
Strengthening the Connection between Science, Society and Environment to Develop Future French and European Bioeconomies: Cutting-Edge Research of VAALBIO Team at UCCS

 ,
,  ,
,  ,
,  ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Recent Works and Results
2.1. Reactions Involving Oxygen
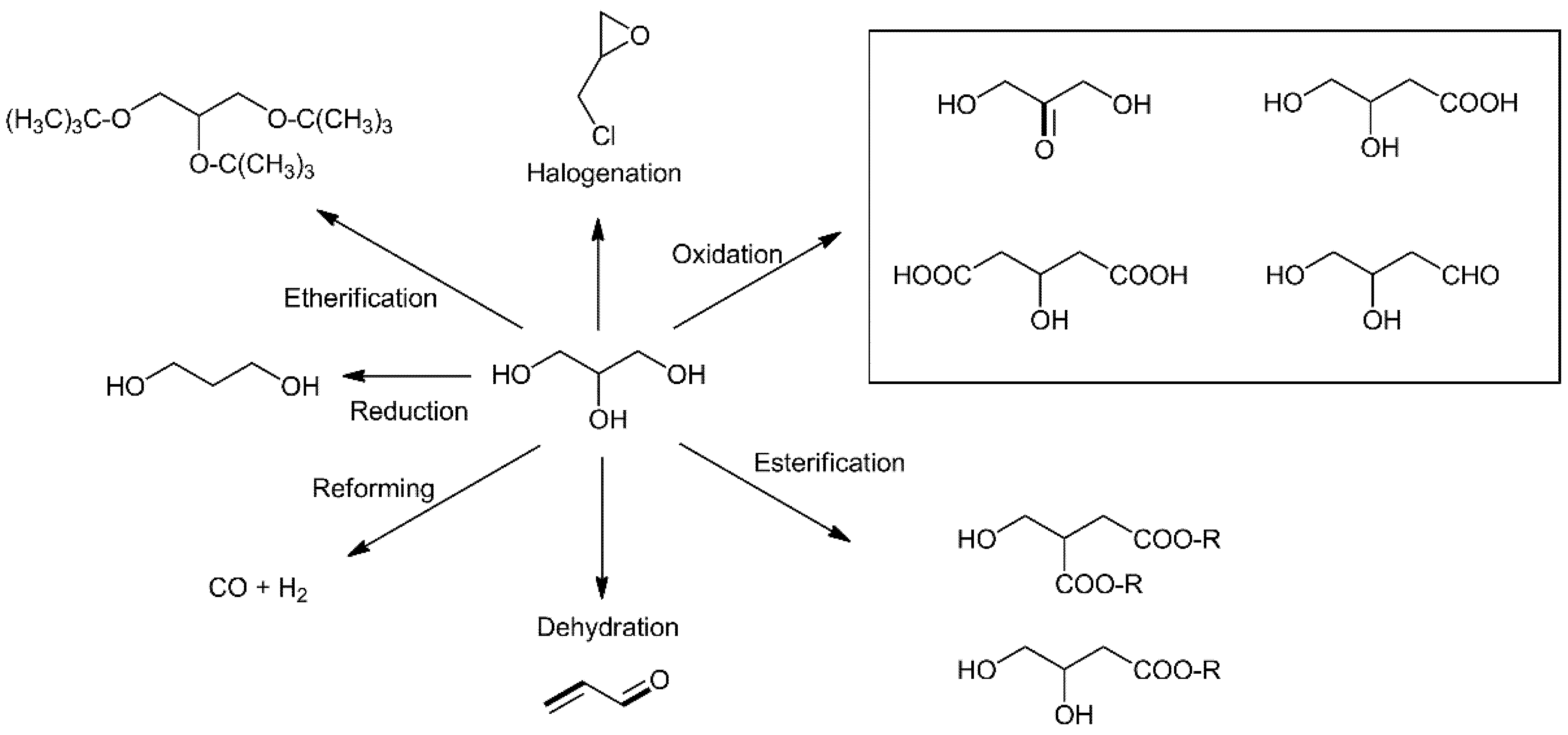
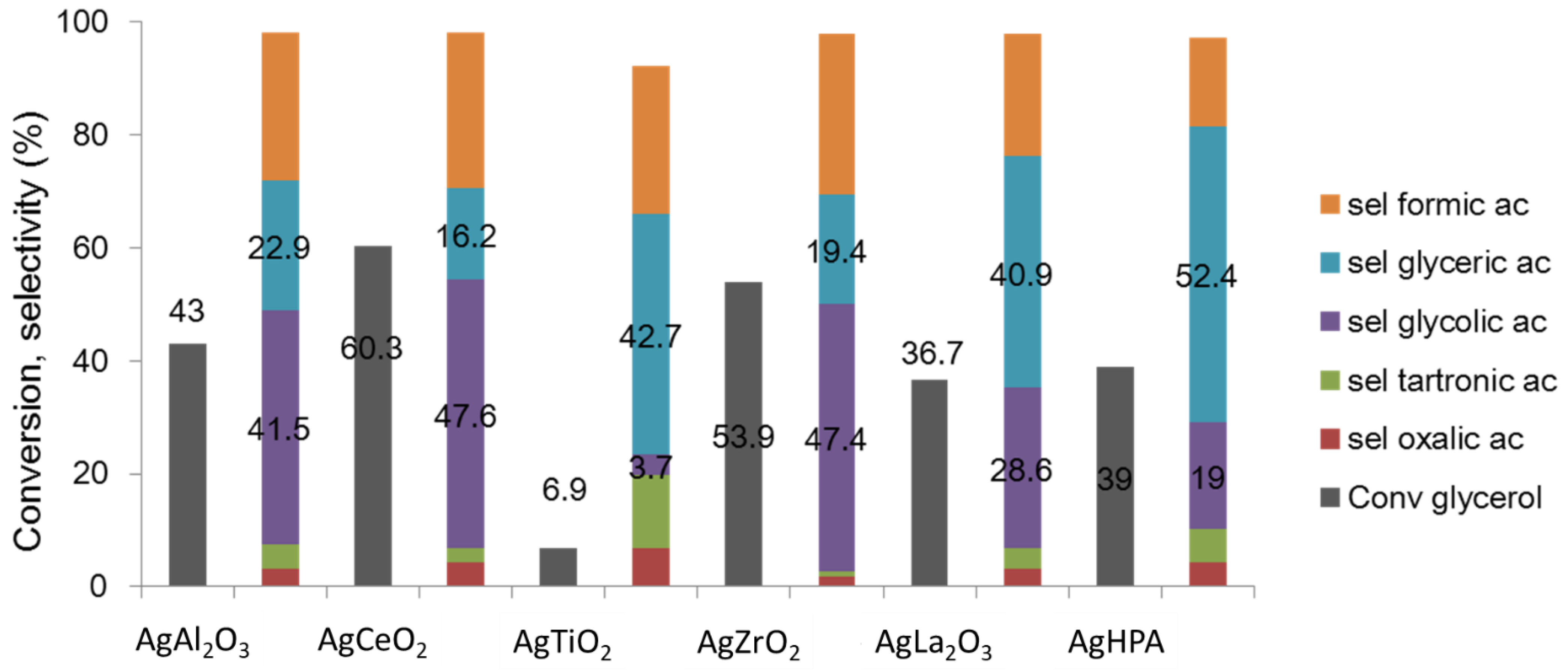
2.1.1. Selective Oxidation of Glycerol
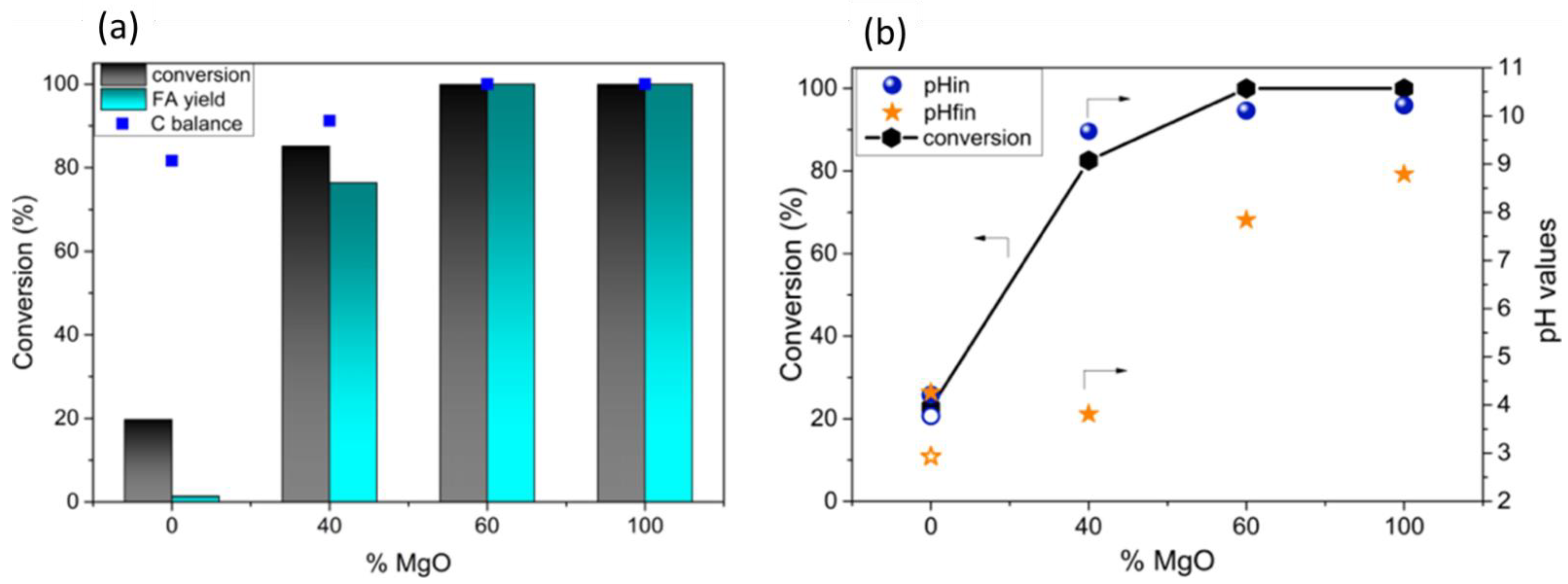
2.1.2. Allyl Alcohol Oxidation to Acrylic Acid
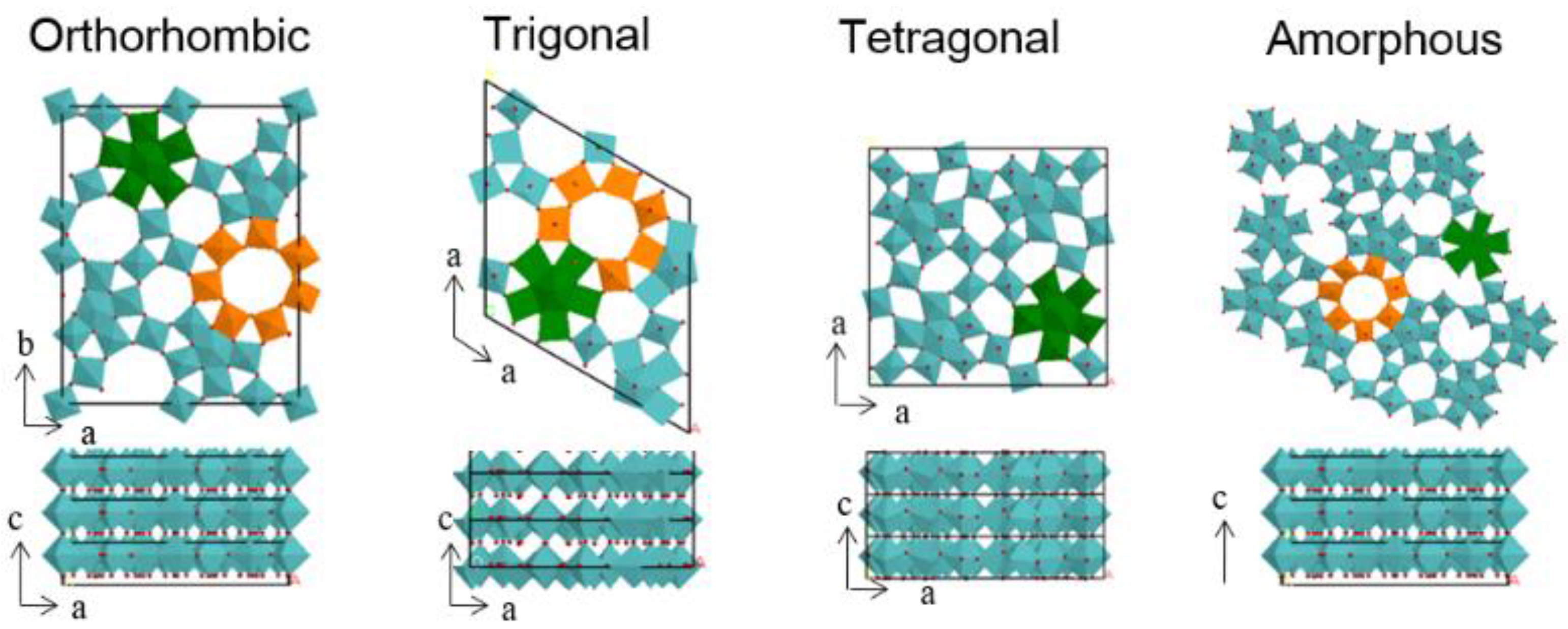
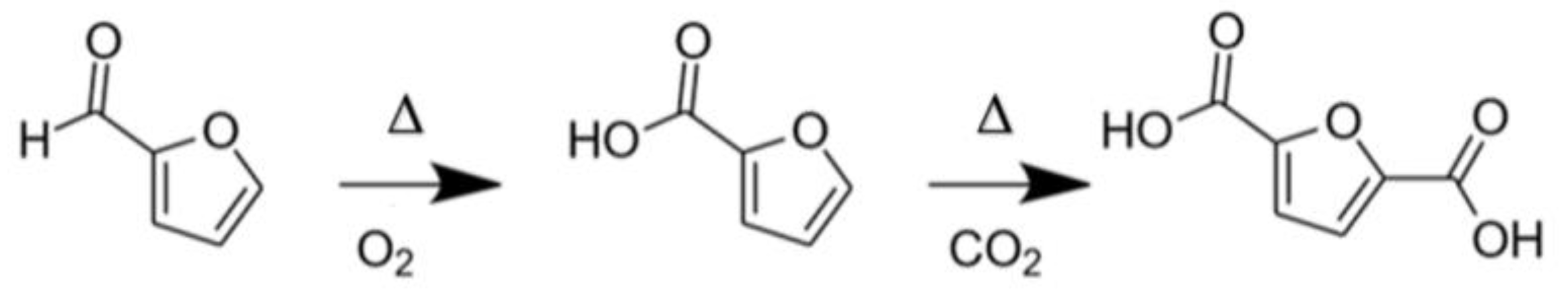
2.1.3. Selective Oxidation of Furanics
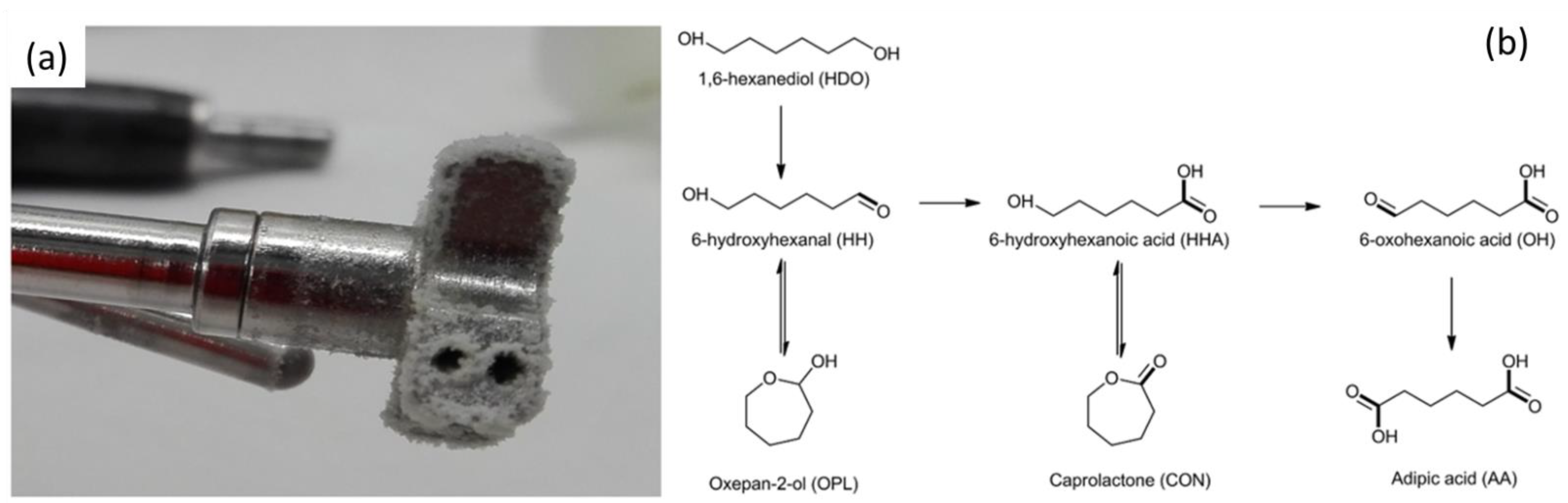
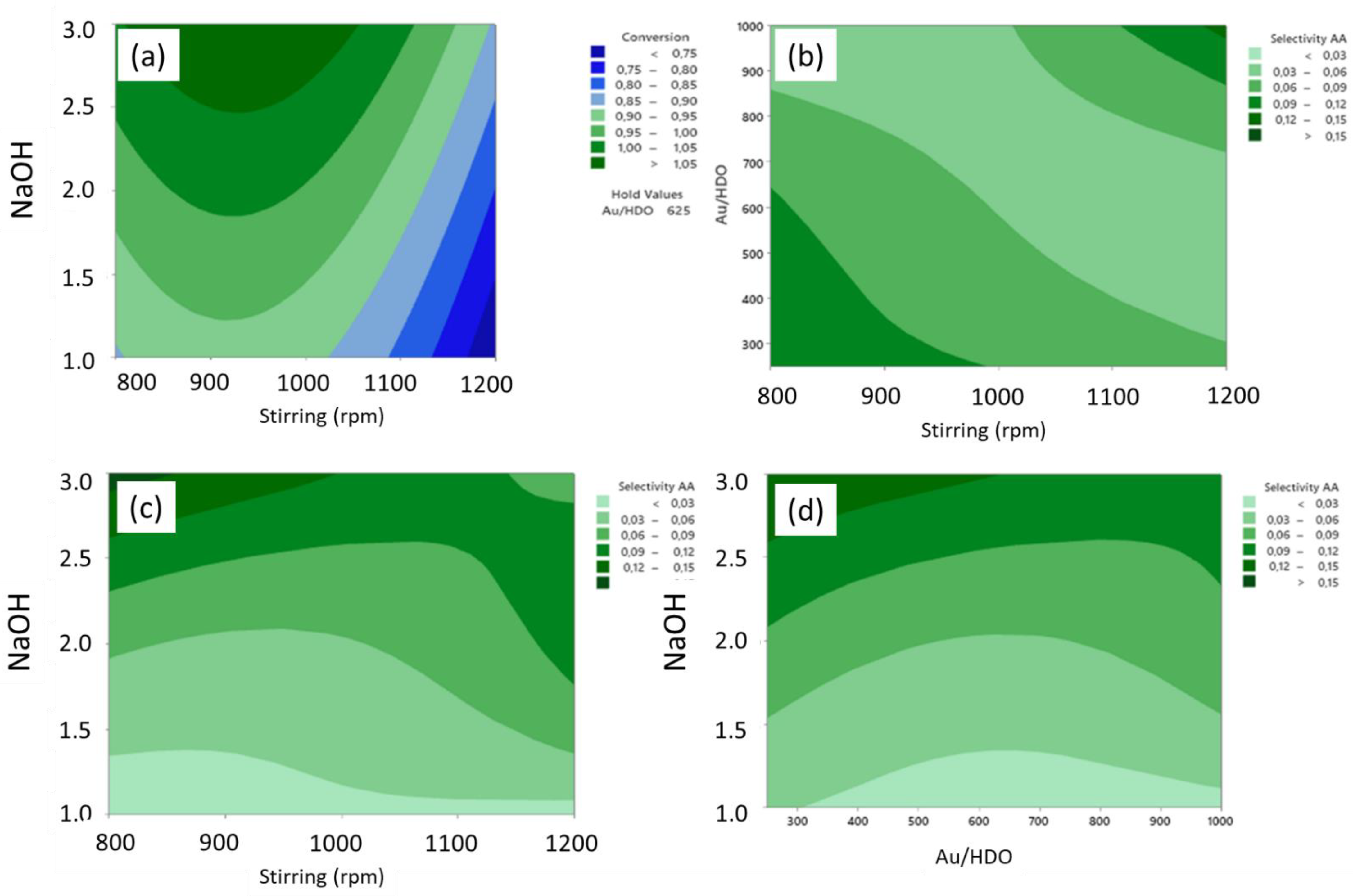
2.1.4. Adipic Acid Synthesis
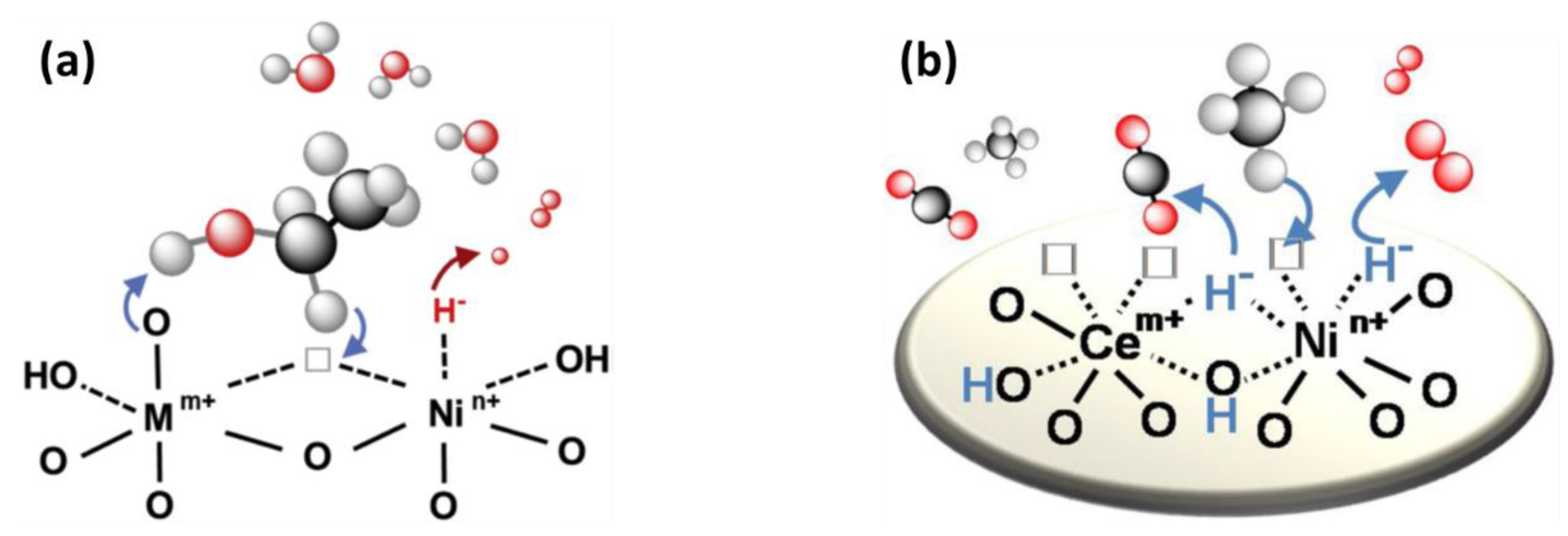
2.1.5. Selective Oxidation of H2S via Chemical Looping
- A high reactivity in both the oxidation and the regeneration reactions;
- A good selectivity for the desired oxidation product, i.e., elemental sulfur;
- A good stability over many redox cycles;
- A good resistance towards sulfur deposition and removal.
2.2. Reactions Involving Hydrogen
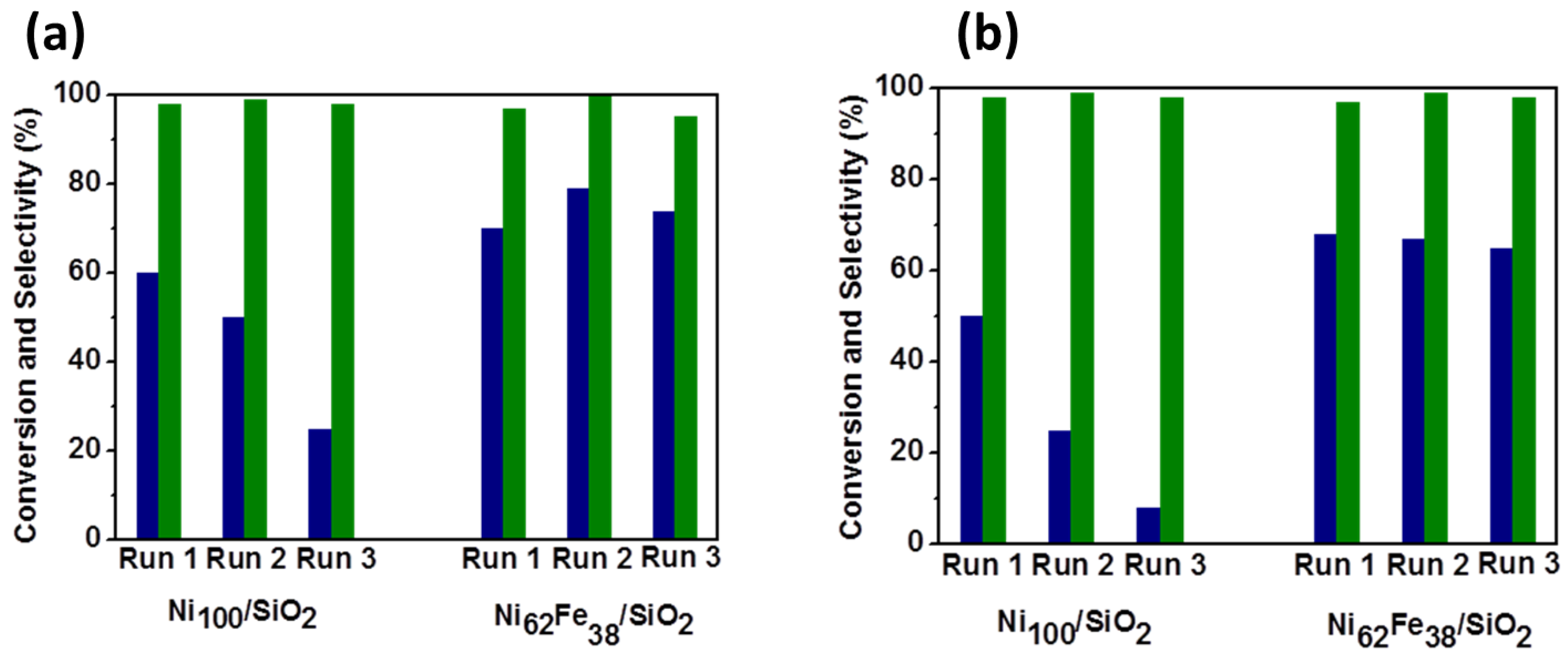
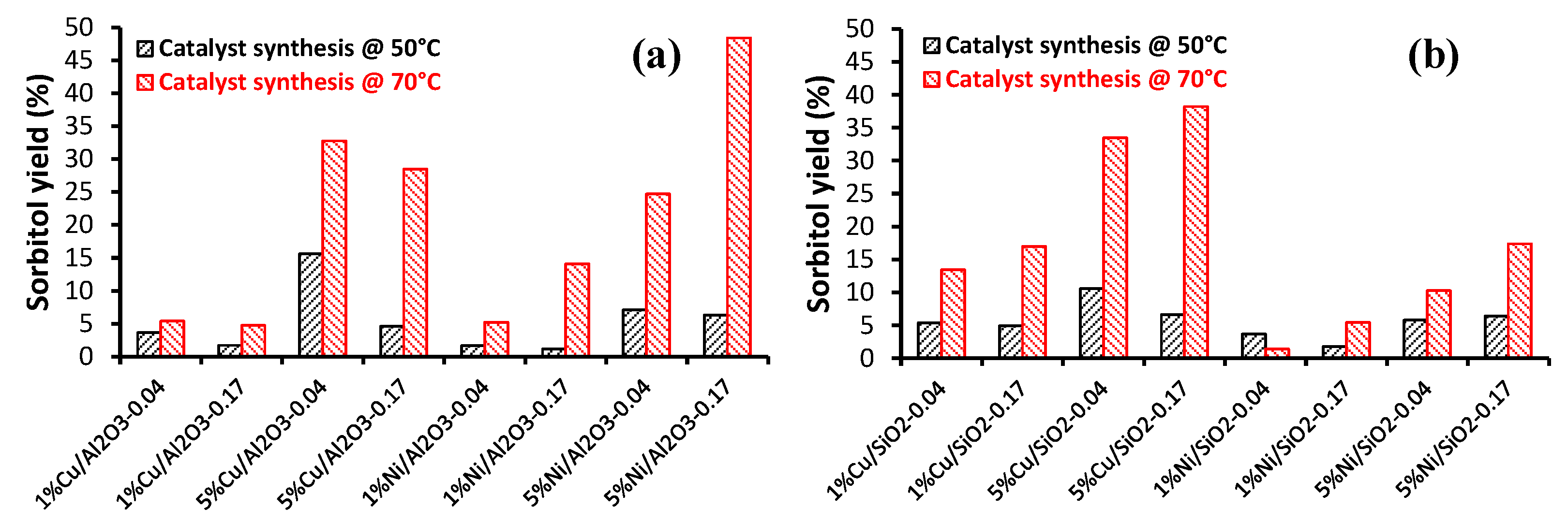
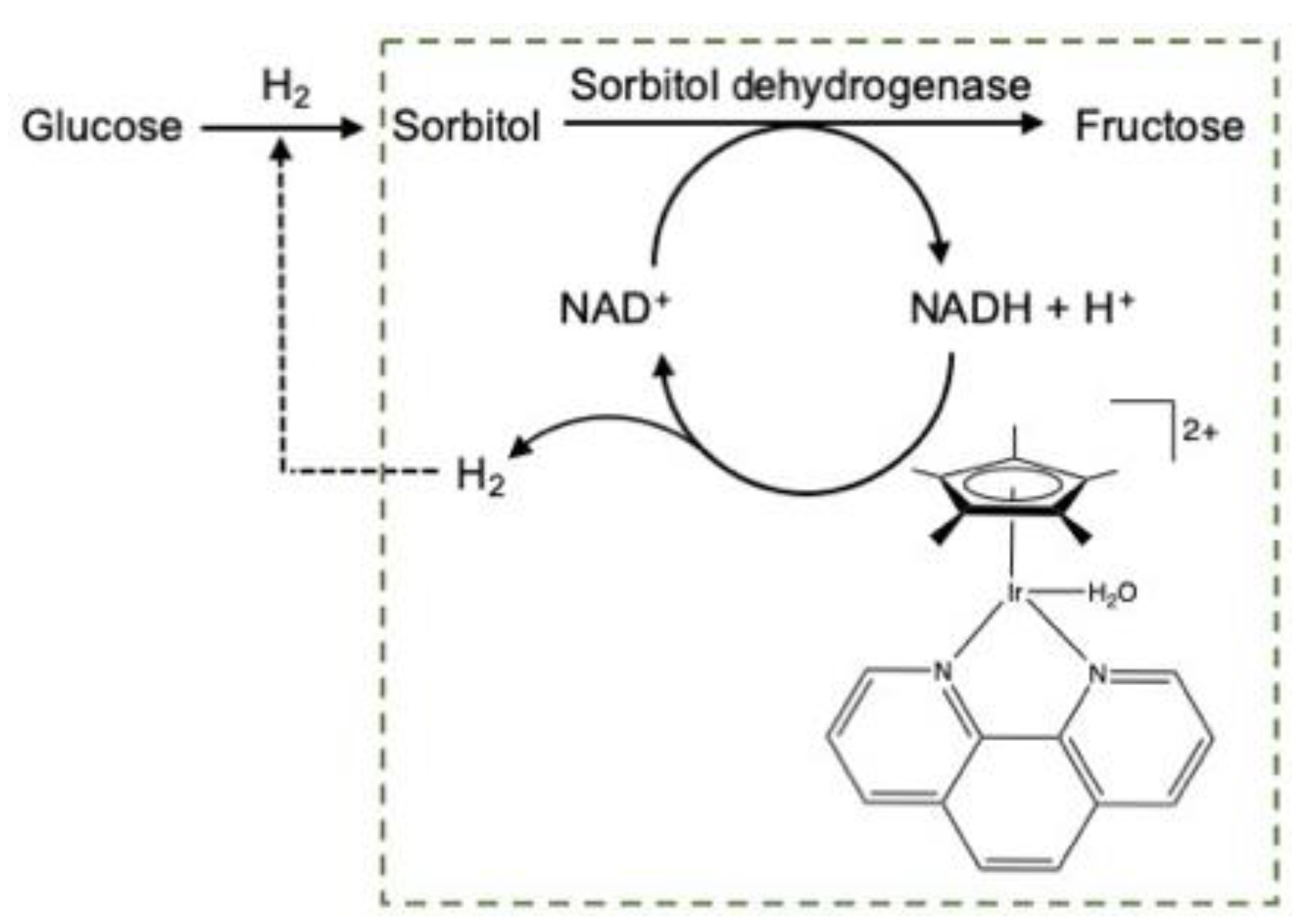
2.2.1. Carbohydrate Hydrogenation
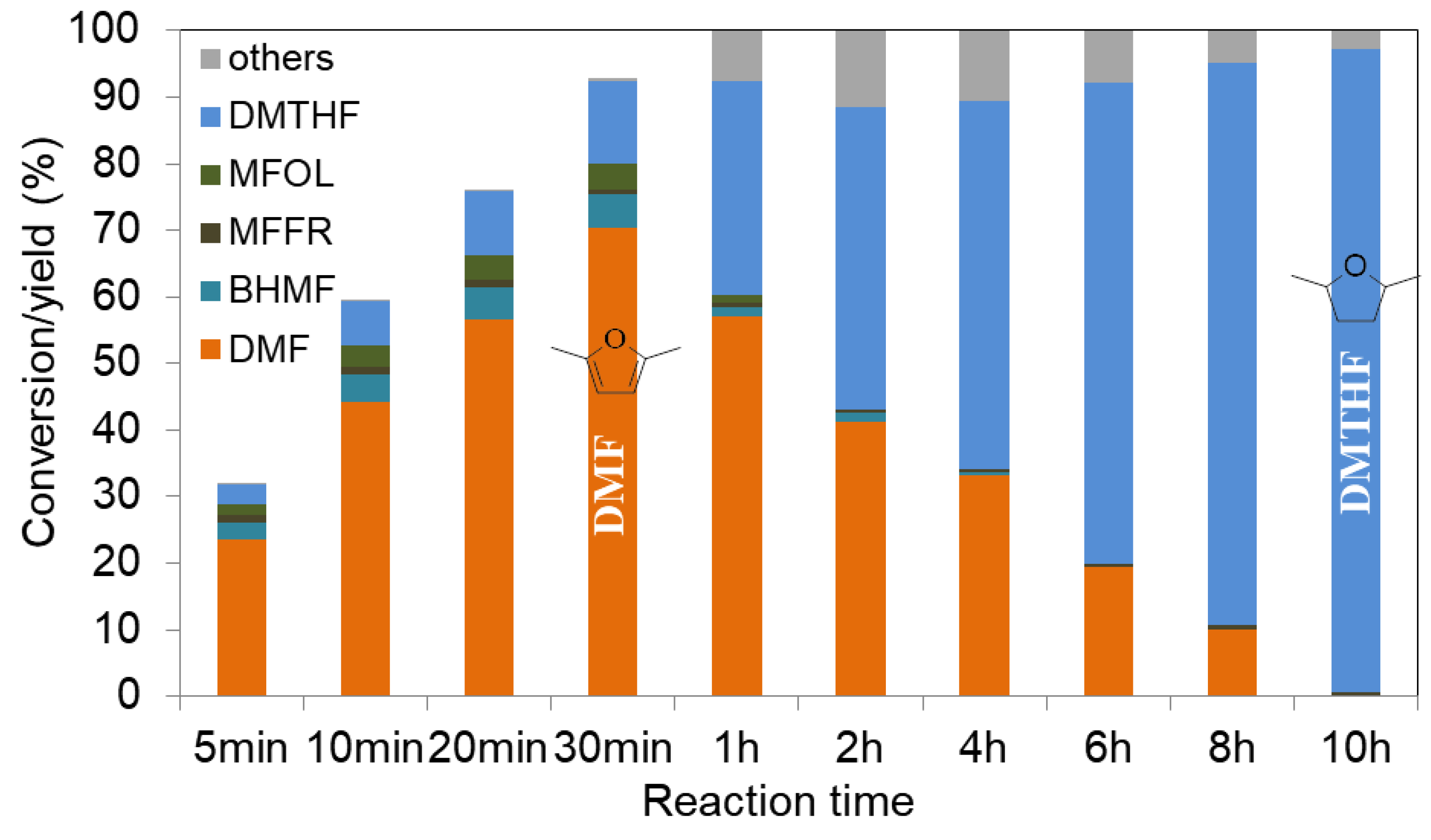
2.2.2. Furanics Hydrogenation
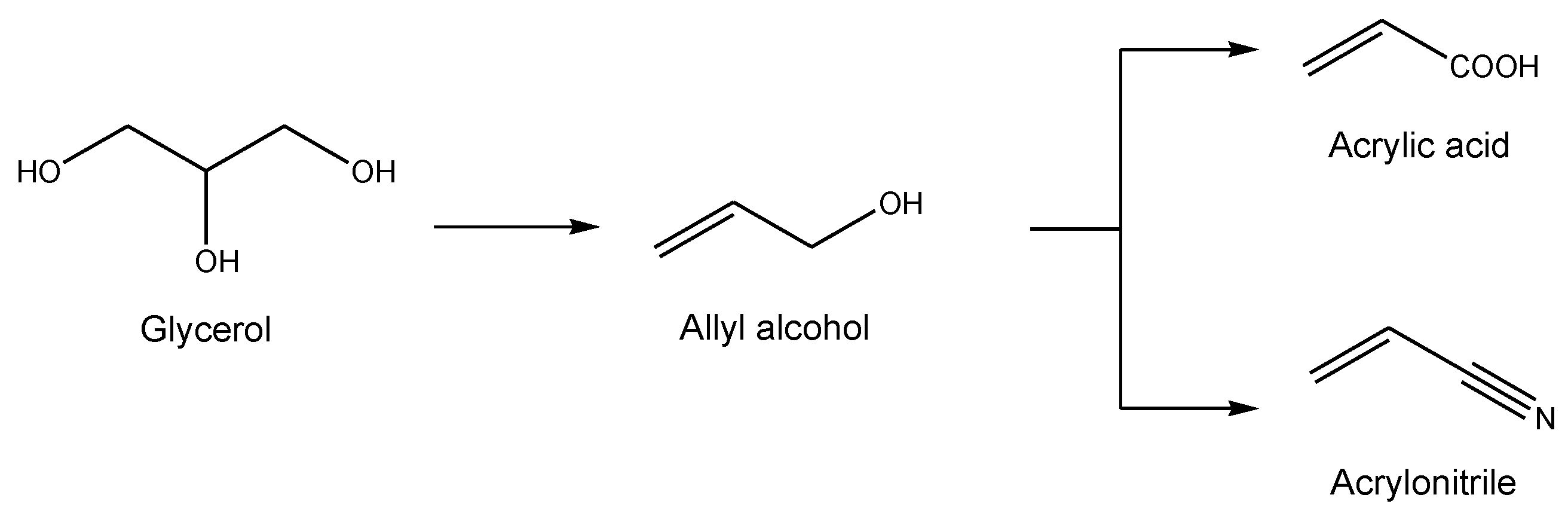
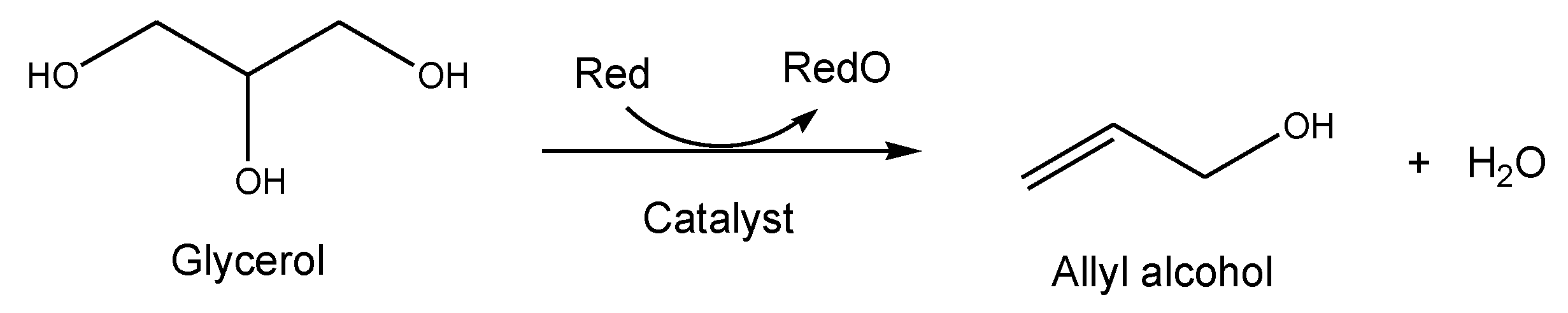
2.2.3. Allyl Alcohol Synthesis from Glycerol
2.3. Reactions under Specific Atmosphere (CO2, NH3 and N2)
2.3.1. Direct Carboxylation
2.3.2. Ammoxidation
2.3.3. Glycerol Polymerization
2.3.4. Butadiene Synthesis from Ethanol
2.4. H2 Production from Bioresources (Biomass and Biogas)
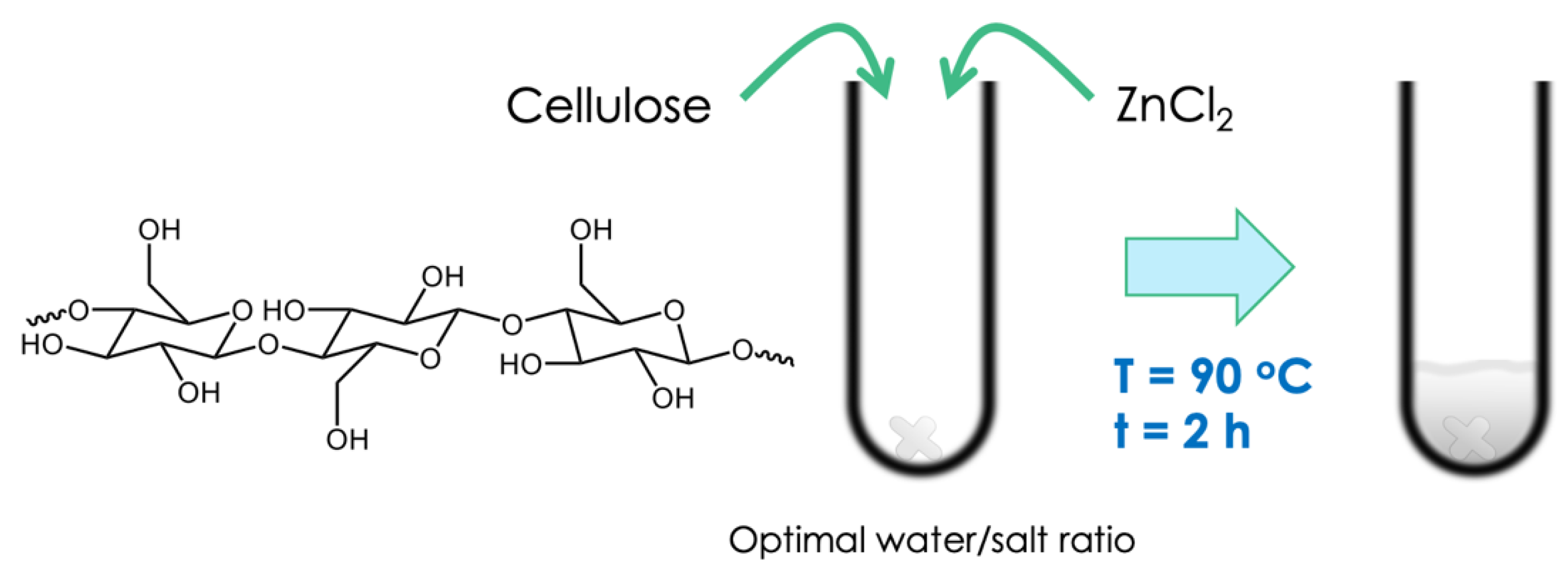
2.5. Biomass Fractionation
2.6. Hybrid Catalysis
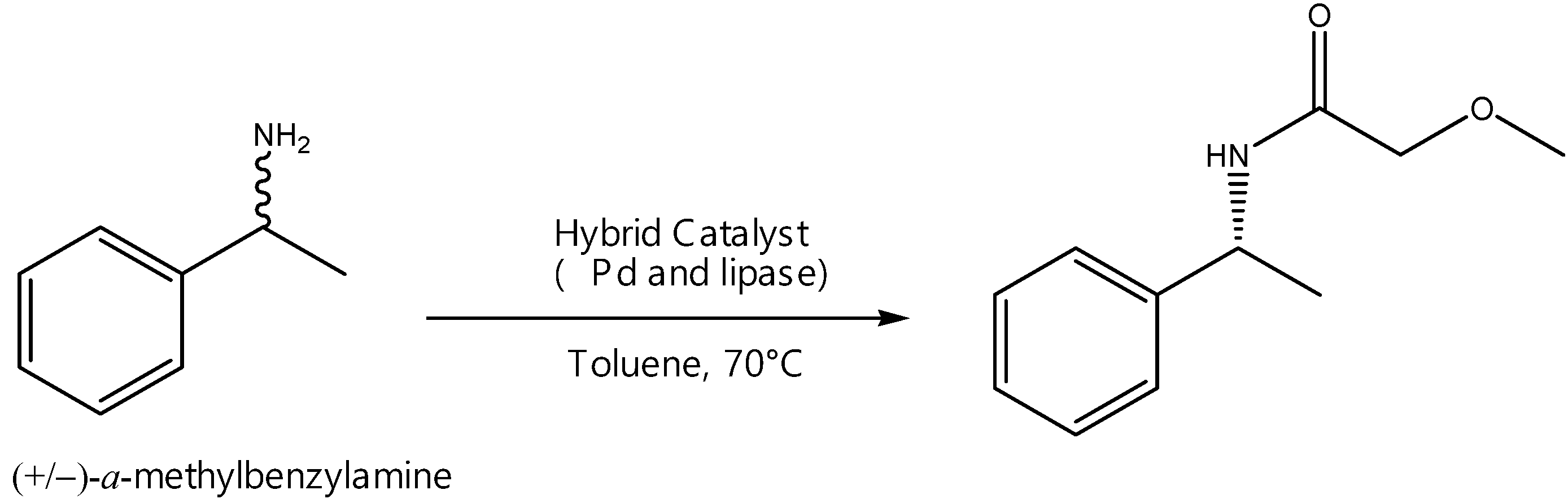
2.6.1. Enhanced Dynamic Kinetic Resolution
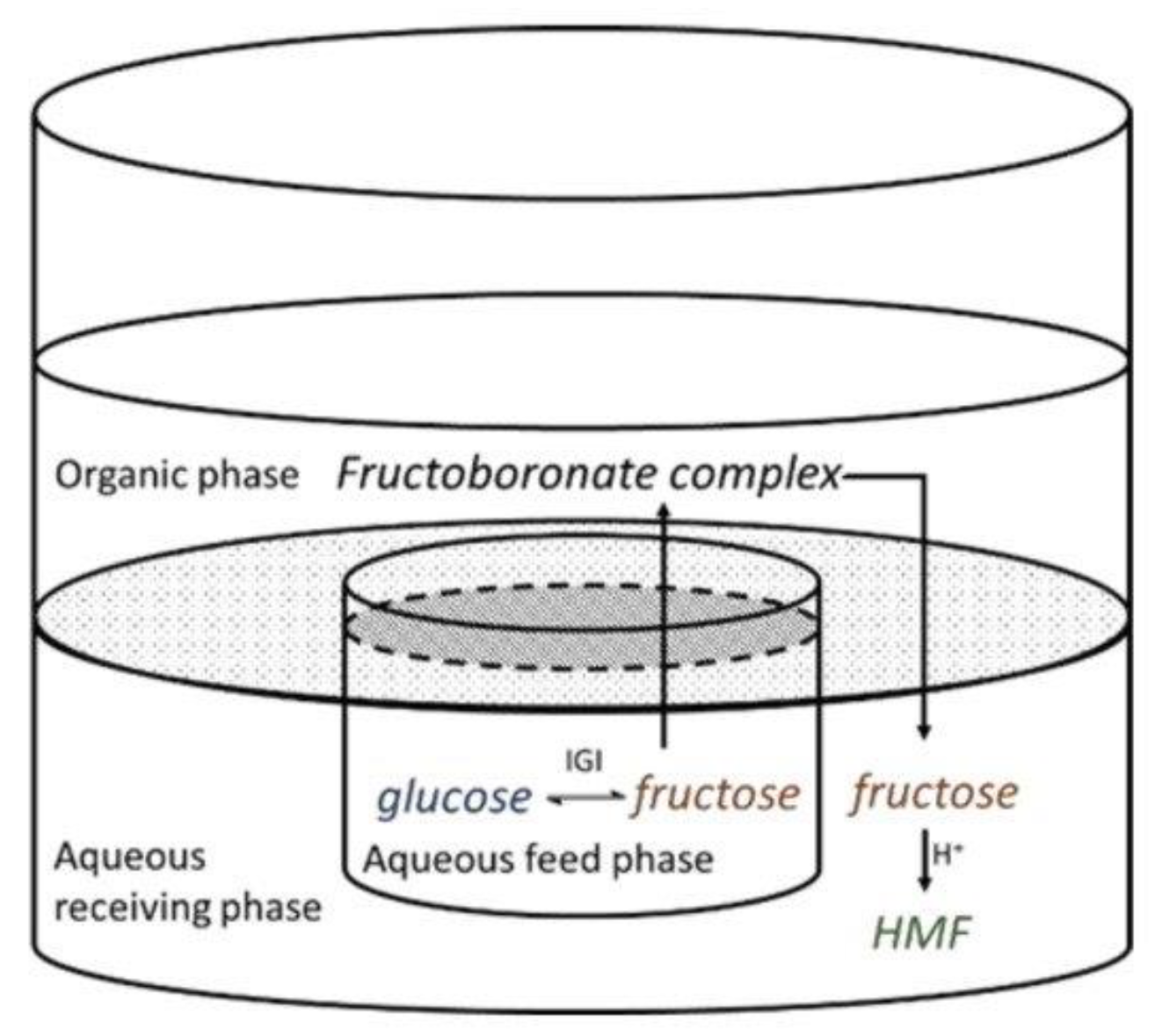
2.6.2. Compartmentalization
2.6.3. Hybrid Isomerization
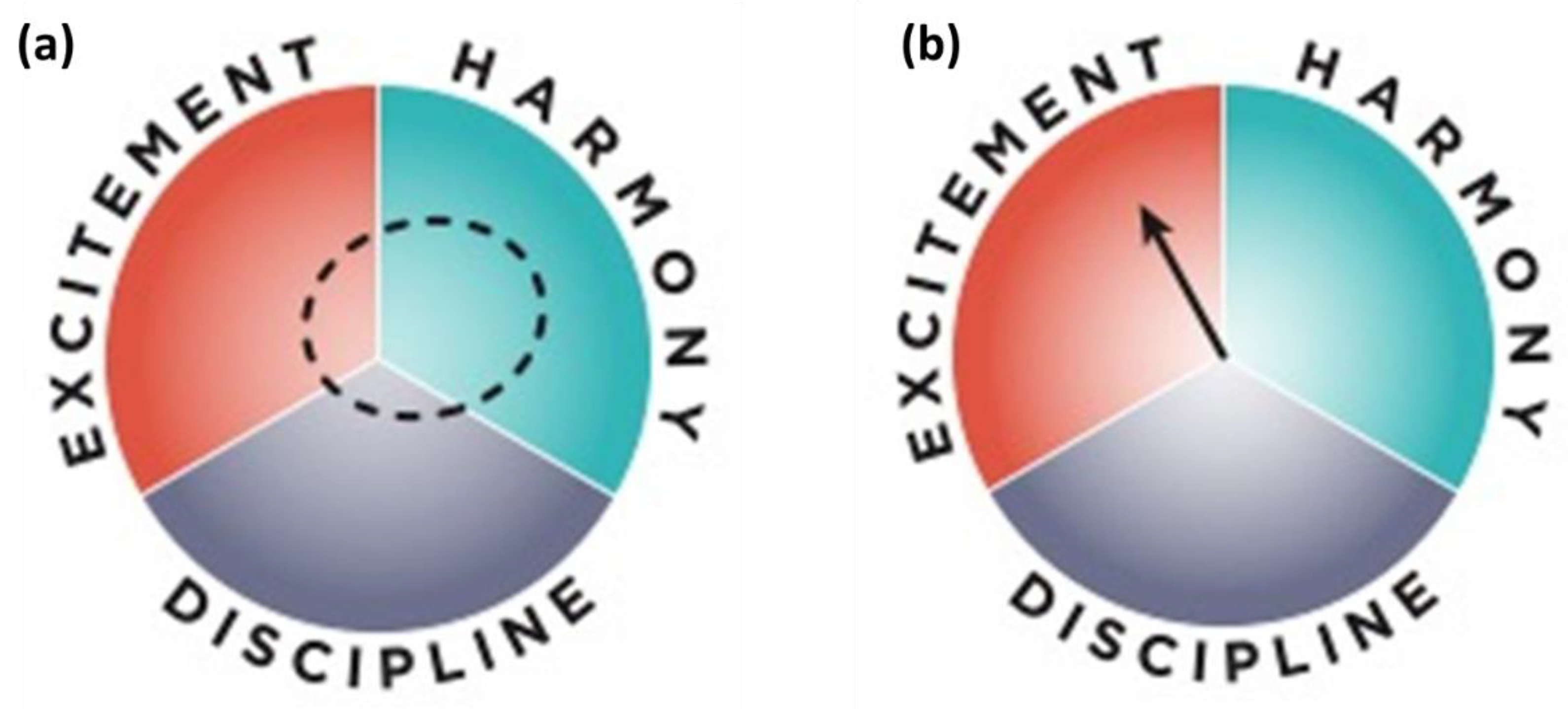
2.7. Solving the Contextual Problem of Implementing Solutions
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dumeignil, F.; Capron, M.; Katryniok, B.; Wojcieszak, R.; Löfberg, A.; Girardon, J.S.; Desset, S.; Araque-Marin, M.; Jalowiecki-Duhamel, L.; Paul, S. Biomass-derived Platform Molecules Upgrading through Catalytic Processes: Yielding Chemicals and Fuels. J. Jpn. Pet. Inst. 2015, 58, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Heuson, E.; Froidevaux, R.; Itabaiana, I., Jr.; Wojcieszak, R.; Capron, M.; Dumeignil, F. Optimisation of Catalysts Coupling in Multi-Catalytic Hybrid Materials: Perspectives for the Next Revolution in Catalysis. Green Chem. 2021, 23, 1942–1954. [Google Scholar] [CrossRef]
- Friend, M. The Institutional Compass: Method, Use and Scope; Series: Methods; Springer Nature: Berlin, Germany, 2022; ISSN 2542-9892. [Google Scholar]
- Kenar, J. Glycerol as a platform chemical: Sweet opportunities on the horizon? Lipid Technol. 2007, 19, 249–253. [Google Scholar] [CrossRef]
- Katryniok, B.; Paul, S.; Capron, M.; Bellière-Baca, V.; Rey, P.; Dumeignil, F. Regeneration of Silica-Supported Silicotungstic Acid as a Catalyst for the Dehydration of Glycerol. ChemSusChem 2012, 5, 1298–1306. [Google Scholar] [CrossRef]
- Skrzyńska, E.; Ftouni, J.; Girardon, J.S.; Capron, M.; Jalowiecki-Duhamel, L.; Paul, J.F.; Dumeignil, F. Quasi-Homogeneous Oxidation of Glycerol by Unsupported Gold Nanoparticles in the Liquid Phase. ChemSusChem 2012, 5, 2065–2068. [Google Scholar] [CrossRef] [PubMed]
- Skrzyńska, E.; Ftouni, J.; Mamede, A.S.; Addad, A.; Trentesaux, M.; Girardon, J.S.; Capron, M.; Dumeignil, F. Glycerol oxidation over gold supported catalysts—“Two faces” of sulphur based anchoring agent. J. Mol. Catal. A Chem. 2014, 382, 71–78. [Google Scholar] [CrossRef]
- Skrzyńska, E.; Zaid, S.; Girardon, J.S.; Capron, M.; Dumeignil, F. Catalytic behaviour of four different supported noble metals in the crude glycerol oxidation. Appl. Catal. A Gen. 2015, 499, 89–100. [Google Scholar] [CrossRef]
- Dimitratos, N.; Villa, A.; Bianchi, C.L.; Prati, L.; Makkee, M. Gold on titania: Effect of preparation method in the liquid phase oxidation. Appl. Catal. A Gen. 2006, 311, 185. [Google Scholar] [CrossRef] [Green Version]
- Díaz, J.A.; Skrzyńska, E.; Girardon, J.S.; Ftouni, J.; Capron, M.; Dumeignil, F.; Fongarland, P. Kinetic modeling of the quasi-homogeneous oxidation of glycerol over unsupported gold particles in the liquid phase. Eur. J. Lipid Sci. Technol. 2016, 118, 72–79. [Google Scholar] [CrossRef]
- Díaz, J.A.; Skrzyńska, E.; Zaid, S.; Girardon, J.S.; Capron, M.; Dumeignil, F.; Fongarland, P. Kinetic modelling of the glycerol oxidation in the liquid phase: Comparison of Pt, Au and Ag As active phases. J. Chem. Technol. Biotechnol. 2017, 92, 2267–2275. [Google Scholar] [CrossRef]
- Carrentin, S.; McMorn, P.; Johnston, P.; Griffin, K.; Hutchings, G. Selective oxidation of glycerol to glyceric acid using a gold catalyst in aqueous sodium hydroxide. Chem. Commun. 2002, 7., 696–697. [Google Scholar] [CrossRef] [PubMed]
- Carrentin, S.; McMorn, P.; Johnston, P.; Griffin, K.; Kiely, J.; Hutchings, G. Oxidation of glycerol using supported Pt, Pd and Au catalysts. Phys. Chem. Chem. Phys. 2003, 5, 1329–1336. [Google Scholar] [CrossRef]
- Li, Y.; Zaera, F. Sensitivity of the glycerol oxidation reaction to the size and shape of the platinum nanoparticles in Pt/SiO2 catalysts. J. Catal. 2015, 326, 116. [Google Scholar] [CrossRef]
- Skrzyńska, E.; El Roz, A.; Paul, S.; Capron, M.; Dumeignil, F. Glycerol Partial Oxidation over Pt/Al2O3 Catalysts under Basic and Base-Free Conditions-Effect of the Particle Size. J. Am. Oil Chem. 2019, 96, 63–74. [Google Scholar] [CrossRef] [Green Version]
- El Roz, A.; Fongarland, P.; Dumeignil, F.; Capron, M. Glycerol to Glyceraldehyde Oxidation Reaction Over Pt-Based Catalysts Under Base-Free Conditions. Front. Chem. 2019, 7, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrzyńska, E.; Zaid, S.; Addad, A.; Girardon, J.-S.; Capron, M.; Dumeignil, F. Performance of Ag/Al2O3 catalysts in the liquid phase oxidation of glycerol—Effect of preparation method and reaction conditions. Catal. Sci. Technol. 2016, 6, 3182–3196. [Google Scholar] [CrossRef]
- Zaid, S.; Skrzyńska, E.; Addad, A.; Nandi, S.; Jalowiecki-Duhamel, L.; Girardon, J.S.; Capron, M.; Dumeignil, F. Development of Silver Based Catalysts Promoted by Noble Metal M (M = Au, Pd or Pt) for Glycerol Oxidation in Liquid Phase. Top. Catal. 2017, 60, 1072–1081. [Google Scholar] [CrossRef]
- Skrzyńska, E.; Capron, M.; Dumeignil, F.; Jalowiecki-Duhamel, L. Process for Preparing Glycolic Acid. WO Patent 2014/199256, 2014. [Google Scholar]
- Tavera Ruiz, C.P.; Dumeignil, F.; Capron, M. Catalytic Production of Glycolic Acid from Glycerol Oxidation: An Optimization Using Response Surface Methodology. Catalysts 2021, 11, 257. [Google Scholar] [CrossRef]
- Ishakawa, S.; Murayama, T.; Katryniok, B.; Dumeignil, F.; Araque, M.; Heyte, S.; Paul, S.; Yamada, Y.; Iwazaki, M.; Noda, N.; et al. Influence of the structure of trigonal Mo-V-M3rd oxides (M3rd = -, Fe, Cu, W) on catalytic performances in selective oxidations of ethane, acrolein, and allyl alcohol. Appl. Catal. A Gen. 2019, 584, 117151. [Google Scholar] [CrossRef]
- Ishakawa, S.; Murayama, T.; Ohmura, S.; Sadakane, M.; Ueda, W. Synthesis of Novel Orthorhombic Mo and V Based Complex Oxides Coordinating Alkylammonium Cation in Its Heptagonal Channel and Their Application as a Catalyst. Chem. Mater. 2013, 25, 2211–2219. [Google Scholar] [CrossRef]
- Chen, S.; Wojcieszak, R.; Dumeignil, F.; Marceau, E.; Royer, S. How Catalysts and Experimental Conditions Determine the Selective Hydroconversion of Furfural and 5-Hydroxymethylfurfural. Chem. Rev. 2018, 118, 11023–11117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palombo Ferraz, C.; Navarro-Jaén, S.; Rossi, L.; Dumeignil, F.; Ghazzal, M.N.; Wojcieszak, R. Enhancing the activity of gold supported catalysts by oxide coating: Towards efficient oxidations. Green Chem. 2021, 23, 8453–8457. [Google Scholar] [CrossRef]
- Al Rawas, H.; Palombo Ferraz, C.; Thuriot Roukos, J.; Heyte, S.; Paul, S.; Wojcieszak, R. Influence of Pd and Pt Promotion in Gold Based Bimetallic Catalysts on Selectivity Modulation in Furfural Base-Free Oxidation. Catalysts 2021, 11, 1226. [Google Scholar] [CrossRef]
- Santarelli, F.; Paul, S.; Dumeignil, F.; Cavani, F.; Wojcieszak, R. Furoic Acid Preparation Method. WO Patent WO2017158106A1, 2017. [Google Scholar]
- Roselli, A.; Carvalho, Y.; Dumeignil, F.; Cavani, F.; Paul, S.; Wojcieszak, R. Liquid Phase Furfural Oxidation under Uncontrolled pH in Batch and Flow Conditions: The Role of in Situ Formed Base. Catalysts 2020, 10, 73. [Google Scholar] [CrossRef] [Green Version]
- Wojcieszak, R.; Palombo Ferraz, C.; Sha, J.; Houda, S.; Rossi, L.; Paul, S. Advances in Base-Free Oxidation of Bio-Based Compounds on Supported Gold Catalysts. Catalysts 2017, 7, 352. [Google Scholar] [CrossRef] [Green Version]
- Palombo Ferraz, C.; Zieliński, M.; Pietrowski, M.; Heyte, S.; Dumeignil, F.; Rossi, L.; Wojcieszak, R. Influence of Support Basic Sites in Green Oxidation of Biobased Substrates Using Au- Promoted Catalysts. ACS Sustain. Chem. Eng. 2018, 6, 16332–16340. [Google Scholar] [CrossRef]
- Palombo Ferraz, C.; Braga, A.; Ghazzal, N.; Zieliński, M.; Pietrowski, M.; Itabaiana, I., Jr.; Dumeignil, F.; Rossi, L.; Wojcieszak, R. Efficient Oxidative Esterification of Furfural Using Au Nanoparticles Supported on Group 2 Alkaline Earth Metal Oxides. Catalysts 2020, 10, 430. [Google Scholar] [CrossRef] [Green Version]
- Palombo Ferraz, C.; da Silva Marques, A.; Rodrigues, T.; Camargo, P.; Paul, S.; Wojcieszak, R. Furfural Oxidation on Gold Supported on MnO2: Influence of the Support Structure on the Catalytic Performances. Appl. Sci. 2018, 8, 1246. [Google Scholar] [CrossRef] [Green Version]
- da Silva Marques, A.; Rodrigues, T.; Candido, E.; de Freitas, I.; da Silva, A.; Fajardo, H.; Balzer, R.; Gomes, J.; Assaf, J.; de Oliveira, D.; et al. Combining active phase and support optimization in MnO2 -Au nanoflowers: Enabling high activities towards green oxidations. J. Coll. Interface Sci. 2018, 530, 282–291. [Google Scholar] [CrossRef]
- Palombo Ferraz, C.; Costa, N.; Teixeira-Neto, E.; Teixeira-Neto, A.; Liria, C.; Thuriot-Roukos, J.; Machini, T.; Froidevaux, R.; Dumeignil, F.; Rossi, L.; et al. 5-Hydroxymethylfurfural and Furfural Base-Free Oxidation over AuPd Embedded Bimetallic Nanoparticles. Catalysts 2020, 10, 75. [Google Scholar] [CrossRef] [Green Version]
- Thuriot-Roukos, J.; Khadraoui, R.; Paul, S.; Wojcieszak, R. Raman Spectroscopy Applied to Monitor Furfural Liquid-Phase Oxidation Catalyzed by Supported Gold Nanoparticles. ACS Omega 2020, 5, 14283–14290. [Google Scholar] [CrossRef] [PubMed]
- Fabien, D.; Snoussi, Y.; Itabaiana, I., Jr.; Wojcieszak, R. The Use of CO2 in the Production of Bioplastics for an Even Greener Chemistry. Sustainability 2021, 13, 11278. [Google Scholar] [CrossRef]
- Mazzi, A.; Paul, S.; Cavani, F.; Wojcieszak, R. Cyclohexane Oxidation to Adipic Acid Under Green Conditions: A Scalable and Sustainable Process. ChemCatChem 2018, 10, 3680–3682. [Google Scholar] [CrossRef]
- Monti, E.; Ventimiglia, A.; Garcia Soto, C.A.; Martelli, F.; Rodríguez-Aguado, E.; Cecilia, J.A.; Sadier, A.; Ospitali, F.; Tabanelli, T.; Albonetti, S.; et al. Effect of the Colloidal Preparation Method for Supported Preformed Colloidal Au Nanoparticles for the Liquid Phase Oxidation of 1,6-Hexanediol to Adipic Acid. Catalysts 2022, 12, 196. [Google Scholar] [CrossRef]
- Mattisson, T.; Keller, M.; Linderholm, C.; Moldenhauer, P.; Ryden, M.; Leion, H.; Lyngfelt, A. Chemical-looping technologies using circulating fluidized bed systems: Status of development. Fuel Process. Technol. 2018, 172, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Corma, A.; Sauvanaud, L. FCC testing at bench scale: New units, new processes, new feeds. Catal. Today 2013, 218-219, 107–114. [Google Scholar] [CrossRef]
- Löfberg, A.; Guerrero-Caballero, J.; Kane, T.; Rubbens, A.; Jalowiecki-Duhamel, L. Ni/CeO2 based catalysts as oxygen vectors for the chemical looping dry reforming of methane for syngas production. Appl. Catal. B Environ. 2017, 212, 159–174. [Google Scholar] [CrossRef]
- Bhavsar, S.; Najera, M.; Veser, G. Chemical Looping Dry Reforming as Novel, Intensified Process for CO2 Activation. Chem. Eng. Technol. 2012, 35, 1281–1290. [Google Scholar] [CrossRef]
- Löfberg, A.; Guerrero-Caballero, J. Plant and Process for Treating a Stream Comprising Hydrogen Sulfide. WO Patent 2016. [Google Scholar]
- Kalinkin, P.; Kovalenko, O.; Lapina, O.; Khabibulin, D.; Kundo, N. Kinetic peculiarities in the low-temperature oxidation of H2S over vanadium catalysts. J. Mol. Catal. A Chem. 2002, 178, 173–180. [Google Scholar] [CrossRef]
- Kane, T.; Guerrero-Caballero, J.; Löfberg, A. H2S chemical looping selective and preferential oxidation to sulfur by bulk V2O5. Appl. Catal. B Environ. 2020, 265, 118566. [Google Scholar] [CrossRef]
- Kane, T.; Guerrero-Caballero, J.; Löfberg, A. Chemical looping selective oxidation of H2S using V2O5 impregnated over different supports as oxygen carriers. ChemCatChem 2020, 12, 2569–2579. [Google Scholar] [CrossRef]
- Kane, T. Selective and Preferential Oxidation of Hydrogen Sulfide by Chemical Looping. Ph.D. Thesis, University of Lille, Lille, France, 4 December 2018. [Google Scholar]
- de Souza, P.; Silvester, L.; da Silva, A.; Fernandes, C.; Rodrigues, T.; Paul, S.; Camargo, P.; Wojcieszak, R. Exploiting the Synergetic Behavior of PtPd Bimetallic Catalysts in the Selective Hydrogenation of Glucose and Furfural. Catalysts 2019, 9, 132. [Google Scholar] [CrossRef] [Green Version]
- Silvester, L.; Ramos, F.; Thuriot-Roukos, J.; Heyte, S.; Araque, M.; Paul, S.; Wojcieszak, R. Fully integrated high-throughput methodology for the study of Ni- and Cu-supported catalysts for glucose hydrogenation. Catal. Today 2019, 338, 72–80. [Google Scholar] [CrossRef]
- Sadier, A.; Shi, S.; Mamede, A.S.; Paul, S.; Marceau, E.; Wojcieszak, R. Selective aqueous phase hydrogenation of xylose to xylitol over SiO2-supported Ni and Ni-Fe catalysts: Benefits of promotion by Fe. Appl. Catal. B Environ. 2021, 298, 120564. [Google Scholar] [CrossRef]
- Sadier, A.; Paul, S.; Marceau, E.; Wojcieszak, R. Ni-Fe alloying enhances the efficiency of the maltose hydrogenation process: The role of surface species and kinetic study. Appl. Catal. B Environ. 2022, 313, 121446. [Google Scholar] [CrossRef]
- Dautzenberg, G.; Gerhardt, M.; Kamm, B. Bio based fuels and fuel additives from lignocellulose feedstock via the production of levulinic acid and furfural. Holzforschung 2011, 65, 439–451. [Google Scholar] [CrossRef]
- Bu, Q.; Lei, H.; Zacher, A.H.; Wang, L.; Ren, S.; Liang, J.; Wei, Y.; Liu, Y.; Tang, J.; Zhang, Q.; et al. A review of catalytic hydrodeoxygenation of lignin-derived phenols from biomass pyrolysis. Bioresour. Technol. 2012, 124, 470–477. [Google Scholar] [CrossRef]
- Besson, M.; Gallezot, P.; Pinel, C. Conversion of biomass into chemicals over metal catalysts. Chem. Rev. 2014, 114, 1827–1870. [Google Scholar] [CrossRef]
- Lee, J.; Xu, Y.; Huber, G.W. High-throughput screening of monometallic catalysts for aqueous-phase hydrogenation of biomass-derived oxygenates. Appl. Catal. B Environ. 2013, 140–141, 98–107. [Google Scholar] [CrossRef]
- Pizzi, R.; van Putten, R.-J.; Brust, H.; Perathoner, S.; Centi, G.; van der Waal, J.C. High-Throughput Screening of Heterogeneous Catalysts for the Conversion of Furfural to Bio-Based Fuel Components. Catalysts 2015, 5, 2244–2257. [Google Scholar] [CrossRef] [Green Version]
- Alamillo, R.; Tucker, M.; Chia, M.; Pagań-Torres, Y.; Dumesic, J. The selective hydrogenation of biomass-derived 5-hydroxymethylfurfural using heterogeneous catalysts. Green Chem. 2012, 14, 1413–1419. [Google Scholar] [CrossRef]
- Shi, S.; Wojcieszak, R.; Paul, S.; Marceau, E. Ni Promotion by Fe: What Benefits for Catalytic Hydrogenation? Catalysts 2019, 9, 451. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Yang, Q.; Peterson, C.; Lamic-Humblot, A.F.; Girardon, J.S.; Griboval-Constant, A.; Stievano, L.; Sougrati, M.; Briois, V.; Bagot, P.; et al. Bimetallic Fe-Ni/SiO2 catalysts for furfural hydrogenation: Identification of the interplay between Fe and Ni during deposition-precipitation and thermal treatments. Catal. Today 2019, 334, 162–172. [Google Scholar] [CrossRef]
- Shi, D.; Sadier, A.; Girardon, J.S.; Mamede, A.S.; Ciotonea, C.; Marinova, M.; Stievano, L.; Sougrati, M.; La Fontaine, C.; Paul, S.; et al. Probing the core and surface composition of nanoalloy to rationalize its selectivity: Study of Ni-Fe/SiO2 catalysts for liquid-phase hydrogenation. Chem. Catal. 2022, in press. [CrossRef]
- Chen, S.; Ciotonea, C.; De Oliveira Vigier, K.; François, J.; Wojcieszak, R.; Dumeignil, F.; Marceau, E.; Royer, S. Hydroconversion of 5-Hydroxymethylfurfural to 2,5-Dimethylfuran and 2,5-Dimethyltetrahydrofuran over Non-promoted Ni/SBA-15. ChemCatChem 2020, 12, 2050–2059. [Google Scholar] [CrossRef] [Green Version]
- Katryniok, B.; Paul, S.; Bellière-Baca, V.; Rey, P.; Dumeignil, F. Glycerol Dehydration to Acrolein in the context of Glycerol new usages. Green Chem. 2010, 12, 2079–2098. [Google Scholar] [CrossRef]
- Katryniok, B.; Kimura, H.; Skrzynska, E.; Girardon, J.S.; Fongerland, P.; Capron, M.; Ducoulombier, R.; Mimura, N.; Paul, S.; Dumeignil, F. Selective catalytic oxidation of glycerol: Perspectives for high value chemicals. Green Chem. 2011, 13, 1960–1979. [Google Scholar] [CrossRef]
- Ebadipour, N.; Paul, S.; Katryniok, B.; Dumeignil, F. Alkaline-based catalysts for glycerol polymerization reaction: A review. Catalysts 2020, 10, 1021. [Google Scholar] [CrossRef]
- Bouriakova, A.; Mendes, P.; Katryniok, B.; De Clercq, H.; Thybaut, J.W. Co-metal induced stabilization of alumina supported copper—Impact on in the hydrogenolysis of glycerol to 1,2-propanediol. Catal. Commun. 2020, 164, 106134. [Google Scholar] [CrossRef]
- Tazawa, S.; Ota, N.; Tamura, M.; Nakagawa, Y.; Okumura, K.; Tomishige, K. Deoxydehydration with Molecular Hydrogen over Ceria-Supported Rhenium Catalyst with Gold Promoter. ACS Catal. 2016, 6, 6393–6397. [Google Scholar] [CrossRef]
- Kon, Y.; Araque, M.; Nakashima, T.; Paul, S.; Dumeignil, F.; Katryniok, B. Direct Conversion of Glycerol to Allyl Alcohol over Alumina-supported Rhenium oxide. Chem. Sel. 2017, 30, 9864. [Google Scholar] [CrossRef]
- Yi, J.; Liu, S.; Abu-Omar, M.M. Rhenium-catalyzed transfer hydrogenation and deoxygenation of biomass-derived polyols to small and useful organics. ChemSusChem 2012, 5, 1401–1404. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, J.R.; Lupp, D.; Oh, B.C.; Fristrup, P. Molybdenum-catalyzed deoxydehydration of vicinal diols. ChemSusChem 2014, 7, 425–428. [Google Scholar] [CrossRef]
- Sandbrink, S.; Klindtworth, E.; Islam, H.U.; Beale, A.M.; Palkovits, R. ReOx/TiO2: A Recyclable Solid Catalyst for Deoxydehydration. ACS Catal. 2016, 6, 677–680. [Google Scholar] [CrossRef]
- Silva, K.; Araque, M.; Katryniok, B. Production D’alcool Allylique à Partir de Glycérol en Phase Liquid. French Patent EP2021057525, 2021. [Google Scholar]
- Kolbe, H.; Lautemann, E. Ueber die Constitution und Basicität der Salicylsäure. Liebigs Ann. Chem. 1860, 115, 157–206. [Google Scholar] [CrossRef] [Green Version]
- Drault, F.; Snoussi, Y.; Paul, S.; Itabaiana, I.; Wojcieszak, R. Recent Advances in Carboxylation of Furoic Acid into 2,5-Furandicarboxylic Acid: Pathways towards Bio-Based Polymers. ChemSusChem 2020, 13, 5164–5172. [Google Scholar] [CrossRef]
- Drault, F.; Snoussi, Y.; Thuriot-Roukos, J.; Paul, S.; Itabaiana, I.; Wojcieszak, R. Study of the Direct CO2 Carboxylation Reaction on Supported Metal Nanoparticles. Catalysts 2021, 11, 326. [Google Scholar] [CrossRef]
- Liebig, C.; Paul, S.; Katryniok, B.; Guillon, C.; Couturier, J.-L.; Dubois, J.-L.; Dumeignil, F.; Hölderich, W.F. Glycerol conversion to acrylonitirle by consecutive dehydration over WO3/TiO2 and ammoxidation over Sb-/Fe,V-O. Appl. Catal. B Environ. 2013, 132–133, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Guillon, C.; Liebig, C.; Paul, S.; Mamede, A.-S.; Hölderich, W.F.; Dumeignil, F.; Katryniok, B. Ammoxidation of allyl alcohol—A sustainable route to acrylonitrile. Green Chem. 2013, 13, 3015–3019. [Google Scholar] [CrossRef]
- Ebadi, N.; Dumeignil, F.; Katryniok, B.; Delevoye, L.; Revel, B.; Paul, S. Investigating the active phase of Ca-based glycerol polymerization catalysts: On the importance of calcium glycerolate. Mol. Catal. 2021, 507, 111571. [Google Scholar] [CrossRef]
- Pomalaza, G.; Capron, M.; Ordomsky, V.; Dumeignil, F. Recent Breakthroughs in the Conversion of Ethanol to Butadiene. Catalysts 2016, 6, 203. [Google Scholar] [CrossRef] [Green Version]
- Pomalaza, G.; Arango, P.; Capron, M.; Dumeignil, F. Butadiene from Ethanol: The Reaction and its catalysts. Catal. Sci. Technol. 2020, 10, 4860–4911. [Google Scholar] [CrossRef]
- Pomalaza, G.; Capron, M.; Dumeignil, F. Improving the Synthesis of Zn-Ta-TUD-1 for the Lebedev Process using the Design of Experiments Methodology. Appl. Catal. A Gen. 2020, 591, 117386. [Google Scholar] [CrossRef]
- Pomalaza, G.; Capron, M.; Dumeignil, F. ZnTa-TUD-1 as Easily Prepared, Highly Efficient Catalyst for the Selective Conversion of Ethanol to 1,3-Butadiene. Green Chem. 2018, 20, 3203–3209. [Google Scholar] [CrossRef]
- Pomalaza, G.; Simon, P.; Addad, A.; Capron, M.; Dumeignil, F. Properties and Activity of Zn-Ta-TUD-1 in the Lebedev Process. Green Chem. 2020, 22, 2558–2574. [Google Scholar] [CrossRef]
- Duhamel, L.; Fang, W.; Paul, S.; Dumeignil, F. Process for Production of Hydrogen. U.S. Patent 9,968,913, 2018. [Google Scholar]
- Pirez, C.; Fang, W.; Capron, M.; Paul, S.; Jobic, H.; Dumeignil, F.; Jalowiecki-Duhamel, L. Steam reforming, partial oxidation and oxidative steam reforming for hydrogen production from ethanol over cerium nickel based oxyhydride catalyst. Appl. Catal. A Gen. 2016, 518, 78–86. [Google Scholar] [CrossRef]
- Fang, W.; Paul, S.; Capron, M.; Biradar, A.V.; Umbarkar, S.B.; Dongare, M.K.; Dumeignil, F.; Jalowiecki-Duhamel, L. Highly loaded well dispersed stable Ni species in NiXMg2AlOY nanocomposites: Application to hydrogen production from bioethanol. Appl. Catal. B Environ. 2015, 166–167, 485–496. [Google Scholar] [CrossRef]
- Fang, W.; Pirez, C.; Paul, S.; Jiménez-Ruiz, M.; Jobic, H.H.; Dumeignil, F.; Jalowiecki-Duhamel, L. Advanced functionalized Mg2AlNiXHZOY nano-oxyhydrides ex-hydrotalcites for hydrogen production from oxidative steam reforming of ethanol. Int. J. Hydr. Energy 2016, 41, 15443–15452. [Google Scholar] [CrossRef]
- Fang, W.; Romani, Y.; Wei, Y.; Jiménez-Ruiz, M.M.; Jobic, H.; Paul, S.; Jalowiecki-Duhamel, L. Steam reforming and oxidative steam reforming for hydrogen production from bioethanol over Mg2AlNiXHZOY nano-oxyhydride catalysts. Int. J. Hydrogen Energy 2018, 43, 17643–17655. [Google Scholar] [CrossRef]
- Löfberg, A.; Kane, T.; Guerrero-Caballero, J.; Jalowiecki-Duhamel, L. Chemical looping dry reforming of methane: Towards shale-gas and biogas valorization. Chem. Eng. Process. Process Intensif. 2017, 122, 523–529. [Google Scholar] [CrossRef]
- Guerrero-Caballero, J.; Kane, T.; Haidar, N.; Jalowiecki-Duhamel, L.; Löfberg, A. Ni, Co, Fe supported on Ceria and Zr doped Ceria as oxygen carriers for chemical looping dry reforming of methane. Catal. Today 2019, 333, 251–258. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, X.; Haidar, N.; Jobic, H.; Paul, S.; Jalowiecki-Duhamel, L. CeNiXAl0.5HZOY nano-oxyhydrides for H2 production by oxidative dry reforming of CH4 without carbon formation. Appl. Catal. A Gen. 2020, 594, 117439. [Google Scholar] [CrossRef]
- Managutti, P.; Tymen, S.; Liu, X.; Hernandez, O.; Prestipino, C.; Le Gal La Salle, A.; Paul, S.; Jalowiecki-Duhamel, L.; Dorcet, V.; Billard, A.; et al. Exsolution of Ni nanoparticles from A-site deficient layered double perovskites for dry reforming of methane and as anode material for solid oxide fuel cell. ACS Appl. Mater. Interfaces 2021, 13, 35719–35728. [Google Scholar] [CrossRef] [PubMed]
- Bi, Z.; Lai, B.; Zhao, Y.; Yan, L. Fast Disassembly of Lignocellulosic Biomass to Lignin and Sugars by Molten Salt Hydrate at Low Temperature for Overall Biorefinery. ACS Omega 2018, 3, 2984–2993. [Google Scholar] [CrossRef] [Green Version]
- Leipner, H.; Fischer, S.; Brendler, E.; Voigt, W. Structural changes of cellulose dissolved in molten salt hydrates. Macromol. Chem. Phys. 2000, 201, 2041–2049. [Google Scholar] [CrossRef]
- Sen, S.; Losey, B.P.; Gordon, E.E.; Argyropoulos, D.S.; Martin, J.D. Ionic Liquid Character of Zinc Chloride Hydrates Define Solvent Characteristics that Afford the Solubility of Cellulose. J. Phys. Chem. B 2016, 120, 1134–1141. [Google Scholar] [CrossRef]
- Quiroz, N.R.; Padmanathan, A.M.D.; Mushrif, S.H.; Vlachos, D.G. Understanding Acidity of Molten Salt Hydrate Media for Cellulose Hydrolysis by Combining Kinetic Studies, Electrolyte Solution Modeling, Molecular Dynamics Simulations, and 13C NMR Experiments. ACS Catal. 2019, 9, 10551–10561. [Google Scholar] [CrossRef]
- Sen, S.; Martin, J.D.; Argyropoulos, D.S. Review of Cellulose Non-Derivatizing Solvent Interactions with Emphasis on Activity in Inorganic Molten Salt Hydrates. ACS Sustain. Chem. Eng. 2013, 1, 858–870. [Google Scholar] [CrossRef]
- Ragg, P.L.; Fields, P.R. The development of a process for the hydrolysis of lignocellulosic waste. Phil. Trans. R. Soc. Land. A 1987, 321, 537–547. [Google Scholar] [CrossRef]
- Fischer, S.; Leipner, H.; Thümmler, K.; Brendler, E.; Peters, J. Inorganic Molten Salts as Solvents for Cellulose. Cellulose 2003, 10, 227–236. [Google Scholar] [CrossRef]
- Deng, W.; Kennedy, J.R.; Tsilomelekis, G.; Zheng, W.; Nikolakis, V. Cellulose Hydrolysis in Acidified LiBr Molten Salt Hydrate Media. Ind. Eng. Chem. Res. 2015, 54, 5226–5236. [Google Scholar] [CrossRef]
- Liu, Q.; Ma, Q.; Sabnis, S.; Zheng, W.; Vlachos, D.G.; Fan, W.; Li, W.; Ma, L. Production of high-yield short-chain oligomers from cellulose via selective hydrolysis in molten salt hydrates and separation. Green Chem. 2019, 21, 5030–5038. [Google Scholar] [CrossRef]
- Ahmed, S.T.; Leferink, N.; Scrutton, N. Chemo-enzymatic routes towards the synthesis of bio-based monomers and polymers. Mol. Catal. 2019, 467, 95–110. [Google Scholar] [CrossRef] [Green Version]
- Wojcieszak, R.; Itabaiana, I., Jr. Engineering the future: Perspectives in the 2,5-furandicarboxylic acid synthesis. Catal. Today 2020, 354, 211–217. [Google Scholar] [CrossRef]
- Dumeignil, F. Hailing the hybrid—Professor Franck Dumeignil, EuroBioref Coordinator at the Université Lille Nord de France turns the spotlight onto a new golden age of catalysis. Public Serv. Rev. Eur. Union 2011, 22, 528. [Google Scholar]
- Dumeignil, F. Chemical Catalysis and Biotechnology: From a Sequential Engagement to a One-Pot Wedding. Chem. Ing. Tech. 2014, 86, 1496. [Google Scholar] [CrossRef]
- Dumeignil, F.; Guehl, M.; Gimbernat, A.; Capron, M.; Lopes Ferreira, N.; Froidevaux, R.; Girardon, J.S.; Wojcieszak, R.; Dhulster, P.; Delcroix, D. From Sequential Chemoenzymatic Synthesis to Integrated Hybrid Catalysis: Taking the Best of Both Worlds to Open up the Scope of Possibilities for a Sustainable Future. Catal. Sci. Technol. 2018, 8, 5708–5734. [Google Scholar] [CrossRef]
- Heuson, E.; Dumeignil, F. The Various Levels of Integration of Chemo- and Bio-Catalysis towards Hybrid Catalysis. Catal. Sci. Technol. 2020, 12, 7082–7100. [Google Scholar] [CrossRef]
- Lee, J.H.; Han, K.; Kim, M.-J.; Park, J. Chemoenzymatic dynamic kinetic resolution of alcohols and amines. Eur. J. Org. Chem. 2010, 6, 999–1015. [Google Scholar] [CrossRef]
- Kamal, A.; Azhar, M.A.; Krishnaji, T.; Malik, M.S.; Azeeza, S. Approaches based on enzyme mediated kinetic to dynamic kinetic resolutions: A versatile route for chiral intermediates. Coord. Chem. Rev. 2008, 252, 569–592. [Google Scholar] [CrossRef]
- De Miranda, A.S.; De Souza, L.; de Souza, R.O.M.A. Lipases: Valuable catalysts for dynamic kinetic resolutions. Biotechnol. Adv. 2015, 33, 372–393. [Google Scholar] [CrossRef] [PubMed]
- Ghislieri, D.; Turner, N. Biocatalytic Approaches to the Synthesis of Enantiomerically Pure Chiral Amines. Top. Catal. 2014, 57, 284–300. [Google Scholar] [CrossRef]
- Itabaiana, I., Jr.; Miranda, L.; de Souza, R. Towards a continuous flow environment for lipase-catalyzed reactions. J. Mol. Catal. B Enzym. 2013, 85–86, 1–9. [Google Scholar] [CrossRef]
- de Souza, S.; Leão, R.; Bassut, J.; Leal, I.; Wang, S.; Ding, Q.; Li, Y.; Leung-Yuk Lam, F.; de Souza, R.; Itabaiana, I., Jr. New Biosilified Pd-lipase hybrid biocatalysts for dynamic resolution of amines. Tetrahedron Lett. 2017, 58, 4849–4854. [Google Scholar] [CrossRef]
- Ferraz, C.; do Nascimento, M.; Almeida, R.; Sergio, G.; Junior, A.; Dalmônico, G.; Caraballo, R.; Finotelli, P.; Leão, R.; Wojcieszak, R.; et al. Synthesis and characterization of a magnetic hybrid catalyst containing lipase and palladium and its application on the dynamic kinetic resolution of amines. Mol. Catal. 2020, 493, 111106. [Google Scholar] [CrossRef]
- van Putten, R.; van der Waal, J.; de Jong, E.; Rasrendra, C.; Heeres, H.; de Vries, J. Hydroxymethylfurfural, A Versatile Platform Chemical Made from Renewable Resources. Chem. Rev. 2013, 113, 1499–1597. [Google Scholar] [CrossRef]
- Guehl, M.; Delcroix, D.; Lopes Ferreira, N.; Desset, S.; Dumeignil, F. Process for the selective transformation of biosourced polyols. FR Patent FR3031983A1, 2016. [Google Scholar]
- Gimbernat, A.; Guehl, M.; Capron, M.; Lopes Ferreira, N.; Froidevaux, R.; Girardon, J.S.; Dhulster, P.; Delcroix, D.; Dumeignil, F. Hybrid Catalysis: A Suitable Concept for the Valorization of Biosourced Saccharides to Value-Added Chemicals. ChemCatChem 2017, 9, 2080–2084. [Google Scholar] [CrossRef]
- Gimbernat, A.; Guehl, M.; Lopes Ferreira, N.; Heuson, E.; Dhulster, P.; Capron, M.; Dumeignil, F.; Delcroix, D.; Girardon, J.S.; Froidevaux, R. From a Sequential Chemo-Enzymatic Approach to a Continuous Process for HMF Production from Glucose. Catalysts 2018, 8, 335. [Google Scholar] [CrossRef] [Green Version]
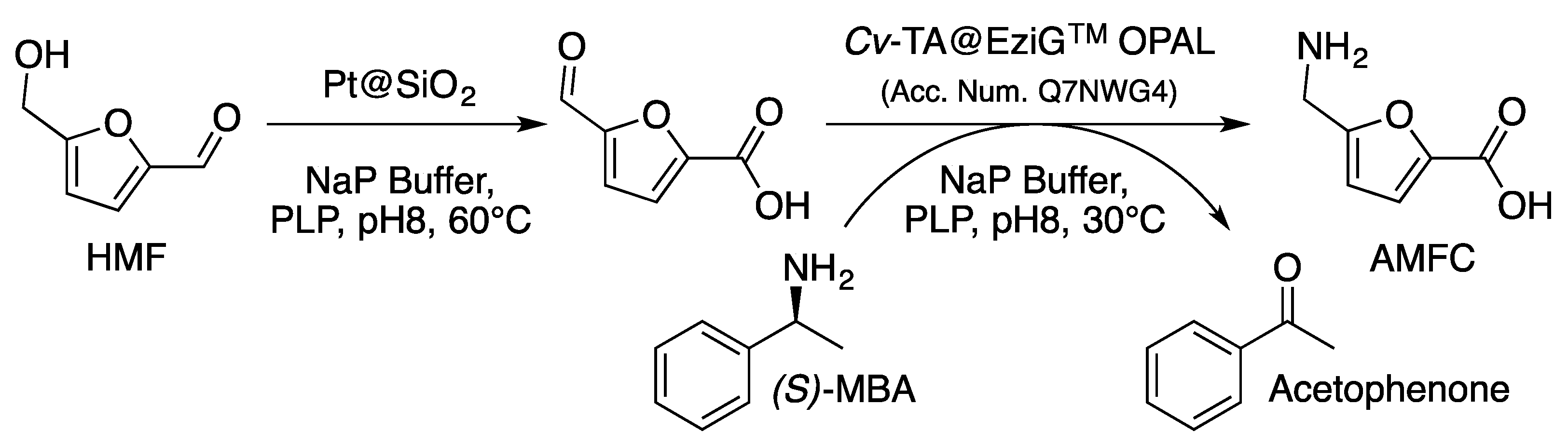
- Lancien, A.; Wojcieszak, R.; Cuvelier, E.; Duban, M.; Dhulster, P.; Paul, S.; Dumeignil, F.; Froidevaux, R.; Heuson, E. Hybrid Conversion of 5-Hydroxymethylfurfural to 5 -Aminomethyl- 2-furancarboxylic acid: Toward New Bio-sourced Polymers. ChemCatChem 2021, 13, 247–259. [Google Scholar] [CrossRef]
- EU Bioeconomy Strategy. A Sustainable Bioeconomy for Europe: Strengthening the Connection Between Economy, Society and the Environment, European Commission B-1049, Brussels. 2018. Available online: https://eur-lex.europa.eu/legal-content/en/ALL/?uri=CELEX%3A52018DC0673 (accessed on 20 May 2022).
- Avelar do Nascimento, M.; Ester Gotardo, L.; Miguez Bastos, E.; Almeida, F.; Leão, R.; Wojcieszak, R.; Itabaiana, I., Jr.; de Souza, R. Regioselective Acylation of Levoglucosan Catalyzed by Candida Antarctica (CaLB) Lipase Immobilized on Epoxy Resin. Sustainability 2019, 11, 6044. [Google Scholar] [CrossRef] [Green Version]
- Itabaiana Junior, I.; Avelar do Nascimento, A.; de Souza, R.; Dufour, D.; Wojcieszak, R. Levoglucosan: A promising platform molecule? Green Chem. 2020, 22, 5859–5880. [Google Scholar] [CrossRef]
- Wojcieszak, R.; Santarelli, F.; Paul, S.; Dumeignil, F.; Cavani, F.; Gonçalves, R. Recent developments in maleic acid synthesis from bio-based chemicals. Sustain. Chem. Proc. 2015, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves Renato, V.; Vono, L.; Wojcieszak, R.; Dias, C.; Wender, H.; Teixeira-Neto, E.; Rossi, L. Selective hydrogenation of CO2 into CO on a highly dispersed nickel catalyst obtained by magnetron sputtering deposition: A step towards liquid fuels. Appl. Catal. B Environ. 2017, 209, 240–246. [Google Scholar] [CrossRef]
- Navarro-Jaén, S.; Bonin, J.V.; Robert, M.; Wojcieszak, R.; Khodakov, A. Highlights and challenges in the selective reduction of carbon dioxide to methanol. Nat. Rev. Chem. 2021, 5, 564–579. [Google Scholar] [CrossRef]
- Navarro-Jaén, S.; Morin, J.C.V.; Thuriot-Roukos, J.; Wojcieszak, R.; Khodakov, A. Hybrid monometallic and bimetallic copper-palladium zeolite catalysts for direct synthesis of dimethyl ether from CO2. New J. Chem. 2022, 46, 3889–3900. [Google Scholar] [CrossRef]
- Navarro-Jaén, S.; Virginie, M.; Thuriot-Roukos, J.; Wojcieszak, R.; Khodakov, A. Structure–performance correlations in the hybrid oxide-supported copper–zinc SAPO-34 catalysts for direct synthesis of dimethyl ether from CO2. J. Mater. Sci. 2022, 57, 3268–3279. [Google Scholar] [CrossRef]
- Barrios, A.; Peron, D.; Chakkingal, A.; Dugulan, A.J.; Moldovan, S.; Nakouri, K.; Thuriot-Roukos, J.; Wojcieszak, R.; Thybaut, J.; Virginie, M.; et al. Efficient Promoters and Reaction Paths in the CO2 Hydrogenation to Light Olefins over Zirconia-Supported Iron Catalysts. ACS Catal. 2022, 12, 3211–3225. [Google Scholar] [CrossRef]
- Shi, D.; Heyte, S.; Capron, M.; Paul, S. Catalytic processes for the direct synthesis of dimethyl carbonate from CO2 and methanol: A review. Green Chem. 2022, 24, 1067–1089. [Google Scholar] [CrossRef]
- Júnior, A.A.d.T.; da Silva França, A.; de Souza, R.O.M.A.; Henrique Moraes, A.; Wojcieszak, R.; Itabaiana, I., Jr.; de Miranda, A.S. Multicatalytic Hybrid Materials for Biocatalytic and Chemoenzymatic Cascades—Strategies for Multicatalyst (Enzyme) Co-Immobilization. Catalysts 2021, 11, 936. [Google Scholar] [CrossRef]
- RECABIO. Available online: http://www.isite-ulne.fr/index.php/fr/recabio/ (accessed on 22 May 2022).
- REALCAT. Available online: https://www.realcat.fr (accessed on 22 May 2022).




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Temperature (°C) | Conversion (%) | FA Selectivity (%) | Carbon Balance (%) |
|---|---|---|---|---|
| Au/HT 2:1 | 110 | 82 | 86 | 89 |
| Au/HT 3:1 | 110 | 76 | 88 | 91 |
| Au/HT 4:1 | 110 | 99 | 100 | 100 |
| Au/HT 5:1 | 110 | 99 | 100 | 100 |
| T a (°C) | t b (h) | X Cha c (%) | Selectivity d (%) | ||||
|---|---|---|---|---|---|---|---|
| Chole | Chone | AA | GA | SA | |||
| 120 | 4 | 1.2 | 84 | 11 | 5 | 0 | 0 |
| 10 | 5.2 | 66 | 17 | 13 | 4 | 0 | |
| 135 | 4 | 3.7 | 65 | 15 | 18 | 2 | 0 |
| 10 | 12 | 13 | 38 | 33 | 11 | 5 | |
| Catalyst | Temperature (°C) | Conversion (%) | STYFDCA (µmol kg−1 h−1) | STYDFF (µmol kg−1 h−1) |
|---|---|---|---|---|
| CdI2 | 200 | 0 | - | - |
| Ag/SiO2 + CdI2 | 200 | 51 | 264 | 951 |
| Ag/SiO2 + CdI2 | 230 | 74 | 145 | - |
| Ag/SiO2 + CdI2 | 260 | 69 | 188 | - |
| Ag/SiO2 | 200 | 20 | 1203 | - |
| Biocatalyst | Conversion (%) | ee Prod | Productivity c |
|---|---|---|---|
| N435 a | 99 | >99 | 0.17 |
| EnzPd_1% b | 84 | >99 | 2.21 |
| EnzPd_5% b | 76 | 98 | 2.00 |
| EnzPd_10% b | 78 | 95 | 2.17 |
| EnzNoPd a | 92 | 82 | 2.36 |
| Entry | Catalyst (Pd + CALBimm) | Reaction Time (h) | Conversion (%) | ee Prod (%) | Selectivity(%) |
|---|---|---|---|---|---|
| 1 | [email protected]%Pd a + MN@CALB | 1 | 95 | >99 | 91 |
| 2 | [email protected]%Pd + N435 b | 4 | >99 | >99 | 96 |
| 3 | Pd/BaSO4 + MN@CALB | 1 | 65 | >99 | 84 |
| 4 | Pd/BaSO4 + N435 | 1 | 72 | 97 | 94 |
| 5 | [email protected]%Pd_CALB | 9 | 65 | >99 | 88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araque-Marin, M.; Bellot Noronha, F.; Capron, M.; Dumeignil, F.; Friend, M.; Heuson, E.; Itabaiana, I., Jr.; Jalowiecki-Duhamel, L.; Katryniok, B.; Löfberg, A.; et al. Strengthening the Connection between Science, Society and Environment to Develop Future French and European Bioeconomies: Cutting-Edge Research of VAALBIO Team at UCCS. Molecules 2022, 27, 3889. https://doi.org/10.3390/molecules27123889
Araque-Marin M, Bellot Noronha F, Capron M, Dumeignil F, Friend M, Heuson E, Itabaiana I Jr., Jalowiecki-Duhamel L, Katryniok B, Löfberg A, et al. Strengthening the Connection between Science, Society and Environment to Develop Future French and European Bioeconomies: Cutting-Edge Research of VAALBIO Team at UCCS. Molecules. 2022; 27(12):3889. https://doi.org/10.3390/molecules27123889
Chicago/Turabian StyleAraque-Marin, Marcia, Fabio Bellot Noronha, Mickäel Capron, Franck Dumeignil, Michèle Friend, Egon Heuson, Ivaldo Itabaiana, Jr., Louise Jalowiecki-Duhamel, Benjamin Katryniok, Axel Löfberg, and et al. 2022. "Strengthening the Connection between Science, Society and Environment to Develop Future French and European Bioeconomies: Cutting-Edge Research of VAALBIO Team at UCCS" Molecules 27, no. 12: 3889. https://doi.org/10.3390/molecules27123889