
Human Serum Amyloid a Impaired Structural Stability of High-Density Lipoproteins (HDL) and Apolipoprotein (Apo) A-I and Exacerbated Glycation Susceptibility of ApoA-I and HDL


Abstract
:1. Introduction
2. Results
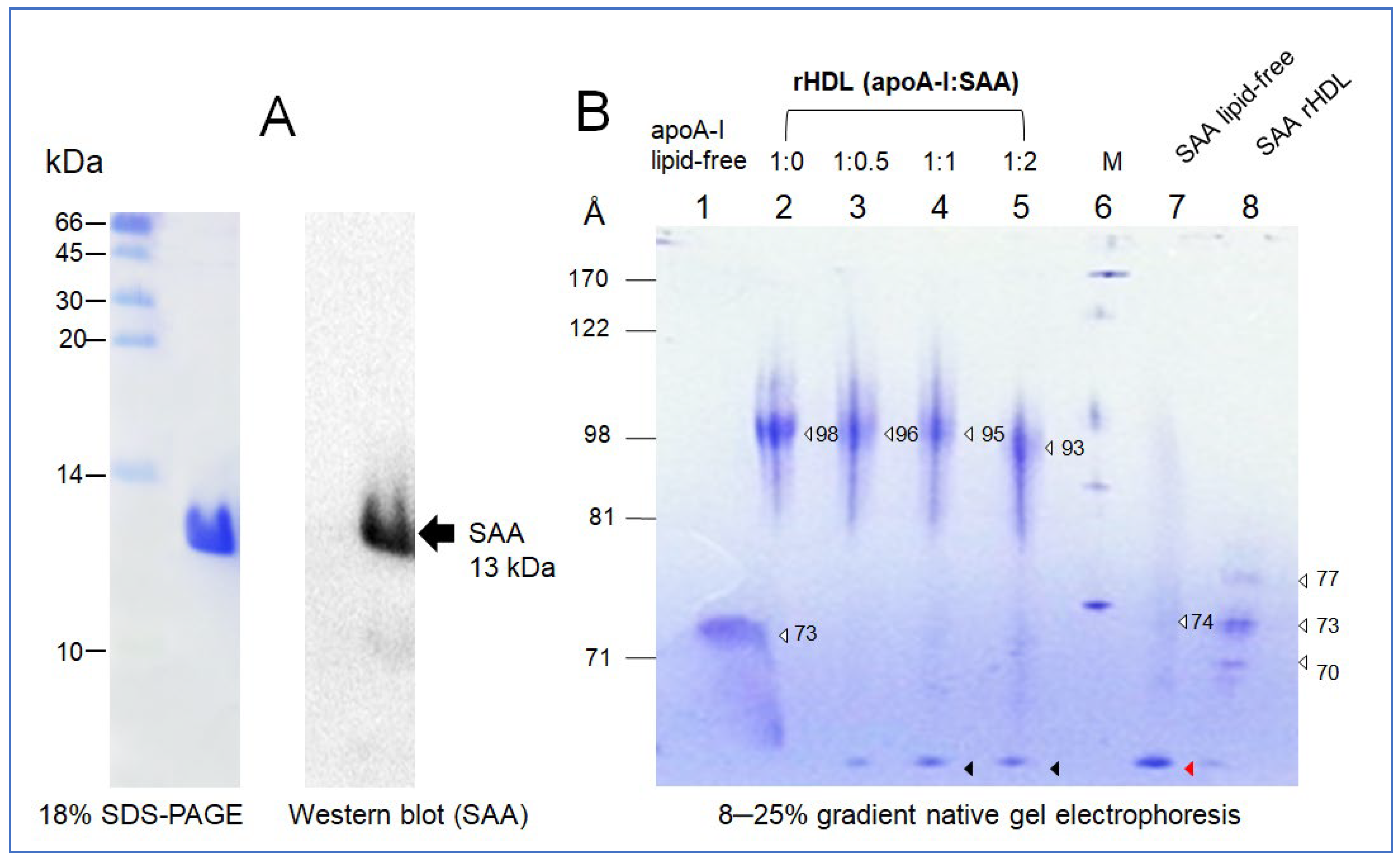
2.1. Purification and Characterization of SAA1
2.2. Synthesis of rHDL with SAA1 and ApoA-I
2.3. Phospholipid-Binding Assay
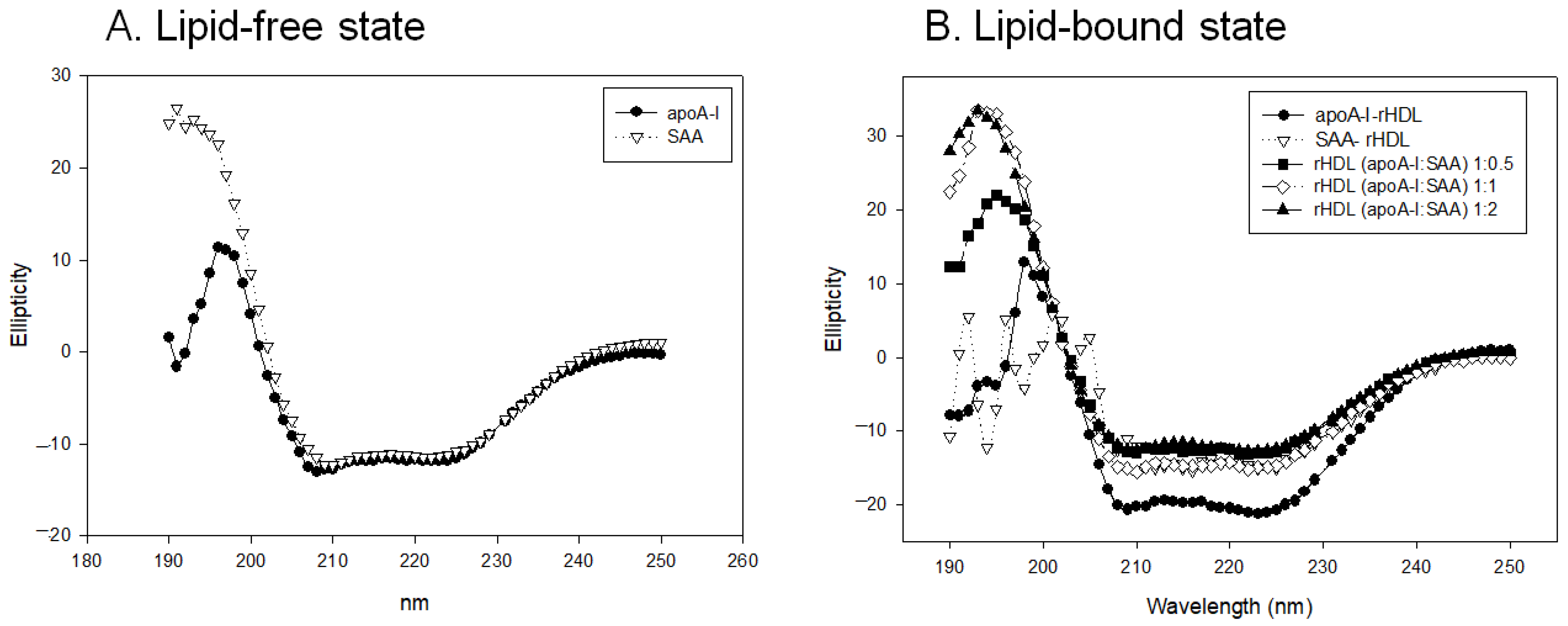
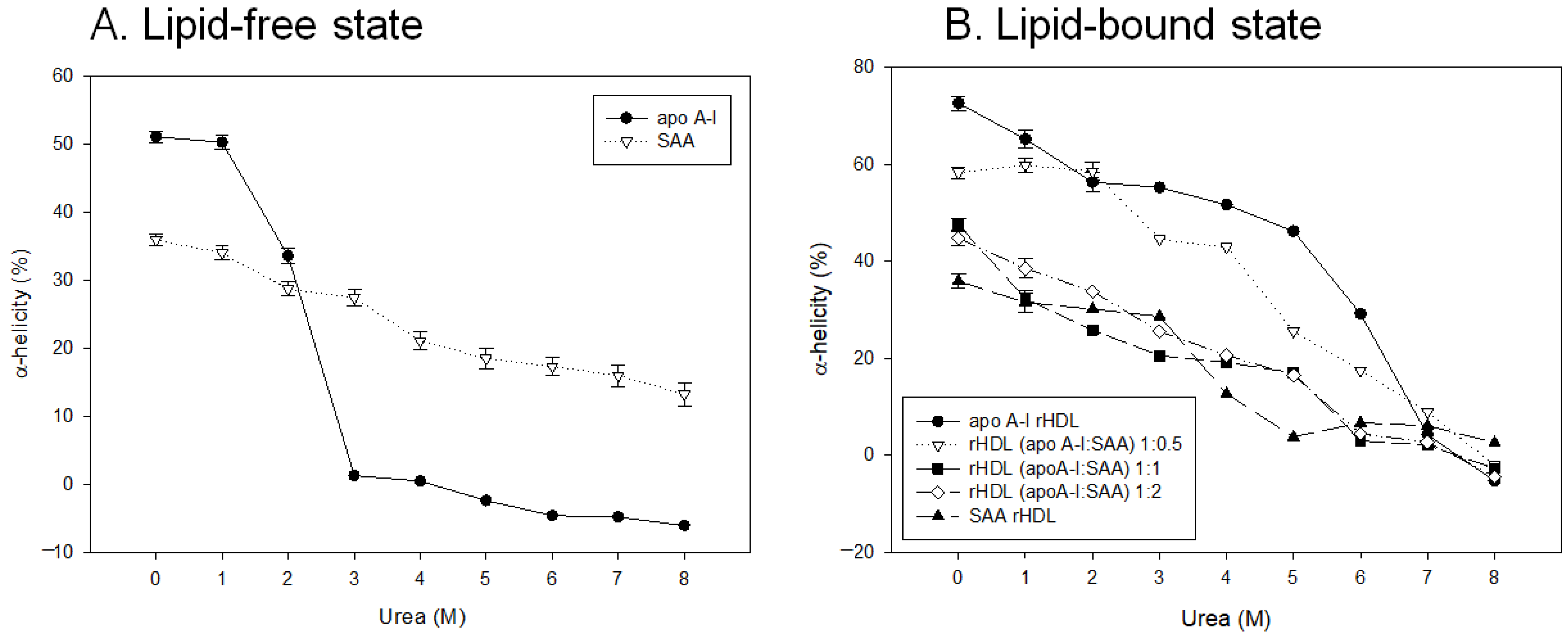
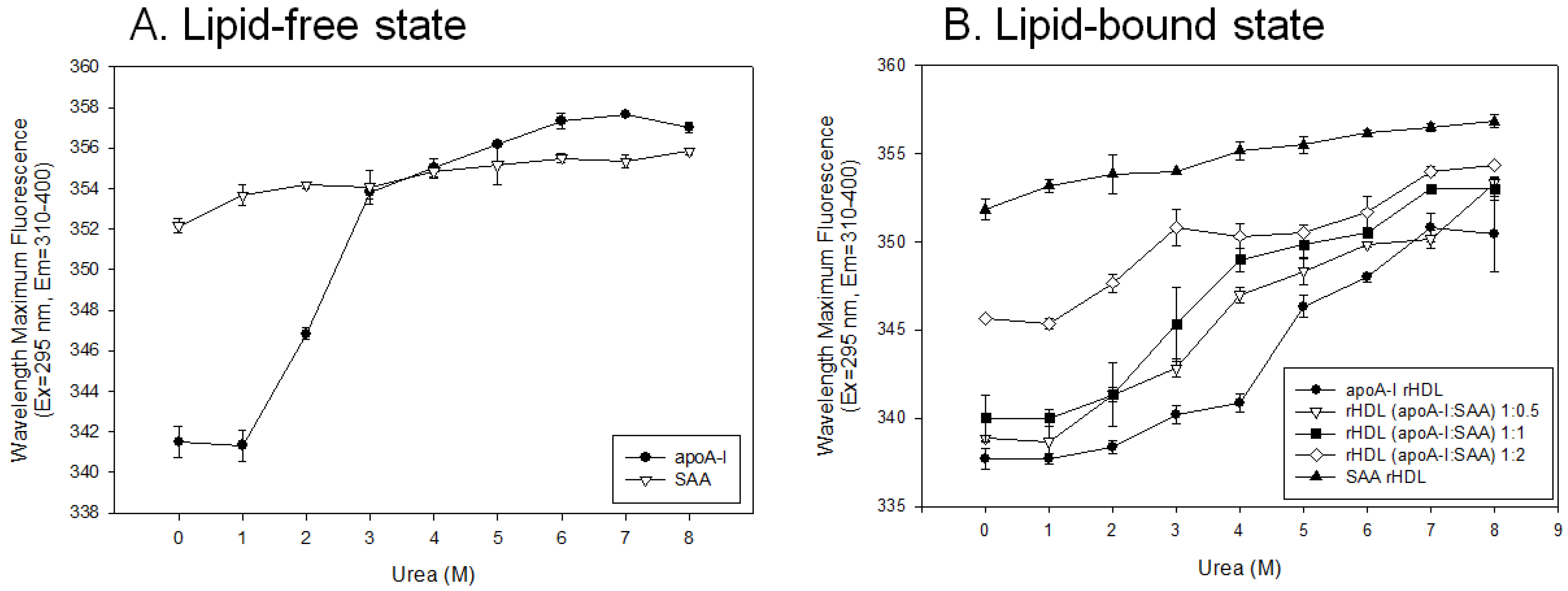
2.4. Change in Secondary Structure and Trp Movement
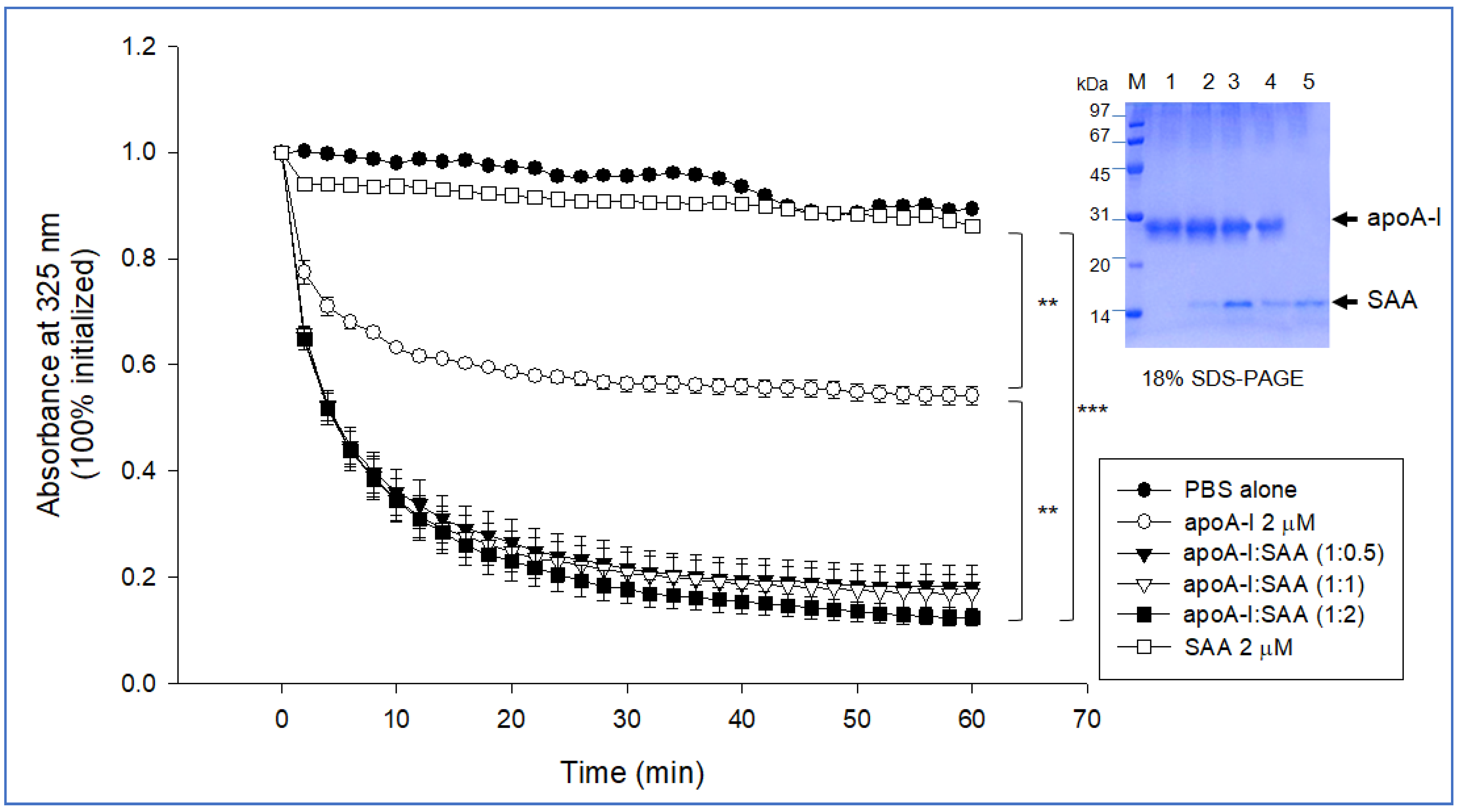
2.5. SAA Accelerated Isothermal Denaturation
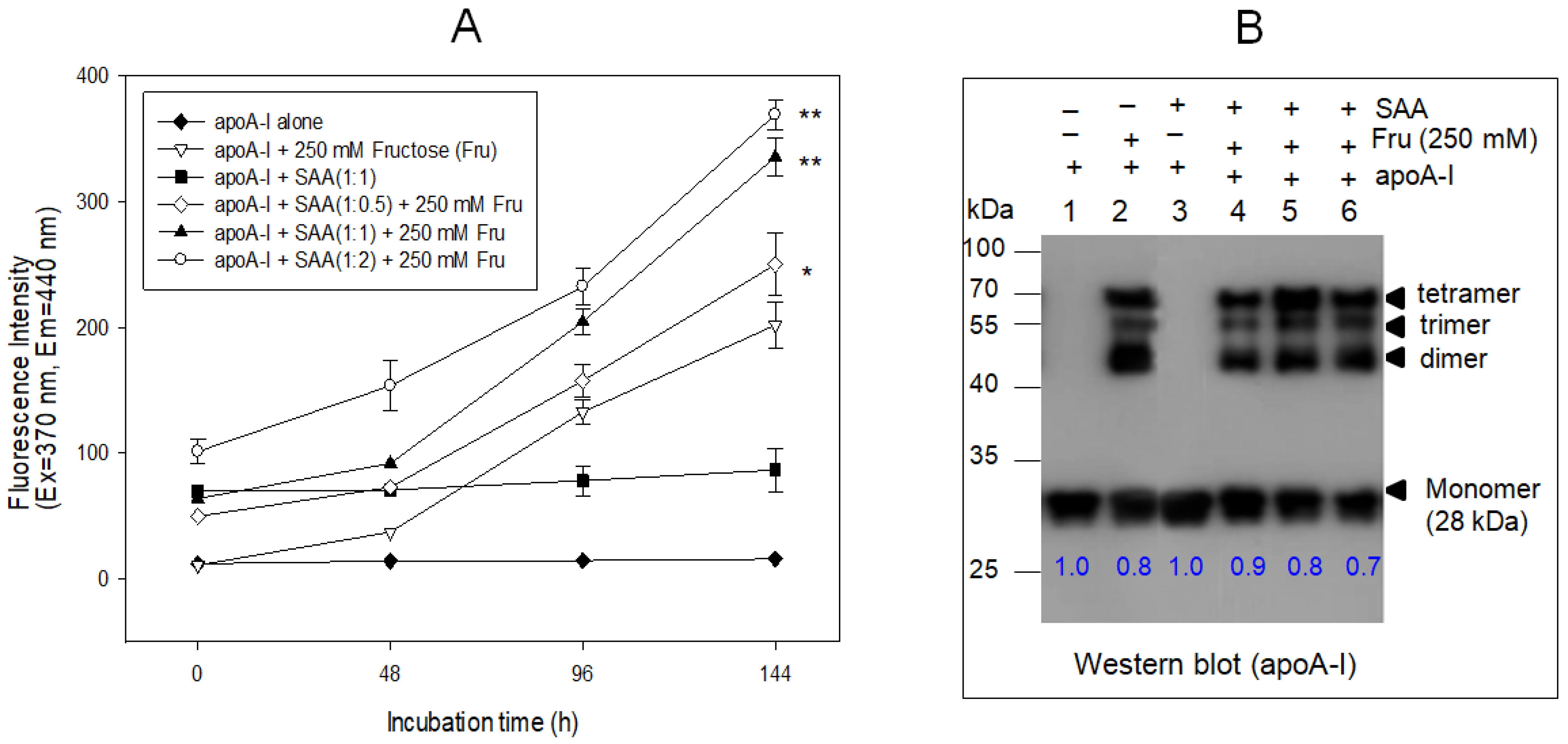
2.6. SAA Accelerated the Glycation of ApoA-I
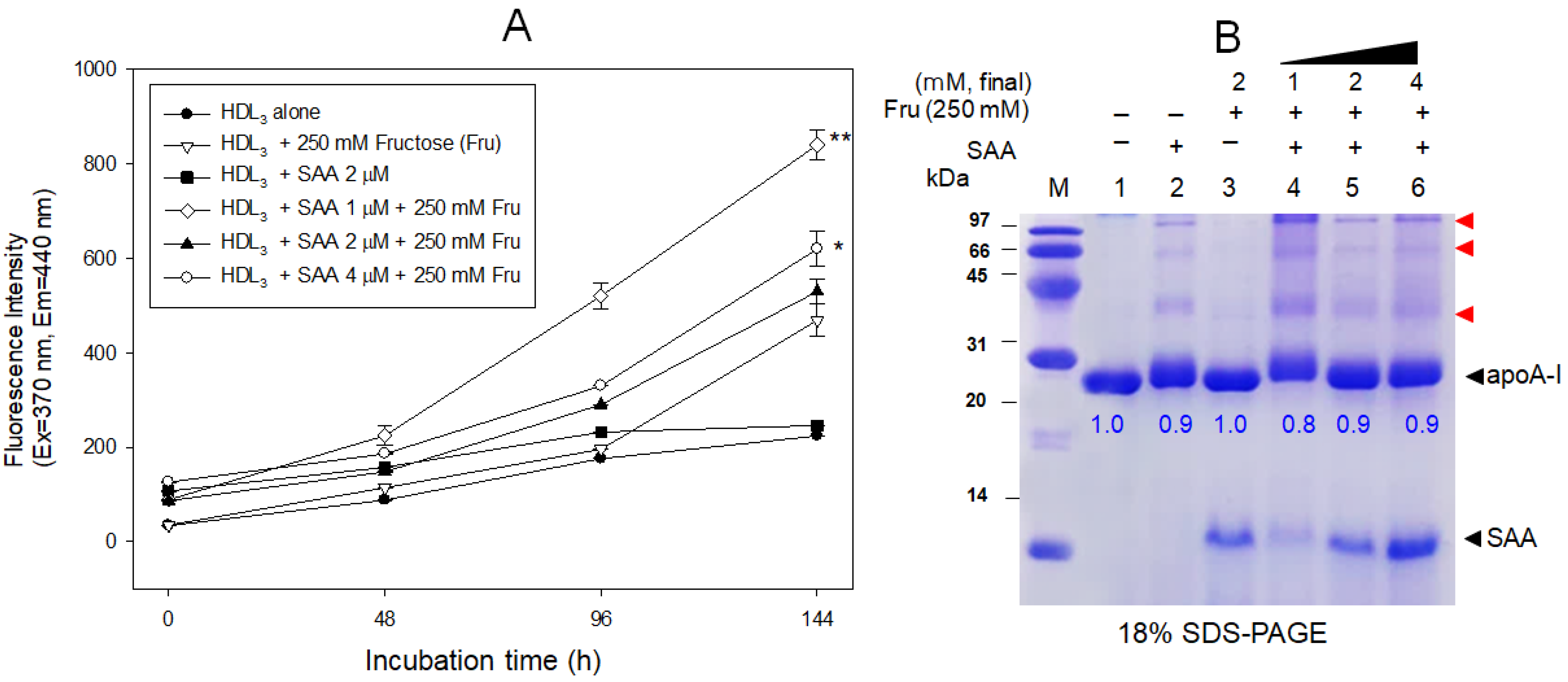
2.7. SAA Accelerated the Glycation of HDL
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Expression and Purification of SAA1
4.3. Protein Sequencing
4.4. Characterization of Secondary Structure
4.5. Characterization of Trp Fluorescence
4.6. Purification of Lipoproteins
4.7. Purification of Human apoA-I
4.8. Synthesis of Reconstituted HDL
4.9. Phospholipid Binding Assay
4.10. Glycation of apoA-I with SAA
4.11. Western Blotting
4.12. Statistical Analysis
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Shridas, P.; Patrick, A.C.; Tannock, L.R. Role of serum amyloid a in abdominal aortic aneurysm and related cardiovascular diseases. Biomolecules 2021, 11, 1883. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.; Franklin, E.C.; Frangione, B.; Pras, M. The amino acid sequence of a major nonimmunoglobulin component of some amyloid fibrils. J. Clin. Investig. 1972, 51, 2773–2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linke, R.P.; Sipe, J.D.; Pollock, P.S.; Ignaczak, T.F.; Glenner, G.G. Isolation of a low-molecular-weight serum component antigenically related to an amyloid fibril protein of unknown origin. Proc. Natl. Acad. Sci. USA 1975, 72, 1473–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, F.J.; Binder, C.J.; Krychtiuk, K.A.; Schillinger, M.; Minar, E.; Hoke, M. The prognostic value of serum amyloid A for long-term mortality among patients with subclinical carotid atherosclerosis. Eur. J. Clin. Investig. 2019, 49, e13095. [Google Scholar] [CrossRef]
- Johnson, B.D.; Kip, K.E.; Marroquin, O.C.; Ridker, P.M.; Kelsey, S.F.; Shaw, L.J.; Pepine, C.J.; Sharaf, B.; Bairey Merz, C.N.; Sopko, G.; et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: The National Heart, Lung, and Blood Institute-sponsored women’s ischemia syndrome evaluation (WISE). Circulation 2004, 109, 726–732. [Google Scholar] [CrossRef] [Green Version]
- Malle, E.; De Beer, F.C. Human serum amyloid A (SAA) protein: A prominent acute-phase reactant for clinical practice. Eur. J. Clin. Investig. 1996, 26, 427–435. [Google Scholar] [CrossRef]
- Benditt, E.P.; Eriksen, N. Amyloid protein SAA is associated with high density lipoprotein from human serum. Proc. Natl. Acad. Sci. USA 1977, 74, 4025–4028. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Ye, R.D. Serum amyloid A1: Structure, function and gene polymorphism. Gene 2016, 583, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Targonska-Stepniak, B.; Majdan, M. Serum amyloid A as a marker of persistent inflammation and an indicator of cardiovascular and renal involvement in patients with rheumatoid arthritis. Med. Inflamm. 2014, 2014, 793628. [Google Scholar] [CrossRef]
- Reisinger, A.C.; Schuller, M.; Sourij, H.; Stadler, J.T.; Hackl, G.; Eller, P.; Marsche, G. Impact of sepsis on high-density lipoprotein metabolism. Front. Cell. Dev. Biol. 2022, 9, 795460. [Google Scholar] [CrossRef]
- Webb, N.R. High-density lipoproteins and serum amyloid A (SAA). Curr. Atheroscler. Rep. 2021, 23, 7. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Torigoshi, T.; Motokawa, S.; Miyashita, T.; Maeda, Y.; Nakamura, M.; Komori, A.; Aiba, Y.; Uemura, T.; Yatsuhashi, H.; et al. Serum amyloid A-induced IL-6 production by rheumatoid synoviocytes. FEBS Lett. 2008, 582, 579–585. [Google Scholar] [CrossRef] [Green Version]
- Zheng, A.; Widmann, C. The interplay between serum amyloid A and HDLs. Curr. Opin. Lipidol. 2020, 31, 300–301. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.G.; Thompson, J.C.; Shridas, P.; McNamara, P.J.; de Beer, M.C.; de Beer, F.C.; Webb, N.R.; Tannock, L.R. Serum amyloid A is an exchangeable apolipoprotein. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1890–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.H. Impacts of Lifestyle, Diseases, and Environmental Stressors on HDL. In High-Density Lipoproteins as Biomarkers and Therapeutic Tools, 1st ed.; Springer: New York, NY, USA, 2019; Volume 1, ISBN 978-981-13-7382-4. [Google Scholar]
- Masana, L.; Correig, E.; Ibarretxe, D.; Anoro, E.; Arroyo, J.A.; Jericó, C.; Guerrero, C.; Miret, M.; Näf, S.; Pardo, A.; et al. Low HDL and high triglycerides predict COVID-19 severity. Sci. Rep. 2021, 11, 7217. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, Q.; Zhao, X.; Dong, H.; Wu, C.; Wu, F.; Yu, B.; Lv, J.; Zhang, S.; Wu, G.; et al. Low high-density lipoprotein level is correlated with the severity of COVID-19 patients: An observational study. Lipids Health Dis. 2020, 19, 204. [Google Scholar] [CrossRef]
- Feingold, K.R. The bidirectional link between HDL and COVID-19 infections. J. Lipid Res. 2021, 62, 100067. [Google Scholar] [CrossRef]
- Cho, K.H.; Kim, J.R.; Lee, I.C.; Kwon, H.J. Native high-density lipoproteins (HDL) with higher paraoxonase exerts a potent antiviral effect against SARS-CoV-2 (COVID-19), while glycated HDL lost the antiviral activity. Antioxidants 2021, 10, 209. [Google Scholar] [CrossRef]
- Li, H.; Xiang, X.; Ren, H.; Xu, L.; Zho, L.; Chen, X.; Long, H.; Wang, Q.; Wu, Q. Serum amyloid A is a biomarker of severe coronavirus disease and poor prognosis. J. Infect. 2020, 80, 646–655. [Google Scholar] [CrossRef]
- Souza Junior, D.R.; Silva, A.R.M.; Rosa-Fernandes, L.; Reis, L.R.; Alexandria, G.; Bhosale, S.D.; Ghilardi, F.R.; Dalçóquio, T.F.; Bertolin, A.J.; Nicolau, J.C.; et al. HDL proteome remodeling associates with COVID-19 severity. J. Clin. Lipidol. 2021, 15, 796–804. [Google Scholar] [CrossRef]
- Kisilevsky, R.; Manley, P.N. Acute-phase serum amyloid A: Perspectives on its physiological and pathological roles. Amyloid. 2012, 19, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Artl, A.; Marsche, G.; Lestavel, S.; Sattler, W.; Malle, E. Role of serum amyloid A during metabolism of acute-phase HDL by macrophages. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 763–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takase, H.; Furuchi, H.; Tanaka, M.; Yamada, T.; Matoba, K.; Iwasaki, K.; Kawakami, T.; Mukai, T. Characterization of reconstituted high-density lipoprotein particles formed by lipid interactions with human serum amyloid A. Biochim. Biophys. Acta 2014, 1842, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H. Structural and functional impairments of reconstituted high-density lipoprotein by incorporation of recombinant β-Amyloid42. Molecules 2021, 26, 4317. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, S.; Haupt, C.; Gursky, O. Thermal transitions in serum amyloid A in solution and on the lipid: Implications for structure and stability of acute-phase HDL. J. Lipid Res. 2015, 56, 1531–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.Y.; Lee, E.Y.; Park, J.K.; Song, Y.W.; Kim, J.R.; Cho, K.H. Patients with rheumatoid arthritis show altered lipoprotein profiles with dysfunctional high-density lipoproteins that can exacerbate inflammatory and atherogenic process. PLoS ONE 2016, 11, e0164564. [Google Scholar] [CrossRef] [Green Version]
- Ji, A.; Wang, X.; Noffsinger, V.P.; Jennings, D.; de Beer, M.C.; de Beer, F.C.; Tannock, L.R.; Webb, N.R. Serum amyloid A is not incorporated into HDL during HDL biogenesis. J. Lipid Res. 2020, 61, 328–337. [Google Scholar] [CrossRef]
- Chiba, T.; Chang, M.Y.; Wang, S.; Wight, T.N.; McMillen, T.S.; Oram, J.F.; Vaisar, T.; Heinecke, J.W.; De Beer, F.C.; De Beer, M.C.; et al. Serum amyloid A facilitates the binding of HDL from mice injected with lipopolysaccharide to vascular proteoglycans. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1326–1332. [Google Scholar] [CrossRef] [Green Version]
- Frame, N.M.; Gursky, O. Structure of serum amyloid A suggests a mechanism for selective lipoprotein binding and functions: SAA as a hub in macromolecular interaction networks. FEBS Lett. 2016, 590, 866–879. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, S.; Sánchez-Quesada, J.L.; Gursky, O. Triglyceride increase in the core of high-density lipoproteins augments apolipoprotein dissociation from the surface: Potential implications for treatment of apolipoprotein deposition diseases. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 200–210. [Google Scholar] [CrossRef]
- McCubbin, W.D.; Kay, C.M.; Narindrasorasak, S.; Kisilevsky, R. Circular-dichroism studies on two murine serum amyloid A proteins. Biochem. J. 1988, 256, 775–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.H. Structural and functional changes of reconstituted high-density lipoprotein (HDL) by incorporation of α-synuclein: A potent antioxidant and anti-glycation activity of α-synuclein and apoA-I in HDL at high molar ratio of α-synuclein. Molecules 2021, 26, 7485. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-H. The Current Status of Research on High-Density Lipoproteins (HDL): A Paradigm Shift from HDL Quantity to HDL Quality and HDL Functionality. Int. J. Mol. Sci. 2022, 23, 3967. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, G.A.; Strachan, A.F.; van der Westhuyzen, D.R.; Hoppe, H.C.; Jeenah, M.S.; de Beer, F.C. Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J. Biol. Chem. 1986, 261, 9644–9651. [Google Scholar] [CrossRef]
- Plochberger, B.; Röhrl, C.; Preiner, J.; Rankl, C.; Brameshuber, M.; Madl, J.; Bittman, R.; Ros, R.; Sezgin, E.; Eggeling, C.; et al. HDL particles incorporate into lipid bilayers—A combined AFM and single molecule fluorescence microscopy study. Sci. Rep. 2017, 7, 15886. [Google Scholar] [CrossRef] [Green Version]
- Gan, C.; Ao, M.; Liu, Z.; Chen, Y. Imaging and force measurement of LDL and HDL by AFM in air and liquid. FEBS Open Bio 2015, 5, 276–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.M.; Jeong, T.S.; Lee, W.S.; Choi, I.; Cho, K.H. Structural and functional properties of V156K and A158E mutants of apolipoprotein A-I in the lipid-free and lipid-bound states. J. Lipid. Res. 2005, 46, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Cho, K.H. A point mutant of apolipoprotein A-I (V156K) showed enhancement of cellular insulin secretion and potent activity of facultative regeneration in zebrafish. Rejuvenation Res. 2012, 15, 313–321. [Google Scholar] [CrossRef]
- Matsudaira, P. Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes. J. Biol. Chem. 1987, 262, 10035–10038. [Google Scholar] [CrossRef]
- Chen, Y.H.; Yang, J.T.; Martinez, H.M. Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry 1972, 11, 4120–4131. [Google Scholar] [CrossRef]
- Cho, K.H.; Jonas, A. A key point mutation (V156E) affects the structure and functions of human Apolipoprotein A-I. J. Biol. Chem. 2000, 275, 26821–26827. [Google Scholar] [CrossRef]
- Cho, K.H.; Durbin, D.M.; Jonas, A. Role of individual amino acids of apolipoprotein A-I in the activation of lecithin:cholesterol acyltransferase and in HDL rearrangements. J. Lipid Res. 2001, 42, 379–389. [Google Scholar] [CrossRef]
- Park, K.H.; Shin, D.G.; Kim, J.R.; Cho, K.H. Senescence-related truncation and multimerization of apolipoprotein A-I in highdensity lipoprotein with an elevated level of advanced glycated end products and cholesteryl ester transfer activity. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 600–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havel, R.J.; Eder, H.A.; Bragdon, J.H. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J. Clin. Investig. 1955, 34, 1345–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brewer, H.B., Jr.; Ronan, R.; Meng, M.; Bishop, C. Isolation and characterization of apolipoproteins A-I, A-II, and A-IV. Methods. Enzymol. 1986, 128, 223–246. [Google Scholar] [CrossRef]
- Cho, K.H. Synthesis of reconstituted high density lipoprotein (rHDL) containing apoA-I and apoC-III: The functional role of apoC-III in rHDL. Mol. Cells 2009, 27, 291–297. [Google Scholar] [CrossRef]
- Pownall, H.J.; Massey, J.B.; Kusserow, S.K.; Gotto, A.M., Jr. Kinetics of lipid—Protein interactions: Effect of cholesterol on the association of human plasma high-density apolipoprotein A-I with L-alpha-dimyristoylphosphatidylcholine. Biochemistry 1979, 18, 574–579. [Google Scholar] [CrossRef]
- Park, K.H.; Jang, W.; Kim, K.Y.; Kim, J.R.; Cho, K.H. Fructated apolipoprotein A-I showed severe structural modification and loss of beneficial functions in lipid-free and lipid-bound state with acceleration of atherosclerosis and senescence. Biochem. Biophys. Res. Commun. 2010, 392, 295–300. [Google Scholar] [CrossRef]
- Park, K.H.; Kim, J.Y.; Choi, I.; Kim, J.R.; Won, K.C.; Cho, K.H. Fructated apolipoprotein A-I exacerbates cellular senescence in human umbilical vein endothelial cells accompanied by impaired insulin secretion activity and embryo toxicity. Biochem Cell Biol. 2016, 94, 337–345. [Google Scholar] [CrossRef]
- McPherson, J.D.; Shilton, B.H.; Walton, D.J. Role of fructose in glycation and cross-linking of proteins. Biochemistry 1988, 27, 1901–1907. [Google Scholar] [CrossRef]
- Markwell, M.A.; Haas, S.M.; Bieber, L.L.; Tolbert, N.E. A modification of the lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal. Biochem. 1978, 87, 206–210. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molar Composition (POPC:FC:apoA-I:SAA) | WMF a (nm) | α-helicity b (%) | Size (Å) c | |
|---|---|---|---|---|
| Lipid free apoA-I | - | 342 ± 1 | 53 ± 2 | - |
| Lipid free SAA | - | 352 ± 1 | 36 ± 1 | - |
| apoA-I-rHDL | 95:5:1:0 | 339 ± 0 | 73 ± 4 | 98 |
| SAA-rHDL | 95:5:0:1 | 350 ± 1 | 36 ± 2 | 77, 73, 70 |
| rHDL (apoA-I:SAA) | 95:5:1:0.5 | 341 ± 1 | 54 ± 3 | 96 |
| 95:5:1:1 | 344 ± 1 | 49 ± 2 | 95 | |
| 95:5:1:2 | 345 ± 1 | 40 ± 3 | 93 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, K.-H. Human Serum Amyloid a Impaired Structural Stability of High-Density Lipoproteins (HDL) and Apolipoprotein (Apo) A-I and Exacerbated Glycation Susceptibility of ApoA-I and HDL. Molecules 2022, 27, 4255. https://doi.org/10.3390/molecules27134255
Cho K-H. Human Serum Amyloid a Impaired Structural Stability of High-Density Lipoproteins (HDL) and Apolipoprotein (Apo) A-I and Exacerbated Glycation Susceptibility of ApoA-I and HDL. Molecules. 2022; 27(13):4255. https://doi.org/10.3390/molecules27134255
Chicago/Turabian StyleCho, Kyung-Hyun. 2022. "Human Serum Amyloid a Impaired Structural Stability of High-Density Lipoproteins (HDL) and Apolipoprotein (Apo) A-I and Exacerbated Glycation Susceptibility of ApoA-I and HDL" Molecules 27, no. 13: 4255. https://doi.org/10.3390/molecules27134255