey, J.; Blosser, W.; Beckmann, R.; Barda, D.; Marshall, M.S. LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther. 2015, 14, 2004–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, S.-B. The expanding role of WEE1. Cell. Signal. 2022, 94, 110310. [Google Scholar] [CrossRef]
- Day, M.; Oliver, A.W.; Pearl, L.H. Phosphorylation-dependent assembly of DNA damage response systems and the central roles of TOPBP1. DNA Repair 2021, 108, 103232. [Google Scholar] [CrossRef] [PubMed]
- Moudry, P.; Watanabe, K.; Wolanin, K.M.; Bartkova, J.; Wassing, I.E.; Watanabe, S.; Strauss, R.; Pedersen, R.T.; Oestergaard, V.; Lisby, M.; et al. TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. J. Cell Biol. 2016, 212, 281–288. [Google Scholar] [CrossRef]
- Wang, J.; Gong, Z.; Chen, J. MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J. Cell Biol. 2011, 193, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.-P.; Yan, X.-B.; Liu, L.-G.; Tian, C.; Han, K.; Zhang, H.; Min, D.-L. TopBP1 promotes malignant progression and correlates with poor prognosis in osteosarcoma. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4022–4031. [Google Scholar]
- Liu, K.; Graves, J.D.; Lin, F.-T.; Lin, W.-C. Overexpression of TopBP1, a canonical ATR/Chk1 activator, paradoxically hinders ATR/Chk1 activation in cancer. J. Biol. Chem. 2021, 296, 100382. [Google Scholar] [CrossRef]
- Cruz-García, A.; López-Saavedra, A.; Huertas, P. BRCA1 Accelerates CtIP-Mediated DNA-End Resection. Cell Rep. 2014, 9, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Makharashvili, N.; Paull, T.T. CtIP: A DNA damage response protein at the intersection of DNA metabolism. DNA Repair 2015, 32, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Przetocka, S.; Porro, A.; Bolck, H.; Walker, C.; Lezaja, A.; Trenner, A.; von Aesch, C.; Himmels, S.-F.; D’Andrea, A.D.; Ceccaldi, R.; et al. CtIP-Mediated Fork Protection Synergizes with BRCA1 to Suppress Genomic Instability upon DNA Replication Stress. Mol. Cell 2018, 72, 568–582.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Geng, X.; Syeda, M.Z.; Huang, Z.; Zhang, C.; Ying, S. Human MUS81: A Fence-Sitter in Cancer. Front. Cell Dev. Biol. 2021, 9, 657305. [Google Scholar] [CrossRef]
- Ward, A.; Khanna, K.K.; Wiegmans, A.P. Targeting homologous recombination, new pre-clinical and clinical therapeutic combinations inhibiting RAD51. Cancer Treat. Rev. 2015, 41, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Budke, B.; Logan, H.L.; Kalin, J.; Zelivianskaia, A.S.; McGuire, W.C.; Miller, L.L.; Stark, J.M.; Kozikowski, A.P.; Bishop, D.K.; Connell, P.P. RI-1: A chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. 2012, 40, 7347–7357. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Mazina, O.M.; Zentner, I.J.; Cocklin, S.; Mazin, A.V. Inhibition of Homologous Recombination in Human Cells by Targeting RAD51 Recombinase. J. Med. Chem. 2012, 55, 3011–3020. [Google Scholar] [CrossRef]
- Cortesi, L.; Rugo, H.S.; Jackisch, C. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target. Oncol. 2021, 16, 255–282. [Google Scholar] [CrossRef]
- del Rivero, J.; Kohn, E.C. PARP inhibitors: The cornerstone of DNA repair-targeted therapies. Oncology 2017, 31, 265–273. [Google Scholar]
- Shkundina, I.; Gall, A.; Dick, A.; Cocklin, S.; Mazin, A. New RAD51 Inhibitors to Target Homologous Recombination in Human Cells. Genes 2021, 12, 920. [Google Scholar] [CrossRef]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Shen, Y.; Aoyagi-Scharber, M.; Wang, B. Trapping Poly(ADP-Ribose) Polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 446–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-H.; Oshige, M.; Durant, S.T.; Rasila, K.K.; Williamson, E.A.; Ramsey, H.; Kwan, L.; Nickoloff, J.A.; Hromas, R. The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc. Natl. Acad. Sci. USA 2005, 102, 18075–18080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Shi, Y.; Mulligan, P.; Gay, F.; Landry, J.; Liu, H.; Lu, J.; Qi, H.; Wang, W.; Nickoloff, J.A.; et al. A YY1–INO80 complex regulates genomic stability through homologous recombination–based repair. Nat. Struct. Mol. Biol. 2007, 14, 1165–1172. [Google Scholar] [CrossRef] [Green Version]
- Chambers, A.L.; Downs, J.A. The RSC and INO80 Chromatin-Remodeling Complexes in DNA Double-Strand Break Repair. Prog. Mol. Biol. Transl. Sci. 2012, 110, 229–261. [Google Scholar] [CrossRef]
- Bao, Y.; Shen, X. Chromatin remodeling in DNA double-strand break repair. Curr. Opin. Genet. Dev. 2007, 17, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Clouaire, T.; Rocher, V.; Lashgari, A.; Arnould, C.; Aguirrebengoa, M.; Biernacka, A.; Skrzypczak, M.; Aymard, F.; Fongang, B.; Dojer, N.; et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol. Cell 2018, 72, 250–262.e6. [Google Scholar] [CrossRef] [Green Version]
- Price, B.D.; D’Andrea, A.D. Chromatin Remodeling at DNA Double-Strand Breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Morrison, A.J. Genome maintenance functions of the INO80 chromatin remodeller. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160289. [Google Scholar] [CrossRef] [Green Version]
- Fournier, L.-A.; Kumar, A.; Stirling, P.C. Chromatin as a Platform for Modulating the Replication Stress Response. Genes 2018, 9, 622. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Oma, Y.; Schleker, T.; Kugou, K.; Ohta, K.; Harata, M.; Gasser, S.M. Ino80 Chromatin Remodeling Complex Promotes Recovery of Stalled Replication Forks. Curr. Biol. 2008, 18, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Mognato, M.; Burdak-Rothkamm, S.; Rothkamm, K. Interplay between DNA replication stress, chromatin dynamics and DNA-damage response for the maintenance of genome stability. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108346. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Grewal, K.; Grewal, K.; Tabbara, I.A. PARP Inhibitors in Prostate Cancer. Anticancer Res. 2021, 41, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Gonzalez-Martin, A.; Matulonis, U.A.; Korach, J.; Mirza, M.R.; Moore, K.N.; Wu, X.; York, W.; Gupta, D.; Lechpammer, S.; Monk, B.J. Niraparib treatment for patients with BRCA-mutated ovarian cancer: Review of clinical data and therapeutic context. Future Oncol. 2022, 18, 2505–2536. [Google Scholar] [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Aubry, A.; Galiacy, S.; Allouche, M. Targeting ALK in Cancer: Therapeutic Potential of Proapoptotic Peptides. Cancers 2019, 11, 275. [Google Scholar] [CrossRef] [Green Version]
- Oh, D.-Y.; Bang, Y.-J. HER2-targeted therapies—a role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Mustachio, L.; Chelariu-Raicu, A.; Szekvolgyi, L.; Roszik, J. Targeting KRAS in Cancer: Promising Therapeutic Strategies. Cancers 2021, 13, 1204. [Google Scholar] [CrossRef] [PubMed]
- Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, E.D.S.; Nogueira, K.A.B.; Fernandes, L.C.C.; Martins, J.R.P.; Reis, A.V.F.; Neto, J.D.B.V.; Júnior, I.J.D.S.; Pessoa, C.; Petrilli, R.; Eloy, J.O. EGFR targeting for cancer therapy: Pharmacology and immunoconjugates with drugs and nanoparticles. Int. J. Pharm. 2021, 592, 120082. [Google Scholar] [CrossRef]
- Woo, E.-J.; Kim, Y.-G.; Kim, M.-S.; Han, W.-D.; Shin, S.; Robinson, H.; Park, S.-Y.; Oh, B.-H. Structural Mechanism for Inactivation and Activation of CAD/DFF40 in the Apoptotic Pathway. Mol. Cell 2004, 14, 531–539. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Jones, D.; Lee, S.-H.; Williamson, E.A.; Hromas, R. Drugging the Cancers Addicted to DNA Repair. J. Natl. Cancer Inst. 2017, 109, djx059. [Google Scholar] [CrossRef]
Figure 1.
DNA-PK, ATM and ATR roles DSB repair by cNHEJ, HR, and replication fork restart. (A) Two-ended (frank) DSBs are shunted to cNHEJ if resection is blocked by 53BP1 (and RIF1), or to HR when resection is promoted by MRN and signaling through ATM. Ku70/80 binds unresected ends and recruits DNA-PKcs (D-PK) which tethers ends, and promotes end alignment, template-dependent and -independent nucleotide addition by Pol λ and Pol μ, respectively, and ligation by Lig4-XRCC4 and XLF to yield indel repair products. ATM phosphorylates/activates Chk2 which promotes cell cycle arrest and repair. ATM stimulates DSB repair by HR by phosphorylating targets that promote limited end resection by CtIP and MRE11, and extensive resection by EXO1 and DNA2-BLM. The resulting 3′ ssDNA extensions are bound by RPA, which is replaced by the HR recombinase RAD51 that catalyzes invasion of homologous duplexes (typically sister chromatids). Eviction of RAD51 allows repair synthesis to extend the invading strand across the DSB. The extended end releases from the donor duplex (grey) and anneals to complementary ssDNA on the opposite side of the DSB. Gap filling and ligation complete accurate HR repair. (B) Replication forks blocked by single-strand lesions or secondary structures, or stalled by nucleotide depletion or DNA polymerase inhibitors, causes decoupling of polymerase from the MCM helicase, which spools out ssDNA that is bound by RPA. ATRIP mediates the interaction between ssDNA-RPA and ATR, which leads to Chk1 activation that phosphorylates targets that effect several DNA damage checkpoint responses. Blocked or stalled forks may be cleaved by nucleases MUS81 or EEPD1 to yield one-ended DSBs that are resected to promote HR-mediated fork restart and prevent genome rearrangements due to cNHEJ rejoining of one-ended DSBs to ends of other DSBs.
Figure 1.
DNA-PK, ATM and ATR roles DSB repair by cNHEJ, HR, and replication fork restart. (A) Two-ended (frank) DSBs are shunted to cNHEJ if resection is blocked by 53BP1 (and RIF1), or to HR when resection is promoted by MRN and signaling through ATM. Ku70/80 binds unresected ends and recruits DNA-PKcs (D-PK) which tethers ends, and promotes end alignment, template-dependent and -independent nucleotide addition by Pol λ and Pol μ, respectively, and ligation by Lig4-XRCC4 and XLF to yield indel repair products. ATM phosphorylates/activates Chk2 which promotes cell cycle arrest and repair. ATM stimulates DSB repair by HR by phosphorylating targets that promote limited end resection by CtIP and MRE11, and extensive resection by EXO1 and DNA2-BLM. The resulting 3′ ssDNA extensions are bound by RPA, which is replaced by the HR recombinase RAD51 that catalyzes invasion of homologous duplexes (typically sister chromatids). Eviction of RAD51 allows repair synthesis to extend the invading strand across the DSB. The extended end releases from the donor duplex (grey) and anneals to complementary ssDNA on the opposite side of the DSB. Gap filling and ligation complete accurate HR repair. (B) Replication forks blocked by single-strand lesions or secondary structures, or stalled by nucleotide depletion or DNA polymerase inhibitors, causes decoupling of polymerase from the MCM helicase, which spools out ssDNA that is bound by RPA. ATRIP mediates the interaction between ssDNA-RPA and ATR, which leads to Chk1 activation that phosphorylates targets that effect several DNA damage checkpoint responses. Blocked or stalled forks may be cleaved by nucleases MUS81 or EEPD1 to yield one-ended DSBs that are resected to promote HR-mediated fork restart and prevent genome rearrangements due to cNHEJ rejoining of one-ended DSBs to ends of other DSBs.

Figure 2.
Fork restart mechanisms. (A) Blocked forks may regress to form 4-strand, Holliday junction-like structure and a one-ended DSB. DNA synthesis (dashed/red arrow) using newly replicated strand as template allows the blocked end to extend beyond the blocking lesion. The fork is restarted once RECQ1 drives fork reversal by branch migration. (B) The one-ended DSB at regressed forks may be protected by RAD51, BRCA2, and other factors. Fork restart is mediated by RAD51-ssDNA filament invasion of the unreplicated duplex DNA. (C) Stalled forks may be cleaved by MUS81-EME2 (or EEPD1, not shown), creating a one-ended DSB. Resection creates ssDNA that is first bound by RPA and then RAD51, and the RAD51-ssDNA filament catalyzes strand invasion to restart the fork, similar to panel (B).
Figure 2.
Fork restart mechanisms. (A) Blocked forks may regress to form 4-strand, Holliday junction-like structure and a one-ended DSB. DNA synthesis (dashed/red arrow) using newly replicated strand as template allows the blocked end to extend beyond the blocking lesion. The fork is restarted once RECQ1 drives fork reversal by branch migration. (B) The one-ended DSB at regressed forks may be protected by RAD51, BRCA2, and other factors. Fork restart is mediated by RAD51-ssDNA filament invasion of the unreplicated duplex DNA. (C) Stalled forks may be cleaved by MUS81-EME2 (or EEPD1, not shown), creating a one-ended DSB. Resection creates ssDNA that is first bound by RPA and then RAD51, and the RAD51-ssDNA filament catalyzes strand invasion to restart the fork, similar to panel (B).


{kind=link}
{kind=link}
{kind=link}
Table 1.
Replication stress targets in preclinical development and clinical trials.
| Target Protein | Inhibitor | Recent/Ongoing Clinical Trials * | Tumor/Cell Targets ** | References |
|---|---|---|---|---|
| Chk1/Chk2 | UCN-01 | Solid tumors, leukemia | [116] | |
| LY2603618 | NCT01341457 | Solid tumors | [117] | |
| GDC-0575 | NCT01564251 | Solid tumors, lymphoma | [118] | |
| MK-8776 | NCT01870596 | Acute myeloid leukemia | [119] | |
| NCT00779584 | Solid tumors, lymphoma | |||
| LY2606368 | NCT02735980 | Lung cancer | [120] | |
| NCT02778126 | Solid/CNS tumors | [121] | ||
| NCT02203513 | TNBC | [122] | ||
| NCT01115790 | Advanced solid tumors | [123] | ||
| Wee1 | MK1775 | NCT01357161 | Ovarian cancer | [124] |
| ATR | BAY1895344 | NCT03188965 | Advanced solid tumors | [125] |
| AZD6738 | NCT02264678 | Advanced solid tumors | [126] | |
| TopBP1 | Calcein AM | Breast, lung xenografts | [127,128] | |
| CtIP | SP(18–28) | Breast cancer cells | [129] | |
| PACMP | siRNA-lnc15.2 | Breast, ovarian, lung, OS, stomach cancer cells | [130] | |
| MUS81 | siRNA-MUS81 | Gastric cancer cells | [131] | |
| EEPD1 | siRNA-MUS81 | Lung, OS, fibrosarcoma, cervical cancer cells | [103,104] | |
| Metnase | Ciprofloxacin | Embryonic kidney cells | [132] | |
| RAD51 | RI-1 | Glioblastoma, glioma cells | [133,134,135] | |
| B02 | Glioblastoma, multiple myeloma cells | [135,136] | ||
| PARP1 | Olaparib | NCT03531840 | Mesothelioma | [137] |
| NCT03402841 | Ovarian cancer | [138] | ||
| NCT03286842 | Breast cancer | [139] | ||
| NCT02983799 | Ovarian cancer | [140] | ||
| NCT02789332 | Breast cancer | [141] | ||
| NCT02734004 | Breast cancer | [142] | ||
| NCT02477644 | Ovarian cancer | [143] | ||
| NCT01513174 | Lung cancer | [144] | ||
| NCT02282020 | Ovarian cancer | [145] | ||
| Rucaparib | NCT02952534 | Prostate cancer | [146] | |
| NCT02042378 | Pancreatic cancer | [147] | ||
| NCT01891344 | Ovarian cancer | [148] | ||
| NCT01074970 | Ovarian, solid tumors | [149] | ||
| NCT01891344 | Ovarian cancer | [150] | ||
| Niraparib | NCT03759600 | Ovarian cancer | [151] | |
| NCT04475939 | Lung cancer | [152] | ||
| NCT02657889 | TNBC | [153] | ||
| NCT02354131 | Ovarian cancer | [154] | ||
| NCT02354586 | Ovarian cancer | [155] | ||
| ATM | AZD0156 | NCT02588105 | Advanced solid tumors | [156] |
| ATM/ATR | AZD1390 | NCT03215381 | Healthy volunteers | [156] |
* Clinical trials ongoing (actively enrolling) or completed since 2015. Most trials include combination therapies with the indicated inhibitor. For PARP1i, only completed Phase II/III trials with peer-reviewed publications since 2015 are listed: data from ClinicalTrials.gov. Several recent reviews provide additional information about the numerous PARP1i clinical trials [157,158,159,160,161,162,163]. ** CNS, central nervous system; TNBC, triple negative breast cancer; OS, osteosarcoma.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nickoloff, J.A. Targeting Replication Stress Response Pathways to Enhance Genotoxic Chemo- and Radiotherapy. Molecules 2022, 27, 4736. https://doi.org/10.3390/molecules27154736
AMA Style
Nickoloff JA. Targeting Replication Stress Response Pathways to Enhance Genotoxic Chemo- and Radiotherapy. Molecules. 2022; 27(15):4736. https://doi.org/10.3390/molecules27154736
Chicago/Turabian StyleNickoloff, Jac A. 2022. "Targeting Replication Stress Response Pathways to Enhance Genotoxic Chemo- and Radiotherapy" Molecules 27, no. 15: 4736. https://doi.org/10.3390/molecules27154736