
Bis-Cinnamamide Derivatives as APE/Ref-1 Inhibitors for the Treatment of Human Melanoma

 , and
, and
Abstract
:
1. Introduction
2. Results
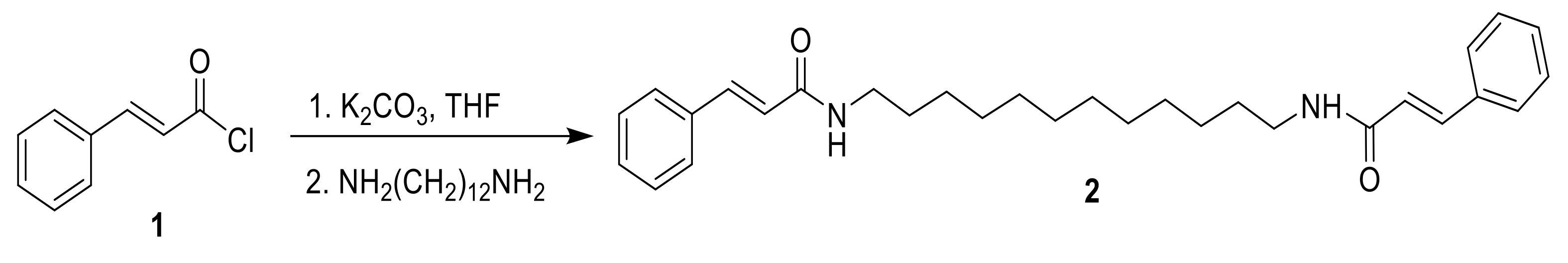
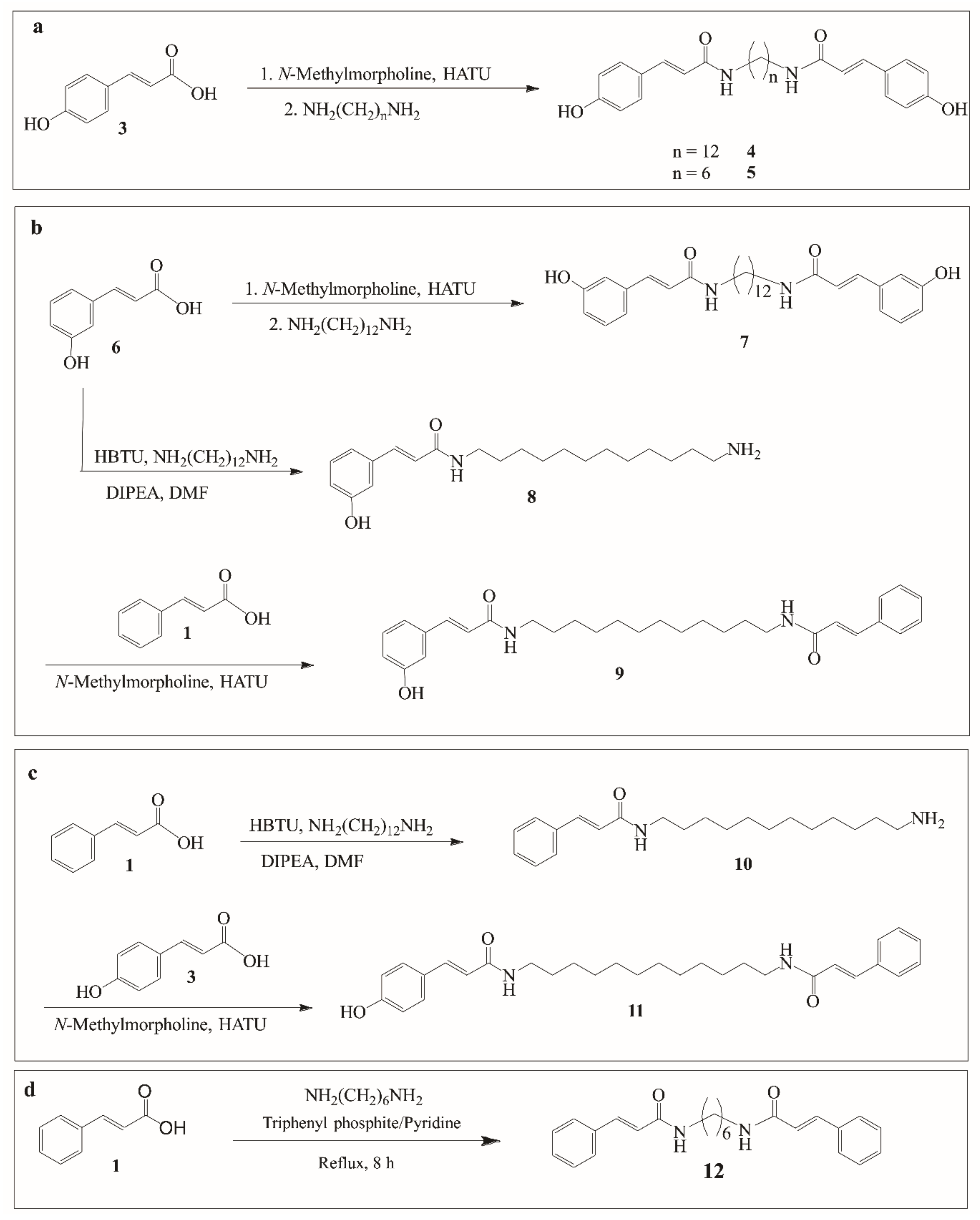
2.1. Chemistry
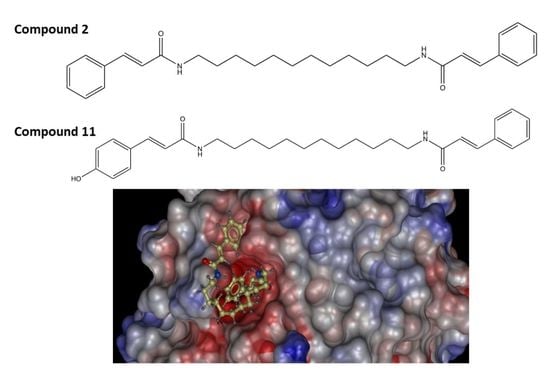
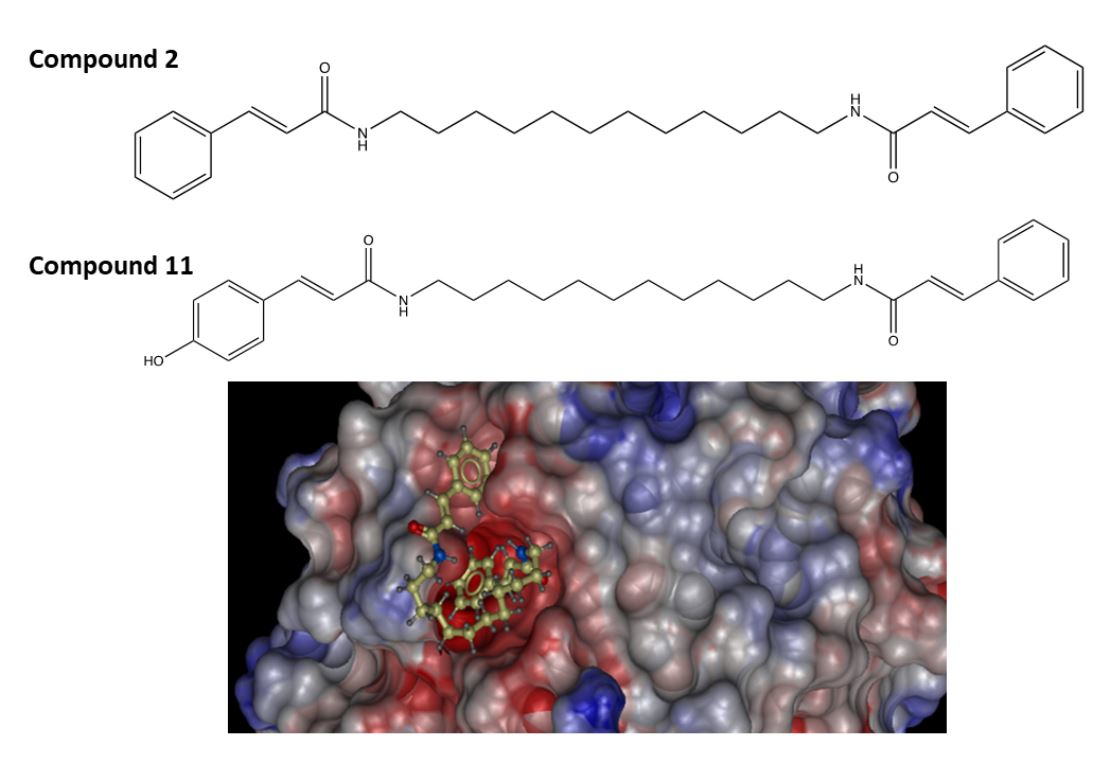
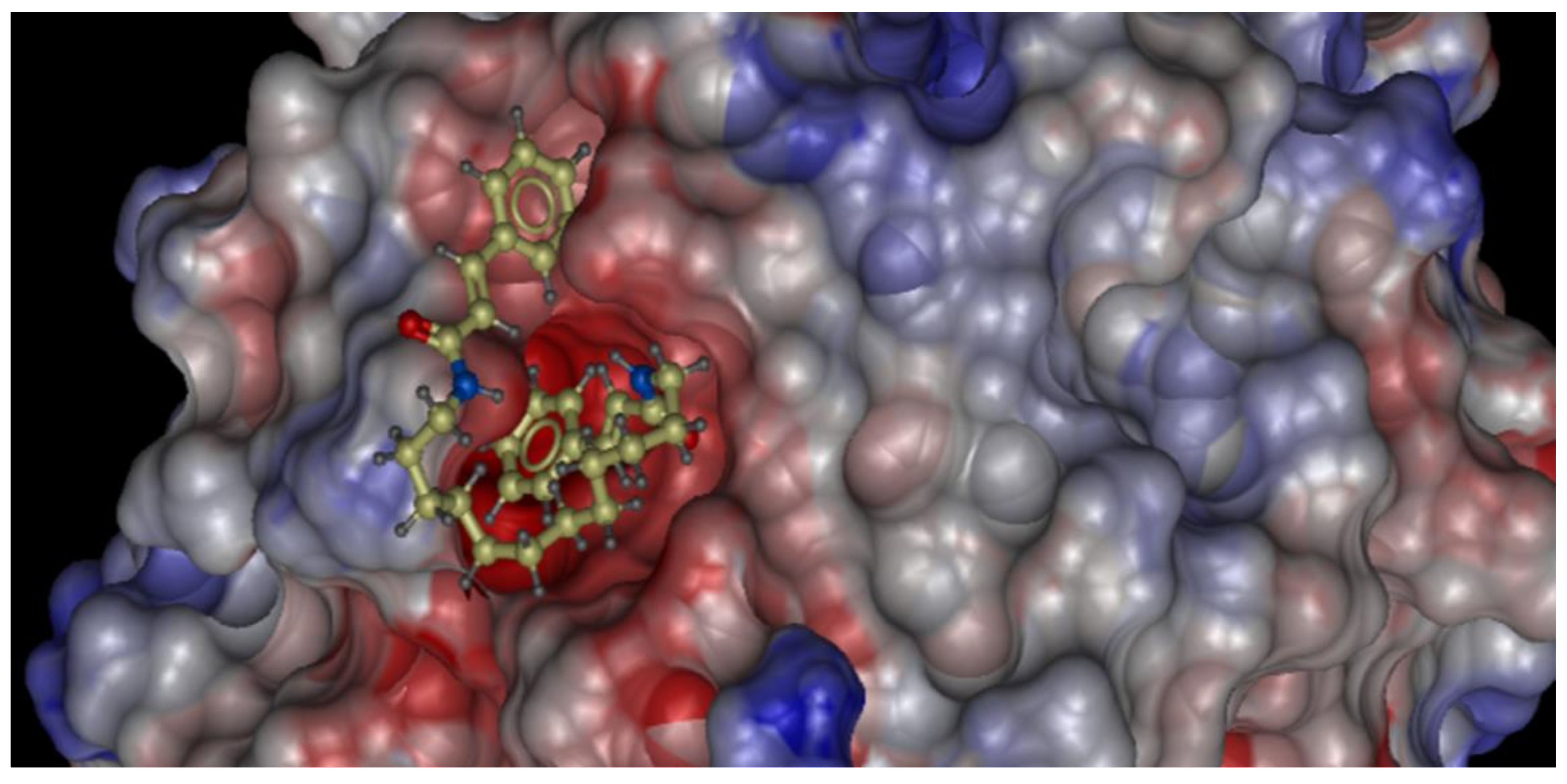
2.2. Molecular Docking Study of the Synthesized Compound Candidates
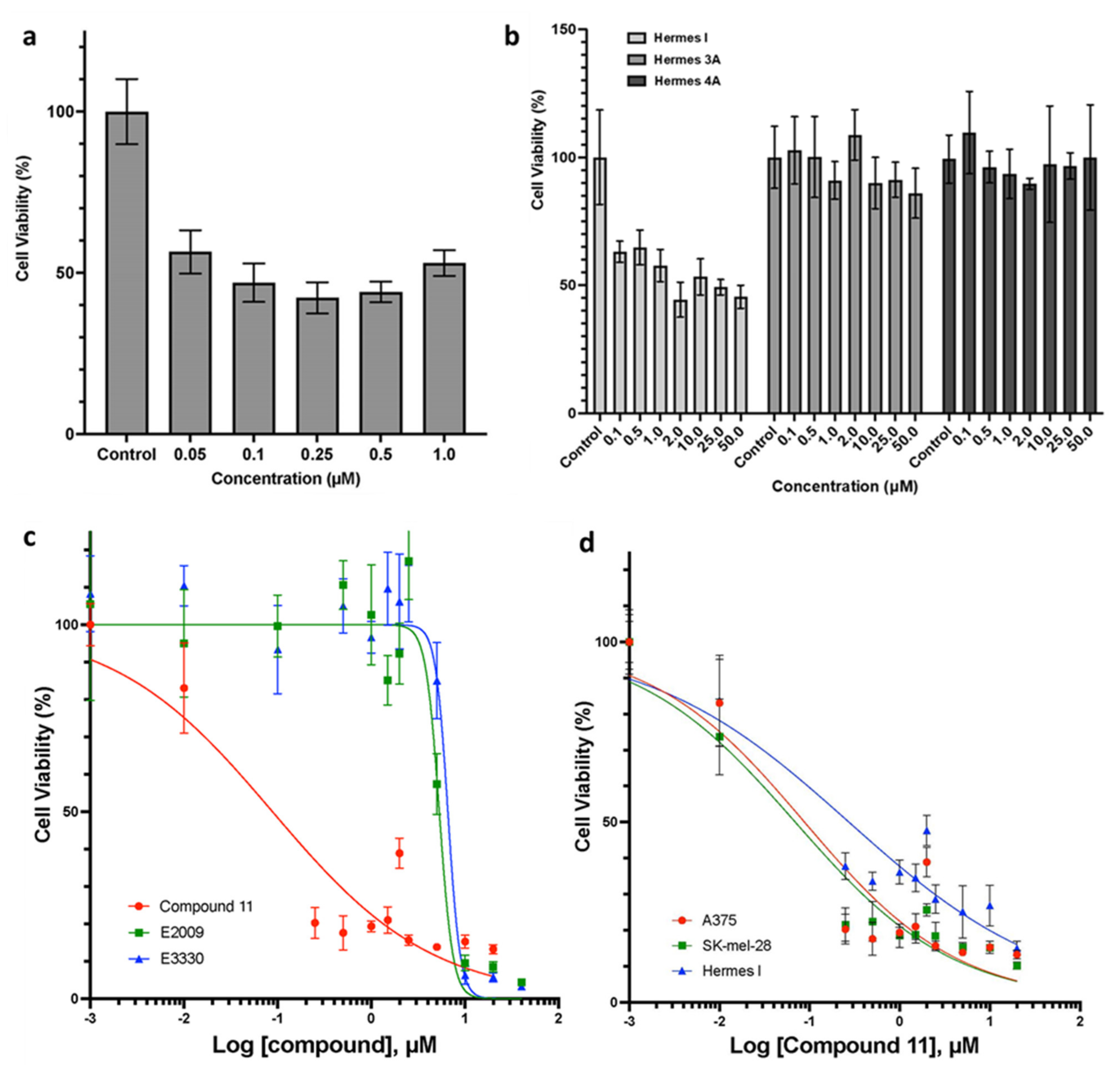
2.3. Effects of Novel APE/Ref-1 Inhibitors on Melanoma Proliferation Using MTT Colorimetric Assay
2.4. Potent and Selective Anti-Tumor Activities of Compound 11 in Human Melanoma Cells
2.5. Compound 2 Suppressed H2O2-Induced AP-1 Transactivation in Human Melanoma Cells
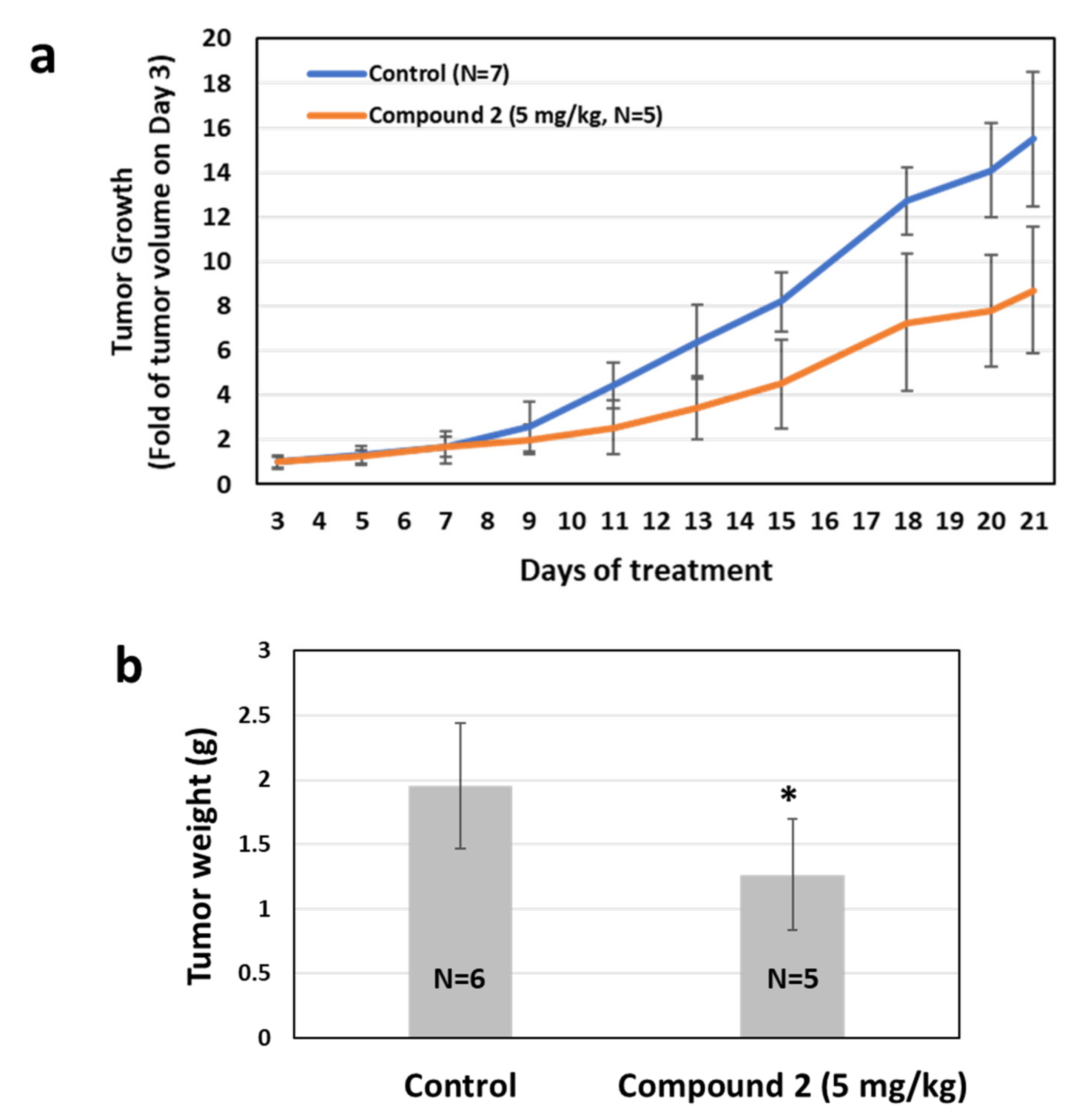
2.6. Effects of APE/Ref-1 Inhibitor on Tumor Growth Using a Melanoma Xenograft Mouse Model
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Chemical Synthesis
4.3. Three-Dimensional Virtual Docking Using Molsoft ICM-Pro System
4.4. Cell Lines and Cell Culture
4.5. In Vitro Anti-Melanoma Activity Screening Using MTT Colorimetric Assay
4.6. Luciferase Assay to Determine AP-1 Transactivation Activities
4.7. In Vivo Xenograft Melanoma
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rastrelli, M.; Tropea, S.; Rossi, C.R.; Alaibac, M. Melanoma: Epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo 2014, 28, 1005–1011. [Google Scholar] [PubMed]
- American Cancer Society. Cancer Facts & Figures 2022; American Cancer Society: Atlanta, GA, USA, 2022. [Google Scholar]
- Romero-Graillet, C.; Aberdam, E.; Clement, M.; Ortonne, J.P.; Ballotti, R. Nitric oxide produced by ultraviolet-irradiated keratinocytes stimulates melanogenesis. J. Clin. Investig. 1997, 99, 635–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, P.A.; Halliday, G.M. Inhibition of nitric oxide and reactive oxygen species production improves the ability of a sunscreen to protect from sunburn, immunosuppression and photocarcinogenesis. Br. J. Dermatol. 2006, 155, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Aitken, G.R.; Henderson, J.R.; Chang, S.C.; McNeil, C.J.; Birch-Machin, M.A. Direct monitoring of UV-induced free radical generation in HaCaT keratinocytes. Clin. Exp. Dermatol. 2007, 32, 722–727. [Google Scholar] [CrossRef]
- Meyskens, F.L., Jr.; Farmer, P.J.; Yang, S.; Anton-Culver, H. New perspectives on melanoma pathogenesis and chemoprevention. Recent Results Cancer Res. 2007, 174, 191–195. [Google Scholar]
- Moan, J.; Grigalavicius, M.; Baturaite, Z.; Dahlback, A.; Juzeniene, A. The relationship between UV exposure and incidence of skin cancer. Photodermatol. Photoimmunol. Photomed. 2015, 31, 26–35. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, S.; Misner, B.J.; Chiu, R.; Liu, F.; Meyskens, F.L., Jr. Nitric oxide initiates progression of human melanoma via a feedback loop mediated by apurinic/apyrimidinic endonuclease-1/redox factor-1, which is inhibited by resveratrol. Mol. Cancer Ther. 2008, 7, 3751–3760. [Google Scholar] [CrossRef] [Green Version]
- Meyskens, F.L., Jr.; McNulty, S.E.; Buckmeier, J.A.; Tohidian, N.B.; Spillane, T.J.; Kahlon, R.S.; Gonzalez, R.I. Aberrant redox regulation in human metastatic melanoma cells compared to normal melanocytes. Free Radic Biol. Med. 2001, 31, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, D.; Paniker, L.; Sanchez, G.; Trono, D.; Nairn, R. The etiology of sunlight-induced melanoma in Xiphophorus hybrid fish. Mol. Carcinog 2007, 46, 679–684. [Google Scholar] [CrossRef]
- Garibyan, L.; Fisher, D.E. How Sunlight Causes Melanoma. Curr. Oncol. Rep. 2010, 12, 319–326. [Google Scholar] [CrossRef]
- Shah, F.; Logsdon, D.; Messmann, R.A.; Fehrenbacher, J.C.; Fishel, M.L.; Kelley, M.R. Exploiting the Ref-1-APE1 node in cancer signaling and other diseases: From bench to clinic. NPJ Precis. Oncol. 2017, 1, 19. [Google Scholar] [CrossRef] [Green Version]
- Evans, A.R.; Limp-Foster, M.; Kelley, M.R. Going APE over ref-1. Mutat Res. 2000, 461, 83–108. [Google Scholar] [CrossRef]
- Yang, S.; Misner, B.J.; Chiu, R.J.; Meyskens, F.L., Jr. Redox effector factor-1, combined with reactive oxygen species, plays an important role in the transformation of JB6 cells. Carcinogenesis 2007, 28, 2382–2390. [Google Scholar] [CrossRef]
- Yang, S.; Irani, K.; Heffron, S.E.; Jurnak, F.; Meyskens, F.L., Jr. Alterations in the expression of the apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE/Ref-1) in human melanoma and identification of the therapeutic potential of resveratrol as an APE/Ref-1 inhibitor. Mol. Cancer Ther. 2005, 4, 1923–1935. [Google Scholar] [CrossRef] [Green Version]
- Vascotto, C.; Cesaratto, L.; Zeef, L.A.; Deganuto, M.; D’Ambrosio, C.; Scaloni, A.; Romanello, M.; Damante, G.; Taglialatela, G.; Delneri, D.; et al. Genome-wide analysis and proteomic studies reveal APE1/Ref-1 multifunctional role in mammalian cells. Proteomics 2009, 9, 1058–1074. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Meyskens, F.L. Apurinic/apyrimidinic endonuclease/redox effector factor-1(APE/Ref-1): A unique target for the prevention and treatment of human melanoma. Antioxid. Redox Signal. 2009, 11, 639–650. [Google Scholar] [CrossRef] [Green Version]
- Caston, R.A.; Gampala, S.; Armstrong, L.; Messmann, R.A.; Fishel, M.L.; Kelley, M.R. The multifunctional APE1 DNA repair-redox signaling protein as a drug target in human disease. Drug Discov. Today 2021, 26, 218–228. [Google Scholar] [CrossRef]
- Angkeow, P.; Deshpande, S.S.; Qi, B.; Liu, Y.X.; Park, Y.C.; Jeon, B.H.; Ozaki, M.; Irani, K. Redox factor-1: An extra-nuclear role in the regulation of endothelial oxidative stress and apoptosis. Cell Death Differ. 2002, 9, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Robertson, K.A.; Bullock, H.A.; Xu, Y.; Tritt, R.; Zimmerman, E.; Ulbright, T.M.; Foster, R.S.; Einhorn, L.H.; Kelley, M.R. Altered expression of Ape1/ref-1 in germ cell tumors and overexpression in NT2 cells confers resistance to bleomycin and radiation. Cancer Res. 2001, 61, 2220–2225. [Google Scholar]
- Li, Q.; Zhou, Z.-W.; Duan, W.; Qian, C.-Y.; Wang, S.-N.; Deng, M.-S.; Zi, D.; Wang, J.-M.; Mao, C.-Y.; Song, G.; et al. Inhibiting the redox function of APE1 suppresses cervical cancer metastasis via disengagement of ZEB1 from E-cadherin in EMT. J. Exp. Clin. Cancer Res. 2021, 40, 220. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, S.; Misner, B.J.; Liu-Smith, F.; Meyskens, F.L. The role of APE/Ref-1 signaling pathway in hepatocellular carcinoma progression. Int. J. Oncol. 2014, 45, 1820–1828. [Google Scholar] [CrossRef] [Green Version]
- Shahda, S.; Lakhani, N.J.; O’Neil, B.; Rasco, D.W.; Wan, J.; Mosley, A.L.; Liu, H.; Kelley, M.R.; Messmann, R.A. A phase I study of the APE1 protein inhibitor APX3330 in patients with advanced solid tumors. J. Clin. Oncol. 2019, 37, 3097. [Google Scholar] [CrossRef]
- Rai, G.; Vyjayanti, V.N.; Dorjsuren, D.; Simeonov, A.; Jadhav, A.; Wilson, D.M., 3rd; Maloney, D.J. Synthesis, biological evaluation, and structure-activity relationships of a novel class of apurinic/apyrimidinic endonuclease 1 inhibitors. J. Med. Chem. 2012, 55, 3101–3112. [Google Scholar] [CrossRef] [Green Version]
- Behrouzi, A.; Xia, H.; Thompson, E.L.; Kelley, M.R.; Fehrenbacher, J.C. Oxidative DNA Damage and Cisplatin Neurotoxicity Is Exacerbated by Inhibition of OGG1 Glycosylase Activity and APE1 Endonuclease Activity in Sensory Neurons. Int. J. Mol. Sci. 2022, 23, 1909. [Google Scholar] [CrossRef]
- Gaiddon, C.; Moorthy, N.C.; Prives, C. Ref-1 regulates the transactivation and pro-apoptotic functions of p53 in vivo. EMBO J. 1999, 18, 5609–5621. [Google Scholar] [CrossRef] [Green Version]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [Green Version]
- Mijit, M.; Caston, R.; Gampala, S.; Fishel, M.L.; Fehrenbacher, J.; Kelley, M.R. APE1/Ref-1—One Target with Multiple Indications: Emerging Aspects and New Directions. J. Cell Signal. 2021, 2, 151–161. [Google Scholar]
- McIlwain, D.W.; Fishel, M.L.; Boos, A.; Kelley, M.R.; Jerde, T.J. APE1/Ref-1 redox-specific inhibition decreases survivin protein levels and induces cell cycle arrest in prostate cancer cells. Oncotarget 2018, 9, 10962–10977. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, P.S.; Corvacho, E.; Costa, J.G.; Saraiva, N.; Fernandes, A.S.; Castro, M.; Miranda, J.P.; Oliveira, N.G. The APE1 redox inhibitor E3330 reduces collective cell migration of human breast cancer cells and decreases chemoinvasion and colony formation when combined with docetaxel. Chem. Biol. Drug Des. 2017, 90, 561–571. [Google Scholar] [CrossRef]
- Kelley, M.R.; Luo, M.; Reed, A.; Su, D.; Delaplane, S.; Borch, R.F.; Nyland, R.L., 2nd; Gross, M.L.; Georgiadis, M.M. Functional analysis of novel analogues of E3330 that block the redox signaling activity of the multifunctional AP endonuclease/redox signaling enzyme APE1/Ref-1. Antioxid. Redox Signal. 2011, 14, 1387–1401. [Google Scholar] [CrossRef]
- Fishel, M.L.; Xia, H.; McGeown, J.; McIlwain, D.W.; Elbanna, M.; Craft, A.A.; Kaimakliotis, H.Z.; Sandusky, G.E.; Zhang, C.; Pili, R.; et al. Antitumor Activity and Mechanistic Characterization of APE1/Ref-1 Inhibitors in Bladder Cancer. Mol. Cancer Ther. 2019, 18, 1947–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavasotto, C.N.; Orry, A.J. Ligand docking and structure-based virtual screening in drug discovery. Curr. Top. Med. Chem. 2007, 7, 1006–1014. [Google Scholar] [CrossRef]
- Orry, A.J.; Abagyan, R.A.; Cavasotto, C.N. Structure-based development of target-specific compound libraries. Drug Discov. Today 2006, 11, 261–266. [Google Scholar] [CrossRef]
- Natala, S.R.; Habas, A.; Stocking, E.M.; Orry, A.; Price, D.L.; Gill, M.B.; Bonhaus, D.W.; Abagyan, R.; Wrasidlo, W. Structure based design and synthesis of novel Toll-like Receptor 2 (TLR 2) lipid antagonists. Bioorganic Med. Chem. Lett. 2021, 40, 127861. [Google Scholar] [CrossRef] [PubMed]
- Zou, G.M.; Maitra, A. Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol. Cancer Ther. 2008, 7, 2012–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray-Schopfer, V.C.; Cheong, S.C.; Chong, H.; Chow, J.; Moss, T.; Abdel-Malek, Z.A.; Marais, R.; Wynford-Thomas, D.; Bennett, D.C. Cellular senescence in naevi and immortalisation in melanoma: A role for p16? Br. J. Cancer 2006, 95, 496–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Wellcome Trust Functional Genomics Cell Bank: Holdings. Pigment. Cell Melanoma Res. 2010, 23, 147–150. [CrossRef]
- Vasanwala, F.H.; Kusam, S.; Toney, L.M.; Dent, A.L. Repression of AP-1 function: A mechanism for the regulation of Blimp-1 expression and B lymphocyte differentiation by the B cell lymphoma-6 protooncogene. J. Immunol. 2002, 169, 1922–1929. [Google Scholar] [CrossRef] [Green Version]
- Dean, E.; Lorigan, P. Advances in the management of melanoma: Targeted therapy, immunotherapy and future directions. Expert Rev. Anticancer. 2012, 12, 1437–1448. [Google Scholar] [CrossRef]
- Kuryk, L.; Bertinato, L.; Staniszewska, M.; Pancer, K.; Wieczorek, M.; Salmaso, S.; Caliceti, P.; Garofalo, M. From Conventional Therapies to Immunotherapy: Melanoma Treatment in Review. Cancers 2020, 12, 3057. [Google Scholar] [CrossRef]
- Barone, A.; Hazarika, M.; Theoret, M.R.; Mishra-Kaly, P.; Chen, H.; He, K.; Sridhara, R.; Subramaniam, S.; Pfuma, E.; Wang, Y.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Patients with Unresectable or Metastatic Melanoma. Clin. Cancer Res. 2017, 23, 5661–5665. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Kelley, M.R. Inhibition of the human apurinic/apyrimidinic endonuclease (APE1) repair activity and sensitization of breast cancer cells to DNA alkylating agents with lucanthone. Anticancer. Res. 2004, 24, 2127–2134. [Google Scholar]
- Zou, G.M.; Karikari, C.; Kabe, Y.; Handa, H.; Anders, R.A.; Maitra, A. The Ape-1/Ref-1 redox antagonist E3330 inhibits the growth of tumor endothelium and endothelial progenitor cells: Therapeutic implications in tumor angiogenesis. J. Cell Physiol. 2009, 219, 209–218. [Google Scholar] [CrossRef]
- Bapat, A.; Glass, L.S.; Luo, M.; Fishel, M.L.; Long, E.C.; Georgiadis, M.M.; Kelley, M.R. Novel small-molecule inhibitor of apurinic/apyrimidinic endonuclease 1 blocks proliferation and reduces viability of glioblastoma cells. J. Pharmacol. Exp. Ther. 2010, 334, 988–998. [Google Scholar] [CrossRef] [Green Version]
- Madhusudan, S.; Smart, F.; Shrimpton, P.; Parsons, J.L.; Gardiner, L.; Houlbrook, S.; Talbot, D.C.; Hammonds, T.; Freemont, P.A.; Sternberg, M.J.; et al. Isolation of a small molecule inhibitor of DNA base excision repair. Nucleic Acids Res. 2005, 33, 4711–4724. [Google Scholar] [CrossRef]
- Nyland, R.L.; Luo, M.; Kelley, M.R.; Borch, R.F. Design and synthesis of novel quinone inhibitors targeted to the redox function of apurinic/apyrimidinic endonuclease 1/redox enhancing factor-1 (Ape1/ref-1). J. Med. Chem. 2010, 53, 1200–1210. [Google Scholar] [CrossRef] [Green Version]
- Shirazi, A.N.; El-Sayed, N.S.; Tiwari, R.K.; Tavakoli, K.; Parang, K. Cyclic Peptide Containing Hydrophobic and Positively Charged Residues as a Drug Delivery System for Curcumin. Curr. Drug Deliv. 2016, 13, 409–417. [Google Scholar] [CrossRef]
- Mozaffari, S.; Salehi, D.; Mahdipoor, P.; Beuttler, R.; Tiwari, R.; Aliabadi, H.M.; Parang, K. Design and application of hybrid cyclic-linear peptide-doxorubicin conjugates as a strategy to overcome doxorubicin resistance and toxicity. Eur. J. Med. Chem. 2021, 226, 113836. [Google Scholar] [CrossRef]
- Schapira, M.; Totrov, M.; Abagyan, R. Prediction of the binding energy for small molecules, peptides and proteins. J. Mol. Recognit. 1999, 12, 177–190. [Google Scholar] [CrossRef]
- Cell Culture Media. Available online: https://www.sgul.ac.uk/about/our-institutes/molecular-and-clinical-sciences/research-centres/cell-biology-research-centre/genomics-cell-bank/cell-culture-media#a8 (accessed on 5 June 2021).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthesized Compounds | Docking Score | Anti-Melanoma Activities |
|---|---|---|
| 2 | −9.23 | Cytotoxicity is not dose-dependent; at 0.1 µM, cell viability reduced to <50% of control in A375 cells. |
| 11 | −16.31 | 0.079 ± 0.013 µM (IC50, detected by MTT assay, average in two human melanoma cell lines: A375 and SK-Mel-28) |
| 4 | −20.14 | 50% inhibition of cell survival not observed up to 10 µM. |
| 5 | −12.91 | 50% inhibition of cell survival not observed up to 50 µM. |
| 7 | −11.74 | 50% inhibition of cell survival not observed up to 10 µM. |
| 8 | −12.51 | 50% inhibition of cell survival not observed up to 25 µM. |
| 9 | −5.97 | 50% inhibition of cell survival not observed up to 10 µM. |
| 10 | −15.44 | 50% inhibition of cell survival not observed up to 25 µM. |
| 12 | −10.06 | 50% inhibition of cell survival not observed up to 10 µM. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhazmi, R.; Tong, S.; Darwish, S.; Khanjani, E.; Khungar, B.; Chawla, S.; Zheng, Z.; Chamberlin, R.; Parang, K.; Yang, S. Bis-Cinnamamide Derivatives as APE/Ref-1 Inhibitors for the Treatment of Human Melanoma. Molecules 2022, 27, 2672. https://doi.org/10.3390/molecules27092672
Alhazmi R, Tong S, Darwish S, Khanjani E, Khungar B, Chawla S, Zheng Z, Chamberlin R, Parang K, Yang S. Bis-Cinnamamide Derivatives as APE/Ref-1 Inhibitors for the Treatment of Human Melanoma. Molecules. 2022; 27(9):2672. https://doi.org/10.3390/molecules27092672
Chicago/Turabian StyleAlhazmi, Razan, Shirley Tong, Shaban Darwish, Elina Khanjani, Bharti Khungar, Swati Chawla, Zhonghui Zheng, Richard Chamberlin, Keykavous Parang, and Sun Yang. 2022. "Bis-Cinnamamide Derivatives as APE/Ref-1 Inhibitors for the Treatment of Human Melanoma" Molecules 27, no. 9: 2672. https://doi.org/10.3390/molecules27092672