
Antioxidant Cardioprotection against Reperfusion Injury: Potential Therapeutic Roles of Resveratrol and Quercetin

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
1.1. Oxidative Stress
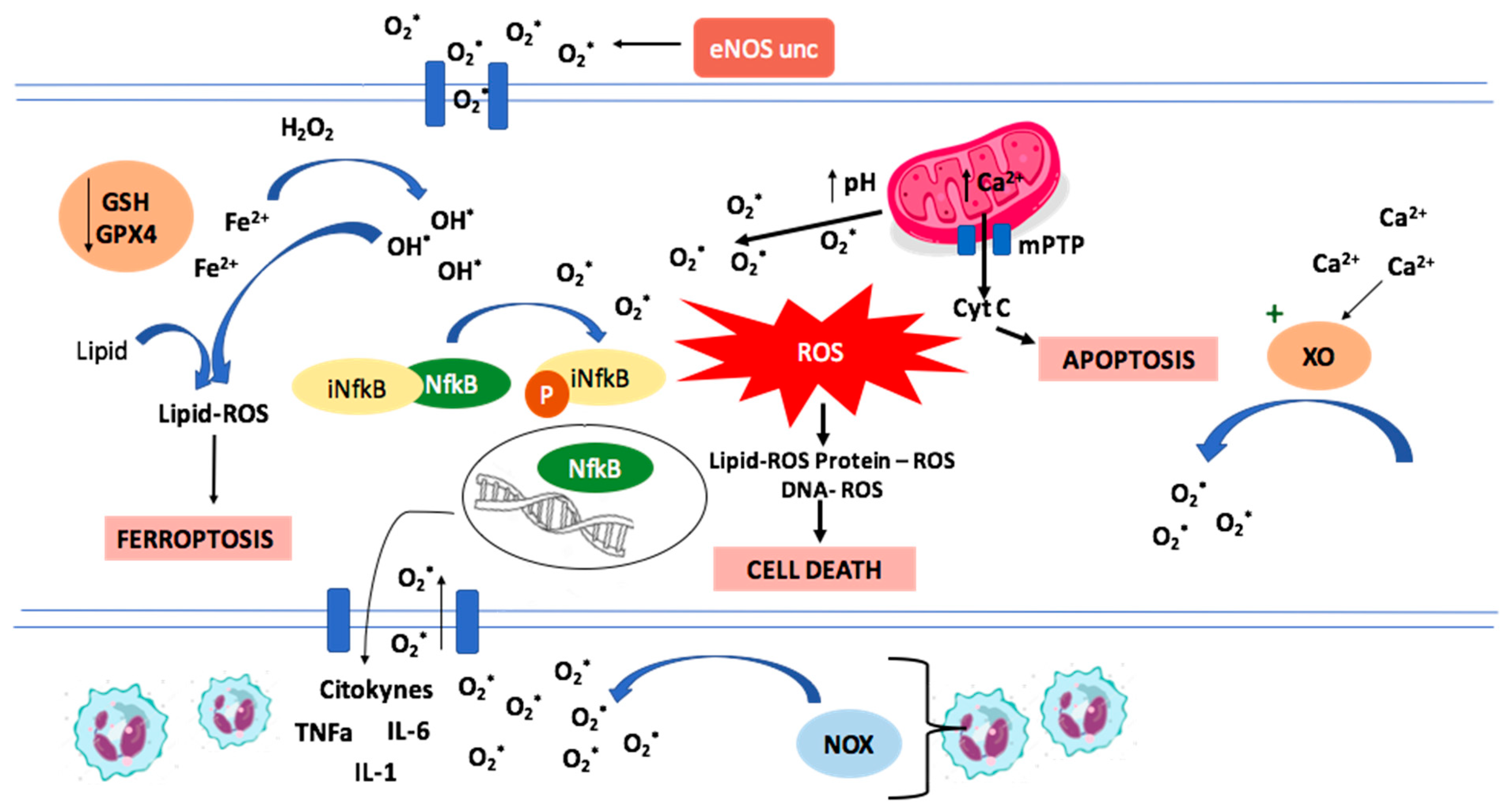
1.1.1. Oxidative Stress and ROS
1.1.2. Mechanisms of Ischemia/Reperfusion Cellular Damage
1.1.3. ROS Production in Ischemia/Reperfusion
1.2. Intracellular Organelle Dysfunctions Associated with Ischemia/Reperfusion Injury
1.2.1. Endoplasmic Reticulum Stress (ERS)
1.2.2. Mitochondrial Dysfunction
1.2.3. Inflammation Mediated Damage
1.3. Mechanisms of Cell Death in Ischemia-Reperfusion
Ferroptosis
1.4. Apoptosis
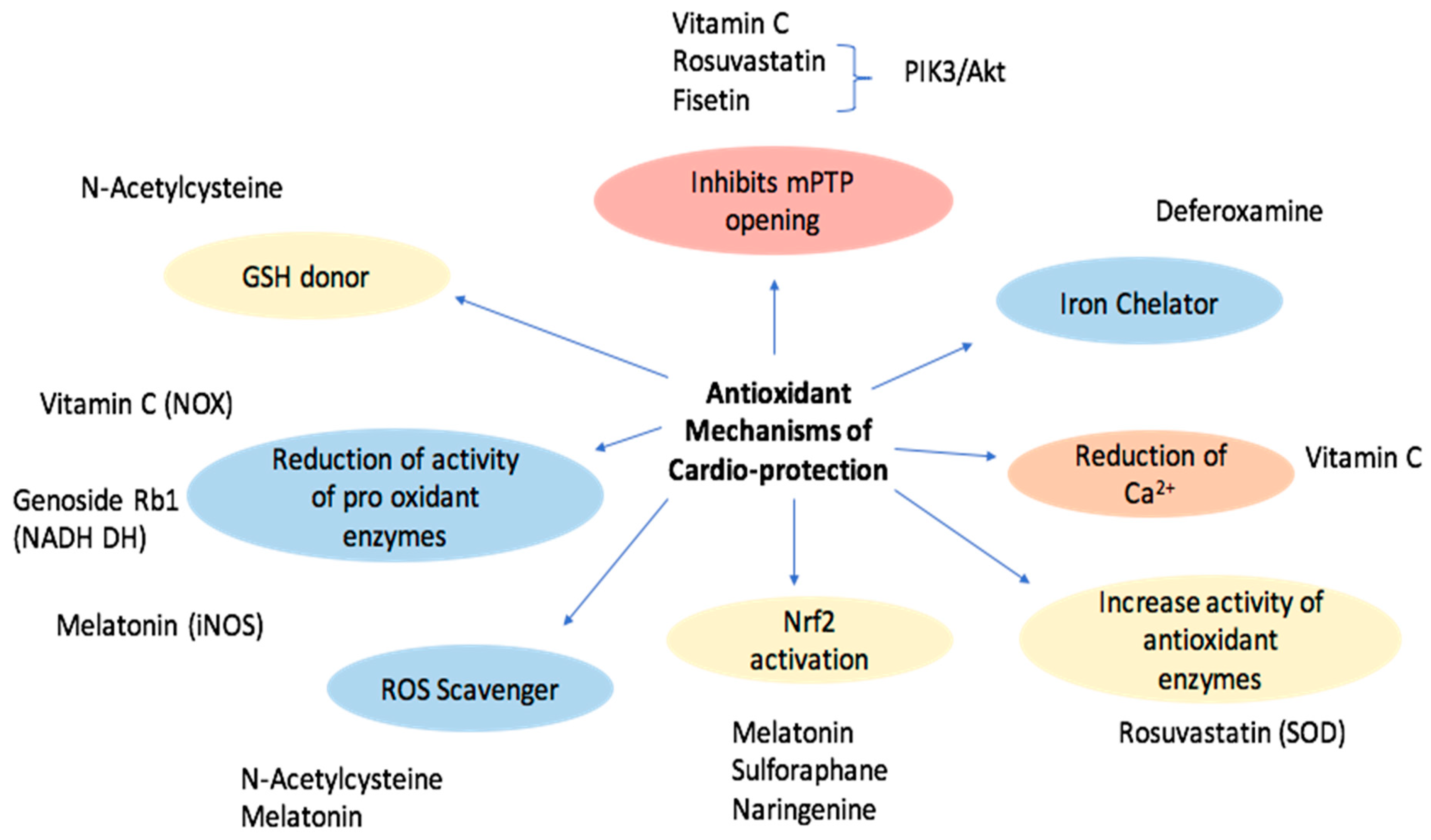
1.5. Defense Mechanisms against Oxidative Stress
1.6. Nrf2 Signalling Pathway
1.7. Exogenous Antioxidants with Reported Cardioprotective Effects
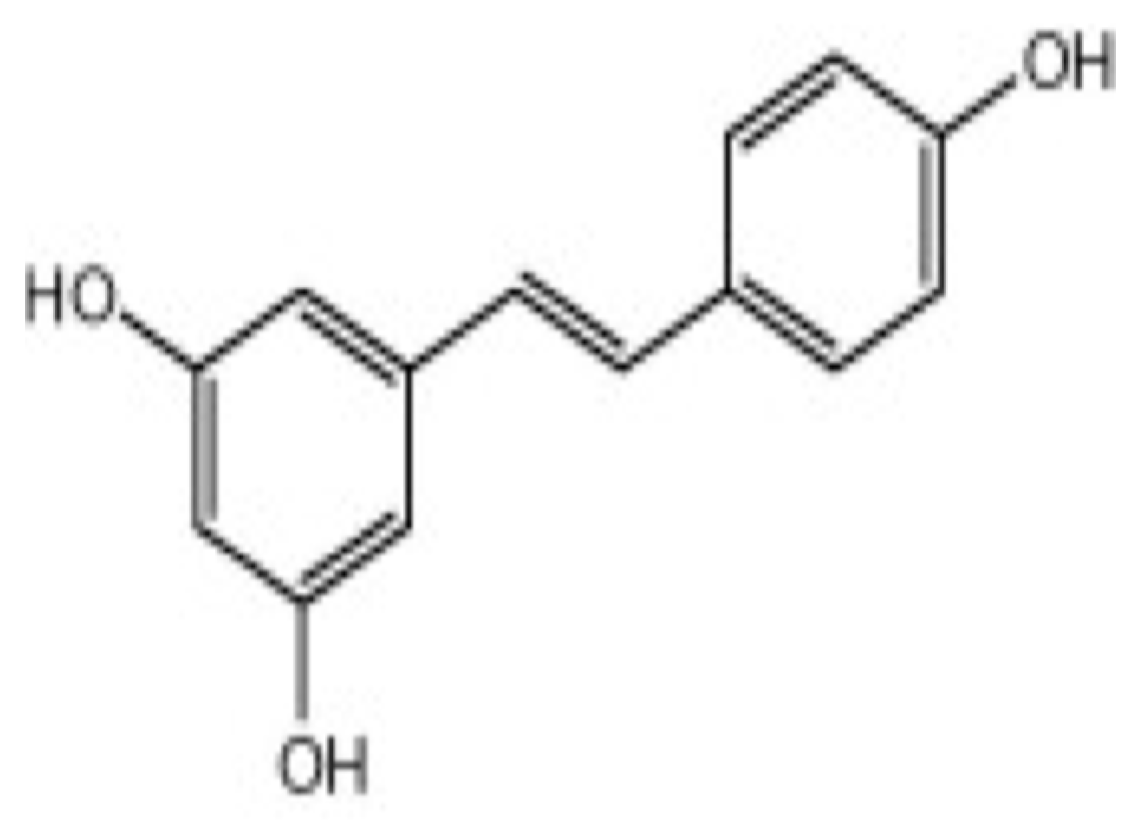
1.8. Resveratrol
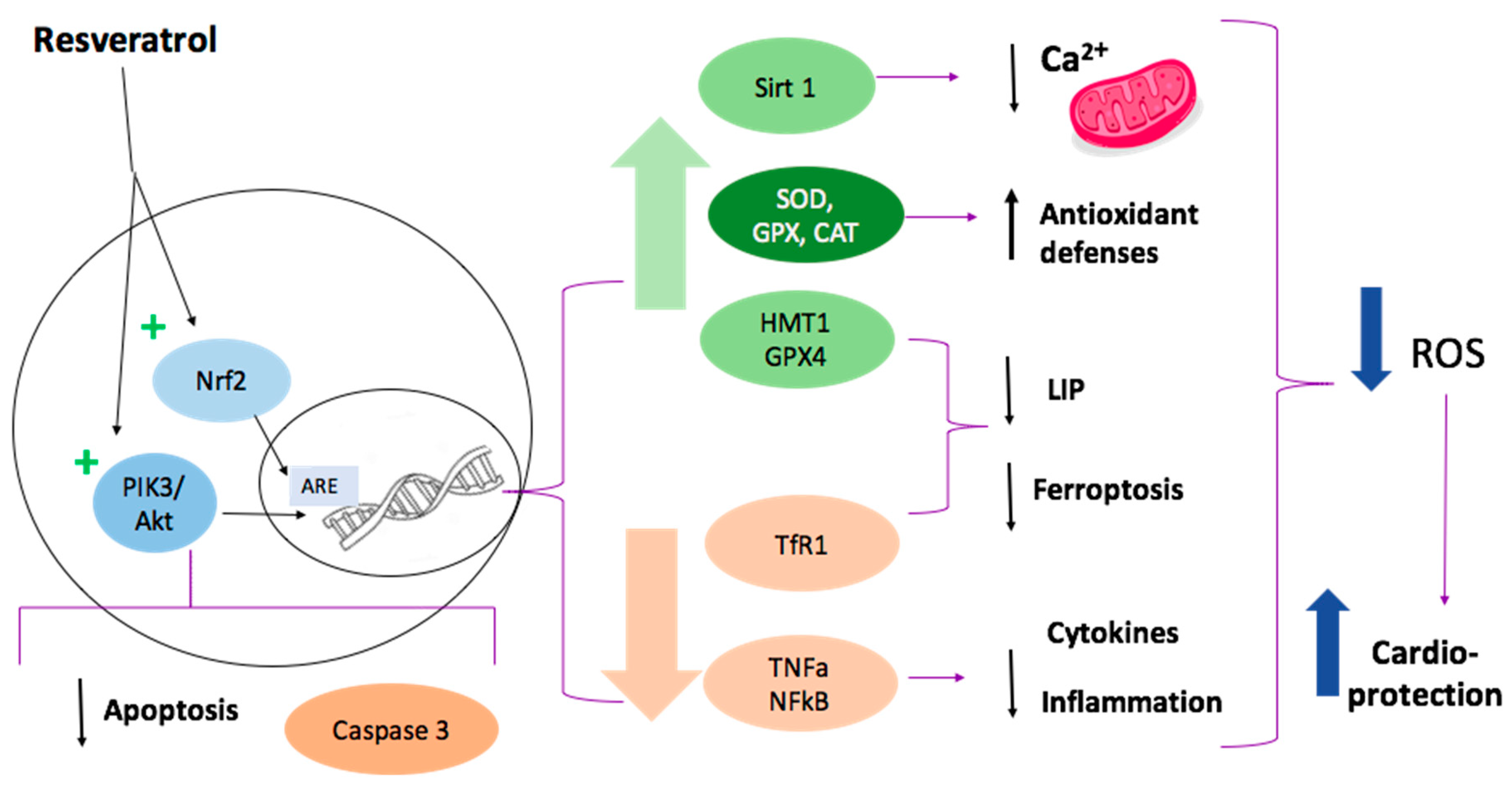
1.9. Resveratrol Cardioprotective Mechanisms
1.9.1. SIRT1 Activation
1.9.2. Ferroptosis Inhibition
1.9.3. Attenuation of Inflammation and Apoptosis
1.9.4. Nrf2 Activation
1.9.5. Reduced Mitochondrial Dysfunction
1.9.6. Other Pathways
1.9.7. Considerations of Safety in the Use of Resveratrol
1.9.8. Associations between Resveratrol and Other Antioxidants
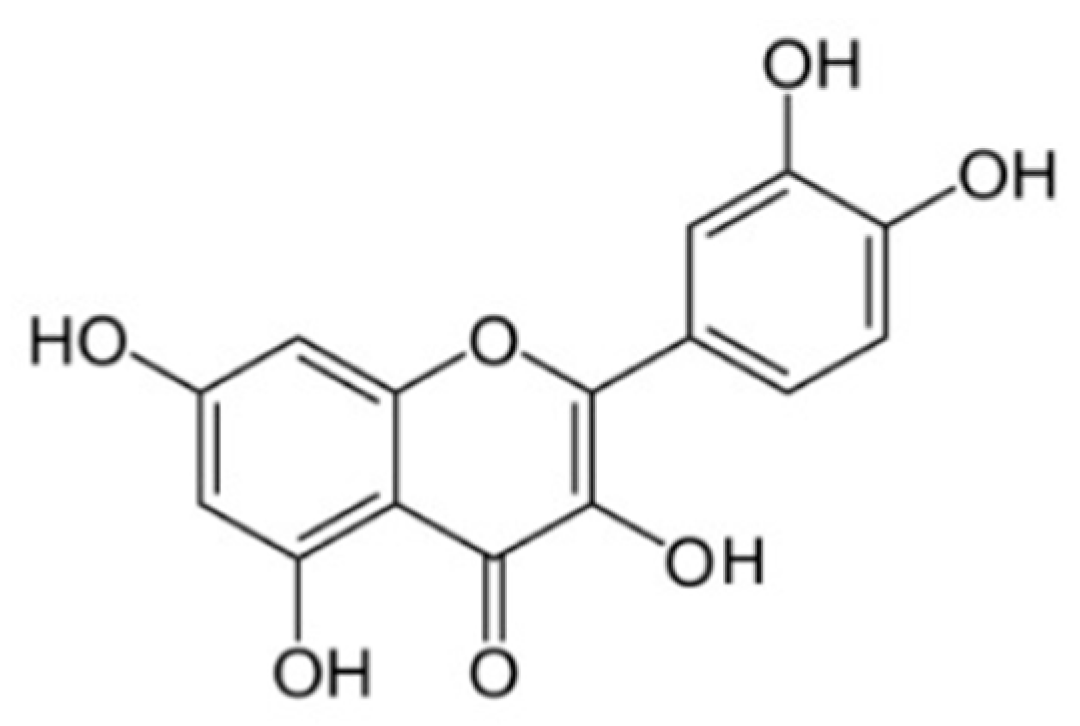
1.10. Quercetin
1.10.1. Properties of Quercetin against Oxidative Stress
1.10.2. Anti-Inflammatory and Anti-Apoptotic Effects
1.10.3. Vasodilatory Effects
1.10.4. Associations between Quercetin and Other Antioxidants
2. Discussion
3. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Cardiovascular Diseases. 2019. Available online: https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_1 (accessed on 10 November 2021).
- Khan, M.A.; Hashim, M.J.; Mustafa, H.; Baniyas, M.Y.; Al Suwaidi, S.K.B.M.; Alkatheeri, R.; Alblooshi, F.M.K.; Almatrooshi, M.E.A.H.; Alzaabi, M.E.H.; Al Darmaki, R.S.; et al. Global Epidemiology of Ischemic Heart Disease: Results from the Global Burden of Disease Study. Cureus 2020, 12, e9349. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef]
- Massalha, S.; Luria, L.; Kerner, A.; Roguin, A.; Abergel, E.; Hammerman, H.; Boulos, M.; Dragu, R.; Kapeliovich, M.R.; Beyar, R.; et al. Heart failure in patients with diabetes undergoing primary percutaneous coronary intervention. Eur. Heart J. Acute Cardiovasc. Care 2016, 5, 455–462. [Google Scholar] [CrossRef]
- Sinning, C.; Westermann, D.; Clemmensen, P. Oxidative stress in ischemia and reperfusion: Current concepts, novel ideas and future perspectives. Biomark Med. 2017, 11, 11031–11040. [Google Scholar] [CrossRef]
- Rodrigo, R.; González-Montero, J.; Sotomayor, C.G. Novel Combined Antioxidant Strategy against Hypertension, Acute Myocardial Infarction and Postoperative Atrial Fibrillation. Biomedicines 2021, 9, 620. [Google Scholar] [CrossRef]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibanez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef]
- Menezes-Rodrigues, F.S.; Errante, P.R.; Araújo, E.A.; Fernandes, M.P.P.; Silva, M.M.D.; Pires-Oliveira, M.; Scorza, C.A.; Scorza, F.A.; Taha, M.O.; Caricati-Neto, A. Cardioprotection stimulated by resveratrol and grape products prevents lethal cardiac arrhythmias in an animal model of ischemia and reperfusion. Acta Cir. Bras. 2021, 36, e360306. [Google Scholar] [CrossRef]
- Huang, J.; Wang, S.; Zhu, M.; Chen, J.; Zhu, X. Effects of genistein, apigenin, quercetin, rutin and astilbin on serum uric acid levels and xanthine oxidase activities in normal and hyperuricemic mice. Food Chem. Toxicol. 2011, 49, 1943–1947. [Google Scholar] [CrossRef]
- Rodrigo, R.; González, J.; Paoletto, F. The role of oxidative stress in the pathophysiology of hypertension. Hypertens. Res. 2011, 34, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef]
- Thompson, R.C.; Allam, A.H.; Lombardi, G.P.; Wann, L.S.; Sutherland, M.L.; Sutherland, J.D.; Soliman, M.A.; Frohlich, B.; Mininberg, D.T.; Monge, J.M.; et al. Atherosclerosis across 4000 years of human history: The Horus study of four ancient populations. Lancet 2013, 381, 1211–1222. [Google Scholar] [CrossRef]
- Sanada, S.; Komuro, I.; Kitakaze, M. Pathophysiology of myocardial reperfusion injury: Preconditioning, postconditioning, and translational aspects of protective measures. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1723–H1741. [Google Scholar] [CrossRef] [Green Version]
- Avkiran, M.; Marber, M.S. Na(+)/H(+) exchange inhibitors for cardioprotective therapy: Progress, problems and prospects. J. Am. Coll. Cardiol. 2002, 39, 747–753. [Google Scholar] [CrossRef]
- Rossi, A.E.; Dirksen, R.T. Sarcoplasmic reticulum: The dynamic calcium governor of muscle. Muscle Nerve 2006, 33, 715–731. [Google Scholar] [CrossRef]
- Sasaki, M.; Joh, T. Oxidative stress and ischemia-reperfusion injury in gastrointestinal tract and antioxidant, protective agents. J. Clin. Biochem. Nutr. 2007, 40, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Raedschelders, K.; Ansley, D.M.; Chen, D.D. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacol. Ther. 2012, 133, 230–255. [Google Scholar] [CrossRef]
- Vásquez-Vivar, J.; Martásek, P.; Whitsett, J.; Joseph, J.; Kalyanaraman, B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: An EPR spin trapping study. Biochem. J. 2002, 362, 733–739. [Google Scholar] [CrossRef]
- Parra, P.; Rodrigo, R. Novel Antioxidant Therapy against Myocardial Ischemia–Reperfusion Injury during Percutaneous Coronary Angioplasty. In Free Radicals and Diseases; Ahmad, R., Ed.; InTech: London, UK, 2016. [Google Scholar]
- Choy, K.W.; Murugan, D.; Mustafa, M.R. Natural products targeting ER stress pathway for the treatment of cardiovascular diseases. Pharmacol. Res. 2018, 132, 119–129. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P.; Vassanelli, S.; Veronese, P.; Colonna, R.; Szabó, I.; Zoratti, M. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J. Biol. Chem. 1992, 267, 2934–2939. [Google Scholar] [CrossRef]
- Griffiths, E.J.; Halestrap, A.P. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem. J. 1995, 307 Pt 1, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. The mitochondrial permeability transition pore: Its fundamental role in mediating cell death during ischaemia and reperfusion. J. Mol. Cell. Cardiol. 2003, 35, 339–341. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Jin, Y.; Lemasters, J.J. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2024–H2034. [Google Scholar] [CrossRef]
- Ong, S.B.; Samangouei, P.; Kalkhoran, S.B.; Hausenloy, D.J. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 2015, 78, 23–34. [Google Scholar] [CrossRef]
- Baines, C.P.; Molkentin, J.D. Adenine nucleotide translocase-1 induces cardiomyocyte death through upregulation of the pro-apoptotic protein Bax. J. Mol. Cell. Cardiol. 2009, 46, 969–977. [Google Scholar] [CrossRef] [Green Version]
- Padmavathi, G.; Ramkumar, K.M. MicroRNA mediated regulation of the major redox homeostasis switch, Nrf2, and its impact on oxidative stress-induced ischemic/reperfusion injury. Arch. Biochem. Biophys. 2021, 698, 108725. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, Y.Y.; Lan, J.N.; Liu, H.M.; Li, W.; Wu, Y.; Leng, Y.; Tang, L.H.; Hou, J.B.; Sun, Q.; et al. Ischemic Postconditioning Alleviates Intestinal Ischemia-Reperfusion Injury by Enhancing Autophagy and Suppressing Oxidative Stress through the Akt/GSK-3β/Nrf2 Pathway in Mice. Oxid. Med. Cell Longev. 2020, 2020, 6954764. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Cabrera-Fuentes, H.A.; Ruiz-Meana, M.; Simsekyilmaz, S.; Kostin, S.; Inserte, J.; Saffarzadeh, M.; Galuska, S.P.; Vijayan, V.; Barba, I.; Barreto, G.; et al. RNase1 prevents the damaging interplay between extracellular RNA and tumour necrosis factor-α in cardiac ischaemia/reperfusion injury. Thromb. Haemost. 2014, 112, 1110–1119. [Google Scholar] [CrossRef]
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-kappaB activation by reactive oxygen species: Fifteen years later. Biochem Pharmacol. 2006, 72, 1493–1505. [Google Scholar] [CrossRef]
- Djavaheri-Mergny, M.; Amelotti, M.; Mathieu, J.; Besançon, F.; Bauvy, C.; Souquère, S.; Pierron, G.; Codogno, P. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J. Biol. Chem. 2006, 281, 30373–30382. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, R.; Prieto, J.C.; Castillo, R. Cardioprotection against ischaemia/reperfusion by vitamins C and E plus n-3 fatty acids: Molecular mechanisms and potential clinical applications. Clin. Sci. 2013, 124, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Vinten-Johansen, J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc. Res. 2004, 61, 481–497. [Google Scholar] [CrossRef] [Green Version]
- Mann, D.L. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef] [Green Version]
- Nian, M.; Lee, P.; Khaper, N.; Liu, P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ. Res. 2004, 94, 1543–1553. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Regulation of the inflammatory response in cardiac repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Lillo-Moya, J.; Rojas-Solé, C.; Muñoz-Salamanca, D.; Panieri, E.; Saso, L.; Rodrigo, R. Targeting Ferroptosis against Ischemia/Reperfusion Cardiac Injury. Antioxidants 2021, 10, 667. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106 (Suppl. 6), 1559S–1566S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eefting, F.; Rensing, B.; Wigman, J.; Pannekoek, W.J.; Liu, W.M.; Cramer, M.J.; Lips, D.J.; Doevendans, P.A. Role of apoptosis in reperfusion injury. Cardiovasc. Res. 2004, 61, 414–426. [Google Scholar] [CrossRef]
- Borutaite VJekabsone AMorkuniene RBrown, G.C. Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction, cytochrome c release and apoptosis induced by heart ischemia. J. Mol. Cell Cardiol. 2003, 35, 357–366. [Google Scholar]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, R.; Libuy, M.; Feliú, F.; Hasson, D. Molecular basis of cardioprotective effect of antioxidant vitamins in myocardial infarction. Biomed. Res. Int. 2013, 2013, 437613. [Google Scholar] [CrossRef] [Green Version]
- Gasparetto, C.; Malinverno, A.; Culacciati, D.; Gritti, D.; Prosperini, P.G.; Specchia, G.; Ricevuti, G. Antioxidant vitamins reduce oxidative stress and ventricular remodeling in patients with acute myocardial infarction. Int. J. Immunopathol. Pharmacol. 2005, 18, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xiao, Z.; Yao, J.; Zhao, G.; Fa, X.; Niu, J. Participation of protein kinase C in the activation of Nrf2 signaling by ischemic preconditioning in the isolated rabbit heart. Mol. Cell. Biochem. 2013, 372, 169–179. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, X.; Shi, J.; Wu, X. Involvement of Nrf2 in myocardial ischemia and reperfusion injury. Int. J. Biol. Macromol. 2019, 125, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Jin, Z.; Zhao, R.; Ren, K.; Deng, C.; Yu, S. Resveratrol attenuates inflammation and oxidative stress induced by myocardial ischemia-reperfusion injury: Role of Nrf2/ARE pathway. Int. J. Clin. Exp. Med. 2015, 8, 10420–10428. [Google Scholar] [PubMed]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: An insight into cullin-ring ubiquitin ligases. Antioxid. Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; León, R. Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Aspects Med. 2011, 32, 234–246. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, L.; Lu, S. Evaluation of antioxidant and immunity activities of quercetin in isoproterenol-treated rats. Molecules 2012, 17, 4281–4291. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Yang, Y.; Mei, J. Protective Effects of L-Malate against Myocardial Ischemia/Reperfusion Injury in Rats. Evid. Based Complement. Altern. Med. 2016, 2016, 3803657. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, L.; Wang, Y.; Zhu, X.; Jiang, M.; Song, E.; Song, Y. New application of the commercial sweetener rebaudioside a as a hepatoprotective candidate: Induction of the Nrf2 signaling pathway. Eur. J. Pharmacol. 2018, 822, 128–137. [Google Scholar] [CrossRef]
- Jain, A.K.; Jaiswal, A.K. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, W.; Lv, C.; Wang, Y.; Ma, B.; Zhang, H.; Fan, Z.; Li, M.; Li, X. A novel biscoumarin compound ameliorates cerebral ischemia reperfusion-induced mitochondrial oxidative injury via Nrf2/Keap1/ARE signaling. Neuropharmacology 2020, 167, 107918. [Google Scholar] [CrossRef] [PubMed]
- López-Bernardo, E.; Anedda, A.; Sánchez-Pérez, P.; Acosta-Iborra, B.; Cadenas, S. 4-Hydroxynonenal induces Nrf2-mediated UCP3 upregulation in mouse cardiomyocytes. Free Radic. Biol. Med. 2015, 88, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Xu, Y.; Liu, Z.Y.; Meng, W.X.; Yang, H. Autophagy assuages myocardial infarction through Nrf2 signaling activation-mediated reactive oxygen species clear. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7381–7390. [Google Scholar] [CrossRef] [PubMed]
- Cebova, M.; Pechanova, O. Protective Effects of Polyphenols against Ischemia/Reperfusion Injury. Molecules 2020, 25, 3469. [Google Scholar] [CrossRef]
- Hao, J.; Li, W.W.; Du, H.; Zhao, Z.F.; Liu, F.; Lu, J.C.; Yang, X.C.; Cui, W. Role of Vitamin C in Cardioprotection of Ischemia/Reperfusion Injury by Activation of Mitochondrial KATP Channel. Chem. Pharm. Bull. 2016, 64, 548–557. [Google Scholar] [CrossRef] [Green Version]
- Pucheu, S.; Coudray, C.; Tresallet, N.; Favier, A.; De Leiris, J. Effect of iron overload in the isolated ischemic and reperfused rat heart. Cardiovasc. Drugs Ther. 1993, 7, 701–711. [Google Scholar] [CrossRef]
- Chan, W.; Taylor, A.J.; Ellims, A.H.; Lefkovits, L.; Wong, C.; Kingwell, B.A.; Natoli, A.; Croft, K.D.; Mori, T.; Kaye, D.M.; et al. Effect of Iron Chelation on Myocardial Infarct Size and Oxidative Stress in ST-Elevation–Myocardial Infarction. Circ. Cardiovasc. Interv. 2012, 5, 270–278. [Google Scholar] [CrossRef] [Green Version]
- Liao, C.L.; Liu, Y.; Huang, M.Z.; Liu, H.Y.; Ye, Z.L.; Su, Q. Myocardial ischemia reperfusion injury is alleviated by curcumin-peptide hydrogel via upregulating autophagy and protecting mitochondrial function. Stem Cell Res. Ther. 2021, 12, 89. [Google Scholar] [CrossRef]
- Duan, W.; Yang, Y.; Yan, J.; Yu, S.; Liu, J.; Zhou, J.; Zhang, J.; Jin, Z.; Yi, D. The effects of curcumin post-treatment against myocardial ischemia and reperfusion by activation of the JAK2/STAT3 signaling pathway. Basic Res. Cardiol. 2012, 107, 263. [Google Scholar] [CrossRef]
- Jiang, L.; Yin, X.; Chen, Y.H.; Chen, Y.; Jiang, W.; Zheng, H.; Huang, F.Q.; Liu, B.; Zhou, W.; Qi, L.W.; et al. Proteomic analysis reveals ginsenoside Rb1 attenuates myocardial ischemia/reperfusion injury through inhibiting ROS production from mitochondrial complex I. Theranostics 2021, 11, 1703–1720. [Google Scholar] [CrossRef]
- Li, C.; Yang, P.; Jiang, Y.; Lin, Z.; Pu, Y.; Xie, L.; Sun, L.; Lu, D. Ginsenoside Rb1 attenuates cardiomyocyte apoptosis induced by myocardial ischemia reperfusion injury through mTOR signal pathway. Biomed. Pharmacother. 2020, 125, 109913. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bao, X.Y.; Zhu, P.C.; Tong, Q.; Zheng, G.Q.; Wang, Y. Ginsenoside Rb1 for Myocardial Ischemia/Reperfusion Injury: Preclinical Evidence and Possible Mechanisms. Oxid. Med. Cell Longev. 2017, 2017, 6313625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugam, K.; Boovarahan, S.R.; Prem, P.; Sivakumar, B.; Kurian, G.A. Fisetin Attenuates Myocardial Ischemia-Reperfusion Injury by Activating the Reperfusion Injury Salvage Kinase (RISK) Signaling Pathway. Front. Pharmacol. 2021, 12, 566470. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Wu, B.; Zhong, B.; Lin, L.; Ding, Y.; Jin, X.; Huang, Z.; Lin, M.; Wu, H.; Xu, D. Naringenin alleviates myocardial ischemia/reperfusion injury by regulating the nuclear factor-erythroid factor 2-related factor 2 (Nrf2) /System xc-/ glutathione peroxidase 4 (GPX4) axis to inhibit ferroptosis. Bioengineered 2021, 12, 10924–10934. [Google Scholar] [CrossRef] [PubMed]
- Ucar, B.I.; Ucar, G.; Saha, S.; Buttari, B.; Profumo, E.; Saso, L. Pharmacological Protection against Ischemia-Reperfusion Injury by Regulating the Nrf2-Keap1-ARE Signaling Pathway. Antioxidants 2021, 10, 823. [Google Scholar] [CrossRef]
- Silva-Palacios, A.; Ostolga-Chavarría, M.; Sánchez-Garibay, C.; Rojas-Morales, P.; Galván-Arzate, S.; Buelna-Chontal, M.; Pavón, N.; Pedraza-Chaverrí, J.; Königsberg, M.; Zazueta, C. Sulforaphane protects from myocardial ischemia-reperfusion damage through the balanced activation of Nrf2/AhR. Free Radic. Biol. Med. 2019, 143, 331–340. [Google Scholar] [CrossRef]
- Bellows, S.D.; Hale, S.L.; Simkhovich, B.Z.; Kay, G.L.; Kloner, R.A. Do antioxidant vitamins reduce infarct size following acute myocardial ischemia/reperfusion? Cardiovasc. Drugs Ther. 1995, 9, 117–123. [Google Scholar] [CrossRef]
- Dodd, S.; Dean, O.; Copolov, D.L.; Malhi, G.S.; Berk, M. N-acetylcysteine for antioxidant therapy: Pharmacology and clinical utility. Expert Opin. Biol. Ther. 2008, 8, 1955–1962. [Google Scholar] [CrossRef]
- Orhan, G.; Yapici, N.; Yuksel, M.; Sargin, M.; Şenay, Ş.; Yalçin, A.; Aykaç, Z.; Aka, S. Effects of N-acetylcysteine on myocardial ischemia-reperfusion injury in bypass surgery. Heart Vessel. 2006, 21, 42–47. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, L.; Shi, X.; Yang, L.; Hua, F.; Ma, J.; Zhu, W.; Liu, X.; Xuan, R.; Shen, Y.; et al. Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway. Aging 2020, 12, 24270–24287. [Google Scholar] [CrossRef]
- Calvert, J.W.; Gundewar, S.; Jha, S.; Greer, J.; Bestermann, W.; Tian, R.; Lefer, D. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes 2008, 57, 696–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Hou, S.A. Protective effects of metformin against myocardial ischemia reperfusion injury via AMPK dependent suppression of NOX4. Mol. Med. Rep. 2021, 24, 712. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.W.; Yang, F.; Cheng, S.Z.; Liu, Y.; Wan, L.H.; Cong, H.L. Rosuvastatin postconditioning protects isolated hearts against ischemia-reperfusion injury: The role of radical oxygen species, PI3K-Akt-GSK-3β pathway, and mitochondrial permeability transition pore. Cardiovasc. Ther. 2017, 35, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.F.; Wang, H.; Song, N.; Jiang, Y.H.; Zhang, J.; Meng, X.W.; Feng, X.M.; Liu, H.; Peng, K.; Ji, F.H. Dexmedetomidine Attenuates Ischemia/Reperfusion-Induced Myocardial Inflammation and Apoptosis Through Inhibiting Endoplasmic Reticulum Stress Signaling. J. Inflamm. Res. 2021, 14, 1217–1233. [Google Scholar] [CrossRef] [PubMed]
- Ibacache, M.; Sanchez, G.; Pedrozo, Z.; Galvez, F.; Humeres, C.; Echevarria, G.; Duaso, J.; Hassi, M.; Garcia, L.; Diaz-Araya, G.; et al. Dexmedetomidine preconditioning activates pro-survival kinases and attenuates regional ischemia/ reperfusion injury in rat heart. Biochim. Biophys. Acta 2012, 1822, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Riquelme, J.A.; Westermeier, F.; Hall, A.R.; Vicencio, J.M.; Pedrozo, Z.; Ibacache, M.; Fuenzalida, B.; Sobrevia, L.; Davidson, S.M.; Yellon, D.M.; et al. Dexmedetomidine protects the heart against ischemia- reperfusion injury by an endothelial eNOS/NO dependent mechanism. Pharmacol. Res. 2016, 103, 318–327. [Google Scholar] [CrossRef] [Green Version]
- Lochner, A.; Marais, E.; Huisamen, B. Melatonin and cardioprotection against ischaemia/reperfusion injury: What’s new? A review. J. Pineal Res. 2018, 65, e12490. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Tan, Y.; Ouyang, S.; He, J.; Liu, L. Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis. Gene 2021, 808, 145968. [Google Scholar] [CrossRef]
- Shaito, A.; Posadino, A.M.; Younes, N.; Hasan, H.; Halabi, S.; Alhababi, D.; Al-Mohannadi, A.; Abdel-Rahman, W.M.; Eid, A.H.; Nasrallah, G.K.; et al. Potential Adverse Effects of Resveratrol: A Literature Review. Int. J. Mol. Sci. 2020, 21, 2084. [Google Scholar] [CrossRef] [Green Version]
- Pastor, R.F.; Restani, P.; Di Lorenzo, C.; Orgiu, F.; Teissedre, P.L.; Stockley, C.; Ruf, J.C.; Quini, C.I.; Garcìa Tejedor, N.; Gargantini, R.; et al. Resveratrol, human health and winemaking perspectives. Crit. Rev. Food Sci. Nutr. 2019, 59, 1237–1255. [Google Scholar] [CrossRef]
- He, Y.; Fu, Y.; Xi, M.; Zheng, H.; Zhang, Y.; Liu, Y.; Zhao, Y.; Xi, J.; He, Y. Zn2+ and mPTP mediate resveratrol-induced myocardial protection from endoplasmic reticulum stress. Metallomics 2020, 12, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zou, L.X.; Lin, Q.Y.; Yan, X.; Bi, H.L.; Xie, X.; Wang, S.; Wang, Q.S.; Zhang, Y.L.; Li, H.H. Resveratrol as a new inhibitor of immunoproteasome prevents PTEN degradation and attenuates cardiac hypertrophy after pressure overload. Redox Biol. 2019, 20, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, M.; Matta, M.J.; Sunderesan, N.R.; Gupta, M.P.; Periasamy, M.; Gupta, M. Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H833–H843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, M.; Wang, D.; He, S.; Xu, R.; Xie, Y. SIRT1 confers protection against ischemia/reperfusion injury in cardiomyocytes via regulation of uncoupling protein 2 expression. Mol. Med. Rep. 2017, 16, 7098–7104. [Google Scholar] [CrossRef]
- Yu, D.; Xiong, J.; Gao, Y.; Li, J.; Zhu, D.; Shen, X.; Sun, L.; Wang, X. Resveratrol activates PI3K/AKT to reduce myocardial cell apoptosis and mitochondrial oxidative damage caused by myocardial ischemia/reperfusion injury. Acta Histochem. 2021, 123, 151739. [Google Scholar] [CrossRef]
- Pompura, S.L.; Dominguez-Villar, M. The PI3K/AKT signaling pathway in regulatory T-cell development, stability, and function. J. Leukoc. Biol. 2018, 103, 1065–1076. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, L.F.; Hua, L.; Feng, H.K.; Shen, B. Resveratrol protects myocardial apoptosis induced by ischemia-reperfusion in rats with acute myocardial infarction via blocking P13K/Akt/e-NOS pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1789–1796. [Google Scholar] [CrossRef]
- Song, H.P.; Chu, Z.G.; Zhang, D.X.; Dang, Y.M.; Zhang, Q. PI3K-AKT pathway protects cardiomyocytes against hypoxia-induced apoptosis by MitoKATP-Mediated mitochondrial translocation of pAKT. Cell. Physiol. Biochem. 2018, 49, 717–727. [Google Scholar] [CrossRef]
- Ge, L.; Li, C.; Wang, Z.; Zhang, Y.; Chen, L. Suppression of Oxidative Stress and Apoptosis in Electrically Stimulated Neonatal Rat Cardiomyocytes by Resveratrol and Underlying Mechanisms. J. Cardiovasc. Pharmacol. 2017, 70, 396–404. [Google Scholar] [CrossRef]
- Hu, Y.; Pan, H.; Peng, J.; He, J.; Tang, M.; Yan, S.; Rong, J.; Li, J.; Zheng, Z.; Wang, H.; et al. Resveratrol inhibits necroptosis by mediating the TNF-α/RIP1/RIP3/MLKL pathway in myocardial hypoxia/reoxygenation injury. Acta Biochim. Biophys. Sin. 2021, 53, 430–437. [Google Scholar] [CrossRef]
- Xing, Z.; He, Q.; Xiong, Y.; Zeng, X. Systematic Pharmacology Reveals the Antioxidative Stress and Anti-Inflammatory Mechanisms of Resveratrol Intervention in Myocardial Ischemia-Reperfusion Injury. Evid. Based Complement. Altern. Med. 2021, 2021, 5515396. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Liu, Z.; Zeng, R.; Shao, J.; Zheng, Y.; Ye, W. Resveratrol Alleviates Vascular Endothelial Damage Caused by Lower-Extremity Ischemia Reperfusion (I/R) through Regulating Keap1/Nrf2 Signaling-Mediated Oxidative Stress. Evid. Based Complement. Altern. Med. 2021, 2021, 5556603. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Zhu, M.; Zhang, Q.; Wang, X.; Wang, Y.; Zhang, J.; Li, J.; Yang, L.; Liu, J.; et al. Resveratrol attenuates myocardial ischemia/reperfusion injury through up-regulation of vascular endothelial growth factor B. Free Radic. Biol. Med. 2016, 101, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Wang, A.B.; Huang, Y.; Liu, D.P.; Wei, C.; Williams, G.M.; Zhang, C.N.; Liu, G.; Liu, Y.Q.; Hao, D.L.; et al. Isorhapontigenin, a new resveratrol analog, attenuates cardiac hypertrophy via blocking signaling transduction pathways. Free Radic. Biol. Med. 2005, 38, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, E.J.; Mattson, M.P.; Calabrese, V. Resveratrol commonly displays hormesis: Occurrence and biomedical significance. Hum. Exp. Toxicol. 2010, 29, 980–1015. [Google Scholar] [CrossRef]
- Amri, A.; Chaumeil, J.C.; Sfar, S.; Charrueau, C. Administration of resveratrol: What formulation solutions to bioavailability limitations? J. Control. Release 2012, 158, 182–193. [Google Scholar] [CrossRef]
- Peng, W.; Xu, S.; Zhang, J.; Zhang, Y. Vitamin C Attenuates Sodium Fluoride-Induced Mitochondrial Oxidative Stress and Apoptosis via Sirt1-SOD2 Pathway in F9 Cells. Biol. Trace Elem. Res. 2019, 191, 189–198. [Google Scholar] [CrossRef]
- Abdullah, M.; Jamil, R.T.; Attia, F.N. Vitamin C (Ascorbic Acid); StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Andres, S.; Pevny, S.; Ziegenhagen, R.; Bakhiya, N.; Schäfer, B.; Hirsch-Ernst, K.I.; Lampen, A. Safety Aspects of the Use of Quercetin as a Dietary Supplement. Mol. Nutr. Food Res. 2018, 62, 1700447. [Google Scholar] [CrossRef]
- Tawani, A.; Mishra, S.K.; Kumar, A. Structural insight for the recognition of G-quadruplex structure at human c-myc promoter sequence by flavonoid Quercetin. Sci. Rep. 2017, 7, 3600. [Google Scholar] [CrossRef] [Green Version]
- Suganthy, N.; Devi, K.P.; Nabavi, S.F.; Braidy, N.; Nabavi, S.M. Bioactive effects of quercetin in the central nervous system: Focusing on the mechanisms of actions. Biomed. Pharmacother. 2016, 84, 892–908. [Google Scholar] [CrossRef]
- Xu, D.; Hu, M.J.; Wang, Y.Q.; Cui, Y.L. Antioxidant Activities of Quercetin and Its Complexes for Medicinal Application. Molecules 2019, 24, 1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Dong, M.; Xu, G.; Tian, Y.; Tang, H.; Wang, Y. Metabolomics Reveals that Dietary Ferulic Acid and Quercetin Modulate Metabolic Homeostasis in Rats. J. Agric. Food Chem. 2018, 66, 1723–1731. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhang, H.; Wu, J.; Zhai, G.; Li, Z.; Luan, Y.; Garg, S. CuS@MOF-Based Well-Designed Quercetin Delivery System for Chemo-Photothermal Therapy. ACS Appl. Mater. Interfaces 2018, 10, 34513–34523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Zhang, Z.Y.; Wang, R.X. Protective Mechanisms of Quercetin Against Myocardial Ischemia Reperfusion Injury. Front. Physiol. 2020, 11, 956. [Google Scholar] [CrossRef]
- Wilson, A.J.; Gill, E.K.; Abudalo, R.A.; Edgar, K.S.; Watson, C.J.; Grieve, D.J. Reactive oxygen species signalling in the diabetic heart: Emerging prospect for therapeutic targeting. Heart 2018, 104, 293–299. [Google Scholar] [CrossRef]
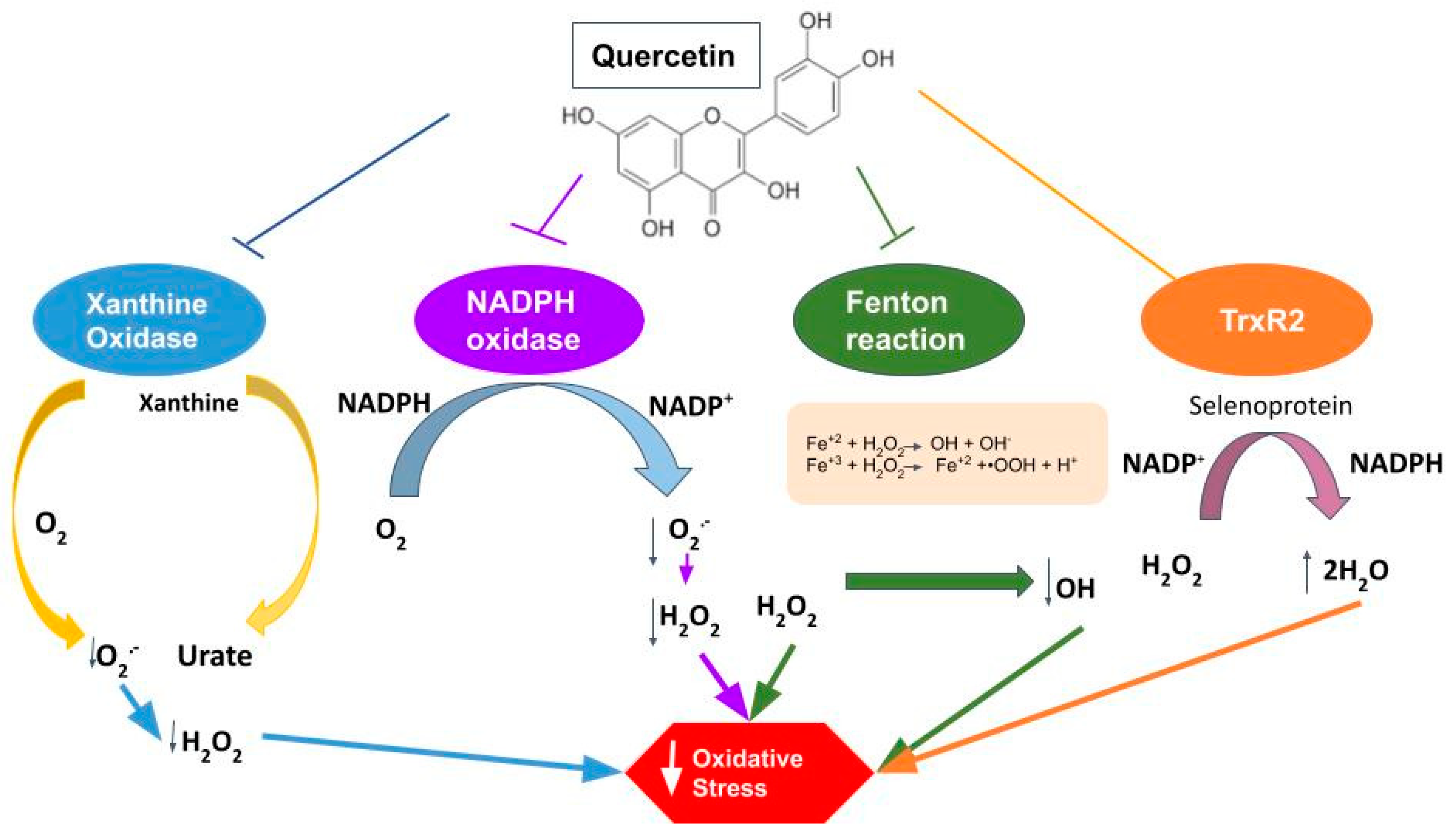
- Malik, N.; Dhiman, P.; Sobarzo-Sanchez, E.; Khatkar, A. Flavonoids and Anthranquinones as Xanthine Oxidase and Monoamine Oxidase Inhibitors: A New Approach Towards Inflammation and Oxidative Stress. Curr. Top. Med. Chem. 2018, 18, 2154–2164. [Google Scholar] [CrossRef]
- Wan, L.L.; Xia, J.; Ye, D.; Liu, J.; Chen, J.; Wang, G. Effects of quercetin on gene and protein expression of NOX and NOS after myocardial ischemia and reperfusion in rabbit. Cardiovasc. Ther. 2009, 27, 28–33. [Google Scholar] [CrossRef]
- Arango, J.C.; Gámez, L.Y.; López, J.A. Sistema NADPH oxidasa: Nuevos retos y perspectivas. Iatreia 2010, 23, 362–372. [Google Scholar]
- Breitenbach, M.; Rinnerthaler, M.; Weber, M.; Breitenbach-Koller, H.; Karl, T.; Cullen, P.; Basu, S.; Haskova, D.; Hasek, J. The defense and signaling role of NADPH oxidases in eukaryotic cells. Wien. Med. Wochenschr. 2018, 168, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Guichard, C.; Pedruzzi, E.; Fay, M.; Ben Mkaddem, S.; Coant, N.; Daniel, F.; Ogier-Denis, E. The Nox/Duox family of ROS-generating NADPH oxidases. Med. Sci. 2006, 22, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Anilkumar, N.; Sirker, A.; Shah, A.M. Redox sensitive signaling pathways in cardiac remodeling, hypertrophy and failure. Front. Biosci. 2009, 14, 3168–3187. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Lawson, M.; Drostinova, L.; Lauro, P.; Poprac, P.; Brezova, V.; Michalik, M.; Lukeš, V.; Valko, M. Protective role of quercetin against copper(II)-induced oxidative stress: A spectroscopic, theoretical and DNA damage study. Food Chem. Toxicol. 2017, 110, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Babenkova, I.V.; Osipov, A.N.; Teselkin, Y.O. The Effect of Dihydroquercetin on Catalytic Activity of Iron (II) Ions in the Fenton Reaction. Bull. Exp. Biol. Med. 2018, 165, 347–350. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Speckmann, B.; Klotz, L.O. Selenoproteins: Antioxidant selenoenzymes and beyond. Arch. Biochem. Biophys. 2016, 595, 113–119. [Google Scholar] [CrossRef]
- Benito, S.; Lopez, D.; Sáiz, M.P.; Buxaderas, S.; Sánchez, J.; Puig-Parellada, P.; Mitjavila, M.T. A flavonoid-rich diet increases nitric oxide production in rat aorta. Br. J. Pharmacol. 2002, 135, 910–916. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, A.; Middya, T.R.; Jana, A.D. A QSAR study of radical scavenging antioxidant activity of a series of flavonoids using DFT based quantum chemical descriptors--the importance of group frontier electron density. J. Mol. Model. 2012, 18, 2621–2631. [Google Scholar] [CrossRef]
- Tang, S.T.; Wang, F.; Shao, M.; Wang, Y.; Zhu, H.Q. MicroRNA-126 suppresses inflammation in endothelial cells under hyperglycemic condition by targeting HMGB1. Vascul. Pharmacol. 2017, 88, 48–55. [Google Scholar] [CrossRef]
- Rohrbach, S.; Troidl, C.; Hamm, C.; Schulz, R. Ischemia and reperfusion related myocardial inflammation: A network of cells and mediators targeting the cardiomyocyte. IUBMB Life 2015, 67, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Akhlaghi, M.; Bandy, B. Mechanisms of flavonoid protection against myocardial ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2009, 46, 309–317. [Google Scholar] [CrossRef]
- Li, J.; Zhang, H.; Zhang, C. Role of inflammation in the regulation of coronary blood flow in ischemia and reperfusion: Mechanisms and therapeutic implications. J. Mol. Cell. Cardiol. 2012, 52, 865–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartekova, M.; Šimončíková, P.; Fogarassyová, M.; Ivanová, M.; Okruhlicová, L.; Tribulova, N.; Dovinová, I.; Barancik, M. Quercetin improves postischemic recovery of heart function in doxorubicin-treated rats and prevents doxorubicin-induced matrix metalloproteinase-2 activation and apoptosis induction. Int. J. Mol. Sci. 2015, 16, 8168–8185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skovsted, G.F.; Kruse, L.S.; Larsen, R.; Pedersen, A.F.; Trautner, S.; Sheykhzade, M.; Edvinsson, L. Heart ischaemia-reperfusion induces local up-regulation of vasoconstrictor endothelin ETB receptors in rat coronary arteries downstream of occlusion. Br. J. Pharmacol. 2014, 171, 2726–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaoukis, A.; Deftereos, S.; Raisakis, K.; Giannopoulos, G.; Bouras, G.; Panagopoulou, V.; Papoutsidakis, N.; Cleman, M.W.; Stefanadis, C. The role of endothelin system in cardiovascular disease and the potential therapeutic perspectives of its inhibition. Curr. Top. Med. Chem. 2013, 13, 95–114. [Google Scholar] [CrossRef]
- Punithavathi, V.R.; Stanely Mainzen Prince, P. The cardioprotective effects of a combination of quercetin and α-tocopherol on isoproterenol-induced myocardial infarcted rats. J. Biochem. Mol. Toxicol. 2011, 25, 28–40. [Google Scholar] [CrossRef]
- Chen, L.; Wu, X.; Wang, W.; Wang, X.; Ma, J. Quercetin with lycopene modulates enzymic antioxidant genes pathway in isoproterenol cardiotoxicity in rats. Libyan J. Med. 2021, 16, 1943924. [Google Scholar] [CrossRef]
- Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Jang, I.A.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Resveratrol, an Nrf2 activator, ameliorates aging-related progressive renal injury. Aging 2018, 10, 83–99. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Tian, R.; Yang, Z.; Peng, Y.Y.; Lu, N. Quercetin suppressed NADPH oxidase-derived oxidative stress via heme oxygenase-1 induction in macrophages. Arch. Biochem. Biophys. 2019, 671, 69–76. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, R.; Zhang, G.; Gong, D. Mechanistic insights into the inhibition of quercetin on xanthine oxidase. Int. J. Biol. Macromol. 2018, 112, 405–412. [Google Scholar] [CrossRef]
- Lucas-Abellán, C.; Fortea, I.; López-Nicolás, J.M.; Nunez-Delicado, E. Cyclodextrins as resveratrol carrier system. Food Chem. 2007, 104, 39–44. [Google Scholar] [CrossRef]
- Das, S.; Lin, H.S.; Ho, P.C.; Ng, K.Y. The impact of aqueous solubility and dose on the pharmacokinetic profiles of resveratrol. Pharm. Res. 2008, 25, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Atanacković, M.; Posa, M.; Heinle, H.; Gojković-Bukarica, L.; Cvejić, J. Solubilization of resveratrol in micellar solutions of different bile acids. Colloids Surf B Biointerfaces 2009, 72, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Hesari, M.; Mohammadi, P.; Khademi, F.; Shackebaei, D.; Momtaz, S.; Moasefi, N.; Farzaei, M.H.; Abdollahi, M. Current Advances in the Use of Nanophytomedicine Therapies for Human Cardiovascular Diseases. Int. J. Nanomed. 2021, 16, 3293–3315. [Google Scholar] [CrossRef] [PubMed]
- Cunico, L.P.; Cobo, A.M.; Al-Hamimi, S.; Turner, C. Solubility and Thermal Degradation of Quercetin in CO2-Expanded Liquids. Molecules 2020, 25, 5582. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Kavuru, P.; Wojtas, L.; Zaworotko, M.J.; Shytle, R.D. Cocrystals of quercetin with improved solubility and oral bioavailability. Mol. Pharm. 2011, 8, 1867–1876. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Cellular Mechanisms | Main Effects | References |
|---|---|---|---|
| Vitamin C | Reduction of ROS burst Down-regulation of NOX activity and prevention of eNOS uncoupling Reduction of intracellular Ca2+ overload Prevention of mitochondrial membrane potential depolarization | Antioxidant | [36,67] |
| Deferoxamine | Iron Chelation | Antioxidant | [68,69] |
| Curcumin | Activation of JAK/STAT3 signaling pathway | Anti-inflammatory Anti-apoptotic Antioxidant | [70,71] |
| Ginsenoside Rb1 | Inhibition of ROS production in mitochondrial complex l (NADH dehydrogenase) Possible activation of Anti-apoptotic mTOR pathway Increasing GSH and NO expression | Antioxidant Anti-inflammatory Anti-apoptotic | [72,73,74] |
| Fisetin | Activation of PI3K/Akt/GSK-3β pathway Reversion of mitochondrial dysfunction | Antioxidant | [75] |
| Naringenin | Regulation of Nrf2 pathway and inhibition of ferroptosis | Ferroptosis Inhibitor | [76] |
| Sulforaphane | Regulation of Nrf2 pathway | Antioxidant | [77,78] |
| Molecule | Cellular Mechanisms | Main Effects | References |
|---|---|---|---|
| N-Acetylcysteine | GSH donation Increase in microvascular blood flow ROS Scavenger | Antioxidant Anti-inflammatory | [79,80,81] |
| Metformin | AMPK-dependent NOX4 suppression and eNOS activation Suppression of NLRP3 inflammasome activation | Antioxidant Anti-inflammatory Anti-apoptotic | [82,83,84] |
| Rosuvastatin | Activation of Akt and GSK-3β Enhancement of superoxide dismutase activity Inhibition of mPTP opening Up-regulation of PPAR-γ and UCP2 | Antioxidant Anti-inflammatory | [85] |
| Dexmedetomidine | Down-regulation of ERS signaling pathways (PERK, CHOP, IREI) Increase of NO production via PI3K signaling | Anti-inflammatory Anti-apoptotic | [86,87,88] |
| Melatonin | Free radical scavenging Activation of Nrf2 pathway Reduction of NFκB binding to DNA Inhibition of iNOS and cyclooxygenase expressions | Antioxidant Anti-inflammatory | [89] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigo, R.; Retamal, C.; Schupper, D.; Vergara-Hernández, D.; Saha, S.; Profumo, E.; Buttari, B.; Saso, L. Antioxidant Cardioprotection against Reperfusion Injury: Potential Therapeutic Roles of Resveratrol and Quercetin. Molecules 2022, 27, 2564. https://doi.org/10.3390/molecules27082564
Rodrigo R, Retamal C, Schupper D, Vergara-Hernández D, Saha S, Profumo E, Buttari B, Saso L. Antioxidant Cardioprotection against Reperfusion Injury: Potential Therapeutic Roles of Resveratrol and Quercetin. Molecules. 2022; 27(8):2564. https://doi.org/10.3390/molecules27082564
Chicago/Turabian StyleRodrigo, Ramón, Catalina Retamal, Denisse Schupper, Diego Vergara-Hernández, Sarmistha Saha, Elisabetta Profumo, Brigitta Buttari, and Luciano Saso. 2022. "Antioxidant Cardioprotection against Reperfusion Injury: Potential Therapeutic Roles of Resveratrol and Quercetin" Molecules 27, no. 8: 2564. https://doi.org/10.3390/molecules27082564