
Deep Learning and Structure-Based Virtual Screening for Drug Discovery against NEK7: A Novel Target for the Treatment of Cancer

 , , ,
, , ,  , , and
, , and
Abstract
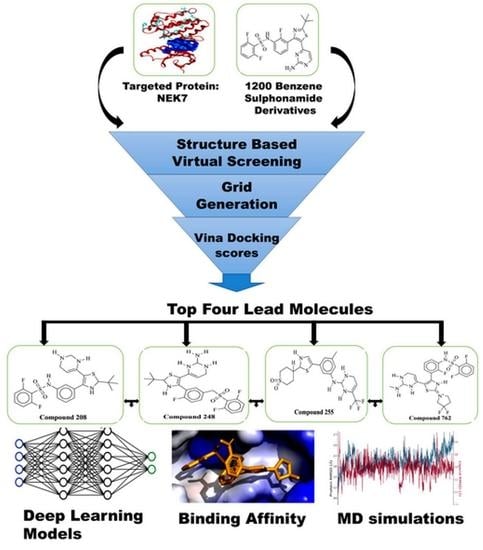
:
1. Introduction
2. Experimental
2.1. Computational Studies
2.1.1. Density Functional Theory Calculation
2.1.2. Structure Based Virtual Screening (SBVS)
2.1.3. Molecular Dynamics Simulation
2.1.4. Prediction of Binding Affinities, pIC50 and ADMET Properties Using Deep Learning Models
3. Results and Discussion
3.1. Density Functional Theory Studies (DFTs)
3.1.1. Optimized Geometries
3.1.2. Frontier Molecular Orbital (FMOs)
3.1.3. Global and Local Reactivity Descriptors
3.2. Structure Based Virtual Screening and Predicted Binding Affinities
3.3. Electrostatic Surface Potential Map
3.4. Buried Surface Area (BSA)
3.5. Molecular Dynamic Simulation
3.5.1. RMSD Analysis of Protein and Protein–Ligand Complexes
3.5.2. Radius of Gyration (Rg) and Solvent-Accessible Surface Area of Protein (SASA)
3.5.3. Principle Component Analysis (PCA)
3.5.4. MM-GBSA Energy Calculations
3.5.5. MM-PBSA Energy Calculations
3.6. ADMET Profile
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Phadke, M.; Remsing Rix, L.L.; Smalley, I.; Bryant, A.T.; Luo, Y.; Lawrence, H.R.; Schaible, B.J.; Chen, Y.A.; Rix, U.; Smalley, K.S. Dabrafenib inhibits the growth of BRAF-WT cancers through CDK16 and NEK9 inhibition. Mol. Oncol. 2018, 12, 74–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Gong, W.; Zhang, Y.; Jia, Z. Physiological and pathological roles of mammalian NEK7. Front. Physiol. 2020, 11, 1608. [Google Scholar] [CrossRef] [PubMed]
- Loncarek, J.; Hergert, P.; Magidson, V.; Khodjakov, A. Control of daughter centriole formation by the pericentriolar material. Nat. Cell Biol. 2008, 10, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.; Nakajima, S.; Wang, Q.; Sun, H.; Xue, J.; Wu, J.; Hellwig, S.; Zeng, X.; Yates, N.A.; Smithgall, T.E.; et al. Nek7 protects telomeres from oxidative DNA damage by phosphorylation and stabilization of TRF1. Mol. Cell 2017, 65, 818–831.e5. [Google Scholar] [CrossRef] [Green Version]
- Haq, T.; Richards, M.W.; Burgess, S.G.; Gallego, P.; Yeoh, S.; O’Regan, L.; Reverter, D.; Roig, J.; Fry, A.M.; Bayliss, R. Mechanistic basis of Nek7 activation through Nek9 binding and induced dimerization. Nat. Commun. 2015, 6, 8771. [Google Scholar] [CrossRef] [Green Version]
- Fry, M.A.; Bayliss, R.; Roig, J. Mitotic regulation by NEK kinase networks. Front. Cell Dev. Biol. 2017, 5, 102. [Google Scholar] [CrossRef] [Green Version]
- Hauwermeiren, V.F.; Lamkanfi, M. The NEK-sus of the NLRP3 inflammasome. Nat. Immunol. 2016, 17, 223–224. [Google Scholar] [CrossRef]
- Xu, J.; Lu, L.; Li, L. NEK7: A novel promising therapy target for NLRP3-related inflammatory diseases. Acta Biochim. Biophys. Sin. 2016, 48, 966–968. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Tsuchiya, Y.; Ohta, M.; Shiratsuchi, G.; Kitagawa, D. NEK7 is required for G1 progression and procentriole formation. Mol. Biol. Cell 2017, 28, 2123–2134. [Google Scholar] [CrossRef]
- Liu, G.; Chen, X.; Wang, Q.; Yuan, L. NEK7: A potential therapy target for NLRP3-related diseases. BioScience Trends 2020, 14, 74–82. [Google Scholar] [CrossRef] [Green Version]
- García-Galán, M.J.; Díaz-Cruz, M.S.; Barceló, D. Identification and determination of metabolites and degradation products of sulfonamide antibiotics. Trends Anal. Chem. 2008, 27, 1008–1022. [Google Scholar] [CrossRef]
- Supuran, C.T.; Casini, A.; Scozzafava, A.J.M. Protease inhibitors of the sulfonamide type: Anticancer, antiinflammatory, and antiviral agents. Med. Res. Rev. 2003, 23, 535–558. [Google Scholar] [CrossRef]
- Scozzafava, A.; Owa, T.; Mastrolorenzo, A.; Supuran, C.T. Anticancer and antiviral sulfonamides. Curr. Med. Chem. 2003, 10, 925–953. [Google Scholar] [CrossRef]
- Reddy, N.S.; Mallireddigari, M.R.; Cosenza, S.; Gumireddy, K.; Bell, S.C.; Reddy, E.P.; Reddy, M.R. Synthesis of new coumarin 3-(N-aryl) sulfonamides and their anticancer activity. Bioorganic Med. Chem. Lett. 2004, 14, 4093–4097. [Google Scholar] [CrossRef]
- Bilbao-Ramos, P.; Galiana-Roselló, C.; Dea-Ayuela, M.A.; González-Alvarez, M.; Vega, C.; Rolón, M.; Pérez-Serrano, J.; Bolás-Fernández, F.; González-Rosende, M.E. Nuclease activity and ultrastructural effects of new sulfonamides with anti-leishmanial and trypanocidal activities. Parasitol. Int. 2012, 61, 604–613. [Google Scholar] [CrossRef]
- Dauvergne, J.; Wellington, K.; Chibale, K. Unprecedented observation of sulfonamides in the transesterification of N-unsubstituted carbamates with sulfonyl chlorides. Tetrahedron Lett. 2004, 45, 43–47. [Google Scholar] [CrossRef]
- Yasuhara, A.; Kameda, M.; Sakamoto, T. Selective monodesulfonylation of N, N-disulfonylarylamines with tetrabutylammonium fluoride. Chem. Pharm. Bull. 1999, 47, 809–812. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, J.F.; Rapoport, H. 1-Benzenesulfonyl-and 1-p-toluenesulfonyl-3-methylimidazolium triflates: Efficient reagents for the preparation of arylsulfonamides and arylsulfonates. J. Org. Chem. 1992, 57, 4775–4777. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Mohapatra, S. Neighbouring group assisted sulfonamide cleavage of Sharpless aminols under acetonation conditions. Tetrahedron Lett. 1998, 39, 695–698. [Google Scholar] [CrossRef]
- Gleckman, R.; Alvarez, S.; Joubert, D.W. Drug therapy reviews: Trimethoprim-sulfamethoxazole. Am. J. Hosp. Pharm. 1979, 36, 893–906. [Google Scholar] [CrossRef]
- Bushby, S.R.M.; Hitchings, G.H. Trimethoprim, a sulphonamide potentiator. Br. J. Pharmacol. Chemother. 1968, 33, 72–90. [Google Scholar] [CrossRef] [Green Version]
- Song, C.M.; Lim, S.J.; Tong, J.C. Recent advances in computer-aided drug design. Briefings Bioinform. 2009, 10, 579–591. [Google Scholar] [CrossRef] [Green Version]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharmacal Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Brief. Bioinform. 2019, 20, 1878–1912. [Google Scholar] [CrossRef]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Sánchez-Linares, I.; Pérez-Sánchez, H.; Cecilia, J.M.; García, J.M. High-throughput parallel blind virtual screening using BINDSURF. BMC Bioinform. 2012, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Imbernón, B.; Cecilia, J.M.; Pérez-Sánchez, H.; Giménez, D. METADOCK: A parallel metaheuristic schema for virtual screening methods. Int. J. High Perform. Comput. Appl. 2018, 32, 789–803. [Google Scholar] [CrossRef]
- Ban, T.A. The role of serendipity in drug discovery. Dialogues Clin. Neurosci. 2022, 8, 335–344. [Google Scholar] [CrossRef]
- Huang, K.; Fu, T.; Khan, D.; Abid, A.; Abdalla, A.; Abid, A.; Glass, L.M.; Zitnik, M.; Xiao, C.; Sun, J. Moldesigner: Interactive design of efficacious drugs with deep learning. arXiv 2020, arXiv:03951. [Google Scholar]
- Huang, K.; Fu, T.; Glass, L.M.; Zitnik, M.; Xiao, C.; Sun, J. DeepPurpose: A deep learning library for drug–target interaction prediction. Bioinformatics 2020, 36, 5545–5547. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864. [Google Scholar] [CrossRef] [Green Version]
- Calais, J.L. Orthonormalization and symmetry adaptation of crystal orbitals. Int. J. Quantum Chem. 1985, 28, 655–667. [Google Scholar] [CrossRef]
- Rodríguez, J.I.; Ayers, P.W.; Götz, A.W.; Castillo-Alvarado, F.D.L. Virial theorem in the Kohn–Sham density-functional theory formalism: Accurate calculation of the atomic quantum theory of atoms in molecules energies. J. Chem. Phys. 2009, 131, 021101. [Google Scholar] [CrossRef]
- Ziegler, T.J.C.R. Approximate density functional theory as a practical tool in molecular energetics and dynamics. Chem. Rev. 1991, 91, 651–667. [Google Scholar] [CrossRef]
- Bartolotti, L.J.; Flurchick, K. An introduction to density functional theory. Rev. Comput. Chem. 1996, 7, 187–260. [Google Scholar]
- Aziz, M.; Ejaz, S.A.; Tamam, N.; Siddique, F.; Riaz, N.; Qais, F.A.; Chtita, S.; Iqbal, J. Identification of potent inhibitors of NEK7 protein using a comprehensive computational approach. Sci. Rep. 2022, 12, 6404. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Person, W.B.; Szczepaniak, K. Properties of Hydrogen-Bonded Complexes Obtained from the B3LYP Functional with 6-31G (d, p) and 6-31+ G (d, p) Basis Sets: Comparison with MP2/6-31+ G (d, p) Results and Experimental Data. J. Phys. Chem. 1995, 99, 10705–10707. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Chemin. 2012, 4, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Azarakhshi, F.; Khaleghian, M.; Farhadyar, N. DFT study and NBO analysis of conformational properties of 2-Substituted 2-Oxo-1, 3, 2-dioxaphosphorinanes and their dithia and diselena analogs. Lett. Org. Chem. 2015, 12, 516–522. [Google Scholar] [CrossRef]
- Aziz, M.; Ejaz, S.A.; Rehman, H.M.; Al-Buriahi, M.S.; Siddique, F.; Somaily, H.H.; Alrowaili, Z.A. Identification of NEK7 Inhibitors: Structure based Virtual Screening, Molecular Docking, Density functional theory calculations and Molecular Dynamics Simulations. Res. Sq. 2022. preprint. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Ricardo, N. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Hildebrand, P.W.; Rose, A.; Tiemann, J. Bringing Molecular Dynamics Simulation Data into View. Trends Biochem. Sci. 2019, 44, 902–913. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, M.A.; Iqbal, M.N.; Saddick, S.; Ali, I.; Khan, F.S.; Kanwal, S.; Ahmed, D.; Ibrahim, M.; Afzal, U.; Awais, M. Identification of lead compounds against Scm (fms10) in Enterococcus faecium using computer aided drug designing. Life 2021, 11, 77. [Google Scholar] [CrossRef]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695. [Google Scholar] [CrossRef] [Green Version]
- Luty, B.A.; Davis, M.E.; Tironi, I.G.; Van Gunsteren, W.F. A comparison of particle-particle, particle-mesh and Ewald methods for calculating electrostatic interactions in periodic molecular systems. Mol. Simul. 1994, 14, 11–20. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, T.J.; Tu, S.; Zhang, Z.H.; Meng, F.H. Identification of Novel Src Inhibitors: Pharmacophore-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations. Molecules 2020, 25, 4094. [Google Scholar] [CrossRef]
- Humphreys, D.D.; Friesner, R.A.; Berne, B.J. A Multiple-Time-Step Molecular Dynamics Algorithm for Macromolecules. J. Phys. Chem. 1994, 98, 6885–6892. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Qu, X.; Latino, D.A.; Aires-De-Sousa, J. A big data approach to the ultra-fast prediction of DFT-calculated bond energies. J. Chemin. 2013, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Challenges for Density Functional Theory. Chem. Rev. 2011, 112, 289–320. [Google Scholar] [CrossRef]
- Ribeiro, J.; Ríos-Vera, C.; Melo, F.; Schüller, A. Calculation of accurate interatomic contact surface areas for the quantitative analysis of non-bonded molecular interactions. Bioinformatics 2012, 35, 3499–3501. [Google Scholar] [CrossRef] [Green Version]
- Azam, F.; Alabdullah, N.H.; Ehmedat, H.M.; Abulifa, A.R.; Taban, I.; Upadhyayula, S. NSAIDs as potential treatment option for preventing amyloid β toxicity in Alzheimer’s disease: An investigation by docking, molecular dynamics, and DFT studies. J. Biomol. Struct. Dyn. 2018, 36, 2099–2117. [Google Scholar] [CrossRef]
- Hospital, A.; Goñi, J.R.; Orozco, M.; Gelpí, J. Molecular dynamics simulations: Advances and applications. Adv. Appl. Bioinform. Chem. AABC 2015, 8, 37. [Google Scholar] [PubMed] [Green Version]
- Choudhary, M.I.; Shaikh, M.; tul-Wahab, A.; ur-Rahman, A. In silico identification of potential inhibitors of key SARS-CoV-2 3CL hydrolase (Mpro) via molecular docking, MMGBSA predictive binding energy calculations, and molecular dynamics simulation. PLoS ONE 2020, 15, e0235030. [Google Scholar] [CrossRef] [PubMed]
- Katari, S.K.; Natarajan, P.; Swargam, S.; Kanipakam, H.; Pasala, C.; Umamaheswari, A. Inhibitor design against JNK1 through e-pharmacophore modeling docking and molecular dynamics simulations. J. Recept. Signal Transduct. 2016, 36, 558–571. [Google Scholar] [CrossRef] [PubMed]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. In Protein Dynamics; Humana Press: Totowa, NJ, USA, 2014; pp. 193–226. [Google Scholar]
- Vijayakumar, B.; Parasuraman, S.; Raveendran, R.; Velmurugan, D. Identification of natural inhibitors against angiotensin I converting enzyme for cardiac safety using induced fit docking and MM-GBSA studies. Pharmacogn. Mag. 2014, 10 (Suppl. S3), S639. [Google Scholar]
- Lyne, P.D.; Lamb, A.M.L.; Saeh, J.C. Accurate Prediction of the Relative Potencies of Members of a Series of Kinase Inhibitors Using Molecular Docking and MM-GBSA Scoring. J. Med. Chem. 2006, 49, 4805–4808. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Optimization Energy (Hartree) | Polarizability (α) (a.u.) | Dipole Moment (Debye) |
|---|---|---|---|
| Compound 208 | −2712.903 | 340.588 | 13.330 |
| Compound 248 | −2391.895 | 345.671 | 7.283 |
| Compound 255 | −2238.320 | 304.820 | 8.827 |
| Compound 762 | −2699.752 | 360.213 | 10.042 |
| Dabrafenib | −2407.203 | 319.254 | 6.682 |
| Compound | EHOMO(eV) | ELUMO(eV) | ∆Egap(eV) | Potential Ionization I (eV) | Affinity A (eV) |
|---|---|---|---|---|---|
| Compound 208 | −0.234 | −0.099 | 0.135 | 0.234 | 0.099 |
| Compound 248 | −0.222 | −0.070 | 0.152 | 0.222 | 0.070 |
| Compound 255 | −0.228 | −0.083 | 0.145 | 0.228 | 0.083 |
| Compound 762 | −0.216 | −0.089 | 0.127 | 0.216 | 0.089 |
| Dabrafenib | −0.233 | −0.074 | 0.159 | 0.233 | 0.074 |
| Compound | Hardness (η) | Softness (S) | Electronegativity (X) | Chemical Potential (μ) | Electrophilicity Index (ω) |
| Compound 208 | 0.067 | 7.433 | 0.167 | −0.167 | 0.207 |
| Compound 248 | 0.076 | 6.566 | 0.147 | −0.147 | 0.141 |
| Compound 255 | 0.072 | 6.901 | 0.156 | −0.156 | 0.167 |
| Compound 762 | 0.063 | 7.880 | 0.153 | −0.153 | 0.184 |
| Dabrafenib | 0.080 | 6.280 [38] | 0.154 | −0.154 | 0.149 |
| Compound | Electrodonating power (ω-) | Electroaccepting power (ω+) | Net Electrophilicity(Δω±) | ||
| Compound 208 | 0.299 | 0.132 | 0.432 | ||
| Compound 248 | 0.224 | 0.077 | 0.301 | ||
| Compound 255 | 0.254 | 0.098 | 0.352 | ||
| Compound 762 | 0.268 | 0.116 | 0.384 | ||
| Dabrafenib | 0.236 | 0.082 | 0.318 [38] | ||
| Compound | Binding Energies (kJ/mol) | Predicted Binding Affinity (IC50) nM | pIC50 (Predicted via Deep Learning Model) |
|---|---|---|---|
| Compound 208 | −33.47 | 206.26 | 6.69 |
| Compound 248 | −35.56 | 268.80 | 6.57 |
| Compound 255 | −35.98 | 283 | 6.55 |
| Compound 762 | −42.67 | 61.74 | 7.21 |
| Dabrafenib | −33.89 | 1-9 (Experimental) [1] | --- |
| Protein-Ligand Complex | Average Protein RMSD (Å) | Average Protein RMSF (Å) | Average Protein-Ligand Complex RMSD (Å) | Average Radius of Gyration (Å) | Average SASA (Residue Wise) (Å2) |
|---|---|---|---|---|---|
| NEK7–Dabrafenib complex | 1.97 | 0.87 | 3.89 | 19.76 | 282.72 |
| Sr No. | Hydrogen Donor | Hydrogen Acceptor |
|---|---|---|
| 1 | ALA114-Main | LIGAND-Side |
| 2 | LIGAND-Side | LEU113-Side |
| 3 | LIGAND-Side | ASP115-Side |
| 4 | ARG121-Side | LIGAND-Side |
| 5 | LIGAND-Side | ASP179-Side |
| 6 | LIGAND-Side | GLU112-Main |
| 7 | GLY117-Main | LIGAND-Side |
| 8 | ASP115-Main | LIGAND-Side |
| 9 | LIGAND-Side | ALA114-Main |
| 10 | LIGAND-Side | ILE40-Main |
| 11 | LIGAND-Side | ARG42-Main |
| 12 | GLY41-Main | LIGAND-Side |
| Binding Free Energy ΔGbind (kcal/mol) | ΔE coulomb (kcal/mol) | ΔE covalent (kcal/mol) | ΔE H-bond (kcal/mol) | ΔE vdW (kcal/mol) | Lipophilic Energy (kcal/mol) | Sol_GB (kcal/mol) | |
|---|---|---|---|---|---|---|---|
| Dabrafenib | −50.44 | 31.05 | 11.71 | −0.18 | −38.88 | −33.98 | −17.74 |
| Binding Free Energy ΔGbind (kcal/mol) | ΔE vdW (kcal/mol) | Eel (kcal/mol) | ENPOLAR (kcal/mol) | EPB (kcal/mol) | EDISPER (kcal/mol) | |
|---|---|---|---|---|---|---|
| Dabrafenib | −56.12 | −38.27 | −17.84 | −26.71 | 32.37 | 47.53 |
| Property Predicted | Compound 208 | Compound 248 | Compound 255 | Compound 762 |
|---|---|---|---|---|
| Solubility | −4.50 log mol/L | −4.05 log mol/L | −3.19 log mol/L | −3.10 log mol/L |
| Lipophilicity | 1.82 (log-ratio) | 1.89 (log-ratio) | 1.40 (log-ratio) | 1.88 (log-ratio) |
| (Absorption) Caco-2 | −5.14 cm/s | −5.20 cm/s | −5.14 cm/s | −5.21 cm/s |
| (Absorption) HIA | 91.18% | 89.62% | 89.69% | 92.89% |
| (Absorption) Pgp | 10.18% | 13.10% | 5.84% | 11.42% |
| (Absorption) Bioavailability F20 | 76.34% | 75.94% | 75.46% | 76.42% |
| (Distribution) BBB | 76.85% | 76.66% | 93.85% | 86.17% |
| (Distribution) PPBR | 79.65% | 77.11% | 63.16% | 80.66% |
| (Metabolism) CYP2C19 | 81.26% | 72.00% | 37.21% | 24.15% |
| (Metabolism) CYP2D6 | 62.01% | 51.90% | 25.60% | 13.64% |
| (Metabolism) CYP3A4 | 74.21% | 56.97% | 60.78% | 22.83% |
| (Metabolism) CYP1A2 | 36.70% | 10.29% | 9.08% | 21.05% |
| (Metabolism) CYP2C9 | 17.86% | 6.99% | 4.13% | 8.60% |
| (Execretion) Half life | 8.06 h | 8.01 h | 7.86 h | 7.88 h |
| (Execretion) Clearance | 8.23 mL/min/kg | 8.26 mL/min/kg | 8.10 mL/min/kg | 8.54 mL/min/kg |
| Clinical Toxicity | 14.92% | 15.59% | 24.33% | 0.28% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aziz, M.; Ejaz, S.A.; Zargar, S.; Akhtar, N.; Aborode, A.T.; A. Wani, T.; Batiha, G.E.-S.; Siddique, F.; Alqarni, M.; Akintola, A.A. Deep Learning and Structure-Based Virtual Screening for Drug Discovery against NEK7: A Novel Target for the Treatment of Cancer. Molecules 2022, 27, 4098. https://doi.org/10.3390/molecules27134098
Aziz M, Ejaz SA, Zargar S, Akhtar N, Aborode AT, A. Wani T, Batiha GE-S, Siddique F, Alqarni M, Akintola AA. Deep Learning and Structure-Based Virtual Screening for Drug Discovery against NEK7: A Novel Target for the Treatment of Cancer. Molecules. 2022; 27(13):4098. https://doi.org/10.3390/molecules27134098
Chicago/Turabian StyleAziz, Mubashir, Syeda Abida Ejaz, Seema Zargar, Naveed Akhtar, Abdullahi Tunde Aborode, Tanveer A. Wani, Gaber El-Saber Batiha, Farhan Siddique, Mohammed Alqarni, and Ashraf Akintayo Akintola. 2022. "Deep Learning and Structure-Based Virtual Screening for Drug Discovery against NEK7: A Novel Target for the Treatment of Cancer" Molecules 27, no. 13: 4098. https://doi.org/10.3390/molecules27134098