
Exploring Natural Product Activity and Species Source Candidates for Hunting ABCB1 Transporter Inhibitors: An In Silico Drug Discovery Study

 ,
,  , , , , , , ,
, , , , , , ,  and
and 
Abstract
:
1. Introduction
2. Results and Discussion
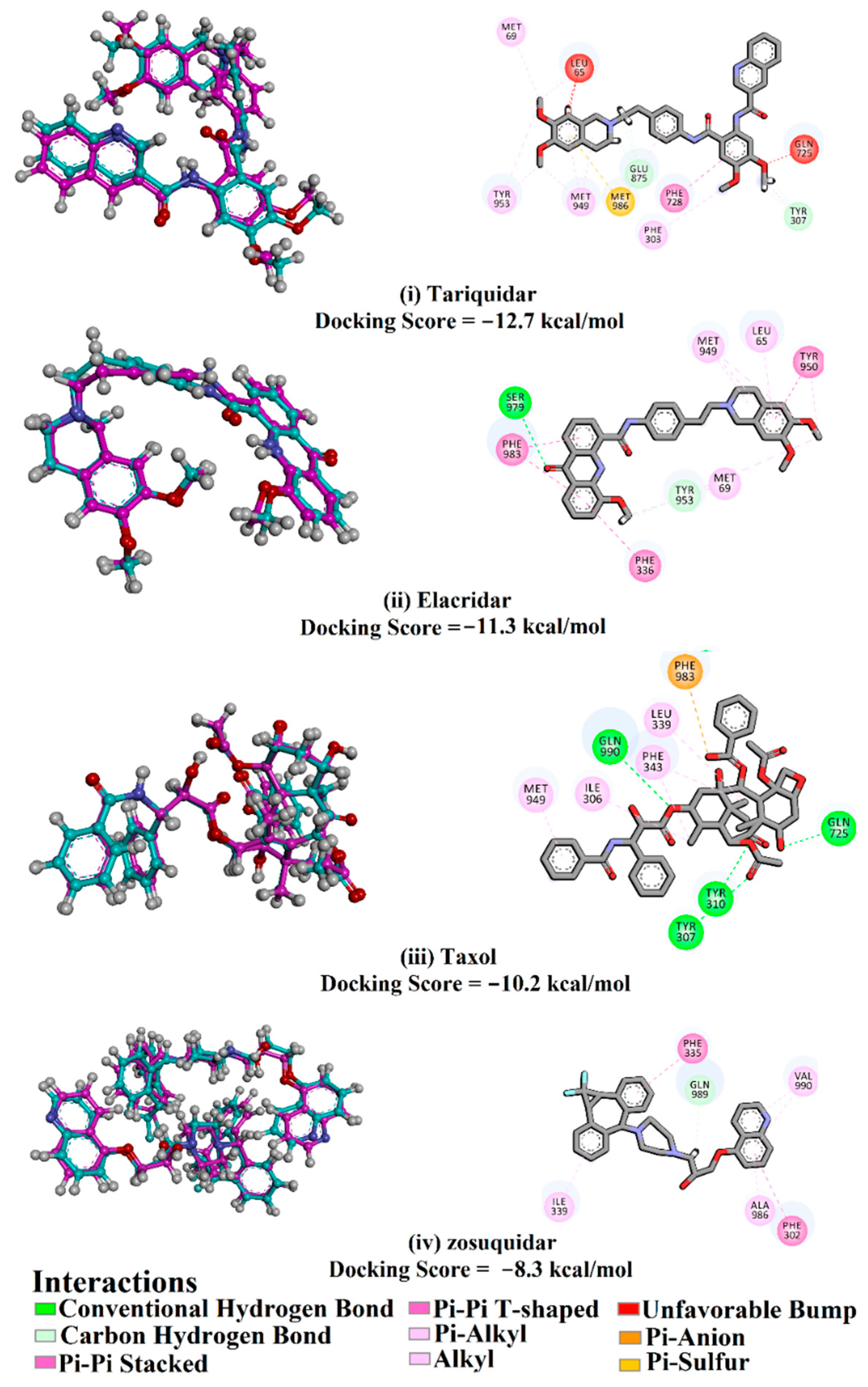
2.1. Assessment of In Silico protocol
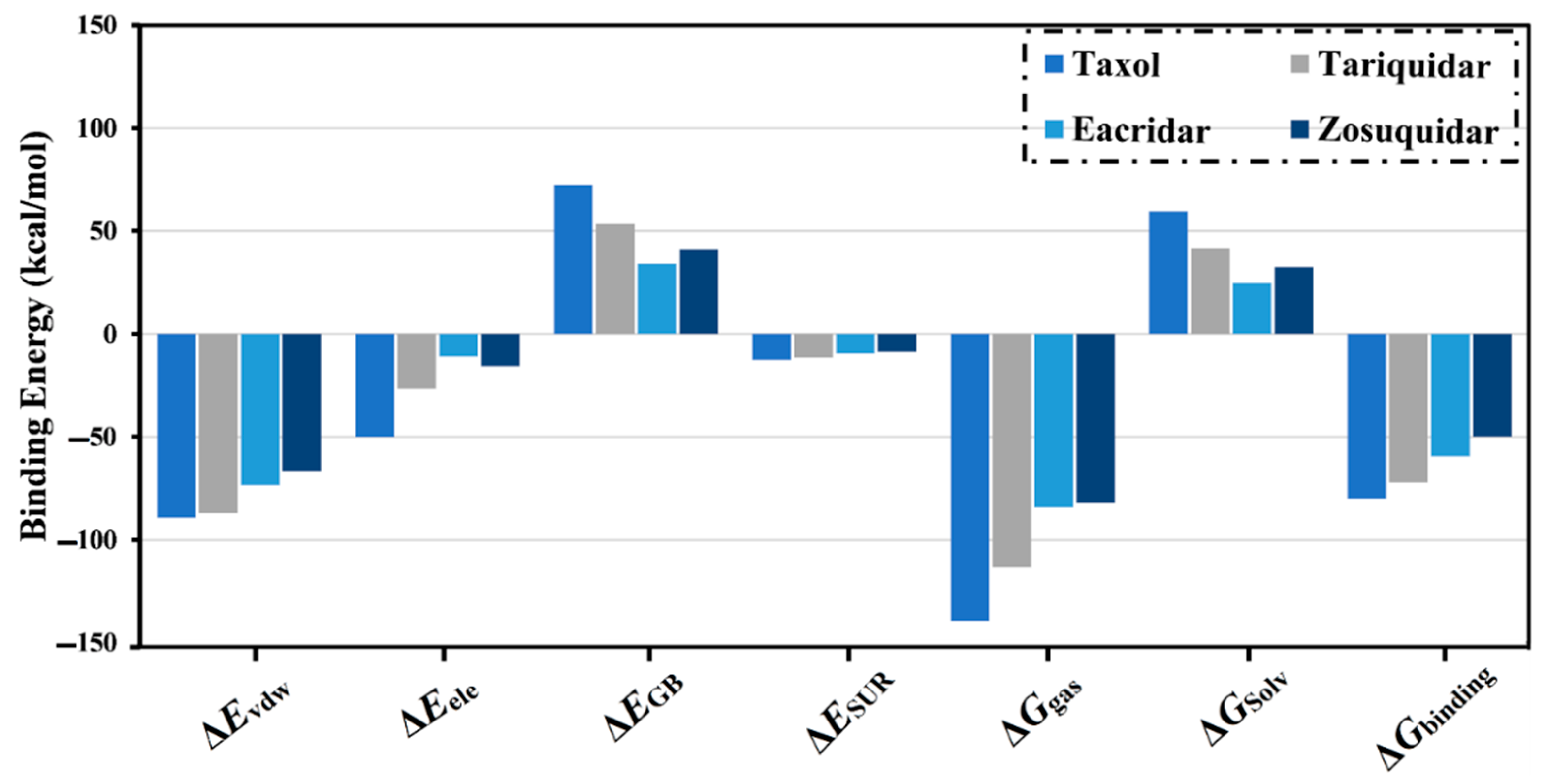
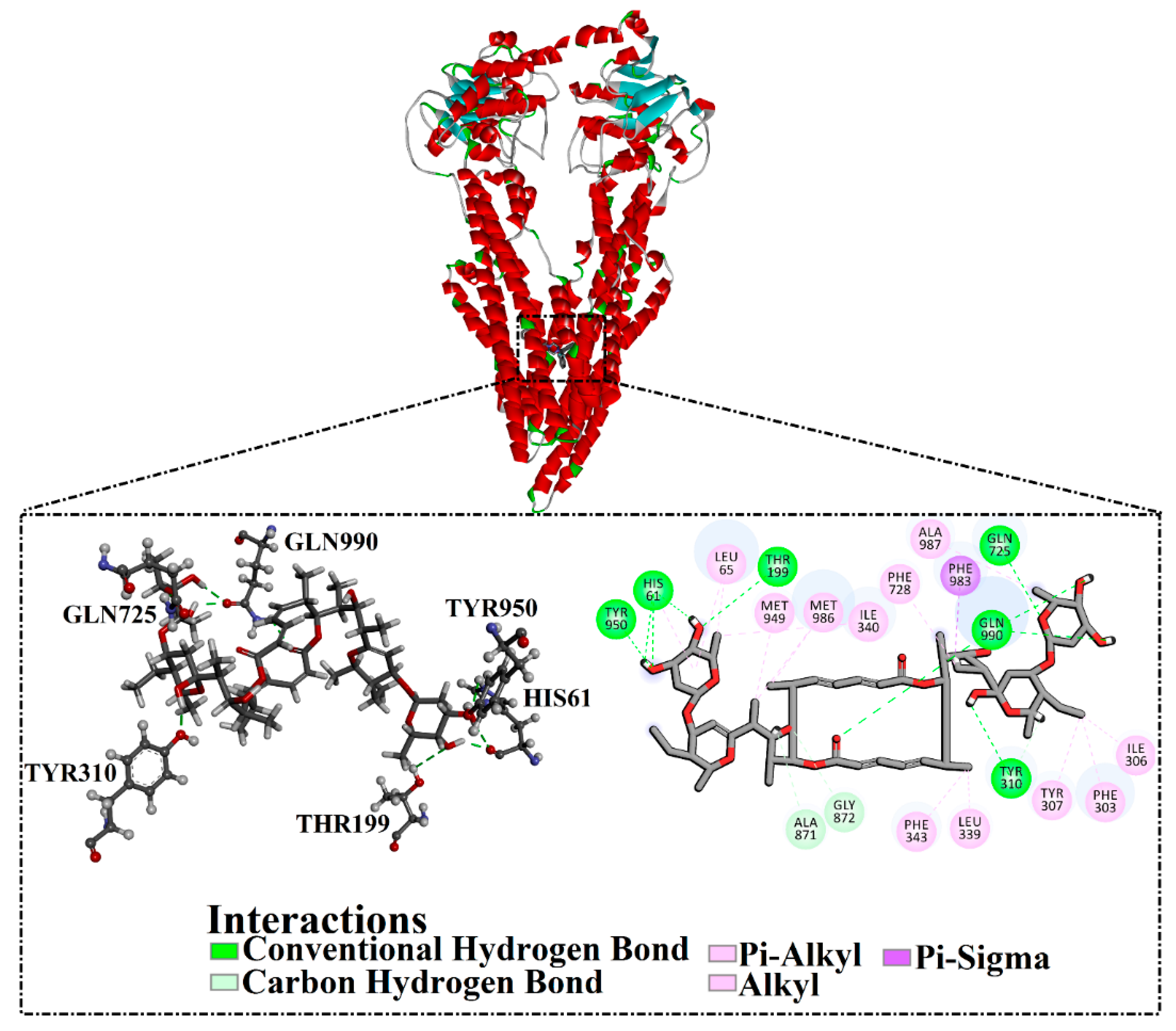
2.2. Molecular Dynamics of Co-Crystallized Ligands
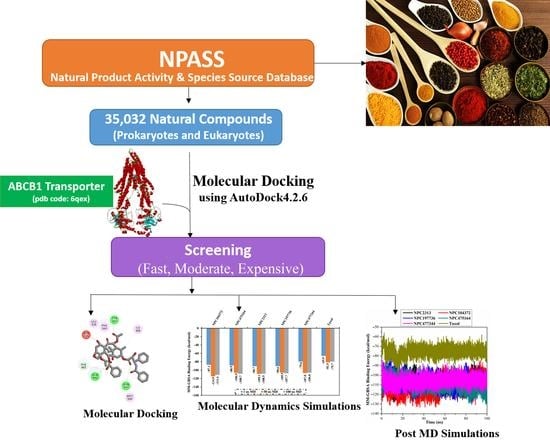
2.3. Virtual Screening of NPASS Database
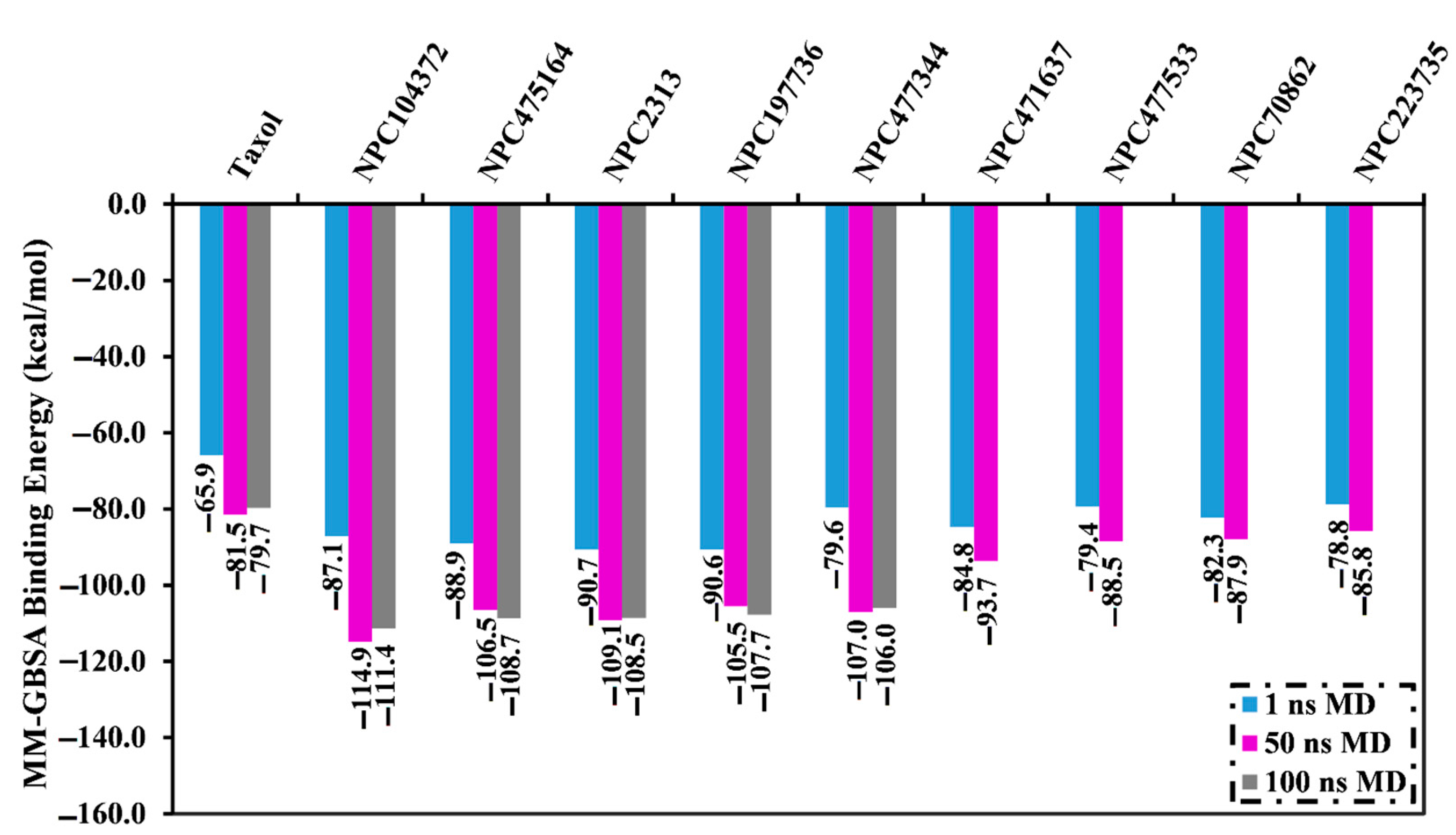
2.4. Molecular Dynamics Simulations
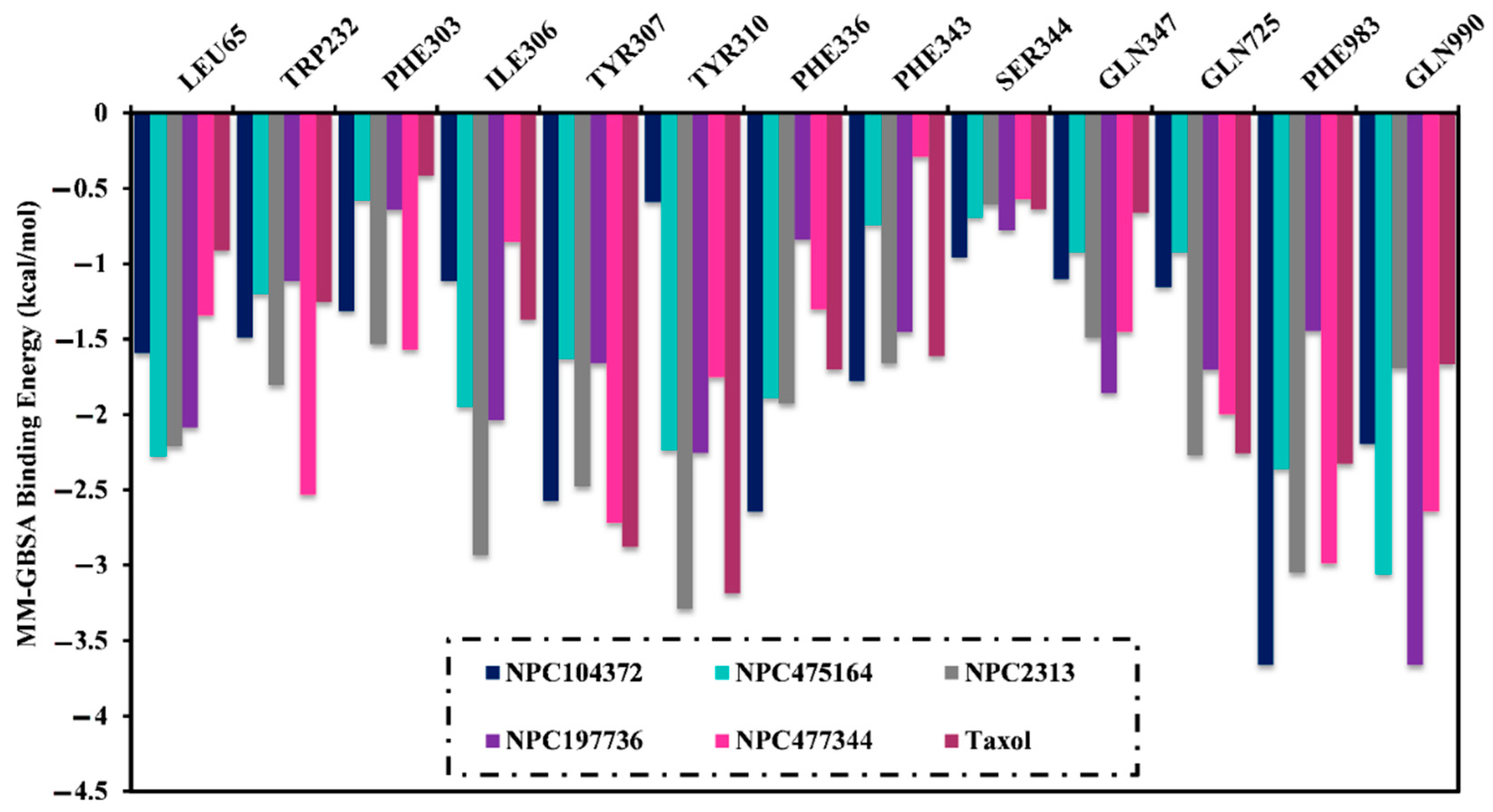
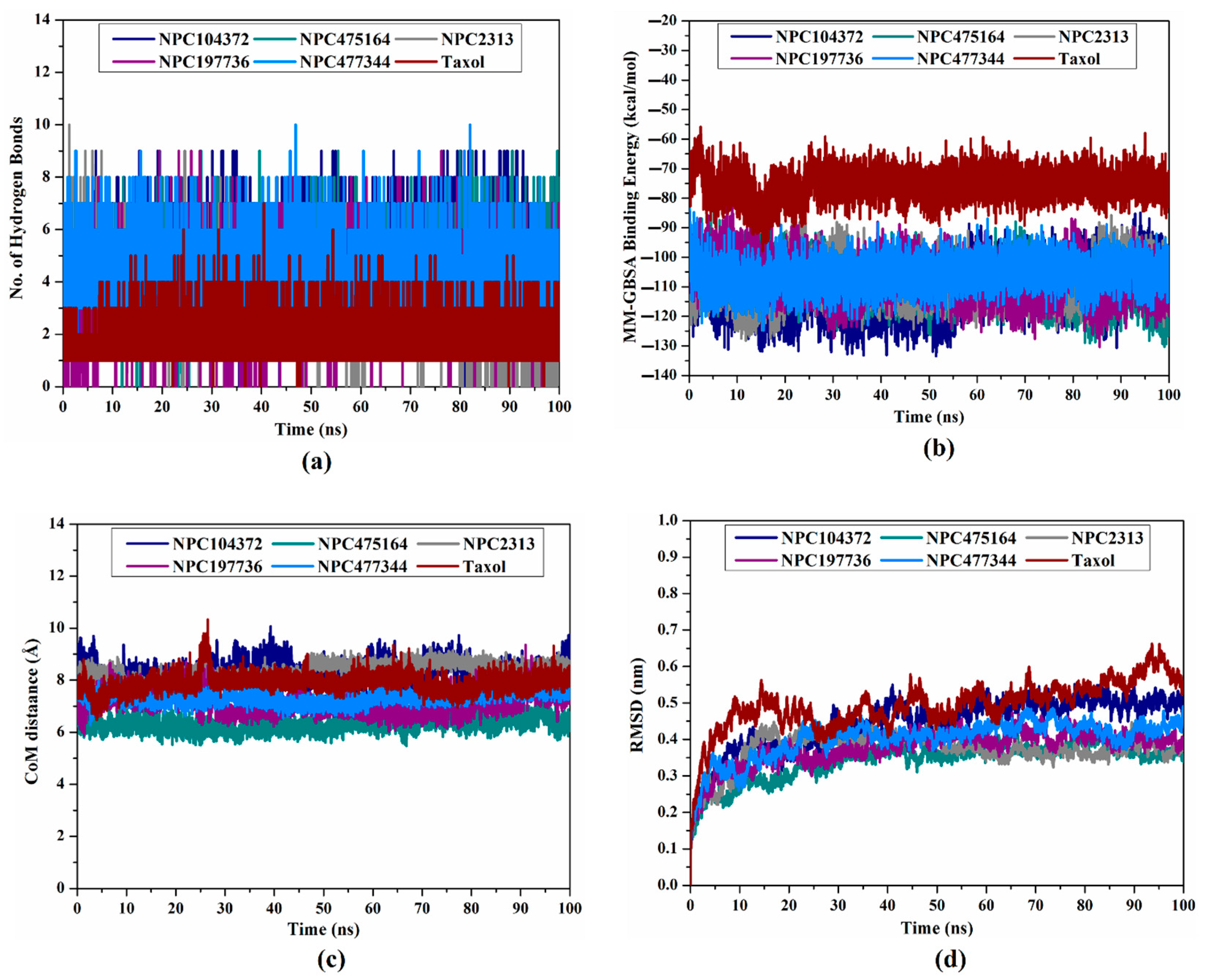
2.5. Post-Dynamics Analyses
2.5.1. Hydrogen Bond Analysis
2.5.2. Binding Energy Per Frame
2.5.3. Center-of-Mass Distance
2.5.4. Root-Mean-Square Deviation
2.6. Water Solubility of the Identified NPs
3. Computational Methodology
3.1. ABCB1 Preparation
3.2. Assessment of Molecular Docking Protocol
3.3. Database Preparation
3.4. Molecular Docking
3.5. Molecular Dynamics Simulations
3.6. MM-GBSA Binding Energy Estimations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Eckford, P.D.; Sharom, F.J. ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 2009, 109, 2989–3011. [Google Scholar] [CrossRef]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef]
- Edwards, J.E.; Alcorn, J.; Savolainen, J.; Anderson, B.D.; McNamara, P.J. Role of P-glycoprotein in distribution of nelfinavir across the blood-mammary tissue barrier and blood-brain barrier. Antimicrob. Agents Chemother. 2005, 49, 1626–1628. [Google Scholar] [CrossRef] [Green Version]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Hodges, L.M.; Markova, S.M.; Chinn, L.W.; Gow, J.M.; Kroetz, D.L.; Klein, T.E.; Altman, R.B. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharm. Genom. 2011, 21, 152–161. [Google Scholar] [CrossRef] [Green Version]
- Crowley, E.; McDevitt, C.A.; Callaghan, R. Generating Inhibitors of P-Glycoprotein: Where to, Now? In Multi-Drug Resistance in Cancer; Zhou, J., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 405–432. [Google Scholar]
- Li, J.W.; Vederas, J.C. Drug discovery and natural products: End of an era or an endless frontier? Science 2009, 325, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Zhu, F.; Qin, C.; Zhang, C.; Xu, F.; Tan, C.Y.; Jiang, Y.Y.; Chen, Y.Z. Nature’s contribution to today’s pharmacopeia. Nat. Biotechnol. 2014, 32, 979–980. [Google Scholar] [CrossRef]
- Gamal-Eldeen, A.M.; Hegazy, M.E. A crystal lapiferin derived from Ferula vesceritensis induces apoptosis pathway in MCF-7 breast cancer cells. Nat. Prod. Res. 2010, 24, 246–257. [Google Scholar] [CrossRef]
- Kurapati, K.R.; Atluri, V.S.; Samikkannu, T.; Garcia, G.; Nair, M.P. Natural Products as Anti-HIV Agents and Role in HIV-Associated Neurocognitive Disorders (HAND): A Brief Overview. Front. Microbiol. 2015, 6, 1444. [Google Scholar] [CrossRef]
- Wells, T.N. Natural products as starting points for future anti-malarial therapies: Going back to our roots? Malar. J. 2011, 10 (Suppl. S1), S3. [Google Scholar] [CrossRef] [Green Version]
- Cragg, G.M.; Newman, D.J.; Snader, K.M. Natural products in drug discovery and development. J. Nat. Prod. 1997, 60, 52–60. [Google Scholar] [CrossRef]
- Wilson, B.A.P.; Thornburg, C.C.; Henrich, C.J.; Grkovic, T.; O’Keefe, B.R. Creating and screening natural product libraries. Nat. Prod. Rep. 2020, 37, 893–918. [Google Scholar] [CrossRef]
- Koehn, F.E. High impact technologies for natural products screening. Prog. Drug Res. Fortschr. Der Arzneim. 2008, 65, 175–210. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [Green Version]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Kerrigan, J.E. Molecular dynamics simulations in drug design. In In Silico Models for Drug Discovery; Kortagere, S., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 95–113. [Google Scholar]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: Importance and enhancement techniques. ISRN Pharm. 2012, 2012, 195727. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Zeng, X.; Zhang, P.; He, W.; Qin, C.; Chen, S.; Tao, L.; Wang, Y.; Tan, Y.; Gao, D.; Wang, B.; et al. NPASS: Natural product activity and species source database for natural product research, discovery and tool development. Nucleic Acids Res. 2018, 46, D1217–D1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- OMEGA 2.5.1.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2013.
- SZYBKI 1.9.0.3; OpenEye Scientific Software: Santa Fe, NM, USA, 2016.
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminform. 2015, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Badr, E.A.A.; Almansour, N.M.; Alzahrani, O.R.; Ahmed, M.N.; Soliman, M.E.S.; Naeem, M.A.; Shawky, A.M.; Sidhom, P.A.; et al. Naturally occurring plant-based anticancerous candidates as prospective ABCG2 inhibitors: An in silico drug discovery study. Mol. Divers. 2022, 1–23. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Badr, E.A.A.; Abdelrahman, A.H.M.; Almansour, N.M.; Mekhemer, G.A.H.; Shawky, A.M.; Moustafa, M.F.; Atia, M.A.M. In Silico targeting human multidrug transporter ABCG2 in breast cancer: Database screening, molecular docking, and molecular dynamics study. Mol. Inform. 2022, 41, e2060039. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Badr, E.A.A.; Abdelrahman, A.H.M.; Almansour, N.M.; Shawky, A.M.; Mekhemer, G.A.H.; Alrumaihi, F.; Moustafa, M.F.; Atia, M.A.M. Prospective drug candidates as human multidrug transporter ABCG2 inhibitors: An in silico drug discovery study. Cell Biochem. Biophys. 2021, 79, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Roux, B.; Simonson, T. Implicit solvent models. Biophys. Chem. 1999, 78, 1–20. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges—The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle—An Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassault Systèmes BIOVIA, B.D.S.V.; Version 2019; Dassault Systèmes BIOVIA: San Diego, CA, USA, 2019.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | NPASS Code | 2D Chemical Structure | Docking Score (kcal/mol) | Binding Features b | ||
|---|---|---|---|---|---|---|
| Fast | Moderate | Expensive | ||||
| Taxol |  | −9.4 | −8.0 | −10.2 | TYR307 (2.27 Å), GLN347 (3.29 Å), GLN725 (1.93 Å), GLN990 (2.91 Å) | |
| 1 | NPC197736 |  | −8.8 | −10.9 | −13.5 | HIS61 (1.90, 2.29, 2.71 Å), THR199 (2.95 Å), TYR310 (1.90 Å), GLN725 (2.34 Å), TYR950 (2.27 Å), GLN990 (1.99, 2.08, 2.36 Å) |
| 2 | NPC477533 |  | −11.1 | −10.9 | −13.3 | GLN990 (2.24 Å) |
| 3 | NPC223735 |  | −9.9 | −11.4 | −12.6 | TYR310 (2.64 Å), GLN347 (2.01, 3.08 Å), GLN725 (2.03 Å), GLN946 (2.05 Å), GLN990 (2.22 Å) |
| 4 | NPC70862 |  | −11.6 | −11.8 | −12.2 | ASN842 (2.57, 2.79 Å), TYR950 (1.74 Å) |
| 5 | NPC471637 |  | −9.7 | −11.4 | −12.2 | THR199 (1.62 Å), TRP232 (2.36 Å), TYR307 (2.03 Å), GLN725 (2.52, 2.59, 2.85 Å), GLU875 (1.86 Å), GLN990 (2.31 Å) |
| 6 | NPC104372 |  | −9.9 | −11.0 | −12.2 | HIS61 (2.15 Å), GLY62 (2.26 Å), GLY64 (2.95 Å), LEU65 (2.30 Å), GLN195 (2.12 Å), THR199 (1.63 Å), SER344 (2.11 Å), GLN946 (3.22 Å) |
| 7 | NPC2313 |  | −9.9 | −12.1 | −12.0 | GLY62 (2.93 Å), LEU65 (2.98 Å), GLN195 (2.20, 2.73 Å), TYR310 (2.96 Å), GLN347 (2.35 Å), GLN946 (2.13 Å), GLN990 (1.76, 2.30 Å) |
| 8 | NPC475164 |  | −9.3 | −11.3 | −11.9 | TYR307 (2.08 Å), TYR310 (2.17 Å), GLN347 (3.20 Å), ASN842 (2.16 Å), GLN946 (1.70 Å), MET986 (1.82 Å), GLN725 (3.00 Å), GLN990 (2.61 Å) |
| 9 | NPC477344 |  | −10.2 | −11.1 | −11.9 | TYR310 (2.86 Å), SER344 (2.21 Å), GLY872 (2.72 Å), GLU875 (1.85 Å), MET986 (1.93 Å), GLN990 (1.89, 2.08, 2.54 Å) |
| NPASS Code | Estimated MM-GBSA Binding Energy (kcal/mol) | ||||||
|---|---|---|---|---|---|---|---|
| ∆Evdw | ∆Eele | ∆EGB | ∆ESUR | ∆Ggas | ∆GSolv | ∆Gbinding | |
| NPC104372 | −110.1 | −83.6 | 98.1 | −15.8 | −193.6 | 82.2 | −111.4 |
| NPC475164 | −120.9 | −64.3 | 93.9 | −17.4 | −185.2 | 76.5 | −108.7 |
| NPC2313 | −119.0 | −65.6 | 92.3 | −16.2 | −184.6 | 76.1 | −108.5 |
| NPC197736 | −120.8 | −60.2 | 89.8 | −16.5 | −181.0 | 73.3 | −107.7 |
| NPC477344 | −111.4 | −76.7 | 98.6 | −16.4 | −188.2 | 82.2 | −106.0 |
| Taxol | −89.4 | −49.9 | 72.2 | −12.6 | −139.3 | 59.6 | −79.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, M.A.A.; Abdeljawaad, K.A.A.; Abdelrahman, A.H.M.; Jaragh-Alhadad, L.A.; Oraby, H.F.; Elkaeed, E.B.; Mekhemer, G.A.H.; Gabr, G.A.; Shawky, A.M.; Sidhom, P.A.; et al. Exploring Natural Product Activity and Species Source Candidates for Hunting ABCB1 Transporter Inhibitors: An In Silico Drug Discovery Study. Molecules 2022, 27, 3104. https://doi.org/10.3390/molecules27103104
Ibrahim MAA, Abdeljawaad KAA, Abdelrahman AHM, Jaragh-Alhadad LA, Oraby HF, Elkaeed EB, Mekhemer GAH, Gabr GA, Shawky AM, Sidhom PA, et al. Exploring Natural Product Activity and Species Source Candidates for Hunting ABCB1 Transporter Inhibitors: An In Silico Drug Discovery Study. Molecules. 2022; 27(10):3104. https://doi.org/10.3390/molecules27103104
Chicago/Turabian StyleIbrahim, Mahmoud A. A., Khlood A. A. Abdeljawaad, Alaa H. M. Abdelrahman, Laila A. Jaragh-Alhadad, Hesham Farouk Oraby, Eslam B. Elkaeed, Gamal A. H. Mekhemer, Gamal A. Gabr, Ahmed M. Shawky, Peter A. Sidhom, and et al. 2022. "Exploring Natural Product Activity and Species Source Candidates for Hunting ABCB1 Transporter Inhibitors: An In Silico Drug Discovery Study" Molecules 27, no. 10: 3104. https://doi.org/10.3390/molecules27103104