
Recent Advances in the Oxone-Mediated Synthesis of Heterocyclic Compounds
by
 and
and
Helen A. Goulart
, 
Daniela R. Araujo
,
Filipe Penteado
, 
Raquel G. Jacob
,
Gelson Perin
* and
Eder J. Lenardão
*
Laboratório de Síntese Orgânica Limpa-LASOL-CCQFA, Universidade Federal de Pelotas-UFPel, P.O. Box 354, Pelotas 96010-900, RS, Brazil
*
Authors to whom correspondence should be addressed.
Molecules 2021, 26(24), 7523; https://doi.org/10.3390/molecules26247523
Submission received: 23 November 2021
/
Revised: 7 December 2021
/
Accepted: 10 December 2021
/
Published: 12 December 2021
(This article belongs to the Special Issue Heterocycles: Synthesis, Biological Activity and Synthetic Applications)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Oxone is a commercially available oxidant, composed of a mixture of three inorganic species, being the potassium peroxymonosulfate (KHSO5) the reactive one. Over the past few decades, this cheap and environmentally friendly oxidant has become a powerful tool in organic synthesis, being extensively employed to mediate the construction of a plethora of important compounds. This review summarizes the recent advances in the Oxone-mediated synthesis of N-, O- and chalcogen-containing heterocyclic compounds, through a wide diversity of reactions, starting from several kinds of substrate, highlighting the main synthetic differences, advantages, the scope and limitations.
1. Introduction
The 12 Principles of Green Chemistry, introduced by Anastas and Warner at the end of the 1990s, set guidelines for the development of sustainable and environmentally friendly chemical processes, aiming to reduce and/or eliminate the chemical pollution by improving design, instead of residue treatment. Among them, Principle #12 (Inherently Safer Chemistry for Accident Prevention) is very important, once it encourages the use of safe substrates and reaction conditions, as well as seeking to eliminate the generation of hazardous substances, reducing the risk of accidents involving explosions, leaks and fires [1,2,3].
To satisfy this premise, the development of selective protocols, employing green oxidant species and circumventing the use of hazardous oxidants (e.g.,: KMnO4, K2Cr2O7, CrO3, OsO4, tBuOCl, m-CPBA, NBS), are welcome. Besides being operationally dangerous, some oxidants also produce toxic organic- and heavy metal-based byproducts, enhancing the environmental and health impact [4]. On the other hand, molecular oxygen (O2) and hydrogen peroxide (H2O2) are, undoubtedly, important green oxidant reagents, due to the formation of non-toxic and environmentally benign byproducts, besides presenting an excellent atom efficiency. However, transportation and handling of these species are hugely faced problems [5].
In this context, Oxone (a white solid, molar mass = 307 g mol−1) plays an important role, being a cheap and commercially available compound, composed of three inorganic salts (KHSO5/0.5KHSO4/0.5K2SO4), among which potassium peroxymonosulfate (KHSO5) is the reactive species. Oxone has been widely used as a green oxidant in organic transformations, generating the nontoxic KHSO4 as the oxidant-derived byproduct. Additionally, it is a very safe species, presenting an impressive bench-stability, which makes its transportation and storage not dangerous [6].
Heterocycles are considered one of the most important class of compounds, widely present in nature. They are also valuable in the manufacturing industry (including pharmaceuticals, agrochemicals and materials), in which they play a pivotal role [7,8]. This importance is evident regarding that around 60% of the small-molecule-approved drugs, registered in the U.S. FDA database, are composed of at least one heterocycle unit [9,10]. For this reason, the search for simple and efficient methods to access these compounds is constant and intense, with hundreds of new strategies emerging each year.
Regarding the synthetic versatility of Oxone as a green oxidant, and the massive search for the development of innovative and efficient methods to prepare heterocyclic compounds, several studies have been published describing the Oxone-mediated synthesis of heterocycles through a wide diversity of strategies (Scheme 1). Thus, this review aims to emerge a comprehensive discussion about the recent advances in this field, covering the relevant works published between 2013 and 2021. For a better understanding, it is divided in four major sections, which involve the synthesis of nitrogen-containing heterocycles, oxygen-containing heterocycles, chalcogen-containing heterocycles and miscellaneous cyclizations.
2. Nitrogen-Containing Heterocycles
N-based heterocycles are the most abundant among this important class of compounds [11]. This importance is closely related to some impressive properties, including a wide range of biological activities [12,13], being present in several worldwide marketed drugs [9] and agrochemicals, and are important in the development of functional materials [14]. As a consequence, there is an increasing search, by the scientific community, for the development of efficient methods to access these compounds. Among the recent advances, several Oxone-promoted protocols have emerged as efficient and greener alternatives to prepare N-based heterocycles.
In 2014, Tanimori and co-workers [15] described a transition metal (TM)-free protocol for the synthesis of N-arylsubstituted 1H-indazole and derivatives 1 (Scheme 2). The optimized reaction condition was reached after stirring a mixture of hydrazones 2, iodobenzene (10 mol%), Oxone (1.5 equiv) and trifluoroacetic acid (TFA), at −10 °C for 0.5 h. The cyclization reaction scope was extended to several substituted hydrazones 2, affording the respective 1H-indazoles 1 at up to an 84% yield. Electron-rich disubstituted hydrazones 2b and 2c (R = Me and OMe), gave the corresponding substituted 1H-indazoles 1b and 1c at a 77% and 71% yield, respectively. Halogen-substituted hydrazones 2d and 2e (R = Cl and F) also reacted smoothly to produce the desired products 1d and 1e in 73% and 71% yield, respectively. It is worth to mention that p-methyl monosubstituted hydrazone 2f provided a 10:1 mixture of the regioisomeric products 1f and 1f’ at a 72% yield. This problem was not observed when the p-methoxy-substituted substrate 2g was reacted, yielding the respective product 1g (69% yield) with high regioselectivity, through the annulative process undergoing in the electron-rich ring. Satisfactorily, p-NO2 and pyridinyl hydrazones 2i and 2j reacted successfully to afford the products 1i and 1j at a 79% and 77% yield, respectively. Nevertheless, limitations were also faced when N-H, N-benzyl and N-tosyl hydrazones 2k m were employed as substrate. In these cases, the desired 1H-indazoles 1k–m could not be obtained, probably due to the low stability of the nitrenium ion, the key intermediate of the reaction. Hydrazones containing CO2Me- and Me groups 2n and 2o were suitable substrates for this reaction; however, the respective indazoles 1n and 1o were obtained in poor yields (Scheme 2).
Based on control experiments, a plausible reaction mechanism was proposed (Scheme 3). Initially, the oxidation of iodobenzene by Oxone affords the hypervalent iodine(III) species A, which reacts with 2 to form the nitrenium ion intermediate B. Then, an intramolecular electrophilic substitution provides the carbocation C, which is finally converted to the aromatic product 1.
In 2015, Nagarajan and co-workers reported the synthesis of triazole-fused heterocycles 3 through a one-pot strategy, starting from aldehydes 4 and heteroarylhydrazines 5. The reaction involves the formation in situ of the hydrazone A, which in the sequence is submitted to an Oxone/CuBr-mediated intramolecular oxidative cyclization (Scheme 4) [16]. Through this simple and selective system, several substituted aldehydes bearing a wide range of functional groups were employed as substrate, affording a diversity of triazole-fused pyridines, pyridazines, pyrimidine and quinolines 3, in good yields and short reaction times.
Additionally, in 2015, Guo and co-workers described the synthesis of oxindoles 6 through an Oxone-mediated reaction between N-arylacrylamides 7 and α-diketones 8, through a Csp2−Csp2 cleavage and the formation of a new Csp2−Csp3 bond (Scheme 5) [17]. The optimized reaction condition was set by using N-arylacrylamina 7, α-diketone 8, and Oxone (2.5 equiv) in THF (3 mL) as solvent. The resulting mixture was stirred at 100 °C under N2 atmosphere for 24 h. Under this condition, a total of ten 3-(2-oxoethyl)indolin-2-ones 6 was obtained in poor to good yields, employing several electron-rich and electron-deficient substrates 7 and 8. It is worth to mention that asymmetric α-diketones, including 1-phenylpropane-1,2-dione 8a and 1-(4-methoxyphenyl)-2-phenylethane-1,2-dione 8b, were also suitable substrates, resulting a mixture of products 6a + 6i (23% and 49% yield) and 6a + 6j (43% and 16% yield), respectively (Scheme 5).
In 2016, Sen and co-workers employed Oxone as an oxidant in the tandem annulation between 2-aminobenzylamines 9 and aldehydes 4, in order to access 2-substituted benzimidazoles 10 (Scheme 6) [18]. The reaction is conducted at room temperature for 8 h, in the presence of Oxone (0.6 equiv), H2O (2 mL) and DMF (10 mL), and was suitable to several aliphatic, aromatic and heteroaromatic aldehydes 4, in the presence of substituted 2-aminobenzylamines 9, giving the products 10 in poor to excellent yields. Limitations were faced when electron-deficient 2-aminobenzylamines (R = 3-NO2) were employed as substrate, affording the products 10c and 10e in 18% and 26%, respectively. The process involves the initial condensation between 2-aminobenzylamine 9 and aldehydes 4, providing the tetrahydroquinazoline intermediate A, which undergoes a radical ring distortion to afford the desired products 10, under oxidative conditions (Scheme 6).
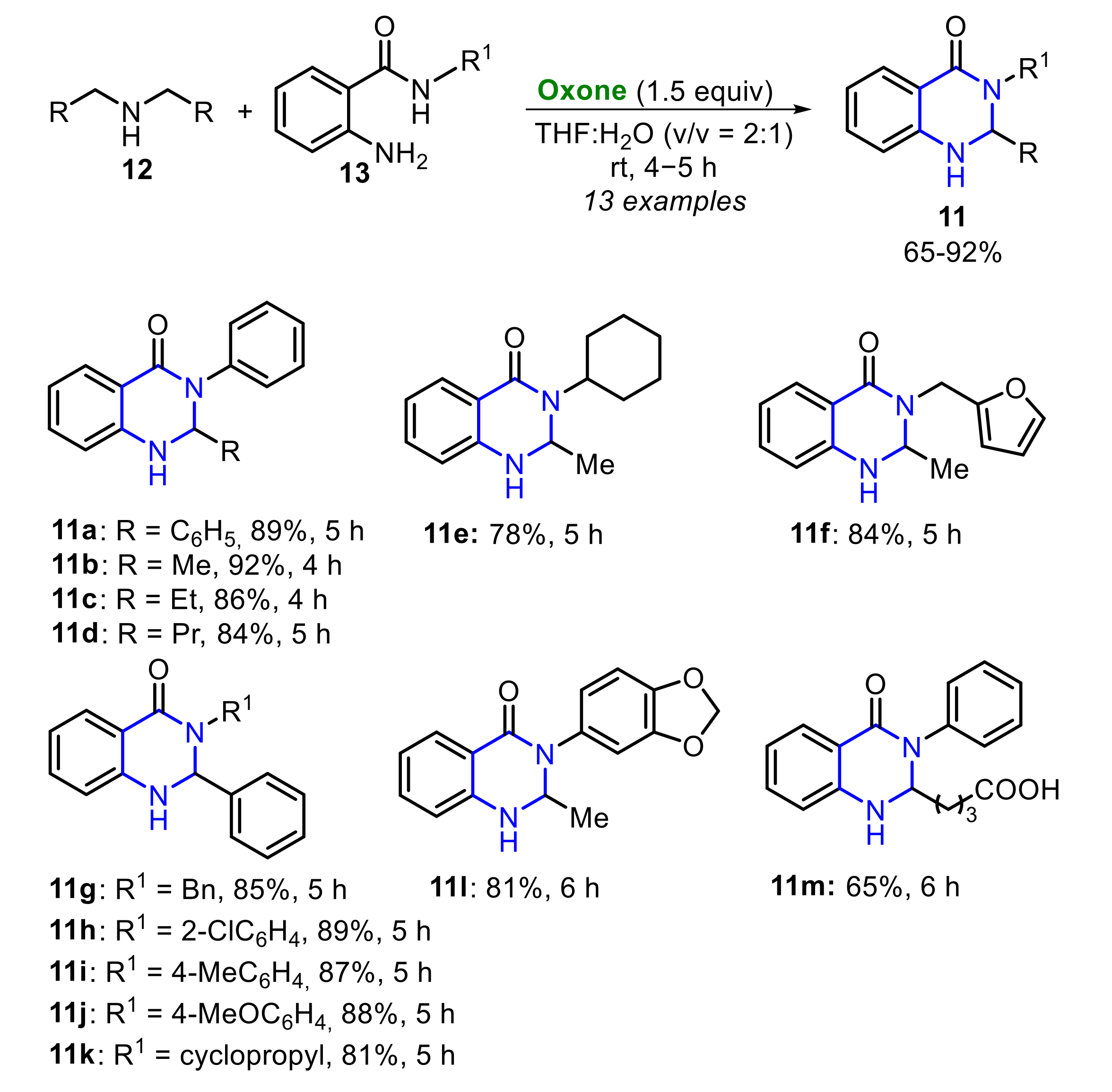
In 2018, Madabhushi and co-workers described an efficient Oxone-mediated protocol for the preparation of 2,3-dihydroquinazolin-4(1H)-ones 11 and 1H-benzimidazoles 10 (Scheme 7) [19]. The reaction involves stirring a mixture of sec-amines 12, via imine-N-oxides, and substituted 2-amino-N-benzamides 13 in the presence of Oxone (3.54 mmol), in a mixture of THF/water (2:1, 10 mL) as solvent, affording the respective 2,3-dihydroquinazoline-4(1H)-ones 11a m in moderate to excellent yields. The reaction scope was satisfactorily expanded, and several alkyl and aryl sec-amines 12 were employed, as well as N-substituted benzamides 13, bearing aliphatic, aromatic and heteroaromatic groups.
Additionally, under the optimized reaction condition, sec-amines 12 reacted with 1,2-diaminobenzenes 9, affording 1H-benzimidazoles 10 in poor to moderate yields (Scheme 8). The reaction scope was also investigated, presenting an acceptable substrate tolerance, including the use of substrates bearing the strong electron-withdrawing nitro group (R1 = 4-NO2).
In 2018, Tang, Zheng and co-workers [20] described an Oxone/NaNO2-mediated oxidative radical cyclization of N-methyl-N-arylpropiolamide 14 to the respective isatins 15 and 16, through the oxidative cleavage of the C≡C bond, affording the desired products in moderate to good yields (Scheme 9). Despite the low reaction selectivity, the present protocol is important, once NO2-containing isatin derivatives 16 are satisfactorily afforded, which is not an easy task, from the synthetic point of view.
The proposed reaction mechanism involves an initial Oxone-promoted oxidation of NaNO2 to the •NO radical species, under thermal condition. Thus, •NO radical is oxidized in the presence of O2 to •NO2, which is trapped by the C≡C triple bond, to afford the intermediate A. Following, a radical isomerization produces the intermediate B, which is quickly converted by electron delocalization to the tertiary radical species C, that undergoes an intramolecular radical annulation to produce the cyclized intermediate D. A hydrogen radical elimination converts the aryl radical D to the intermediate E, which after a decarbonylation, affords the intermediate F. The intermediate D can also be trapped by 1 equivalent of •NO2 radical to produce the intermediate G, which is subsequently decarbonylated to generate the intermediate H. Finally, the intermediates F and H are hydrolyzed and converted to the respective products 15 and 16 (Scheme 10).
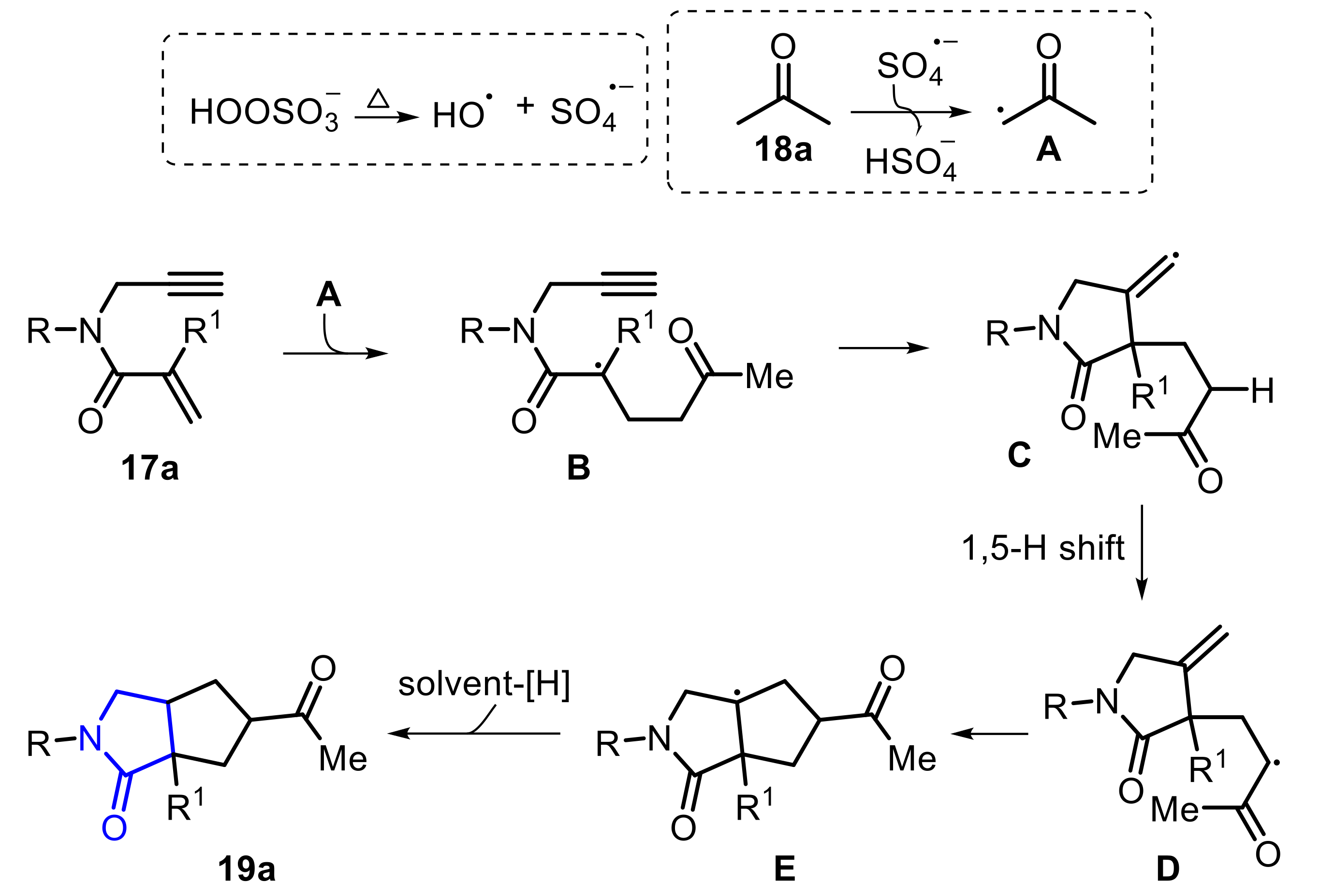
In 2019, Wei and co-workers reported an Oxone-mediated base-free radical bicyclization of 1,6-enynes 17, in the presence of ketones 18, affording several functionalized polycyclic γ-lactams 19 (Scheme 11) [21]. Under the optimal conditions, aliphatic ketones, such as acetone 18a and cyclobutanone 18b, reacted satisfactorily with 1,6-enyne 17a, yielding the respective products 19a and 19b in 82% and 56% yield, respectively. On the other hand, acetophenone 18c and ethyl acetate 18d were not suitable substrates, which may be presumably attributed to the inertness of the α-Csp3−H bond to generate the corresponding α-carbonyl radical species. Additionally, a range of 1,6-enynes were well tolerated as the substrate, giving the respective polycyclic γ-lactams 19e–i at up to an 88% yield (Scheme 11).
The reaction mechanism to prepare the γ-lactams 19 is depicted in Scheme 12. Initially, the α-carbonyl radical A, which is formed after heating the ketone 18a in the presence of Oxone, is added to the terminal C≡C double bond (of 1,6-enyne 17), giving the alkyl radical intermediate B. After an intramolecular cyclization, the tertiary radical B is converted to the vinyl radical intermediate C, which undergoes an 1,5-H shift, to be converted to the intermediate D. Finally, the second cyclization process takes place, generating the intermediate E, that through a homolytic hydrogen abstraction is converted to the desired product 19.
Additionally, in 2019, some of us reported the electrophilic cyclization of β-alkynyl hydrazones 20, promoted by the system Oxone/RSeSeR 21, to obtain 4-organoselanyl-1H-pyrazoles 22 (Scheme 13) [22]. The reaction scope was studied employing several substituted diorganyl diselenide 21 and hydrazones 20, affording the desired products 22 in poor to excellent yields. A limitation was faced when the α,β-alkynyl hydrazone 20k, bearing an electron-deficient ring (R3 = 2,4-F2C6H3), was employed as substrate, providing the product 22k at only a 40% yield after 24 h. Better results were obtained when electron-rich α,β-alkynyl hydrazones 20 (R3 = 3-MeC6H4 and 2,4-Me2C6H3) were reacted, yielding the products 22i and 22j at 97%, after 11 and 8 h of reaction, respectively (Scheme 13).
During control experiments, the in situ generation of Se-based electrophilic species was identified by 77Se NMR and HRMS, which were the key intermediates to disclose the annulation pathway. The proposed mechanism starts with a SET from diphenyl diselenide 21 to the HSO5- species, followed by the cleavage of the Se–Se bond, providing the Se-based electrophiles PhSeOSO3- I and PhSeOH II, which promote the electrophilic annulation of the α,β-alkynyl hydrazone 20, to afford the desired products 22. It is worth to mention that species I and II, in the presence of MeOH and Oxone, can be converted to the methyl benzeneseleninate V and benzeneseleninic acid VI, respectively (Scheme 14).
In the same year, Jacob and co-workers also reported the synthesis of 4-organoselanyl-1H-pyrazoles 22 through a multicomponent reaction between hydrazines 23, 1,3-diketones 24 and diorganyl diselenides 21, in the presence of Oxone (Scheme 15) [23]. In general, the protocol presented a good substrate tolerance, allowing the synthesis of twelve substituted 4-organylselanylpyrazoles 22 in moderate to excellent yields, after short reaction times. When the unsymmetrical 1-phenyl-1,3-butanedione 24c was employed as substrate, two regioisomers 22q and 22q’ in a 96:4 ratio were obtained at 89% yield. Authors attributed the high selectivity to the steric hinderance of ketone 24c and also to the conjugative effect of the aromatic ring, that can contribute to stabilize the enol tautomer, increasing the regioselectivity of this cyclization towards the formation of product 22q. The reactivity of N-aryl hydrazines 23 was affected by electronic effect. The electron-rich system (R = Me) afforded the respective product 22r, at 69% yield, whereas the electron-deficient one (R = Cl) gave 22s at 44% yield, requiring a temperature increase from 50 °C to 100 °C. However, despite this significant difference, in comparison with the absence of substituents (product 22o), the reaction efficiency was remarkably decreased. On the other hand, the results suggest that the process is not sensitive to the electronic effect in the diaryl diselenides 21, and the products 22u-x were obtained in good to excellent yields. Additionally, dibutyl and 2,2′-dipyridyl diselenides 21 reacted smoothly to produce the products 22y and 22z at a 90% and 58% yield, respectively (Scheme 15).
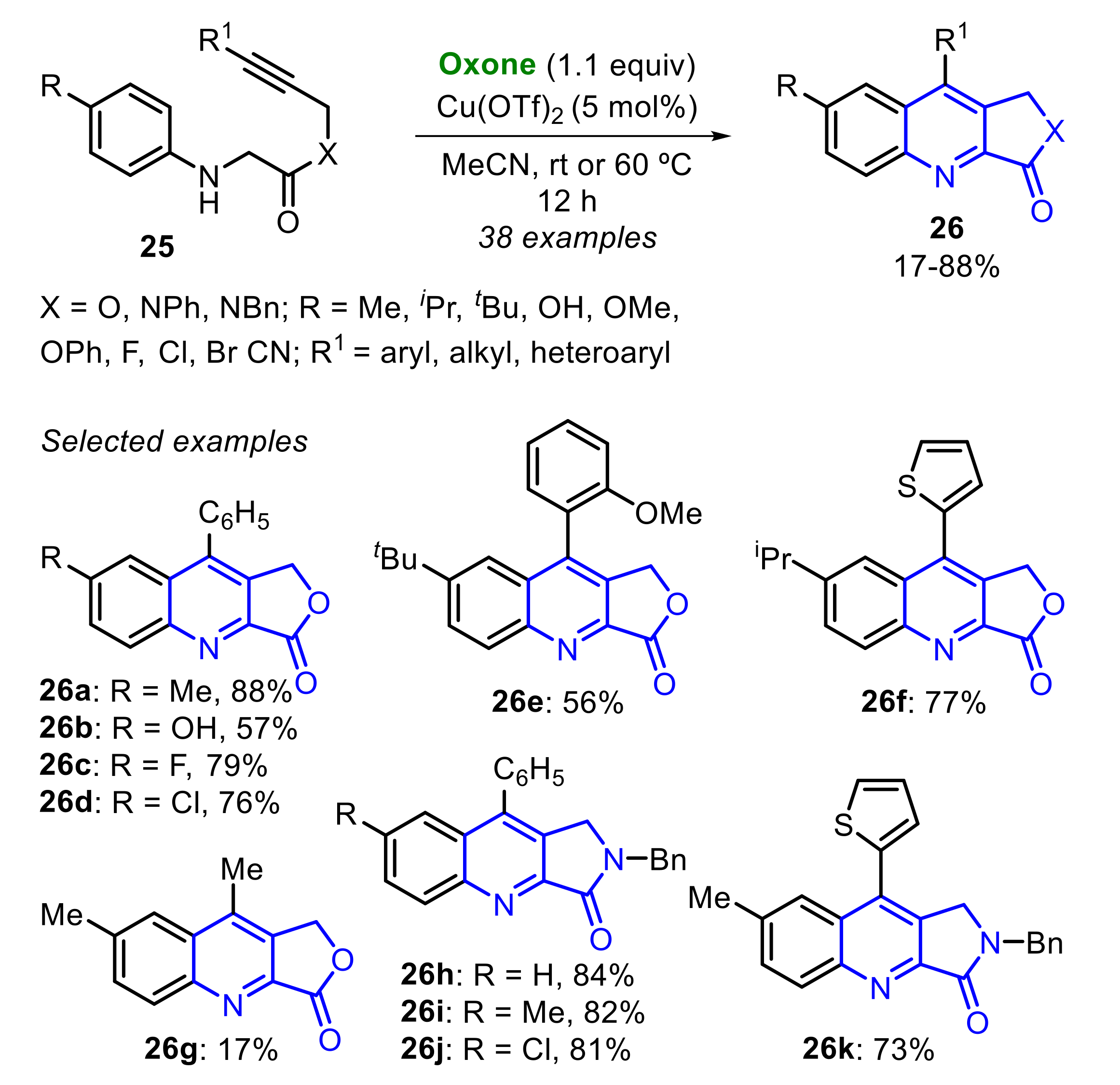
Still in 2019, Muthukrishnan and co-workers reported an Oxone-promoted intramolecular dehydrogenative Povarov cyclization of alkyne tethered N-aryl glycine esters and amides 25 (Scheme 16) [24]. Under the optimal conditions (Oxone (1.1 equiv), Cu(OTf)2 (5 mol%) in MeCN at room temperature or 60 °C for 12 h), a library of thirty-eight functionalized quinoline-fused lactones and lactams 26 were accessed at up to 88% yield.
This methodology was used to construct complex structures, such as the antibiotic Uncialamycin 26l (Scheme 17A). Moreover, the synthesized products 26h and 26i were easily converted to 26m and 26n, which are analogues of the cytotoxic alkaloid Luotonin A (Scheme 17B).
Liu, Qiu, and co-workers reported the ZnBr2/Oxone-mediated radical ipso-cyclization of N-(3-arylprop-2-yn-1-yl)aniline 27 to prepare 1-azaspiro[4.5]deca-3,6,9-trien-8-ones 28 (Scheme 18) [25]. The reactions were carried out in the presence of ZnBr2 (1 equiv), Oxone (2 equiv) and a mixture of acetonitrile and H2O (v/v = 4:1) as solvent at room temperature. In general, electron-rich and electron-deficient aryl groups attached to the Csp–Csp bond (R = CN, Me, COMe and CO2Me) were well tolerated under the optimized condition, affording the products 28a–d in good yields. However, limitations were faced by employing the 2-iodo-N-tosyl-N-(but-2-yn-1-yl)aniline 27e as substrate, and the product 28e was observed only in trace amounts. Regarding the pendent N-aryl group, the presence of both electron donor and electron withdrawing substituents (R = Me and Cl) and (R1 = Me and Br) were tolerated, affording the products 28g–j in good yields. A good result was obtained when the N-protecting group R2 = 4-BrC6H4 was used, and product 28k was obtained at a 72% yield (Scheme 18).
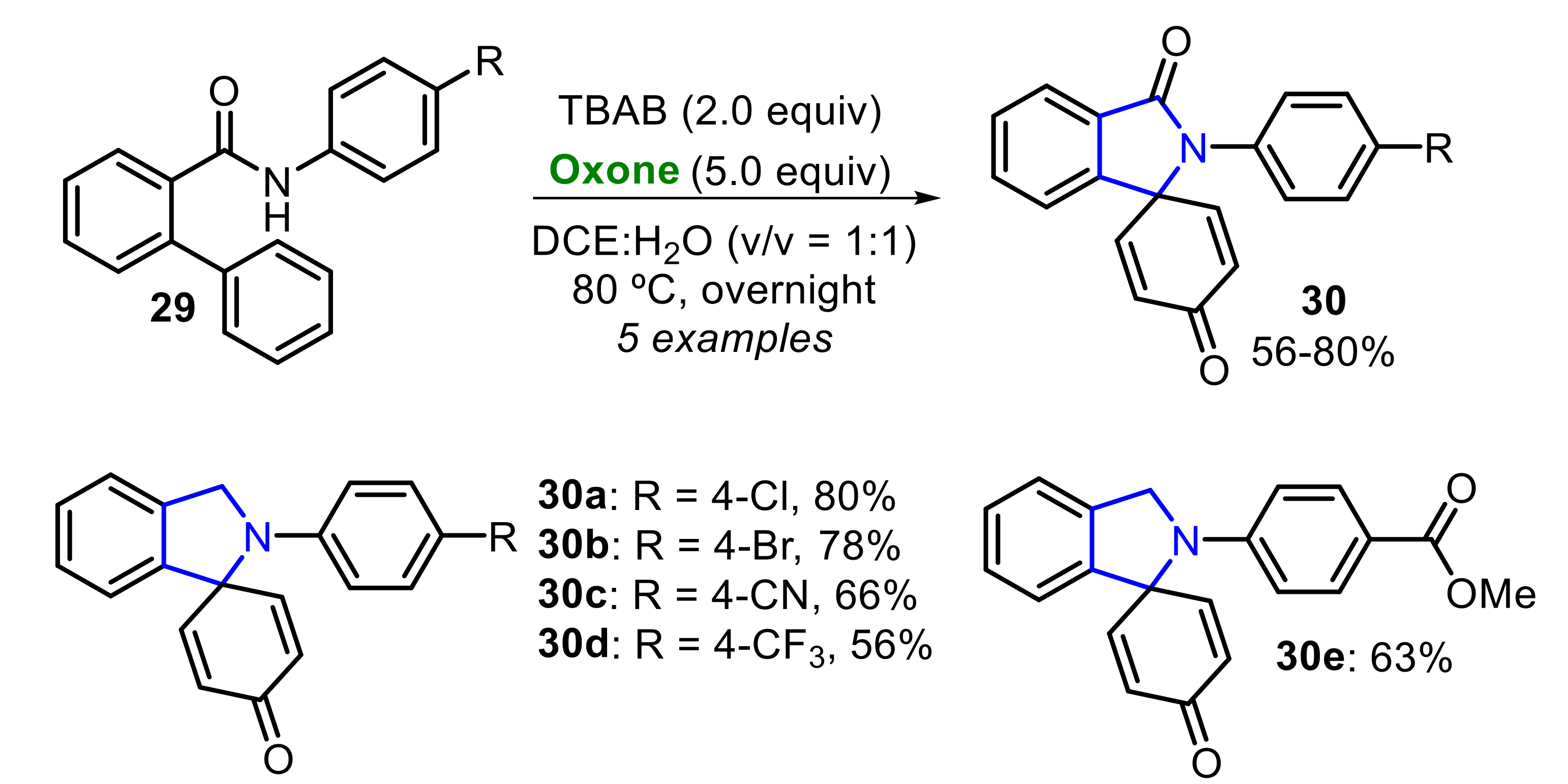
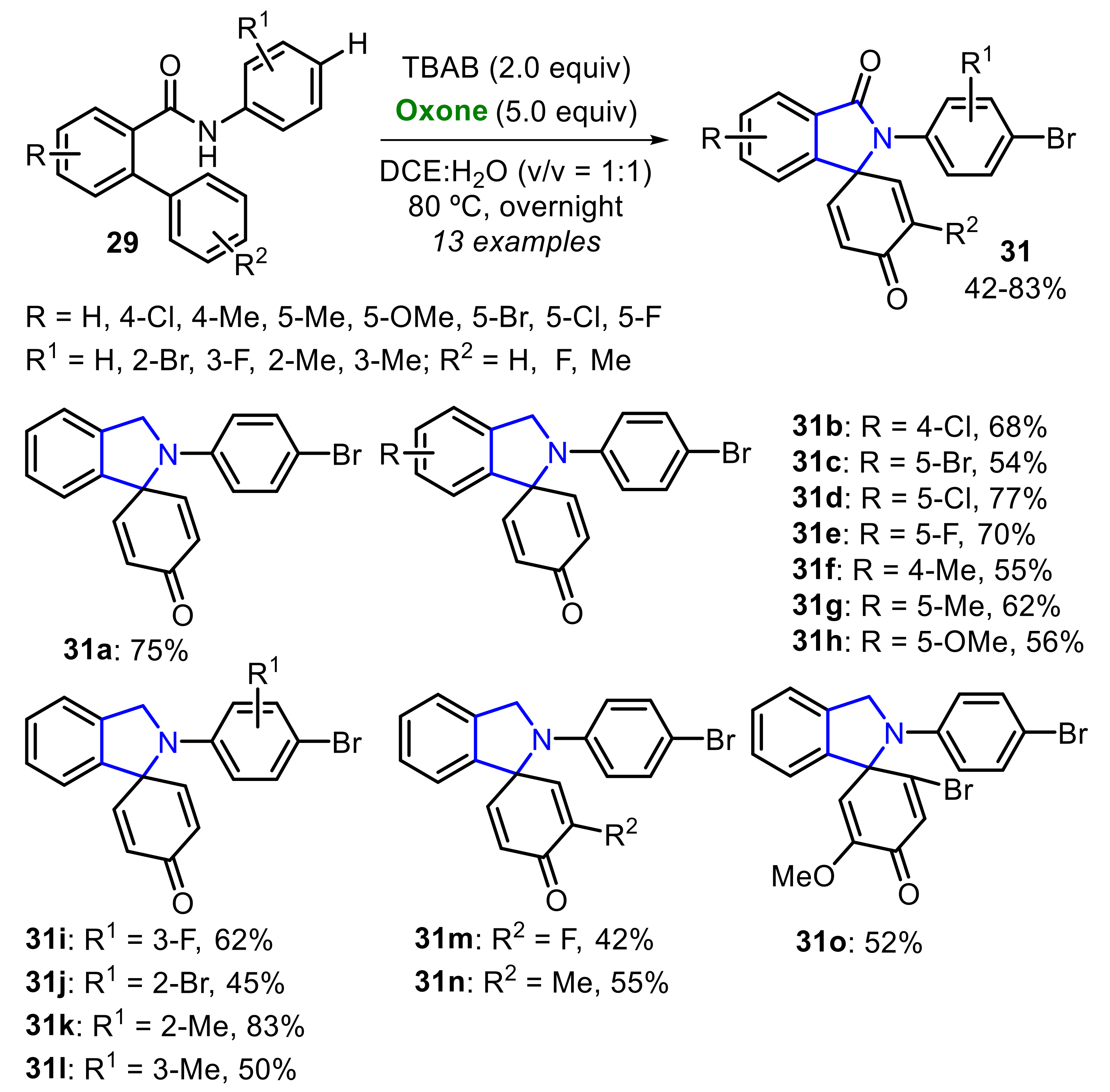
Zhou and co-workers reported an Oxone/TBAB-mediated oxidative regioselective cyclization of N-aryl-2-arylbenzamides 29 to prepare spiro[cyclohexane-1,1′-isoindoline]-2,5-diene-3′,4-diones 30 (Scheme 19) [26]. A total of five para-substituted benzamides 29 (R = Cl, Br, CN, CF3, CO2Me) were employed as substrate, affording the respective products 30a–e in moderate to good yields.
In the absence of a substituent at the para-position of the N-aryl-2-phenylbenzamide 29, the bromination of this position occurred, and the product 31a was afforded in 75% yield. Considering the synthetic versatility of the aryl bromide core, the authors expanded the reaction scope to prepare several brominated products 31 in moderate to good yields (Scheme 20). Interestingly, the presence of a methoxy group in the ortho-phenyl ring (R2 = 3-OMe) caused the formation of the dibrominated product 31o at a 52% yield.
An Oxone/TBAB-mediated synthesis of 3-bromo-1,2-dihydroquinoline derivatives 32 through a radical 6-endo-trig ortho-cyclization of 2-alkynylbenzamide 33 was described in 2019 by Liu and co-workers (Scheme 21) [27]. Several substituted N-alkynyl-benzenesulfonamides 33 were used under optimal conditions, giving a range of dihydroquinoline derivatives 32 in moderate to good yields. 2-Alkynylbenzamides containing electron-rich benzene ring (R2 = Me and OMe) attached to the alkynyl portion were satisfactorily employed as substrate. Among them, the steric hindrance promoted by the methyl group (ortho-, meta- or para-position) slightly affected the reaction efficiency, giving the products 32b–d in similar yields. On the other hand, the presence of the methoxy group at the para-position activated the alkynyl portion, affording the product 32e at a 75% yield, remarkably increasing the reaction efficiency. The protocol presented an interesting halogen tolerance, affording the desired products 32f (R = 4-F) and 32g (R = 4-Cl) at 71% and 68%, respectively. Heteroarene- and alkyl-substituted alkynylbenzamides (R2 = 2-thienyl and Me), as well as N-mesyl, were suitable substrates, affording the respective products 32h, 32i and 32j at 67%, 72% and 56% yields (Scheme 21).
Based on control experiments, a plausible reaction mechanism was proposed, which starts with an Oxone-promoted oxidation of bromide anion (Br–) to the bromo radical species (Br•) through a SET process. Following on from this, a radical addition of the bromo radical to the substrate 33 produces the intermediate A, which undergoes a radical 6-endo-trig cyclization, to be converted to the intermediate B. Finally, the intermediate B undergoes oxidation, followed by a deprotonation, to be converted to the desired dihydroquinoline 32 (Scheme 22).
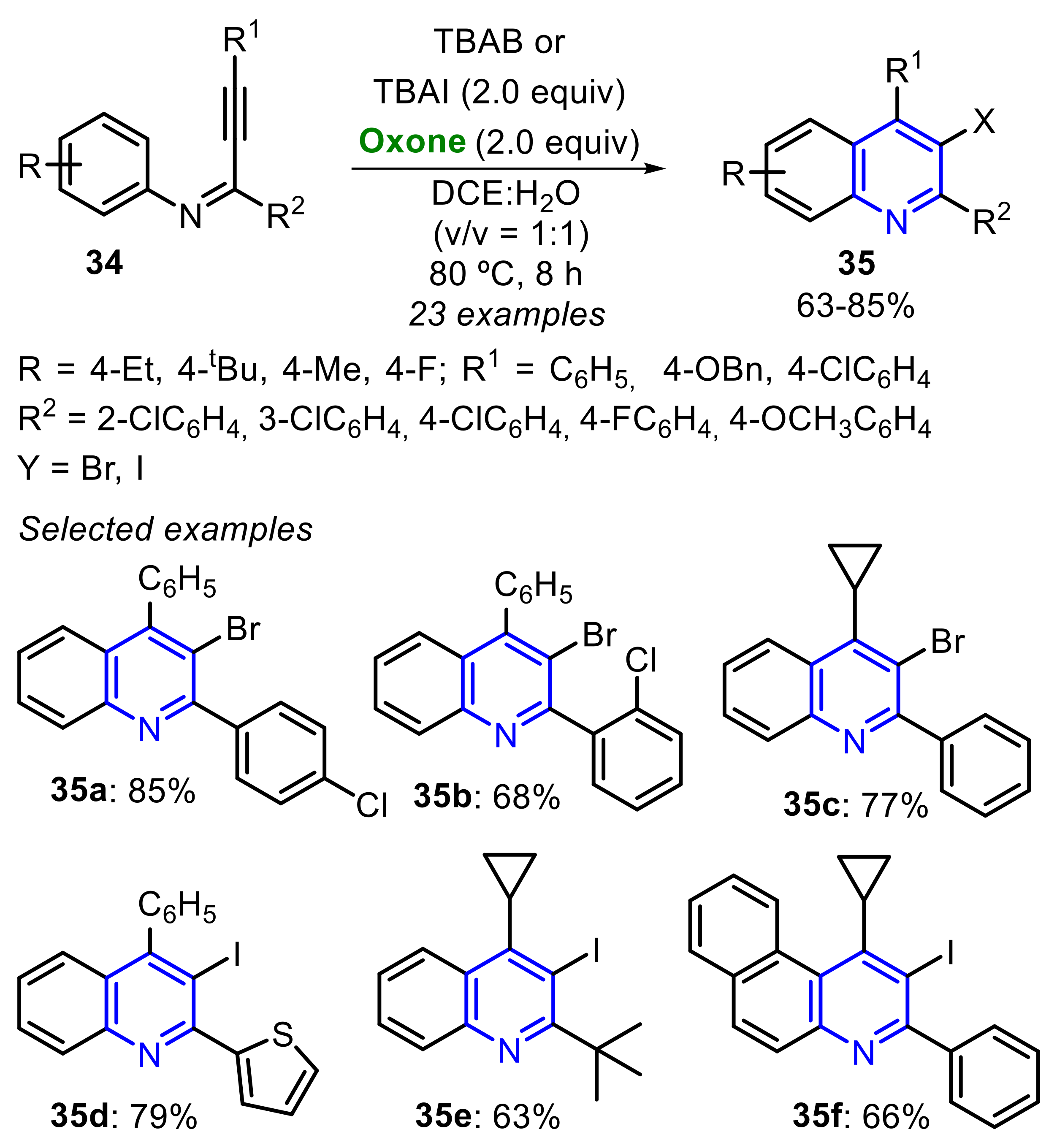
In 2020, Qiu and co-workers described an Oxone/TBAX-promoted (X = Br or I) tandem annulative radical halogenation of alkynyl imines 34 to access 3-haloquinolines 35 regioselectively (Scheme 23) [28]. Comparing TBAB and TBAI acting as the halogen-source, any remarkable difference in the reaction efficiency was observed, with TBAB affording 3-bromoquinolines 35 and TBAI delivering 3-iodoquinolines 35 in good to very good yields.
The reaction mechanism initiates with the Oxone-promoted oxidation of the halogen ion (X–) to the halogen radical species (X•), that is subsequently added to the C≡C bond, to produce the vinyl radical intermediate A. In the sequence, the cyclized intermediate B is obtained via a 6-endo-trig cyclization, followed by the oxidation to the intermediate C, which is finally aromatized to the product 35 (Scheme 24).
In the same year, Wei, Liu, Lei and co-workers reported an Oxone/Cu(NO3)2-promoted radical annulative nitration of 1,6-enynes 17 (Scheme 25) [29]. Under the optimized reaction conditions, several N-substituted 1,6-enynes 17, bearing different functional groups in the alkynyl and alkenyl portions, reacted satisfactorily to produce nitro-containing pyrrolidin-2-ones 36 in good to excellent yields. It is worth to mention that the protocol was satisfactorily scalable to 10 mmol, affording 3-methyl-4-methylene-3-(nitromethyl)-1-phenylpyrrolidin-2-one 36a at a 82% yield.
Qiu, Shan, Chen and co-workers also reported a protocol based on the generation of halogen radical species (X•), employing the system Oxone/TBAX (X = Br and I), which disclosed the radical annulation of para-methoxyl alkynyl-N-phenylimines 37, to access 3-halospirocyclohexadienones 38 (Scheme 26) [30]. Instead of the expected 6-endo-trig cyclization (Scheme 24), a radical 5-exo-trig cyclization occurred, disclosing a dearomative aza-spirocyclization, driving to the spirocyclic system 38. Through an optimization study, the best reaction condition was set reacting the substrate 36 in DCE:H2O (v/v = 1:1), at 80 °C for 8 h, in the presence of Oxone (2.0 equiv) and TBAB or TBAI (2.0 equiv). In general, several para-methoxyl alkynyl-N-phenylimines 37 were satisfactorily submitted to the optimized reaction conditions, tolerating a wide range of substituents, including aryl systems bearing electron-donating and -withdrawing groups, heteroaryl and alkyl (Scheme 26).
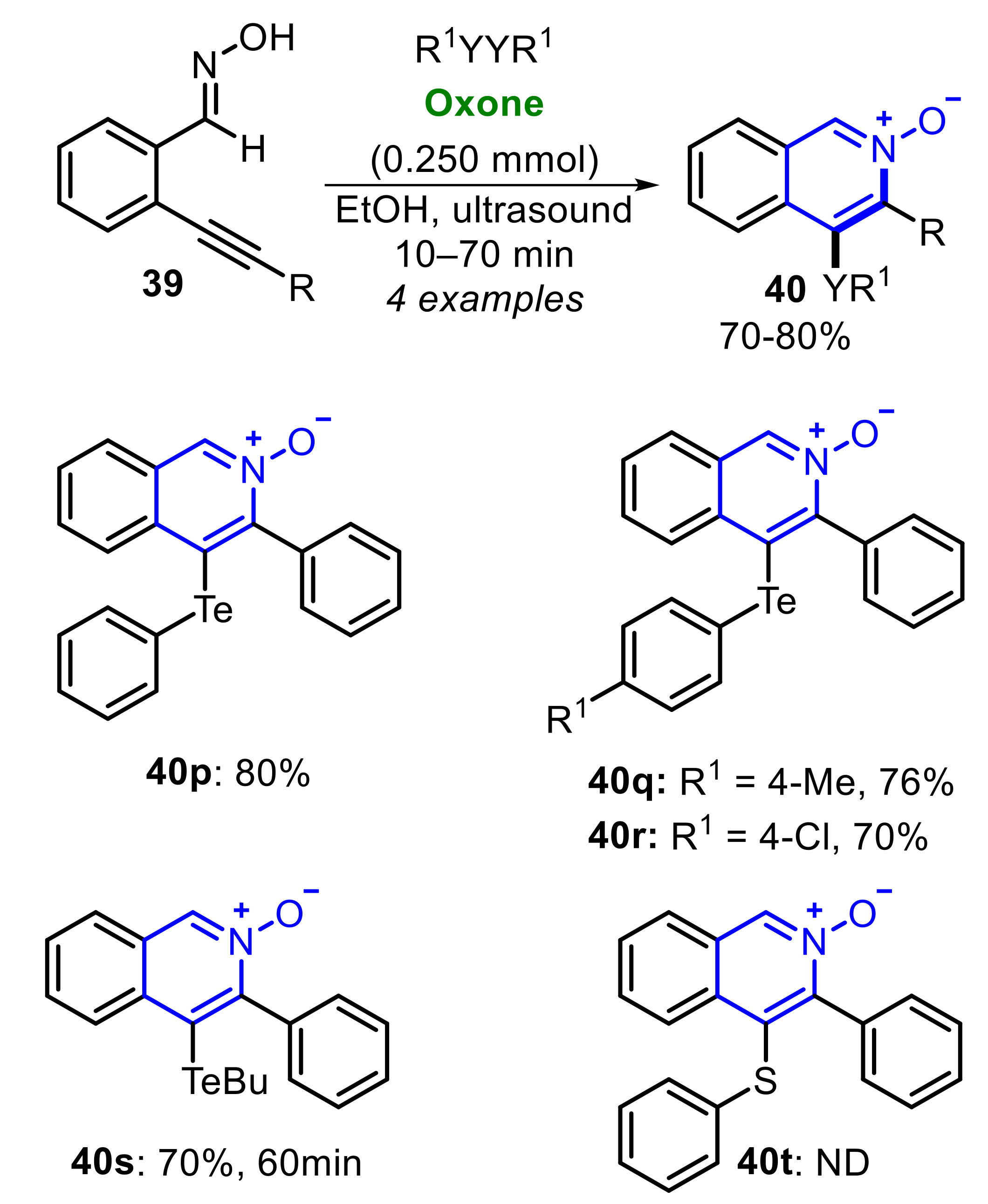
In 2021, some of us reported the Oxone/RSeSeR-promoted electrophilic 6-endo-dig cyclization of alkynylbenzaldoximes 39, to access 3-organyl-4-(organylchalcogenyl)isoquinoline-2-oxides 40 (Scheme 27) [31]. The process was conducted under ultrasound irradiation in the presence of EtOH as promoting reaction medium, affording twenty-one Se-containing N-oxides 40 at good to excellent yields, within just a few minutes of reaction. Diaryl diselenides bearing electron-donating (R1 = 4-MeOC6H4) and -withdrawing groups (R1 = 4-ClC6H4) reacted smoothly to produce the products 40b and 40c at excellent yields. Dialkyl (R1 = nBu) and the sulfonamide-derivative diselenide (R1 = SO2NH2C6H4) were suitable substrates, affording the respective isoquinoline-2-oxides 40d and 40e in 82% and 71% yield. In general, several electron-rich and electron-deficient alkynylbenzaldoximes 39 were satisfactorily submitted to the optimal conditions. However, limitations were found when an alkyl group (R = tBu) was attached in the C≡C bond, and the conversion to the expected product 40j was not observed An interesting result was obtained when the alkynylbenzaldoxime 39g, bearing the TMS group attached to the C≡C bond (R = Si(Me)3) was employed as substrate. In this case, the expected product 40k was not afforded, and the reaction was driven to the C3-free product 40l, obtained in 88% yield. The same product 40l could be accessed from the unprotected terminal alkynylbenzaldoxime 39h at an 85% yield (Scheme 27).
The protocol was satisfactorily expanded to diaryl ditellurides 41a–c and dibutyl ditelluride 41d, affording tellurium-functionalized isoquinoline-2-oxides 40p-s in good to very good yields (Scheme 28). No reaction was observed using diphenyl disulfide 42a, and 4-(phenylthio)isoquinoline-2-oxide 40t, was not obtained, even after 60 min under sonication.
3. Oxygen-Containing Heterocycles
Oxygen-containing heterocycles are an important class of compounds, which are abundantly found in nature, and are especially important in the industry, being widely used as flavor and fragrances [32]. Furthermore, these compounds present an important range of biological activities, including anticancer [33], immunosuppressive and hypolipidemic [34]. As a consequence of the importance of these compounds, several innovative, simple and efficient Oxone-based protocols have been recently developed to synthesize oxygen-containing heterocycles.
In 2018, Qiu and co-workers reported the Oxone/TBAB-promoted 5-exo-dig oxy-cyclization of 2-alkynylbenzamide 43 to access isobenzofuran-1-imines 44 and isobenzofuran 45 selectively, in the presence of K2CO3 (Scheme 29A,B) [35]. Under the optimized reaction conditions, the product 44 is mostly formed at good to very good yields. However, the product 45 could be accessed at moderate to very good yields using an additional HCl-based hydrolysis step (Scheme 29).
In 2019, Liu and co-workers described the TBAB-catalyzed Oxone-mediated 6-endo-dig oxidative annulation of 2-alkynylbenzamide 43 to access several substituted isocoumarin-1-imines 46 (Scheme 30) [36]. Differently substituted 2-alkynylbenzamides 43, bearing electron-rich and electron-deficient aryl and alkyl groups, were suitably employed as substrates, affording the respective products 46 regioselectively, in moderate do very good yields. Limitations were found when 2-ethynyl-N-phenylbenzamide 43l was employed as substrate, and the respective isocoumarin-1-imine 46l was not formed.
During their studies, the authors noticed that when the N-phenyl 2-trimethylsilylethynylbenzamides 47 was submitted to the optimal reaction conditions, isobenzofuran-1-imines 48 were obtained as the major product, through an oxidative 5-exo-dig annulation (Scheme 31). Inspired by this result, they decided to investigate the reaction scope, employing several meta-substituted N-phenyl 2-trimethylsilylethynylbenzamides 47, bearing electron-donating and electron-withdrawing groups (R = H, Me, F, Br and Cl), and the respective products 48 were obtained in very good yields.
In the same year, Wang, Sun, and co-workers reported the Pd(II)-catalyzed Oxone-mediated oxidative sequential addition-annulation of 1,2-naphthofuroquinones 49 with diaryl alkynes 50, to prepare functionalized naphthofuroquinones 51 (Scheme 32) [37]. The method was efficiently applied to several functionalized 2-hydroxy-1,4-naphthoquinones 49, bearing electron-neutral, -donating, and -withdrawing aryl substituents. Regarding the diaryl alkyne counterpart 50, symmetrical and unsymmetrical, as well as electron-rich and electron-deficient derivatives could be converted to the respective naphthofuroquinone 51 in poor to very good yields. The biological potential of the synthesized products was evaluated, and the naphthofuroquinone 51a presented a potent endothelial protective activity.
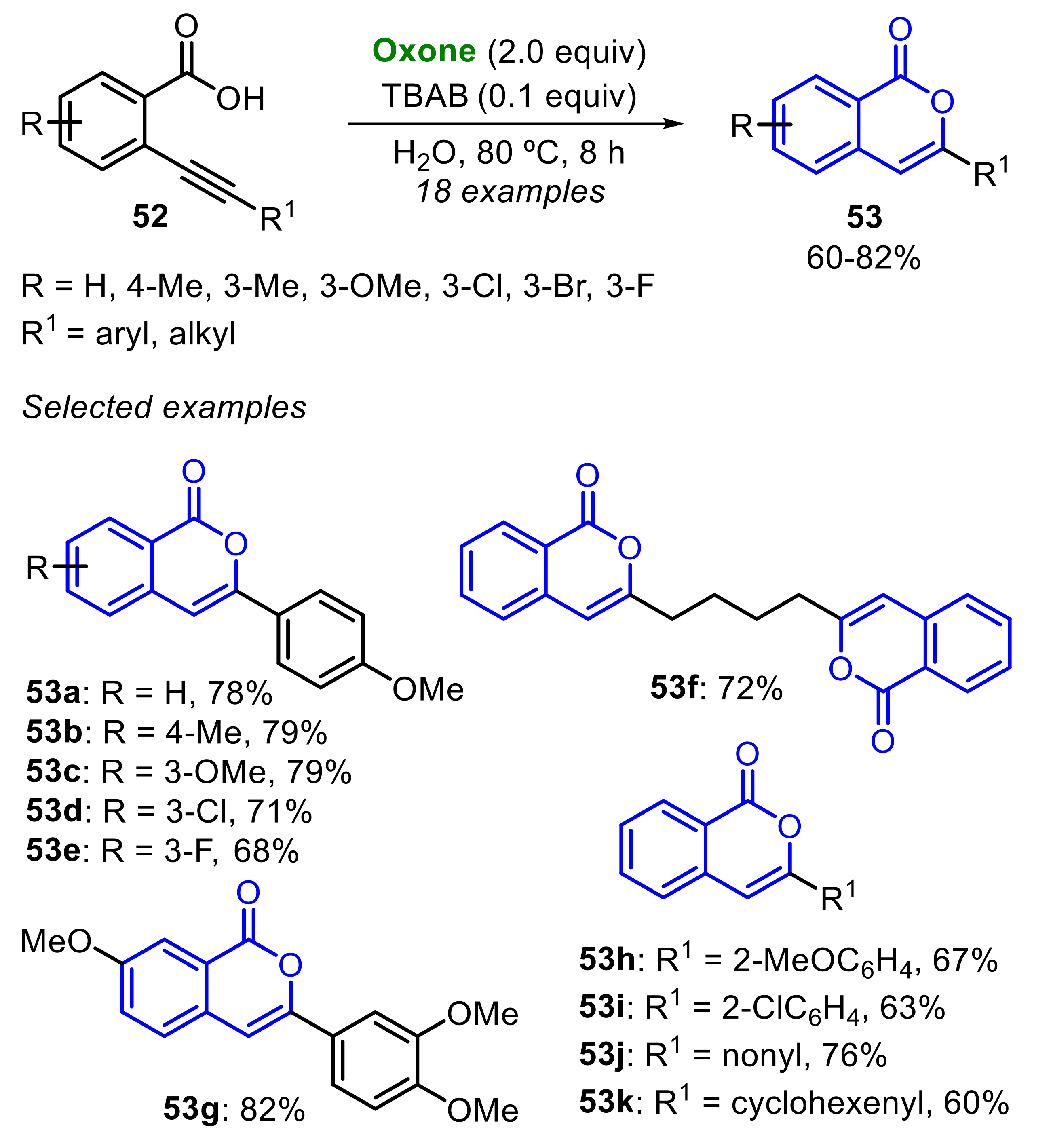
Recently, Liu, Zhou, Qiu and co-workers described the Oxone/TBAB-promoted radical cyclization of 2-alkynylbenzoic acids 52, to construct isocoumarins 53, employing catalytic amounts of TBAB (10 mol%) (Scheme 33) [38]. Substituted 2-alkynylbenzoic acids 52 bearing electron-donating and -withdrawing groups were satisfactorily employed, affording a total of eighteen functionalized isocoumarins 53 in moderate to very good yields. Regarding the alkynylbenzoic aryl moiety, when the system was deactivated (R = 3-Cl and 3-F), a slight loss in the reaction efficiency to access the products 53d and 53e was faced, whereas electron-donating groups (R = 4-Me and 3-OMe) did not affect the reactivity remarkably. Interestingly, the method was extended to the bis-functionalized substrate 2,2’-(octa-1,7-diyne-1,8-diyl)dibenzoic 52f, giving the corresponding isocoumarin 53f in 72% yield. Additionally, 2-alkynylbenzoic acids 52 bearing vinyl and alkyl groups attached in the C≡C bond, reacted smoothly to produce the products 53j–k at good yields (Scheme 33).
The first step of the reaction mechanism is the formation of the bromo radical (Br•) species, as proposed in Scheme 21 and Scheme 23. In the sequence, the 2-alkynylbenzoic acid 52 is oxidized, affording the oxygen-centered radical species A, which quickly undergoes a 6-endo-dig radical annulation to produce the isocoumarin radical B. Finally, after a radical hydrogen abstraction, the intermediate B is converted to the isocoumarin 53 (Scheme 34).
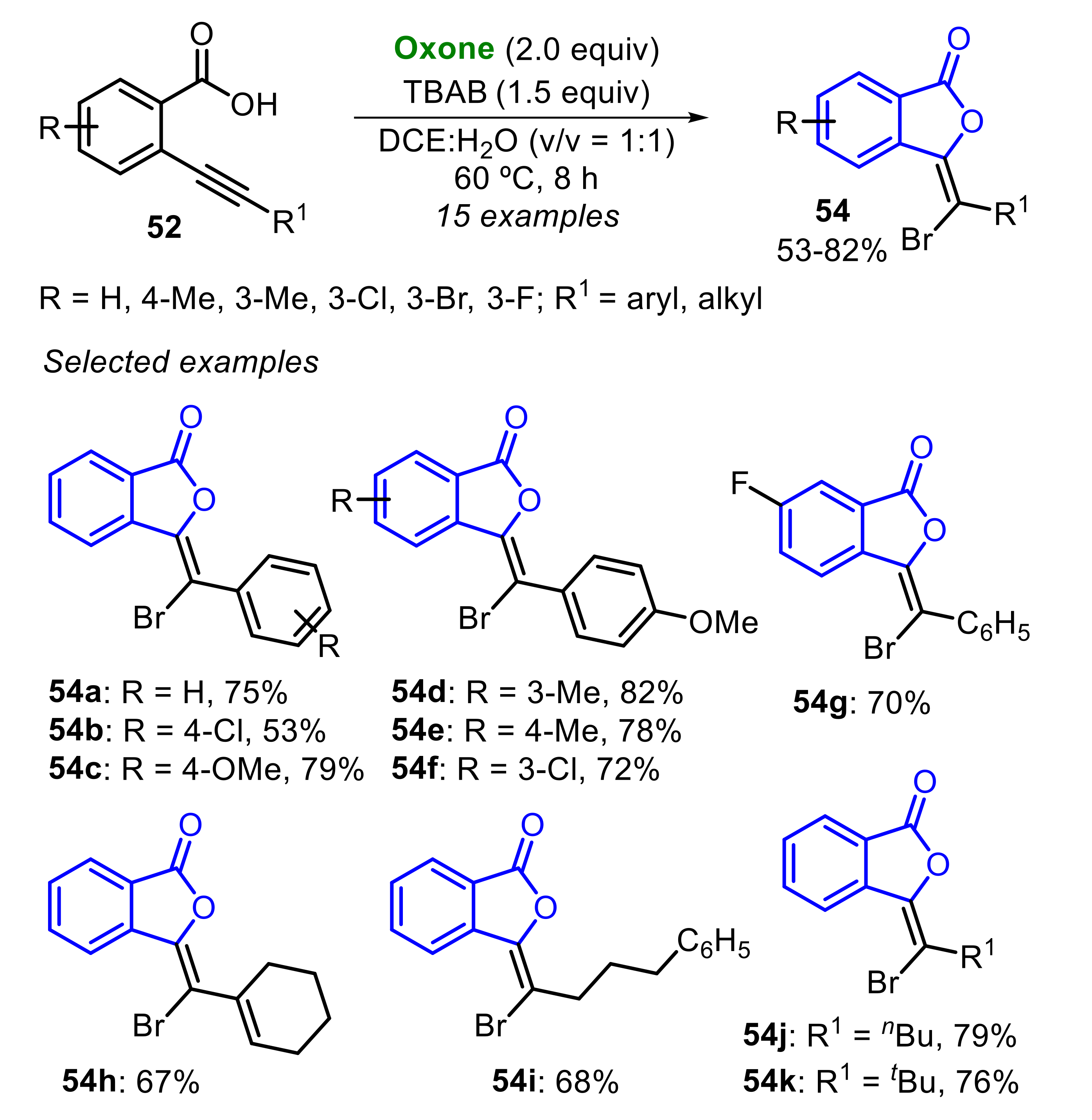
The system TBAB/Oxone was also employed in the 5-exo-dig bromocyclization of 2-alkynylbenzoic acids 52, aiming the synthesis of 3-(bromomethylene)isobenzofuran-1(3H)-ones 54, employing overstoichiometric amounts of TBAB (1.5 equiv) (Scheme 35) [39]. A total of fifteen 3-(bromomethylene)isobenzofuran-1(3H)-ones 54 were obtained in moderate to very good yields, starting from alkynyl-substituted substrate 52 bearing electron-rich and -deficient aryl groups. Good results were obtained using the vinyl derivative 52h (R1 = cyclohex-1-enyl), which reacted smoothly to yield the product 54h at a 67% yield. Equally good outcomes were observed using Csp-substituted with alkyl groups (R1 = phenylpropanyl, nBu and tBu), and the respective products 54i–k were obtained at good yields (Scheme 35).
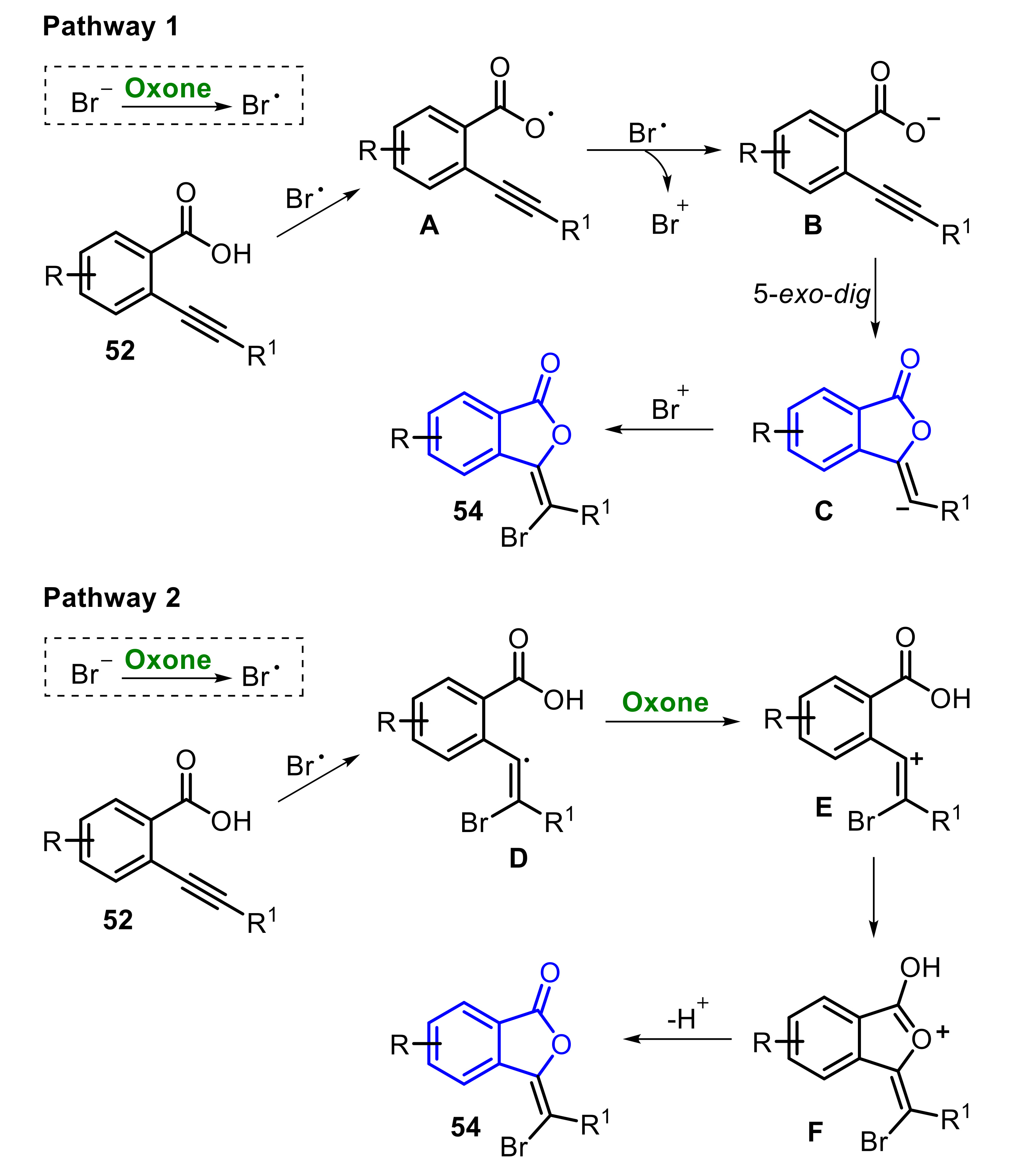
The proposed mechanism of the formation of 54 from 52 has two possible pathways (Scheme 36). Initially, an Oxone-promoted SET process affords the bromo radical intermediate, which discloses the radical process. Thus, considering the pathway 1, the substrate 52 is initially oxidized by the bromo radical (Br•), releasing HBr and generating the 2-alkynylbenzoic acid radical A. Once formed, A is reduced by another equivalent of Br•, to produce the alkynylbenzoic acid anion B and a bromonium ion (Br+). Following on from this, the intermediate B undergoes a 5-exo-dig cyclization to form the isobenzofuran-1-one anion C, which combines with the bromonium ion, to afford the respective product 54. On the other hand, in the pathway 2, the bromo radical (Br•) firstly adds to the C≡C bond, giving the intermediate D, which is oxidized by Oxone, to afford the vinyl cation intermediate E. Subsequently, an annulative process drives the reaction toward the intermediate F, which is finally converted to the desired product 54 by deprotonation (Scheme 36).
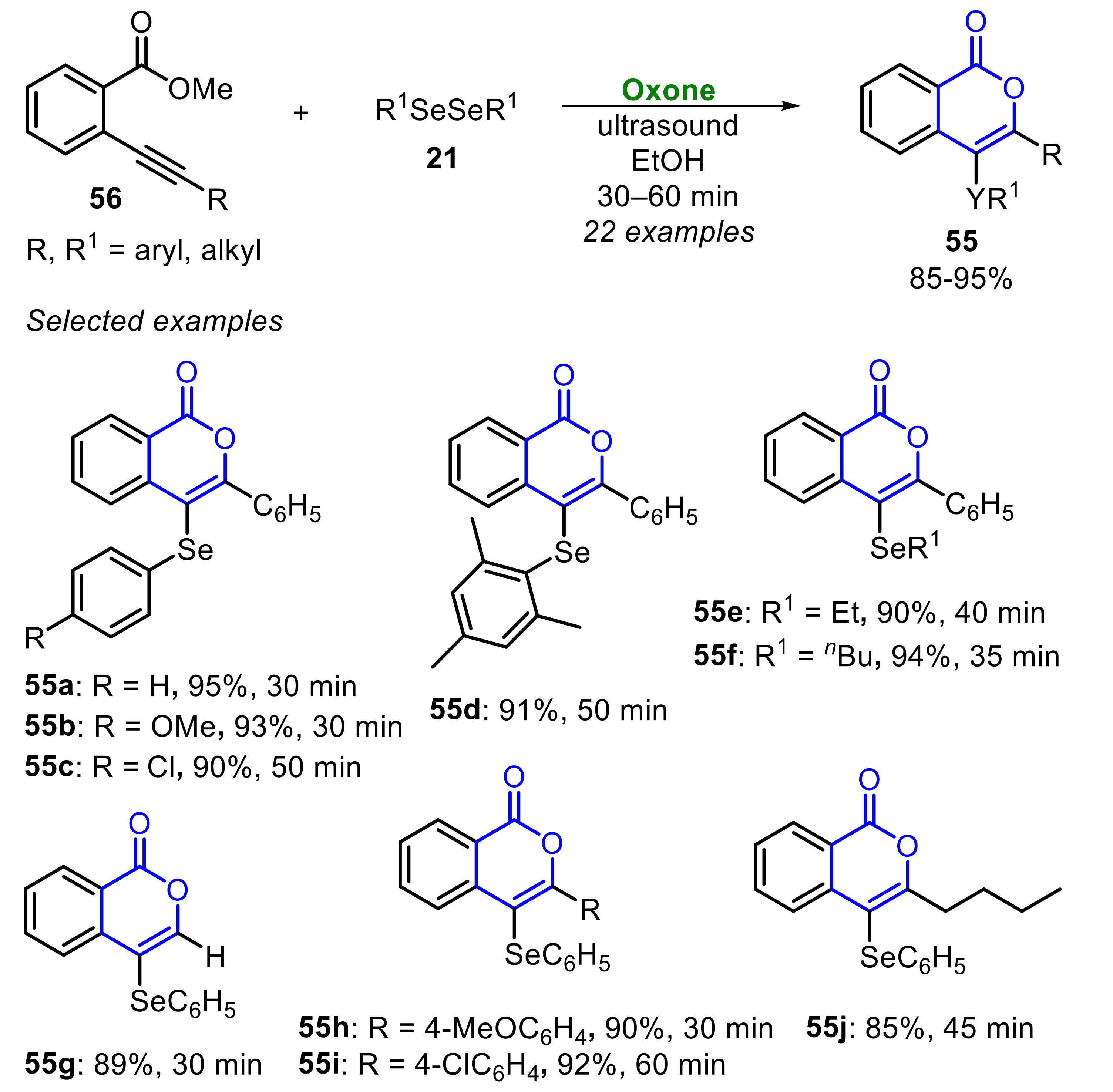
Very recently, some of us [40] described a simple and general Oxone/diselenide-promoted ultrasound-assisted protocol to prepare 4-chalcogenyl-1H-isochromen-1-ones 55, through a 6-endo-dig electrophilic cyclization of 2-alkynylaryl esters 56 (Scheme 37) [40]. By using EtOH as solvent, the reaction scope to access Se-containing isochromen-1-ones 55 was investigated using several diorganyl dichalcogenides 21, 41, 42 and 2-alkynylaryl esters 56. Diaryl and dialkyl diselenides 21 were suitable substrates, yielding the products 55a-f in excellent yields, with the reaction time ranging between 30 and 50 min. 2-Alkynylaryl esters 56 bearing a diversity of alkynyl-substituted groups (R groups) reacted smoothly to produce the products 55g–j in very good to excellent yields, in a reaction time ranging from 30 to 60 min. For instance, some examples can be highlighted, including (1) the synthesis of C3-free isochromen-1-ones 55g, which may be derivatized through a TM-catalyzed cross-coupling strategy, as well as (2) the synthesis of the C3-alkyl derivative 55j, both in high yields (Scheme 37).
The reaction scope was expanded to tellurium-containing isochromen-1-ones 55k–n, employing glycerol as solvent. Neutral and electron-rich aryl ditellurides 41 reacted smoothly to produce the respective products 55k–m in very good yields, under sonication for 1 h. Surprisingly, 4-chlorophenyl ditelluride and dibutyl ditelluride were not able to afford the respective tellurium-containing isochromen-1-ones 55; the formation of the 3-phenyl-1H-isochromen-1-one 55n was observed in these cases. Moreover, a limitation was also found when diphenyl disulfide 42 was employed as substrate, and the expected sulfur-containing derivative 55o could not be obtained (Scheme 38).
In 2019, Ishihara and co-workers [41] described the Oxone-promoted dearomatization of functionalized arenols in the presence of quaternary ammonium hypoiodite. In this study, the authors proposed the use of two chiral quaternary ammonium hypoiodites (Figure 1), and carried out the oxidation of phenols, 1- and 2-naphthols.
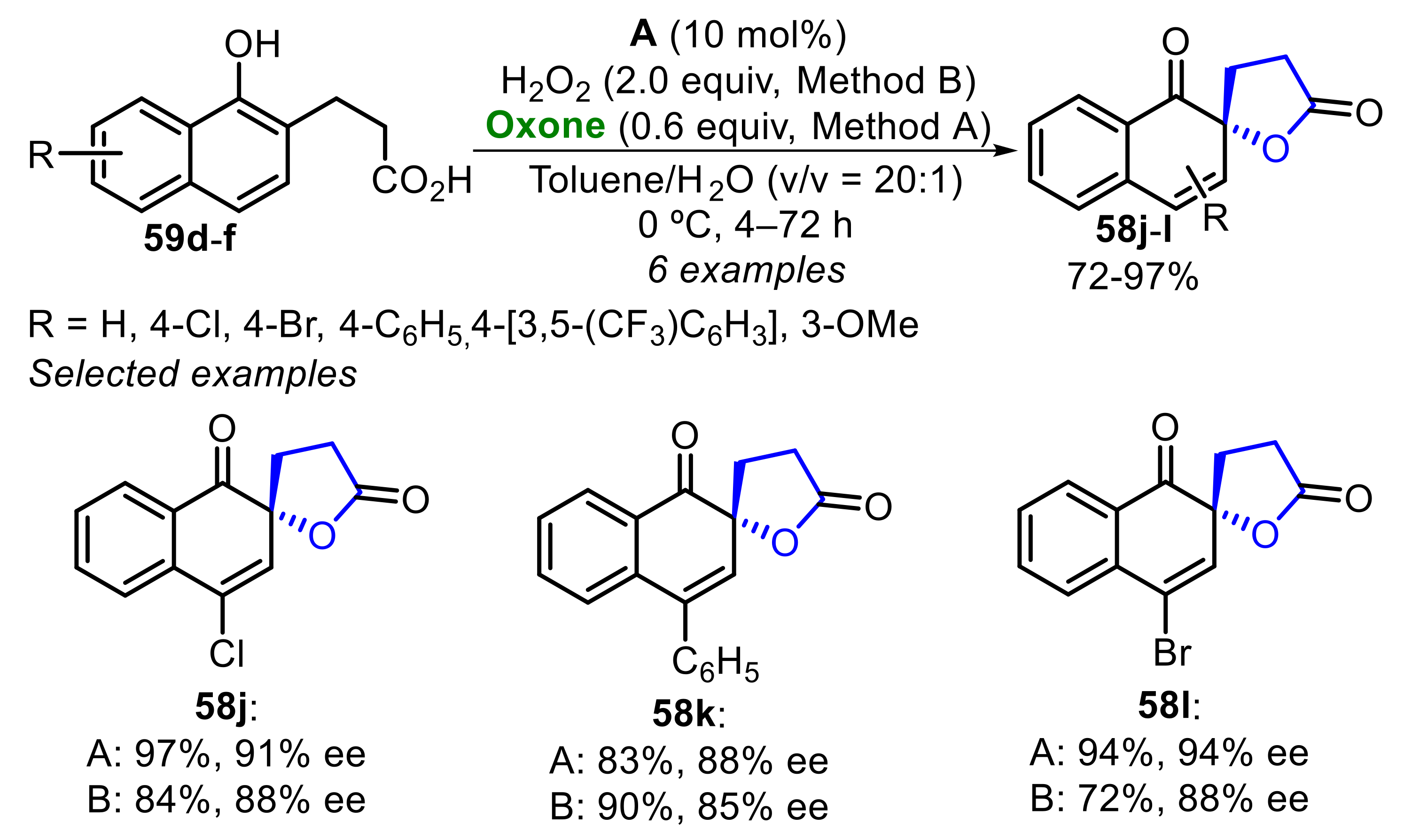
Initially, several phenols substituted with aliphatic groups were used for the enantioselective oxidative spirolactonization using chiral ammonium hypoiodite A or B (Scheme 39). When using the phenols 57a–f, the corresponding spirolactones 58a–f were obtained at moderate to good yields (73–95%) and with good to high enantioselectivity (73–93% ee) after 16–48 h. It was observed that when naphthols with bulky substituents in the ortho-position were used, a decrease in the enantioselectivity of the respective compounds 58a, 58b and 58e was observed (Scheme 39).
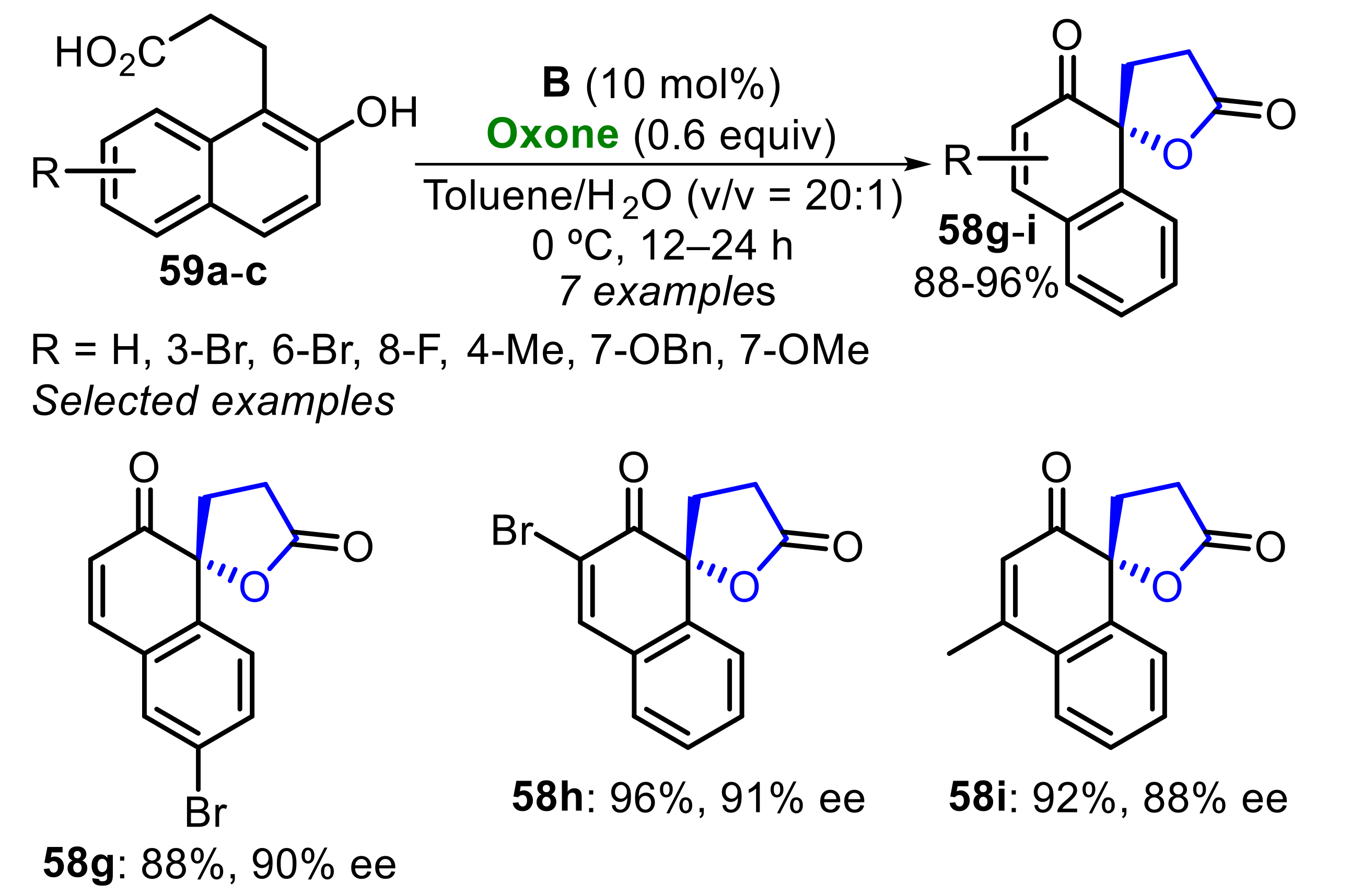
Concomitantly, under the optimized conditions, several 2-naphthol 59a–c derivatives were oxidized to the corresponding spirolactones 58g–i in very good to excellent yields (88–96%), and with high enantioselectivity (85–91% ee), regardless of the electronic nature and position of the substituents (Scheme 40). The best hypoiodite in this reaction was the derivative B.
On the other hand, the oxidation of 1-naphthols 59d–f was examined using hypoiodite A/Oxone catalysis under two conditions: the optimal one described above (method A), and for comparison the reactions were carried out using 2 equiv. of H2O2 (method B), this condition was tested in the reaction optimization step. Thus, the corresponding spirolactones 58j–l were obtained at higher chemical yields and enantioselectivity, and in shorter reaction times when using method A. These results again demonstrated the good efficiency of hypoiodite/Oxone catalysis (Scheme 41).
4. Organochalcogen-Containing Heterocycles
Chalcogenophenes are a valuable class of heterocyclic compounds, which play an important role as building blocks in organic synthesis [42], in the development of new materials [43,44], and in the prospection of new drugs in the pharmaceutical industry [45,46]. Due to these important features, the search for the development of simple and efficient methods to access chalcogenophenes has attracted interest during the last decades. In this context, the use of the system Oxone/dichalcogenide has demonstrated to be a very efficient strategy to access chalcogenophenes through chalcogen-based electrophilic cyclization processes.
In 2020, some of us reported an Oxone/dichalcogenide-promoted synthesis of 3-selanyl-chalcogenophenes and 3-tellanylchalcogenophenes 60–62, through the electrophilic cyclization of (Z)-chalcogenoenynes 63–65, under ultrasound irradiation (Scheme 42) [47]. The annulation of (Z)-1-butylselen-1-en-3-ynes 63 was conducted in the presence of several diorganyl diselenides 21 and diphenyl ditelluride 41a, affording the respective 3-arylselanyl- and 3-aryltellanylselenophenes 60 in poor to very good yields. Initially, by employing diphenyl diselenide 21a, the respective product 60a was obtained in 87% yield. On the other hand, the reaction was remarkably affected by the presence of substituents attached to the Se-phenyl ring, and the products 60b-d were obtained in just 40–54% yield. Heteroaryl diselenides 21 (R = 2-naphtyl and 2-thienyl) were smoothly submitted to the optimized reaction conditions to afford the corresponding products 60e and 60f in 59% and 42% yield, respectively. It is worth to mention that dibutyl diselenide 21 reacted efficiently to produce the selenophene 60g in 60% yield. Finally, as already observed in other Oxone-promoted oxidative cleavage of the chalcogen–chalcogen bond, diphenyl disulfide 42a failed to access the expected product 60h, whereas diphenyl ditelluride 41a reacted satisfactorily to be converted to the tellurium-functionalized selenophene 60i at a 50% yield (Scheme 42).
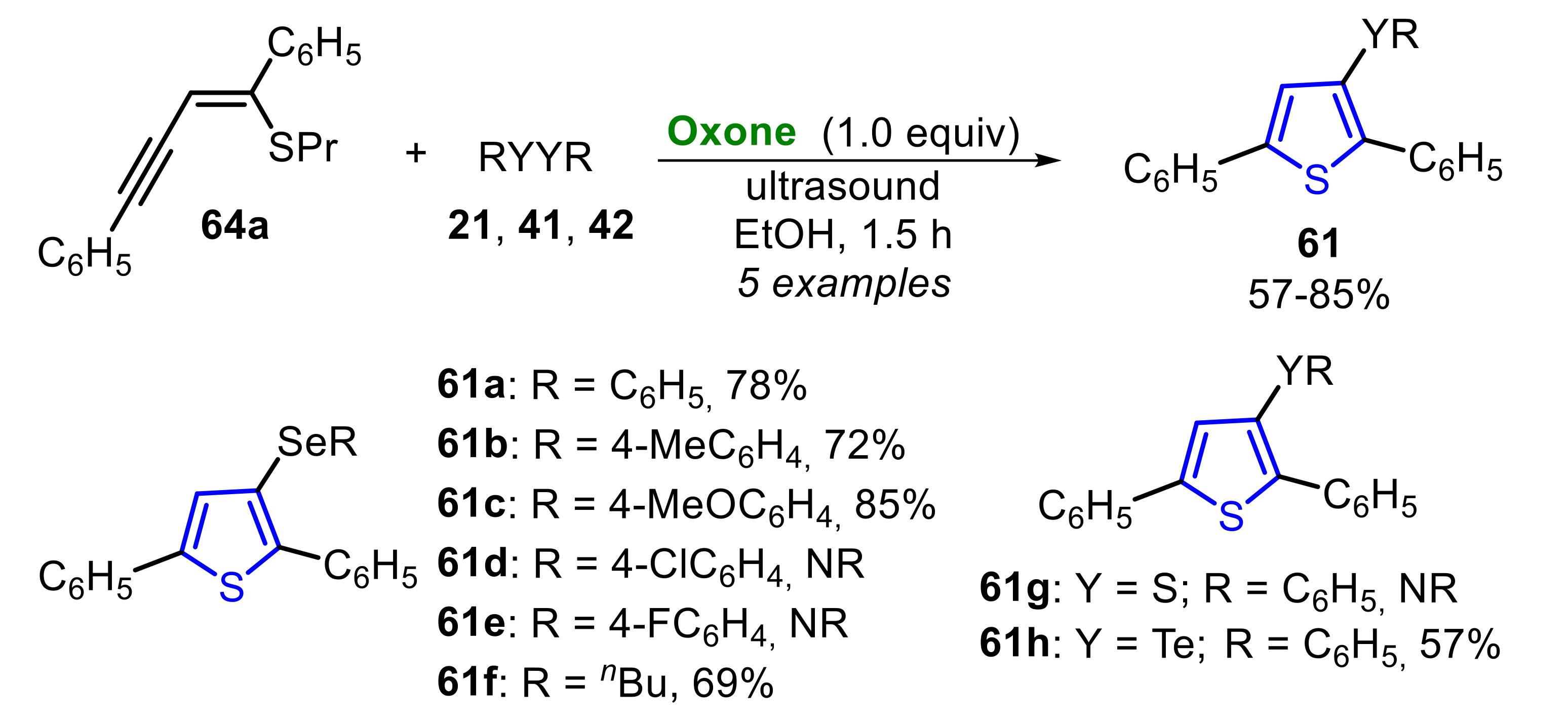
The reaction scope was expanded to (Z)-(1,4-diphenylbut-1-en-3-yn-1-yl)(propyl)sulfane 64a at the same optimal conditions, aiming to access a library of chalcogen-functionalized thiophenes 61 (Scheme 43). The authors observed that neutral and electron-rich diaryl diselenides 21 were suitable electrophile precursors, affording the respective 3-selanylthiophenes 61a–c in 72% to 85% yield. However, the presence of halogen at the para-position of the Se-phenyl ring of the diaryl diselenide (R = 4-FC6H4 and 4-ClC6H4), prevented the formation of the respective thiophenes 61d and 61e. Dibutyl diselenide 21f reacted smoothly to produce 3-(butylselanyl)-2,5-diphenylthiophene 61f at a 69% yield. Additionally, in this case, diphenyl disulfide 42a did not react with enyne 64a to produce 3-sulfanylthiophene 61g, whereas diphenyl ditelluride 41a was efficient in the synthesis of 3-(phenyltellanyl)-2,5-diphenylthiophene 61h, obtained at a 57% yield (Scheme 43).
Additionally, the protocol was expanded for the synthesis of tellurophenes 62, using (Z)-butyl(1,4-diphenylbut-1-en-3-yn-1-yl)tellane 65a in the reaction with Oxone/dichalcogenide, in the presence of glycerol as solvent. In this case, the reaction temperature could be increased to up 100 °C under ultrasound, which was necessary to allow the cyclization (Scheme 44). Under this condition, electron-rich and electron-deficient diaryl diselenides 21 reacted adequately to produce the products 62a–c at moderate yields, whereas dibutyl diselenide 21f afforded the tellurophene 62d in 37% yield. As previously observed, the protocol was not suitable to diphenyl disulfide 42a, whereas 3-(phenyltellanyl)-2,5-diphenyltellurophene 62f was obtained in only 28% by the reaction between 65a and diphenyl ditelluride 41a under the optimal conditions (Scheme 44).
The proposed reaction mechanism for the electrophilic cyclizations follows an ionic and a radical step simultaneously (Scheme 45). Initially, the reactive intermediates A and B are obtained via anionic or radical pathways, by the reaction between the diorganyl dichalcogenide 21 or 41 and potassium peroxymonosulfate (KHSO5, the active component of Oxone). Once formed, the intermediate B can be protonated to afford the strongest electrophile B′. In the presence of chalcogenoenynes 63, 64 or 65, the intermediates A and B′ are converted to the chalcogenironium intermediate C, that undergoes an intramolecular annulation to form the intermediate D. Finally, an alkyl group displacement, promoted by a nucleophilic species from the reaction medium, yields the desired chalcogenophenes 60–63 (Scheme 45).
In 2021, some of us reported an Oxone/dibutyl diselenide-promoted 5-endo-dig electrophilic cyclization of 1,3-diynes 66, to access selectively 3,4-bis(butylselanyl)selenophenes 67 and 4-alkoxyselenophenes 68 (Scheme 46) [48]. The selectivity in the formation of the products 67 and 68 was achieved by controlling the solvent (MeCN or ROH) and the dibutyl diselenide 21f amount (2 equiv or 1.5 equiv). Thus, by employing 2 equiv of dibutyl diselenide 21f in the presence of MeCN as solvent, 3,4-bis(butylselanyl)selenophenes 67 were prepared in moderate to good yields, after being stirred at 80 °C. Neutral and electron-rich 1,3-diynes 66 (R = H, OMe and Me) reacted adequately to yield the products 64a, 67b and 67c in 78%, 50% and 70% yield, respectively. On the other hand, the electron-deficient 1,4-bis(4-chlorophenyl)buta-1,3-diyne (66d, R = 4-ClC6H4) could not be converted to the expected product 67d, even after 72 h of reaction. By employing dodeca-5,7-diyne (66e, R = nBu) as substrate, a complex mixture of products was obtained. Interestingly, 1,4-di(naphthalen-2-yl)buta-1,3-diyne 66f allowed the formation of the selenophene 67f in 40% yield, after 48 h. Finally, dibutyl ditelluride 41d and dimethyl disulfide 42b were submitted to the optimized reaction conditions; however, both were not able to produce the expected products 67g and 67h, even after 72 h of reaction (Scheme 46).
On the other hand, by employing 1.5 equiv of dibutyl diselenide 21f and aliphatic alcohols as solvent/nucleophiles under reflux, the reaction was driven selectively to 3-(butylselanyl)-4-alkoxyselenophenes 68, that were afforded in low to very good yields (Scheme 47). Initially, several alcohols (EtOH, MeOH, iPrOH and tBuOH) were employed as solvent in the presence of the 1,3-diyne 66a and dibutyl diselenide 21f. Among them, EtOH and MeOH were able to conduct the reaction adequately, affording the products 68a and 68b at a 70% and 75% yield, respectively. On the other hand, in the presence of the sterically hindered iPrOH and tBuOH, a remarkable loss of efficiency was faced, with product 65c (R1 = iPr) being obtained at a 35% yield, whereas 68d (R1 = tBu) could not be obtained. Different symmetrical 1,3-diynes 66 where employed as substrate in the reaction with dibutyl diselenide 21f in the presence of EtOH, affording the products 68e–i. For instance, the electron-rich methoxy-derivative diyne (R = 4-MeOC6H4) afforded 68e in just 35% yield, whereas the p-tolyl analogue (R = 4-MeC6H4) was a more suitable substrate, affording 68f in 80% yield. On the other hand, no product 68g was observed when the para-chlorophenyl ring was attached to the 1,3-diyne (R = 4-ClC6H4). When the sterically hindered ortho-substituted diynes 66e (R = 2-MeC6H4) and 66f (R = 2-ClC6H4) were used, the respective 4-ethoxyselenophenes 68h and 68i were obtained both at a 15% yield after 72 h. Alkyl-substituted 1,3-diyne (66e, R = nBu) and hexa-2,4-diyne-1,6-diol (66h, R = CH2OH) afforded a complex mixture of products, not yielding the respective selenophenes 68j and 68k. As observed above, in the reactions in acetonitrile (Scheme 42), dibutyl telluride 41d and dimethyl disulfide 42b were not suitable dichalcogenides in this reaction, and the formation of products 68l and 68m was not observed, even after 48 h (Scheme 47).
The same strategy was used by some of us to access selectively selenophene-fused benzo[b]chalcogenophenes 70, through the intramolecular cyclization of ortho-1,3-diynyl phenyl chalcogenides 69 (Scheme 48) [49]. The optimal conditions were efficiently applied to substituted 1,3-diynes 69 bearing electron-neutral and electron-rich aryl groups, to produce the products 70a–d at excellent yields. Good results were obtained even in the presence of a chlorine a the ortho-position, giving the product 70d was obtained in 85% yield. Interesting, symmetric ortho-thiosubstituted 1,3-diyne 69e was a suitable substrate, being satisfactorily converted to 3,3′-bis(butylselanyl)-2,2′-dibenzo[b]thiophene 70e in 77% yield, after 3 h. Additionally, unsymmetric [2-(phenylbuta-1,3-diin-1-yl)phenyl](propyl)sulfide 69f reacted smoothly to produce the respective selenophene 70f in 85% yield, after 1 h. The electron-rich substrate 69g (R1 = 4-MeC6H4) was more reactive in comparison with the electron-deficient one 69h (R1 = 4-ClC6H4), and the respective products 70g and 70h were obtained at a 80% and 70% yield, after 2 and 3 h, respectively (Scheme 48).
In 2019, some of us reported the Oxone/diselenide-promoted electrophilic cyclization of 2-functionalized chalcogenoalkynes 71, allowing the access to benzo[b]chalcogenophenes 72, in moderate to excellent yields (Scheme 49) [50]. It is important to highlight the high substrate tolerance presented by the method, allowing the use of alkynes with different alkyl, or electron-rich and electron-deficient aryl substituents.
5. Miscellaneous Cyclizations
Besides the several important transformations discussed above, many other Oxone-promoted cyclization processes have been described during the last few years, to prepare a wide variety of compounds.
In 2014, Punniyamurthy and co-workers described the use of Oxone as a green oxidant in the organocatalytic synthesis of 2-arylbenzoxazoles 73 and 2-arylbenzothiazoles 74 (Scheme 50) [51]. In the presence of 1-iodo-4-nitrobenzene (20 mol%) as catalyst, Oxone (1.5 equiv), TfOH (3.0 equiv) and hexafluoro-2-propanol (HFIP, 2.5 mL) as solvent, N-4-tolylbenzamides 75 were converted to benzoxazoles 73 in poor to excellent yields, under room temperature.
In parallel, several electron-rich and electron-deficient N-phenylthiobenzamides 76 were employed as substrate to access 2-arylbenzothiazoles 74, under similar conditions, at room temperature (Scheme 51). The protocol showed an excellent substrate tolerance, allowing the use of different N-phenylthiobenzamides 76 to prepare a total of twenty-seven substituted benzothiazoles 74 in poor to excellent yields.
The proposed reaction mechanism starts by the generation of the hypervalent iodine(III) species II, by reaction of aryl iodide I, TfOH and Oxone (Scheme 52). The species II is able to catalyze the oxidative annulation of substrates 75 or 76 to produce the intermediate III, that can be stabilized by HFIP. The intramolecular cyclization of III affords the cationic intermediate IV, releasing iodobenzene I to the reaction medium, which is eventually converted to II to restart the process. Finally, the intermediate IV is easily converted to the aromatic products 73 and 74 by deprotonation.
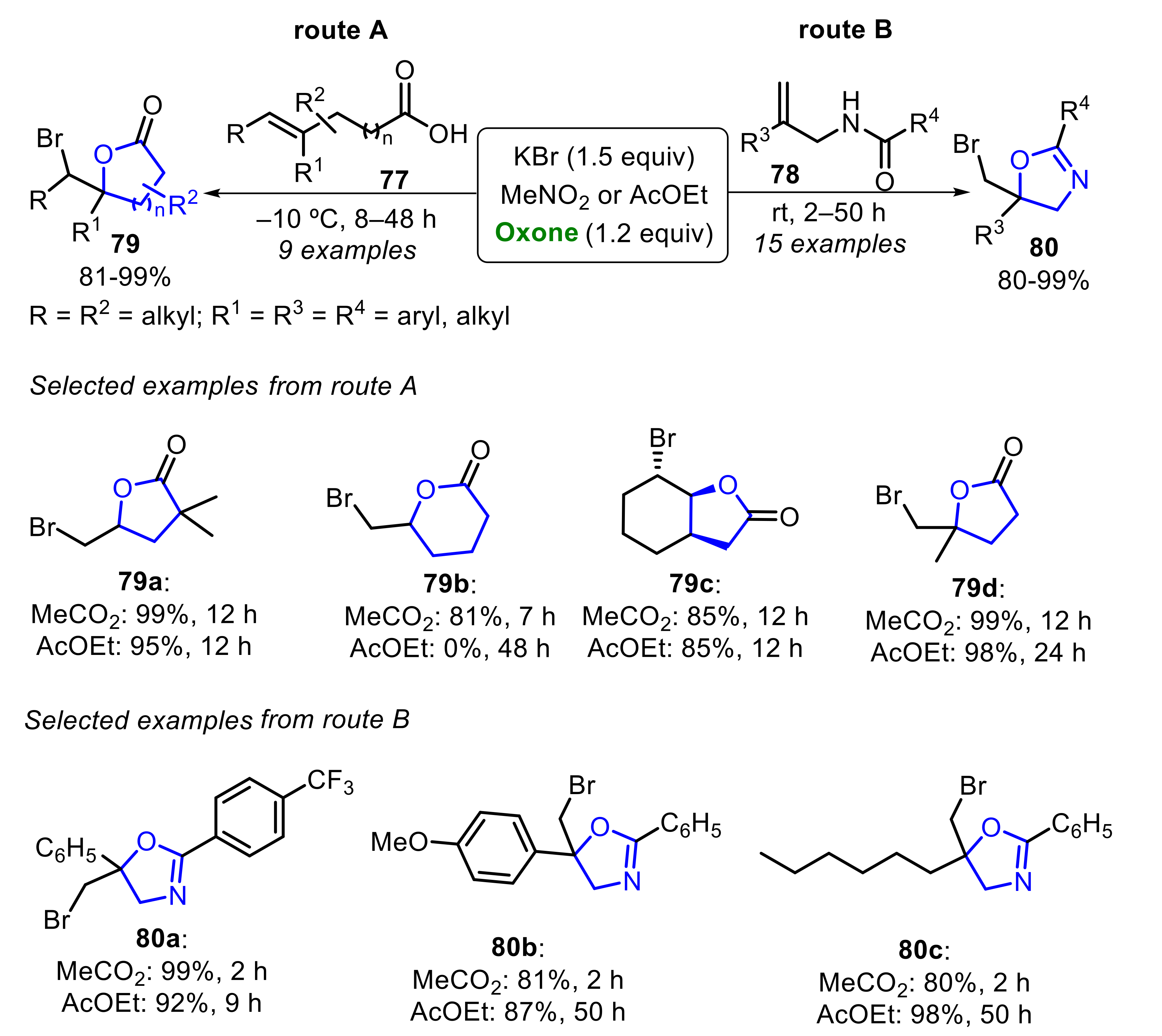
Togo and co-workers described, in 2015, the Oxone/KBr-promoted bromo-lactonization of alkenyl carboxylic acids 77 and bromo-cyclization of N-allyl amides 78 to provide dihydrofurans 79 and dihydrooxazoles 80, respectively (Scheme 53) [52]. The reaction optimization study revealed that MeNO2 and AcOEt, acting as solvent, remarkably increased the bromo-lactonization diastereoselectivity. In the presence of MeNO2, several substrates α,α-disubstituted 77 (gem- and vic-disubstituted alkenes) were converted to the corresponding products 79 in very good to excellent yields (Scheme 53, route A). Additionally, the six-membered ring cyclization proceeded efficiently to produce δ-lactone 79b at a 81% yield. The bromo-lactonization in AcOEt presented similar behavior, except to access the six-membered ring derivative 79b, in which no product was observed. Subsequently, the bromo-lactonization of α-substituted N-allyl amides 78 was investigated, and both MeNO2 and AcOEt, were able to produce the desired products 80 at very good to excellent yields (Scheme 53route B).
The Oxone/NBS-promoted synthesis of 2-aminobenzimidazoles 81 and 2-aminobenzoxazoles 82 was reported in 2016, by Kumar and co-workers, starting from cyclohexanone 83 and guanidine/urea 84 as substrates (Scheme 54) [53]. The Oxone/NBS system promoted the in situ halogenation of cyclohexanone 83, which reacted with electron-rich and electron-deficient guanidine/urea 84, affording the respective products 81 and 82 at good to excellent yields. The whole features of this strategy make it a simple and economical approach to prepare 2-aminobenzimidazoles 81 and 2-aminobenzoxazoles 82.
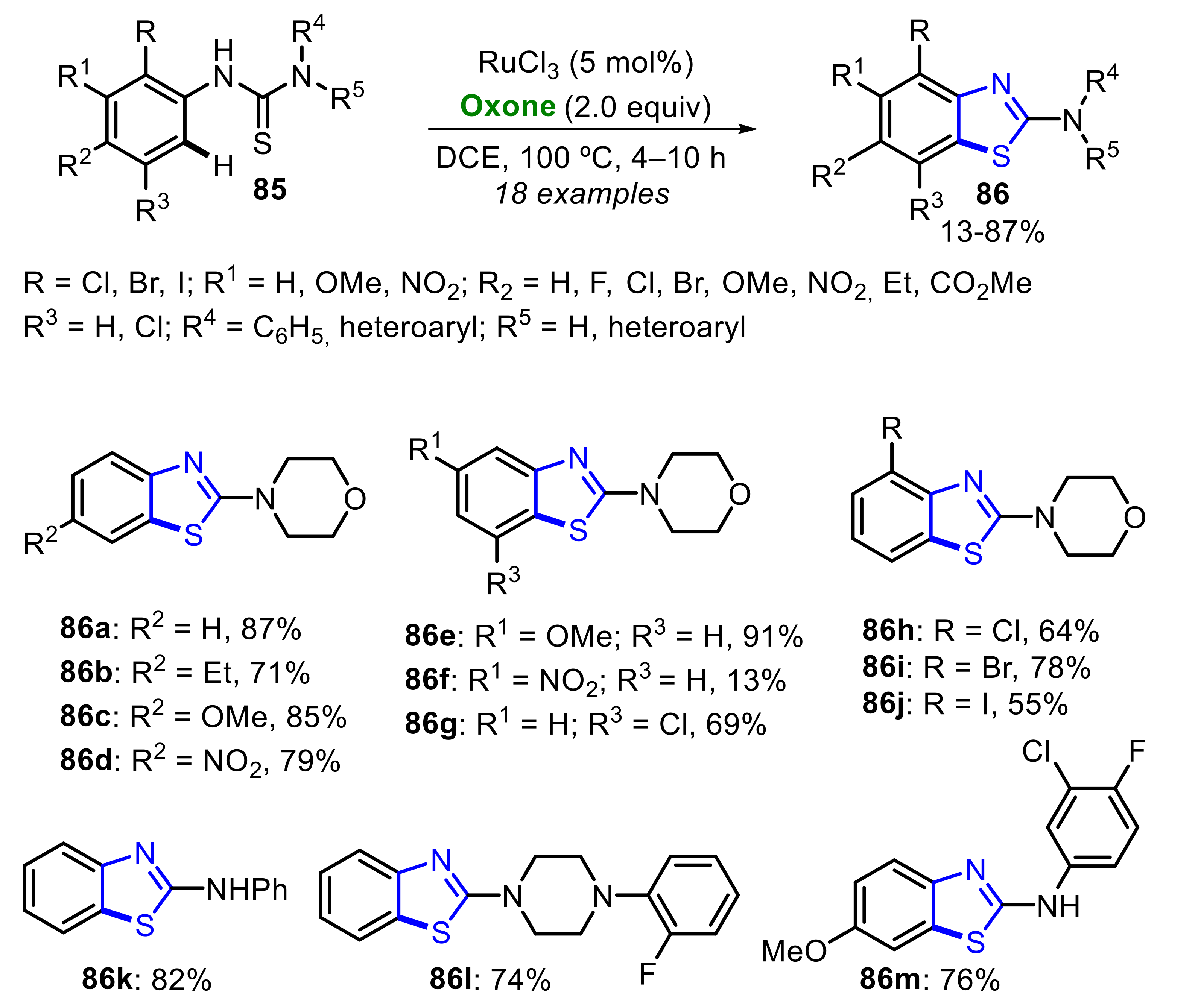
In 2016, Sawant and co-workers have reported a Ru-catalyzed Oxone-mediated intramolecular C−S coupling of N-arylthioureas 85, aiming to access 2-aminobenzothiazoles 86 (Scheme 55) [54]. A good substrate tolerance was achieved, allowing the synthesis of a wide library of 2-aminobenzothiazoles 86 in poor to excellent yields. In general, electron-rich N-arylthioureas 85 were more reactive in relation to the electron-poor ones. An example of this remarkable difference can be observed by comparing the access to the products 86e (R1 = OMe) and 86f (R1 = NO2), which were obtained at a 91% and 13% yield, respectively. This specific reactivity indicates the involvement of an electrophilic ruthenation mechanistic pathway, which is supported by the observed inverse secondary kinetic isotopic effect (KIE) effect and by the computational studies.
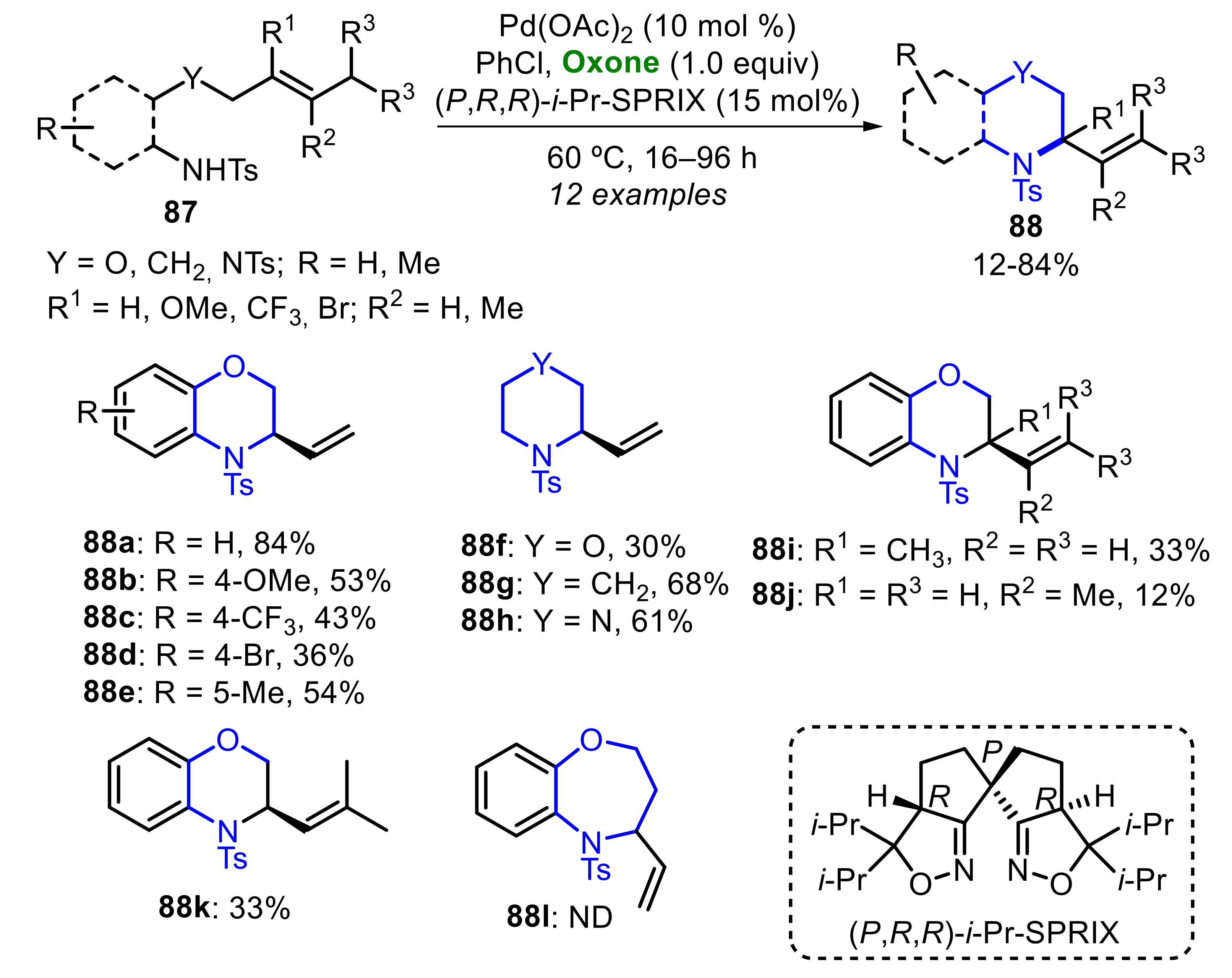
In 2018, Sasai and co-workers developed a Pd(II)-catalyzed Oxone-mediated enantioselective aza-Wacker type reaction of alkenyl sulfonamides 87, to afford several substituted heterocyclic systems 88, including morpholines, piperazines and piperidines, as well as their benzo-fused derivatives (Scheme 56) [55]. Electron-rich and electron-deficient alkenyl sulfonamides 87 were adequately employed as substrates to access the products 88b-e, and any remarkable difference on reactivity was observed. The presence of the phenylene binder in the starting material was not mandatory, and the products 88f–h were satisfactorily obtained. Limitations were found when trisubstituted alkenes 87 were used, and the respective products 88i–k were obtained at just a 33%, 12% and 33% yield. The attempts in preparing the benzo[b][1,4]oxazepine 88l were not successful (Scheme 56).
In 2018, Imai and Togo reported an Oxone-mediated synthesis of 3-arylisoxazole-4,5-dicarboxylates 89, through the one-pot reaction between benzylic chlorides 80 or alkyl 4-tosylates 91, and acetylenedicarboxylate 50p (Scheme 57) [56]. The substrates 90 and 91 were treated with N-methylmorpholine N-oxide (NMO, 4 equiv), and then with hydroxylamine hydrochloride (1.2 equiv) and potassium carbonate (0.6 equiv), followed by the addition of acetylenedicarboxylate 50p, to generate desired products 89. The method efficiency was proved by accessing a library of twenty-three new isoxazoles 89 at up to a 97% yield and in short reaction times.
The study was extended to different acetylene derivatives 50 in the reaction with 4-chlorobenzyl chloride 90a, and the desired isoxazoles 89h–m were obtained at moderate to good yields (Scheme 58). As previously observed, a good substrate tolerance was afforded, allowing the application of electron-rich and electron-deficient acetylenes 50.
Bhatt and co-workers reported, in 2019, a Cu(II)-catalyzed Oxone-mediated one-pot synthesis of 3,5-diarylisoxazoles 92, starting from α,β-unsaturated ketones 93 and hydroxylamine hydrochloride, via an oxidative annulation reaction (Scheme 59) [57]. The protocol was successfully applied to a variety of electron-rich and electron-deficient chalcones 93 affording a wide collection of compounds 92a–o in good to excellent yields, just after a few minutes under thermal conditions (refluxing EtOH).
In 2019, some of us reported an Oxone/dialkyl diselenide-promoted synthesis of 5H-selenopheno[3,2-c]isochromen-5-ones 94, through a double intramolecular cyclization of methyl 2-(organyl-1,3-diynyl)benzoate 95 (Scheme 60) [58]. Dialkyl diselenides 21 (R2 = Et, Bu and Oct) were satisfactorily employed, giving the corresponding isochromenones 94a–c in good yields, whereas dibenzyl diselenide, bis(2-naphthylmethyl)diselenide, dibutyl ditelluride and dimethyl disulfide were not suitable to produce the products 94d, 94e, 94f and 94g, respectively. Additionally, several substituted 1,3-diynes were efficiently employed as substrates, affording the products 94h–k at moderate to good yields (Scheme 60).
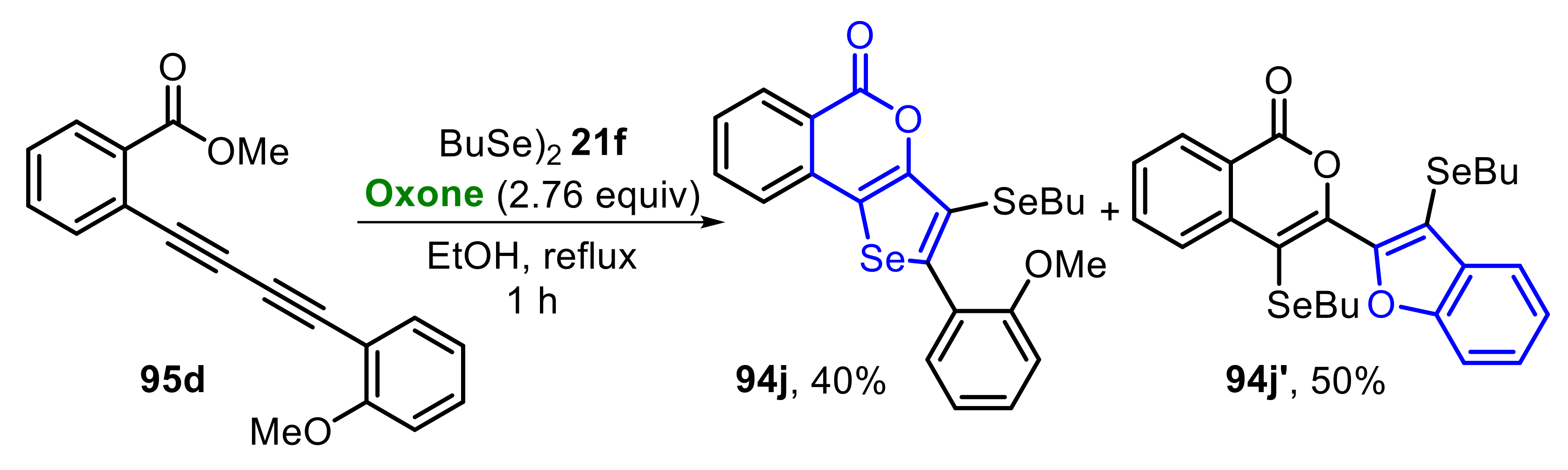
Curiously, when 2-(2-methoxyphenylbuta-1,3-diynyl)benzoate 95d was employed as substrate, in the presence of dibutyl diselenide 21f, the expected 5H-selenopheno[3,2-c]isochromen-5-one 94j was isolated in only 40% yield. The low efficiency toward 94j is associated with the parallel formation of benzofuran derivative 94j′, that was obtained at a 50% yield (Scheme 61). This behavior is due to two competing reactions (Se-cyclization vs. O-cyclization) of the intermediate IX, generated in the first step of the transformation (see Scheme 62, for the reaction mechanism).
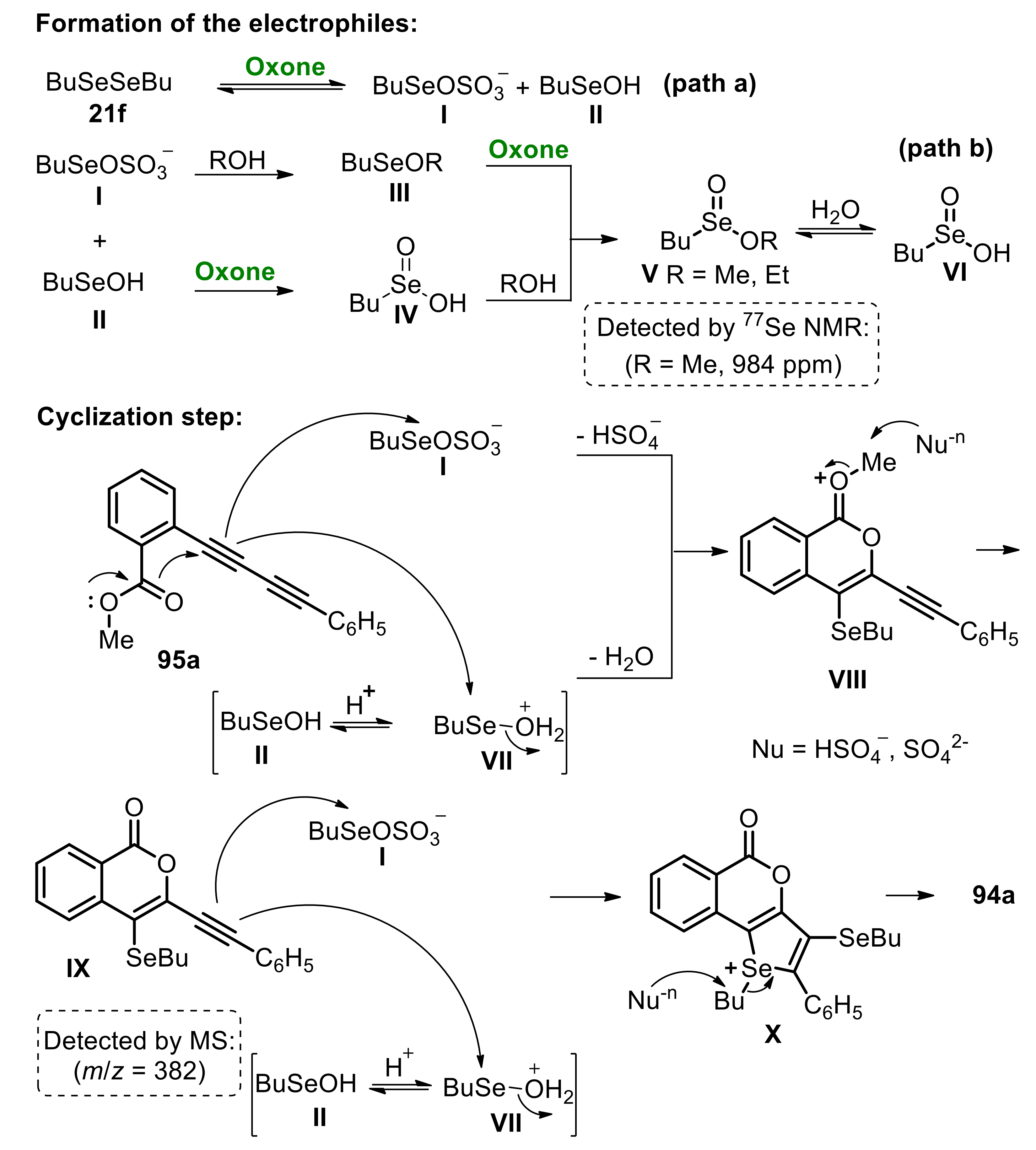
Based on control experiments, which were monitored by 77Se NMR and GC/MS analysis, a possible mechanism for the reaction was proposed (Scheme 62). Initially, dibutyl diselenide 21f reacts with Oxone to form two intermediates: BuSeOSO3– I and BuSeOH II (Scheme 62, path a). The formation of species I and II was confirmed by the formation of seleninic ester V and acid VI (Scheme 62, path b). The species II can be protonated by the reaction medium, leading to BuSeOH2+ VII. Then, the 1,3-diyne 95a reacts with I and VII to form the cyclic intermediate VIII, releasing HSO4- and H2O to the reaction medium. Following, the displacement of the methyl group from VIII, by a nucleophile (HSO4- and/or SO42-) affords the intermediate IX (detected by mass spectrometry), that reacts similarly with I and VII, to produce the fused-selenophene cation intermediate X. The displacement of the butyl group finally affords the expected product 94a (Scheme 62).
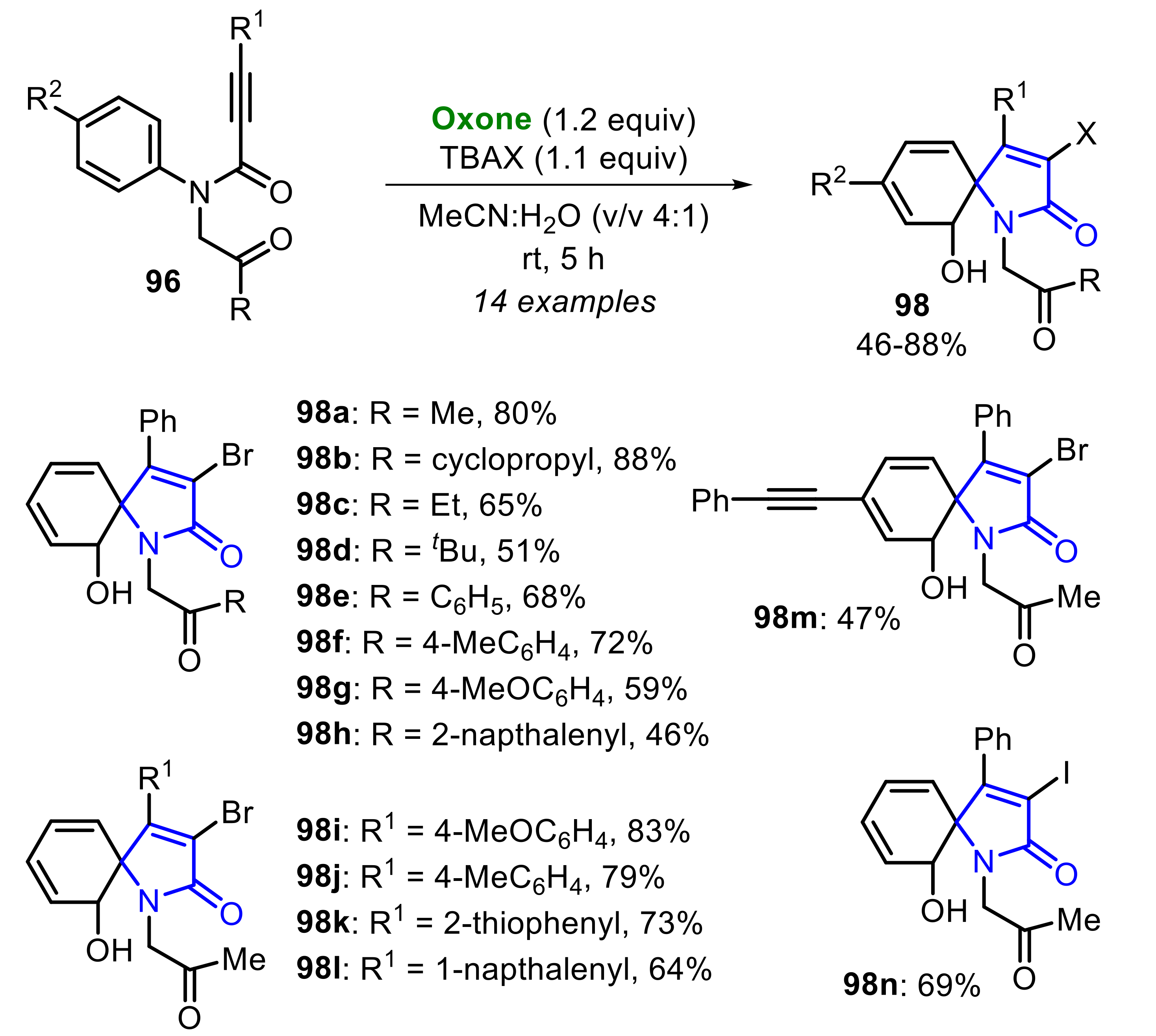
In 2020, Qiu, Liu and co-workers developed the Oxone-mediated ipso-cyclization of N-arylpropiolamide 96 to access the spirocyclo derivatives 97 and 98 (Scheme 63). The reaction was conducted in the presence of MeCN:H2O (v/v 4:1) as reaction medium, and the selectivity was achieved by employing different halogen sources (ZnX2 and TBAX) [59]. In the presence of ZnX2, the spiro-tricycle 97 was formed, through an ortho-hydroxylative process, in moderate to good yields. By employing N-(2-oxopropyl)-N,3-diphenylpropiolamide 96a, the formation of a mixture of the products 97a and 98a was observed. When para-substituted substrates 96 (R = C6H5 and Cl) were used, the expected products 97b–c were obtained at good yields, without formation of the ring-opened derivatives. Interestingly, the fluoro-substituted substrate 96 was suitable to the reaction conditions, giving the respective product 97e at a 61% yield (Scheme 63).
Due to the poor stability of spiro-tricycles 97, authors have focused their attention on the synthesis of ring-opened products 98, that were accessed by employing TBAX as halogen source, and the protocol was extended to N-arylpropiolamide 96 bearing different R, R1 and R2 substituents (Scheme 64). The R group can be replaced by a cyclopropyl ketone, ethyl ketone, tbutyl ketone, and several aryl ketones, giving a range of ortho-hydroxy spirocycles 98a h in 46–88% yields. Additionally, the substituent R1 can be easily replaced by aryl or heteroaryl groups, leading to the respective products 98i–l in good yields. By employing the para-alkynyl N-arylpropiolamide 96, the product 98m was obtained at only a 47% yield. Moreover, TBAI has also demonstrated to be efficient, allowing the synthesis of the compound 98n at a 69% yield.
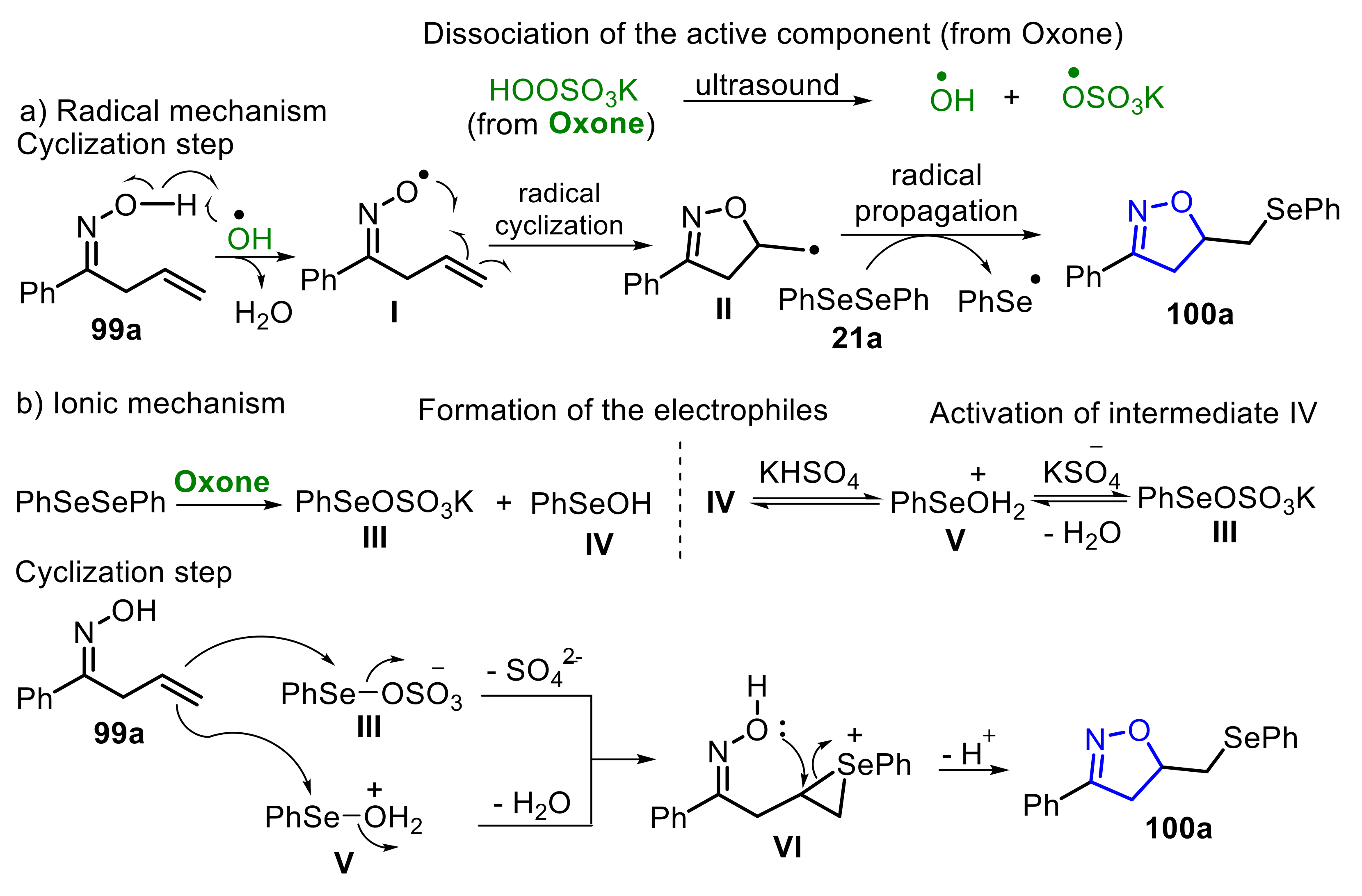
In 2020, some of us described the Oxone/diselenide-promoted ultrasound-assisted selective cyclization of unsaturated oximes 99, to access 5-methylselanyl-4,5-dihydroisoxazoles 100 (Scheme 65) [60]. The sonication was crucial to drive the process selectively, in short reaction times, giving the products 100 in moderate to excellent yields. Several oximes 99 and diselenides 21, bearing electron-donating or electron-withdrawing groups, were satisfactorily employed, proving the applicability and substrate tolerance of the method.
A comparison study, employing different energy sources (conventional heating, microwave and ultrasound irradiation) was conducted by the authors. Under ultrasound irradiation the process followed a radical pathway, as verified by control experiments. On the other hand, by employing conventional heating (65 °C) or microwave irradiation, the transformation followed an ionic pathway. Regarding the radical pathway, the first step is the formation of the hydroxyl radical (HO•) and the radical species •OSO3K, through the US-promoted Oxone dissociation. Once formed, the hydroxyl radical reacts with oxime 99a to form H2O and the O-centered radical intermediate I, which undergoes a radical cyclization, delivering the 5-methyl-4,5-dihydroisoxazole radical II. Following, the intermediate II reacts with diphenyl diselenide 21a to yield the desired product 100a, while releasing a Se-centered radical species, that is recovered as diselenide 21 (Scheme 66a). Regarding the ionic mechanism, the first step is the Oxone-promoted formation of the Se-based electrophilic species III and IV, by the Se–Se bond oxidation. The species IV is protonated to produce the most electrophilic species V. Then, the oxime 99a reacts with III and/or V to be converted to the seleniranium intermediate VI, which undergoes an intramolecular cyclization process, followed by deprotonation and formation of the desired product 100a (Scheme 66b).
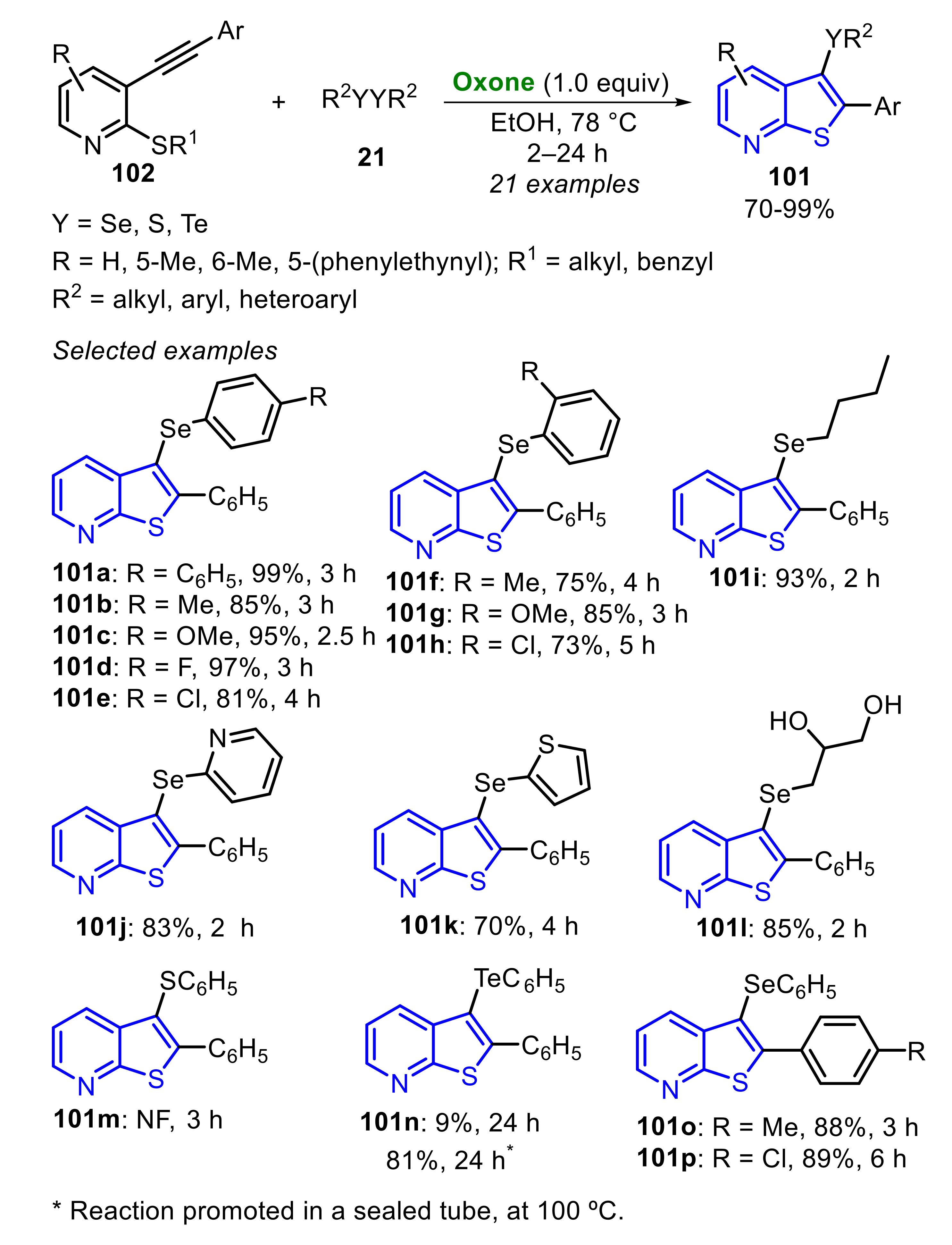
In 2021, some of us described the Oxone/diselenide-promoted synthesis of 2-aryl-(3-organocalcogenyl)thieno[2,3-b]pyridines 101 through the electrophilic cyclization of 3-(arylethynyl)-2-(alkylthio)pyridines 102 (Scheme 67) [61]. A number of electron-rich and electron-deficient diaryl diselenides 21 were adequately employed as substrate, affording the products 101a–h at good to excellent yields. Additionally, alkyl and heteroaryl diselenides 21 were suitable substrates, giving the respective products 101i–k at good to excellent yields. When the glycerol-derived diselenide was used, the unprotected glycol 101l was obtained at an 85% yield, due to the Oxone-promoted ketal deprotection ability. No reaction was observed using diphenyl disulfide as chalcogen source, whereas diphenyl ditelluride was discretely converted to 101n at only a 9% yield after 24 h. Aiming to improve this resulted, the reaction was conducted in a sealed tube at 100 °C, yielding the tellurium-containing product 101n at 81%, after 24 h. Regarding the 3-(arylethynyl)-2-(propylthio)pyridines 102 counterpart, electron-rich (R = 4-MeC6H4) and electron-deficient (R = 4-ClC6H4) substrates were satisfactorily used, giving the products 101o and 101p at a 88% and 89% yield, respectively (Scheme 67).
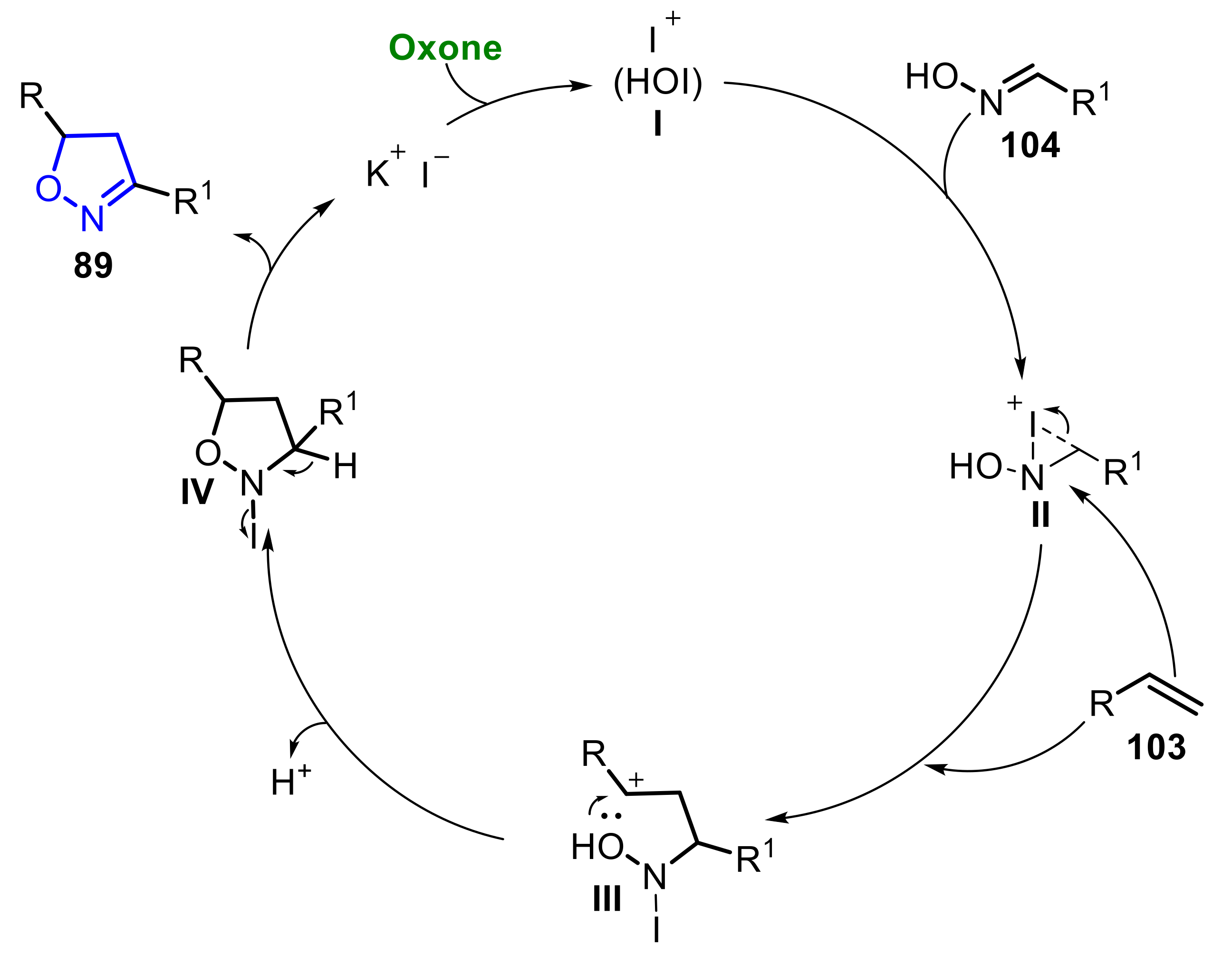
In 2013, Zhdankin and co-workers [62] described the reaction between alkenes 103 and aldoximes 104 using KI/Oxone catalysis to obtain isoxazolines 89n–s (Scheme 68). The reaction took place at room temperature, using a mixture of MeOH/H2O (v/v = 20:1), and after 12 h of reaction, the expected isoxazolines 89n–s were obtained in yields ranging from 13% to 92%. The method was extended to several alkenes and aldoximes containing electron donating and electron withdrawing substituents attached to the aromatic ring. It was observed that when the aliphatic aldoxime 104d was used, the desired product 89q was obtained at only 13% of the yield (Scheme 68).
The proposed reaction mechanism starts with the formation of the active species of iodine (hypoiodous acid) I, from the reaction of KI (0.1 equiv) and Oxone (1.5 equiv). This species reacts with aldoxime 104, forming the iodonium ion intermediate II, which is then opened by the alkene 103 to produce the N-iodohydroxy intermediate III. Finally, a cyclization occurs, forming the N-iodo-isoxazolidine intermediate IV, which undergoes a β-elimination to afford the isoxazoline 89 of interest (Scheme 69).
In 2020, Ishihara and co-workers developed an oxidative spiroetherification and spiroamination of phenol derivatives 105 and 106 using I+/Oxone catalysis (Scheme 70) [63]. In the presence of TBAI (10 mol%), Oxone (0.6 equiv), and a mixture CH3CN/H2O as solvent, arenols tethered to a hydroxy group 105 or a secondary amido group 106 were converted to corresponding spiro adducts 107 or 108 at moderate to excellent yields, at room temperature. The protocol was successfully applied to a variety of arenols 105 and 106, including 1- and 2-naphthols and phenols, tethered to hydroxy or sulfonamido groups. The oxidative cyclization of secondary and tertiary alcohols 105g and 105h was also demonstrated and the corresponding spiroethers 107g and 107h were obtained in good to excellent yields. Interesting, six-membered spiroether 107i could be synthesized at a 68% yield (Scheme 70).
In 2013, Yoshimura, Zhdankin and co-workers reported the cyclization of aldoxime 104 and alkene 103 or alkyne 50 using catalytic hypervalent iodine, Oxone as a terminal oxidant, and a mixture of MeOH:HFIP:H2O (v/v = 10:10:1) as solvent at room temperature for 24 h [64]. The reaction involves the initial formation of nitrile oxides, which react with alkenes and alkynes to produce the corresponding isoxazolines 89t-ad (Scheme 71) and isoxazoles 92p–w (Scheme 72). The protocol was expanded to aldoximes 104 and alkenes 103 or alkynes 50 containing several electron-rich and electron-deficient aryl groups and alkyl groups. By this procedure, a total of sixteen isoxazolines 89 and eight isoxazoles 92 were obtained in poor to excellent yields.
Based on control experiments, a plausible catalytic cyclization mechanism was proposed (Scheme 73). Initially, the activation of iodoarene by Oxone in aqueous HFIP affords the [ArI(OH)]+ species I, which reacts with aldoxime 104 to form the hypervalent alkoxy iodane II, via ligand exchange. Then, the intermediate II undergoes reductive elimination of iodoarene to produce the nitrile oxide III, which finally reacts with alkene 103 or alkyne 50 to produce the corresponding isoxazoline 89 or isoxazole 92. The regenerated iodoarene continues the next catalytic cycle.
6. Conclusions
This review highlights the recent advances in the Oxone-mediated synthesis of heterocyclic compounds, bringing mechanistic insights, protocol particularities, and a comprehensive discussion about scope and limitations. Among the synthetic approaches discussed herein, several examples are conducted under mild reaction conditions, an environmentally friendly reaction medium and by using alternative energy sources. However, despite the impressive advances delivered so far, many improvements are still required. The finds reached until now are the base to develop innovative approaches in coming years, employing Oxone as a green oxidant to promote the formation of a diversity of chemical bonds, to construct important structure cores. Additionally, we also see good prospects in the application of ultrasound irradiation as a low-demand energy source, driving the application of Oxone in synthetic chemistry to new horizons by triggering innovative activation pathways. In this context, we hope that this review can be a solid source of information, inspiring those interested to devote their studies to the application of Oxone in organic synthesis, aiming to deliver more efficient, robust and greener synthetic approaches.
Author Contributions
Conceptualization, G.P. and E.J.L.; methodology, G.P. and R.G.J.; writing—original draft preparation, H.A.G., D.R.A. and F.P.; writing—review and editing, F.P., G.P., R.G.J. and E.J.L.; supervision, G.P. and E.J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001; FAPERGS, grant number 19/2551-0001867-3; CNPq and FINEP.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef]
- Lenardão, E.J.; Freitag, R.A.; Dabdoub, J.M.; Batista, A.C.F.; Silveira, C.C. Green Chemistry: The 12 Principles of Green Chemistry and It Insertion in the Teach and Research Activities. Quim. Nova. 2003, 26, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Liang, L.; Zheng, H.; Chi, Y.R.; Tong, R. Green Oxidation of Indoles Using Halide Catalysis. Nat. Commun. 2019, 10, 4754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noyori, R.; Aoki, M.; Sato, K. Green Oxidation with Aqueous Hydrogen Peroxide. Chem. Commun. 2003, 1977–1986. [Google Scholar] [CrossRef]
- Hussain, H.; Green, I.R.; Ahmed, I. Journey Describing Applications of Oxone in Synthetic Chemistry. Chem. Rev. 2013, 113, 3329–3371. [Google Scholar] [CrossRef]
- Quin, L.D.; Tyrell, J.A. Fundamentals of Heterocyclic Chemistry; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Alvarez-Builla, J.; Vaquero, J.J.; Barluenga, J. Modern Heterocyclic Chemistry; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Vardanyan, R.; Hruby, V. Synthesis of Best-Seller Drugs; Academic Press: London, UK, 2016. [Google Scholar]
- Kaur, N. Microwave-Assisted Synthesis of Fused Polycyclic Six-Membered N-Heterocycles. Synth. Commun. 2014, 45, 273–299. [Google Scholar] [CrossRef]
- Wei, Z.-Y.; Chi, K.-Q.; Wang, K.-S.; Hu, J.; Liu, L.-P.; Piao, H.-R. Design, synthesis, evaluation, and molecular docking of ursolic acid derivatives containing a nitrogen heterocycle as anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2018, 28, 1797–1803. [Google Scholar] [CrossRef]
- Grande, F.; Occhiuzzi, M.A.; Ioele, G.; Ragno, G.; Garofalo, A. Benzopyroloxazines containing a bridgehead nitrogen atom as promising scaffolds for the achievement of biologically active agentes. Eur. J. Med. Chem. 2018, 151, 121–144. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, C.-Y.; Wang, X.-M.; Yang, Y.-H.; Zhu, H.-L. 1,3,4-Thiadiazole: Synthesis, reactions, and applications in medicinal, agricultural, and materials chemistry. Chem. Rev. 2014, 114, 5572–5610. [Google Scholar] [CrossRef] [PubMed]
- Kashiwa, M.; Sonoda, M.; Tanimori, S. Facile Access to 1H-Indazoles through Iodobenzene-Catalyzed C–H Amination under Mild, Transition-Metal-Free Conditions. Eur. J. Org. Chem. 2014, 4720–4723. [Google Scholar] [CrossRef]
- Srinivasan, R.; Ruso, J.S.; Nagarajan, N.S.; Kumaran, R.S.; Manickam, G. A Convenient One-Pot Synthesis of Triazolopyridine and Related Heterocycle Fused-Triazole Analogs Through Copper Catalyzed Oxidative Cyclization Strategy. J. Heterocycl. Chem. 2016, 53, 606–614. [Google Scholar] [CrossRef]
- Zhang, M.-Z.; Ji, P.-Y.; Liu, Y.-F.; Guo, C.-C. Transition-Metal-Free Synthesis of Carbonyl-Containing Oxindoles from N-Arylacrylamides and α-Diketones via TBHP- or Oxone-Mediated Oxidative Cleavage of C(sp2)−C(sp2) Bonds. J. Org. Chem. 2015, 80, 10777–10786. [Google Scholar] [CrossRef] [PubMed]
- Hati, S.; Dutta, P.K.; Dutta, S.; Munshi, P.; Sen, S. Accessing Benzimidazoles via a Ring Distortion Strategy: An Oxone Mediated Tandem Reaction of 2-Aminobenzylamines. Org. Lett. 2016, 16, 3090–3094. [Google Scholar] [CrossRef] [PubMed]
- Sriramoju, V.; Kurva, S.; Madabhushi, S. Oxone-mediated annulation of 2-aminobenzamides and 1,2-diaminobenzenes with sec-amines via imine-N-oxides: New syntheses of 2,3-dihydroquinazolin-4(1H)-ones and 1H-benzimidazoles. New J. Chem. 2018, 42, 3188–3191. [Google Scholar] [CrossRef]
- Liao, Y.-Y.; Gao, Y.-C.; Zheng, W.; Tang, R.-Y. Oxidative Radical Cyclization of N-methyl-N-arylpropiolamide to Isatins via Cleavage of the Carbon-carbon Triple Bond. Adv. Synth. Catal. 2018, 360, 3391–3400. [Google Scholar] [CrossRef]
- Meng, Y.-N.; Kang, Q.-Q.; Cao, T.-T.; Song, S.-Z.; Ge, G.-P.; Li, Q.; Wei, W.-T. Oxone-Mediated Radical Bicyclization of 1,6-Enynes through Dual α-C(sp3)−H Functionalization of Ketones under Catalyst- and Base-Free Conditions. ACS Sustain. Chem. Eng. 2019, 7, 18738–18743. [Google Scholar] [CrossRef]
- Perin, G.; Nobre, P.C.; Mailahn, D.H.; Silva, M.S.; Barcellos, T.; Jacob, R.G.; Lenardão, E.J.; Santi, C.; Roehrs, J.A. Synthesis of 4-Organoselanyl-1H-pyrazoles: Oxone®-Mediated Electrophilic Cyclization of α,β-Alkynyl Hydrazones by Using Diorganyl Diselenides. Synthesis 2019, 51, 2293–2304. [Google Scholar] [CrossRef]
- Jacob, R.G.; Oliveira, D.H.; Peglow, T.J.; Nascimento, J.E.R.; Bartz, R.H. Oxone®-Promoted One-Pot Synthesis of 1-Aryl-4-(organylselanyl)-1H-pyrazoles. J. Braz. Chem. Soc. 2019, 30, 2144–2152. [Google Scholar] [CrossRef]
- More, D.A.; Shinde, G.H.; Shaikh, A.C.; Muthukrishnan, M. Oxone promoted dehydrogenative Povarov cyclization of N-aryl glycine derivatives: An approach towards quinoline fused lactones and lactams. RSC Adv. 2019, 9, 30277–30291. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Li, J.-N.; Qiu, G.; Xie, W.; Liu, J.-B. ZnBr2/Oxone-mediated ipso-cyclization of N-(3-phenylprop-2-yn-1-yl)aniline. RSC Adv. 2019, 9, 33460–33464. [Google Scholar] [CrossRef]
- Qiu, G.; Chen, Z.-F.; Xie, W.; Zhou, H. TBAB-Mediated Radical 5-exo-trig ipso-Cyclization of 2-Arylbenzamide for the Synthesis of Spiro[cyclohexane-1,1′-isoindoline]-2,5-diene-3′,4-dione. Eur. J. Org. Chem. 2019, 2019, 4327–4333. [Google Scholar] [CrossRef]
- Yang, M.; Hu, X.; Ouyang, B.; Xie, W.; Liu, J.-B. TBAB-mediated radical 6-endo-trig ortho-cyclization of N-aryl-N-(prop-2-yn-1-yl)benzenesulfonamide for the synthesis of 3-bromo-1,2-dihydroquinoline. Tetrahedron 2019, 75, 3516–3522. [Google Scholar] [CrossRef]
- Chen, D.; Li, J.; Cui, P.; Shan, Y.; Zhao, Y.; Qiu, G. Tandem Oxidative Radical Halogenated Addition of Alkynyl Imines: Regioselective Synthesis of 3-Haloquinolines. Eur. J. Org. Chem. 2020, 2020, 169–175. [Google Scholar] [CrossRef]
- Song, S.-Z.; Kang, Q.-Q.; Cao, T.-T.; Lei, K.-W.; Liu, Y.-Y.; Li, Q.; Wei, W.-T. Cu(NO3)2/Oxone-Mediated Radical Nitration Cyclization of 1,6-Enynes. ChemistrySelect 2019, 4, 13380–13383. [Google Scholar] [CrossRef]
- Chen, D.; Li, J.; Shan, Y.; Cui, P.; Zhao, Y.; Tian, L.; Qiu, G. Halogen-Radical-Promoted Dearomative Aza-Spirocyclization of Alkynylimines: An Efficient Approach to 3-Halo-Spirocyclohexadienones. Synthesis 2020, 52, 609–618. [Google Scholar] [CrossRef]
- Araujo, D.R.; Goulart, H.A.; Barcellos, A.M.; Cargnelutti, R.; Lenardão, E.J.; Perin, G. Oxone-Promoted Synthesis of 4-(Chalcogenyl)isoquinoline-N-oxides from Alkynylbenzaldoximes and Diorganyl Dichalcogenides. J. Org. Chem. 2021, 86, 1721–1729. [Google Scholar] [CrossRef]
- Pisani, L.; Superchi, S.; D’Elia, A.; Scafato, P.; Rosini, C. Synthetic approach toward cis-disubstituted γ- and δ-lactones through enantioselective dialkylzinc addition to aldehydes: Application to the synthesis of optically active flavors and fragrances. Tetrahedron 2012, 68, 5779–5784. [Google Scholar] [CrossRef]
- Kaur, P.; Arora, R.; Gill, N.S. Review on oxygen heterocycles. Indo Am. J. Phar. Res. 2013, 3, 2231–6876. [Google Scholar]
- Cossy, J.; Guérinot, A. Natural products containing oxygen heterocycles—Synthetic advances between 1990 and 2015. Adv. Heterocycl. Chem. 2016, 119, 107–142. [Google Scholar]
- Wang, R.-X.; Yuan, S.-T.; Liu, J.-B.; Wu, J.; Qiu, G. Regioselective 5-exo-dig oxy-cyclization of 2-alkynylbenzamide for the synthesis of isobenzofuran-1-imines and isobenzofuran. Org. Biomol. Chem. 2018, 16, 4501–4508. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Ouyang, B.; Qiu, G.; Zhou, H.; Liu, J.-B. Oxidative oxy-cyclization of 2-alkynylbenzamide enabled by TBAB/Oxone: Switchable synthesis of isocoumarin-1-imines and isobenzofuran-1-imine. Org. Biomol. Chem. 2019, 17, 4335–4341. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Liu, H.; Kang, J.; Luo, Y.; Gong, T.; Dong, Z.; Sun, G.; He, C.; Sun, X.; Wang, L. Palladium-catalyzed enol/enolate directed oxidative annulation: Functionalized naphthofuroquinone synthesis and bioactivity evaluation. Chem. Commun. 2019, 55, 14729–14732. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Qiu, G.; Zhou, H.; Xie, W.; Liu, J.-B. Regioselective cyclization of 2-alkynylbenzoic acid in water for the synthesis of isocoumarin. Tetrahedron 2019, 75, 3850–3855. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, M.; Qiu, G.; Xie, W.; Wu, J. Synthesis of 3-(Bromomethylene)isobenzofuran-1(3H)-ones through regioselective 5-exo-dig bromocyclization of 2-alkynylbenzoic acids. Tetrahedron 2019, 75, 1663–1668. [Google Scholar] [CrossRef]
- Goulart, H.A.; Neto, J.S.S.; Barcellos, A.M.; Silva, K.B.; de Moraes, M.C.; Jacob, R.G.; Lenardão, E.J.; Barcellos, T.; Perin, G. Synthesis of 4-Selanyl- and 4-Tellanyl-1H-isochromen-1-ones Promoted by Diorganyl Dichalcogenides and Oxone. J. Org. Chem. 2021, 86, 14016–14027. [Google Scholar] [CrossRef]
- Uyanik, M.; Kato, T.; Sahara, N.; Katade, O.; Ishihara, K. High-Performance Ammonium Hypoiodite/Oxone Catalysis for Enantioselective Oxidative Dearomatization of Arenols. ACS Catal. 2019, 9, 11619–11626. [Google Scholar] [CrossRef]
- Hellwig, P.S.; Peglow, T.J.; Penteado, F.; Bagnoli, L.; Perin, G.; Lenardão, E.J. Recent Advances in the Synthesis of Selenophenes and Their Derivatives. Molecules 2020, 25, 5907–6010. [Google Scholar] [CrossRef]
- Pander, P.; Motyka, R.; Zassowski, P.; Lapkowski, M.; Swist, A.; Data, P. Electrochromic Properties of Novel Selenophene and Tellurophene Derivatives Based on Carbazole and Triphenylamine Core. J. Phys. Chem. C 2017, 121, 11027–11036. [Google Scholar] [CrossRef] [Green Version]
- Nakano, M.; Mori, H.; Shinamura, S.; Takimiya, K. Naphtho[2,3-b:6,7-b′]dichalcogenophenes: Syntheses, Characterizations, and Chalcogene Atom Effects on Organic Field Effect Transistor and Organic Photovoltaic Devices. Chem. Mater. 2012, 24, 190–198. [Google Scholar] [CrossRef]
- Mahmoud, A.B.A.; Kirsch, G.; Peagle, E. Biologically Active Selenophenes and Benzo[b]selenophenes. Curr. Org. Synth. 2017, 14, 1091–1101. [Google Scholar]
- Costea, T.; Bădiceanu, C.D.; Nuță, D.M.; Gîrd, C.E.; Limban, C.; Karampelas, O.; Avram, S. Antioxidant Activity And Drug Profile Of Several Thiourea Derivatives Of 2-Thiophene Carboxylic Acid. Farmacia 2020, 68, 1047–1054. [Google Scholar] [CrossRef]
- Soares, L.K.; Barcellos, A.M.; Neto, J.S.S.; Alves, D.; Lenardão, E.J.; Rosati, O.; Santi, C.; Perin, G. Dichalcogenides/Oxone®-Mediated Cyclization of (Z)-Chalcogenoenynes under Ultrasound Irradiation. ChemistrySelect 2020, 5, 9813–9819. [Google Scholar] [CrossRef]
- Hellwig, P.S.; Guedes, J.S.; Barcellos, A.M.; Perin, G.; Lenardão, E.J. Synthesis of 3,4-Bis(Butylselanyl)Selenophenes and 4-Alkoxyselenophenes Promoted by Oxone®. Molecules 2021, 26, 2378–2392. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, P.S.; Guedes, J.S.; Barcellos, A.M.; Jacob, R.G.; Silveira, C.C.; Lenardão, E.J.; Perin, G. Synthesis of benzo[b]chalcogenophenes fused to selenophenes via intramolecular electrophilic cyclization of 1,3-diynes. Org. Biomol. Chem. 2021, 19, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Perin, G.; Soares, L.K.; Hellwig, P.S.; Silva, M.S.; Neto, J.S.S.; Roehrs, J.A.; Barcellos, T.; Lenardão, E.J. Synthesis of 2,3-bis-organochalcogenylbenzo[b]chalcogenophenes promoted by Oxone. New J. Chem. 2019, 43, 6323–6331. [Google Scholar] [CrossRef]
- Alla, S.K.; Sadhu, P.; Punniyamurthy, T. Organocatalytic Syntheses of Benzoxazoles and Benzothiazoles using Aryl Iodide and Oxone via C−H Functionalization and C−O/S Bond Formation. J. Org. Chem. 2014, 79, 7502–7511. [Google Scholar] [CrossRef]
- Moriyama, K.; Nishinohara, C.; Sugiue, T.; Togo, H. Oxidative oxygen-nucleophilic bromo-cyclization of alkenyl carbonyl compounds without organic wastes using alkali metal reagents in green solvent. RSC Adv. 2015, 5, 85872–85878. [Google Scholar] [CrossRef]
- Daswani, U.; Dubey, N.; Sharma, P.; Kumar, A. A new NBS/Oxone promoted one-pot cascade synthesis of 2-aminobenzimidazoles/2-aminobenzoxazoles: A facile approach. New J. Chem. 2016, 40, 8093–8099. [Google Scholar] [CrossRef]
- Sharma, S.; Pathare, R.S.; Maurya, A.K.; Gopal, K.; Roy, T.K.; Sawant, D.M.; Pardasani, R.T. Ruthenium Catalyzed Intramolecular C−S Coupling Reactions: Synthetic Scope and Mechanistic Insight. Org. Lett. 2016, 18, 356–359. [Google Scholar] [CrossRef]
- Sen, A.; Takenaka, K.; Sasai, H. Enantioselective Aza-Wacker-Type Cyclization Promoted by Pd-SPRIX Catalyst. Org. Lett. 2018, 20, 6827–6831. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Togo, H. One-Pot Preparation of 3-Arylisoxazole-4,5-dicarboxylates from Benzylic Chlorides and Acetylenedicarboxylates. Eur. J. Org. Chem. 2018, 2018, 1377–1383. [Google Scholar] [CrossRef]
- Bhatt, A.; Singh, R.K.; Kant, R. A convenient one-pot synthesis of 3,5-diarylisoxazoles via oxidative cyclisation using catalytic CuBr2 and oxone. Tetrahedron Lett. 2019, 60, 1143–1147. [Google Scholar] [CrossRef]
- Goulart, H.A.; Neto, J.S.S.; Barcellos, A.M.; Barcellos, T.; Silva, M.S.; Alves, D.; Jacob, R.G.; Lenardão, E.J.; Perin, G. Synthesis of 5H-Selenopheno[3,2-c]isochromen-5-ones Promoted by Dialkyl Diselenides and Oxone®. Adv. Synth. Catal. 2019, 361, 3403–3411. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Liu, J.-B.; Zhou, H.; Xie, W.; Rojsitthisak, P.; Qiu, G. Ortho-Hydroxylative ipso-Cyclization of N-arylpropiolamide. J. Org. Chem. 2020, 85, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Araujo, D.R.; Lima, Y.R.; Barcellos, A.M.; Silva, M.S.; Jacob, R.G.; Lenardão, E.J.; Bagnoli, L.; Santi, C.; Perin, G. Ultrasound-Promoted Radical Synthesis of 5-Methylselanyl-4,5-dihydroisoxazoles. Eur. J. Org. Chem. 2020, 586–592. [Google Scholar] [CrossRef]
- Peglow, T.J.; Bartz, R.H.; Barcellos, T.; Schumacher, R.F.; Cargnelutti, R.; Perin, G. Synthesis of 2-Aryl-(3-Organochalcogenyl)Thieno[2,3-b]Pyridines Promoted by Oxone®. Asian J. Org. Chem. 2021, 10, 1198–1206. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhu, C.; Middleton, K.R.; Todora, A.D.; Kastern, B.J.; Maskaev, A.V.; Zhdankin, V.V. Hypoiodite mediated synthesis of isoxazolines from aldoximes and alkenes using catalytic KI and Oxone as the terminal oxidant. Chem. Commun. 2013, 49, 4800–4802. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, M.; Sahara, N.; Katade, O.; Ishihara, K. Chemoselective Oxidative Spiroetherification and Spiroamination of Arenols Using I+/Oxone Catalysis. Org. Lett. 2020, 22, 560–564. [Google Scholar] [CrossRef]
- Yoshimura, A.; Middleton, K.R.; Todora, A.D.; Kastern, B.J.; Koski, S.R.; Maskaev, A.V.; Zhdankin, V.V. Hypervalent Iodine Catalyzed Generation of Nitrile Oxides from Oximes and their Cycloaddition with Alkenes or Alkynes. Org. Lett. 2013, 15, 4010–4013. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Oxone-mediated synthesis of heterocyclic compounds.

Scheme 2.
Oxone/PhI-promoted annulation of N-substituted hydrazones 2.

Scheme 3.
Plausible reaction mechanism for the Oxone/PhI-promoted annulation process.

Scheme 4.
Oxone/CuBr-mediated synthesis of triazole-fused heterocycles 3.

Scheme 5.
Oxone-mediated synthesis of oxindoles 6.

Scheme 6.
Oxone-mediated synthesis of 2-substituted benzimidazoles 10.

Scheme 7.
Oxone-promoted synthesis of 2,3-dihydroquinazoline-4(1H)-ones 11a–m.

Scheme 8.
Oxone-promoted synthesis of 1H-benzimidazoles 10.

Scheme 9.
Oxone/NaNO2-promoted oxidative radical cyclization of the propylamide 14.

Scheme 10.
Reaction mechanism for the Oxone/NaNO2-promoted oxidative annulation.

Scheme 11.
Oxone-mediated radical bicyclization of 1,6-enynes 17 and ketones 18.

Scheme 12.
Reaction mechanism for the Oxone-promoted bicyclization of 1,6-enynes 17.

Scheme 13.
Oxone/RSeSeR-promoted electrophilic cyclization of α,β-alkynyl hydrazones 20.

Scheme 14.
Reaction mechanism for the Oxone/RSeSeR-promoted electrophilic cyclization of α,β-alkynyl hydrazones 20.
Scheme 14.
Reaction mechanism for the Oxone/RSeSeR-promoted electrophilic cyclization of α,β-alkynyl hydrazones 20.

Scheme 15.
Oxone-mediated multicomponent synthesis of substituted 4-organylselanylpyrazoles 22.

Scheme 16.
Oxone-promoted intramolecular dehydrogenative Povarov cyclization.

Scheme 17.
Synthetic application to construct complex structures, such as (A) the antibiotic Uncialamycin 26l and (B) the analogues of the cytotoxic alkaloid Luotonin A 26m and 26n.
Scheme 17.
Synthetic application to construct complex structures, such as (A) the antibiotic Uncialamycin 26l and (B) the analogues of the cytotoxic alkaloid Luotonin A 26m and 26n.

Scheme 18.
ZnBr2/Oxone-mediated radical ipso-cyclization of alkynyl anilines 27.

Scheme 19.
Oxone/TBAB-mediated oxidative cyclization of N-aryl-2-arylbenzamides 29.

Scheme 20.
Oxone/TBAB-mediated synthesis of brominated products 31.

Scheme 21.
Oxone/TBAB-mediated synthesis of 3-bromo-1,2-dihydroquinoline derivatives 32.

Scheme 22.
Plausible mechanism for the radical 6-endo-trig cyclization of the substrate 33.

Scheme 23.
Oxone/TBAX-promoted tandem annulative radical halogenation of alkynyl imines.

Scheme 24.
Reaction mechanism for the Oxone/TBAX-promoted tandem annulative radical halogenation.

Scheme 25.
Oxone/Cu(NO3)2-promoted radical annulative nitration of 1,6-enynes 17.

Scheme 26.
Oxone/TBAX-promoted spirocyclization of p-methoxyl alkynyl-N-phenylimines 37.

Scheme 27.
Oxone/RSeSeR-promoted electrophilic cyclization of alkynylbenzaldoximes 39.

Scheme 28.
Oxone/RTeTeR-promoted electrophilic cyclization of alkynylbenzaldoximes 39.

Scheme 29.
Oxone/TBAB-promoted 5-exo-dig oxy-cyclization of 2-alkynylbenzamide 43. (A) Synthesis of isobenzofuran-1-imines 44. (B) Synthesis of isobenzofuran 45.
Scheme 29.
Oxone/TBAB-promoted 5-exo-dig oxy-cyclization of 2-alkynylbenzamide 43. (A) Synthesis of isobenzofuran-1-imines 44. (B) Synthesis of isobenzofuran 45.

Scheme 30.
TBAB-catalyzed Oxone-mediated 6-endo-dig oxidative annulation of 2-alkynylbenzamide 43.

Scheme 31.
TBAB-catalyzed Oxone-mediated synthesis of isobenzofuran-1-imines 48.

Scheme 32.
Pd(II)-catalyzed Oxone-mediated oxidative annulation of 1,2-naphthofuroquinone 49 and diaryl alkynes 50.
Scheme 32.
Pd(II)-catalyzed Oxone-mediated oxidative annulation of 1,2-naphthofuroquinone 49 and diaryl alkynes 50.

Scheme 33.
Oxone/TBAB-promoted radical cyclization of 2-alkynylbenzoic acids 52.

Scheme 34.
Reaction mechanism of the Oxone/TBAB-promoted radical cyclization of 2-alkynylbenzoic acids 52.
Scheme 34.
Reaction mechanism of the Oxone/TBAB-promoted radical cyclization of 2-alkynylbenzoic acids 52.

Scheme 35.
Oxone/TBAB-promoted radical cyclization of 2-alkynylbenzoic acids 52.

Scheme 36.
Plausible mechanism of the Oxone/TBAB-promoted radical cyclization of 2-alkynylbenzoic acids 52.
Scheme 36.
Plausible mechanism of the Oxone/TBAB-promoted radical cyclization of 2-alkynylbenzoic acids 52.

Scheme 37.
Oxone/diselenide-promoted electrophilic cyclization of 2-alkynylaryl esters 56.

Scheme 38.
Oxone/ditelluride-promoted electrophilic cyclization of 2-alkynylaryl esters 56.

Figure 1.
Structures of quaternary ammonium hypoiodite A and B.

Scheme 39.
Enantioselective oxidation of phenols 57a–f.

Scheme 40.
Enantioselective oxidation of 2-naphthols 59a–c.

Scheme 41.
Enantioselective Oxidation of 1-Naphthols 59d–f.

Scheme 42.
Oxone/dichalcogenide-promoted annulation of (Z)-1-butylselen-1-en-3-ynes 63.

Scheme 43.
Oxone/dichalcogenide-promoted annulation of (Z)-(1,4-diphenylbut-1-en-3-yn-1-yl)(propyl)sulfane 64a.
Scheme 43.
Oxone/dichalcogenide-promoted annulation of (Z)-(1,4-diphenylbut-1-en-3-yn-1-yl)(propyl)sulfane 64a.

Scheme 44.
Oxone/dichalcogenide-promoted annulation of (Z)-butyl(1,4-diphenylbut-1-en-3-yn-1-yl)tellane 65a.
Scheme 44.
Oxone/dichalcogenide-promoted annulation of (Z)-butyl(1,4-diphenylbut-1-en-3-yn-1-yl)tellane 65a.

Scheme 45.
Reaction mechanism for the Oxone/dichalcogenide-promoted cyclization of chalcogenoenynes.
Scheme 45.
Reaction mechanism for the Oxone/dichalcogenide-promoted cyclization of chalcogenoenynes.

Scheme 46.
Oxone/dibutyl diselenide-promoted synthesis of thiophenes 67.

Scheme 47.
Oxone/dibutyl diselenide-promoted synthesis of thiophenes 68.

Scheme 48.
Oxone/dibutyl diselenide-promoted intramolecular cyclization of ortho-1,3-diynyl phenyl chalcogenide 69.
Scheme 48.
Oxone/dibutyl diselenide-promoted intramolecular cyclization of ortho-1,3-diynyl phenyl chalcogenide 69.

Scheme 49.
Oxone/diselenide-promoted electrophilic cyclization of 2-functionalized chalcogenoalkynes 71.
Scheme 49.
Oxone/diselenide-promoted electrophilic cyclization of 2-functionalized chalcogenoalkynes 71.

Scheme 50.
Oxone-mediated synthesis of 2-arylbenzoxazoles 73.

Scheme 51.
Oxone-mediated synthesis of 2-arylbenzothiazoles 74.

Scheme 52.
Reaction mechanism for the synthesis of 73 and 74.

Scheme 53.
Oxone/KBr-promoted bromo-cyclization of alkenyl carboxylic acids 77 (A) and N-allyl amides 78 (B).
Scheme 53.
Oxone/KBr-promoted bromo-cyclization of alkenyl carboxylic acids 77 (A) and N-allyl amides 78 (B).

Scheme 54.
Oxone/NBS-promoted synthesis of 2-aminobenzimidazoles 81 and 2-aminobenzoxazoles 82.

Scheme 55.
Ru-catalyzed Oxone-mediated intramolecular C−S coupling of N-arylthioureas 85.

Scheme 56.
Pd(II)-catalyzed Oxone-mediated enantioselective aza-Wacker type reaction.

Scheme 57.
Oxone-mediated synthesis of 3-arylisoxazole-4,5-dicarboxylates 89, through a one-pot process.
Scheme 57.
Oxone-mediated synthesis of 3-arylisoxazole-4,5-dicarboxylates 89, through a one-pot process.

Scheme 58.
Oxone-mediated synthesis of 3-arylisoxazole-4,5-dicarboxylates 89, through a one-pot process.
Scheme 58.
Oxone-mediated synthesis of 3-arylisoxazole-4,5-dicarboxylates 89, through a one-pot process.

Scheme 59.
Cu(II)-catalyzed Oxone-mediated one-pot synthesis of 3,5-diarylisoxazoles 92.

Scheme 60.
Oxone/dialkyl diselenide-promoted synthesis of 5H-selenopheno[3,2-c]isochromen-5-ones 94.
Scheme 60.
Oxone/dialkyl diselenide-promoted synthesis of 5H-selenopheno[3,2-c]isochromen-5-ones 94.

Scheme 61.
Competition between Se-cyclization and O-cyclization of the substrate 95d.

Scheme 62.
Plausible reaction mechanism for the formation of the products 94.

Scheme 63.
Oxone-mediated ipso-cyclization of N-arylpropiolamide 93 to access the spirocyclo derivatives 97.
Scheme 63.
Oxone-mediated ipso-cyclization of N-arylpropiolamide 93 to access the spirocyclo derivatives 97.

Scheme 64.
Oxone-mediated ipso-cyclization of N-arylpropiolamide 96 to access the spirocyclo derivatives 98.
Scheme 64.
Oxone-mediated ipso-cyclization of N-arylpropiolamide 96 to access the spirocyclo derivatives 98.

Scheme 65.
Oxone/diselenide-promoted ultrasound-assisted selective cyclization of unsaturated oximes 99.
Scheme 65.
Oxone/diselenide-promoted ultrasound-assisted selective cyclization of unsaturated oximes 99.

Scheme 66.
Plausible reaction mechanism for the Oxone/diselenide-promoted ultrasound-assisted selective cyclization of unsaturated oximes 99. a: Radical mechanism. b: Ionic mechanism.
Scheme 66.
Plausible reaction mechanism for the Oxone/diselenide-promoted ultrasound-assisted selective cyclization of unsaturated oximes 99. a: Radical mechanism. b: Ionic mechanism.

Scheme 67.
Oxone/diselenide-promoted synthesis of 2-aryl-(3-organocalcogenyl)thieno[2,3-b]pyridines 101.
Scheme 67.
Oxone/diselenide-promoted synthesis of 2-aryl-(3-organocalcogenyl)thieno[2,3-b]pyridines 101.

Scheme 68.
Catalytic cyclization of alkenes 103 with aldoximes 104.

Scheme 69.
Mechanism proposed for the synthesis of isoxazoles 89n–s.

Scheme 70.
I+/Oxone catalyzed oxidative spiroetherification and spiroamination of phenols 105 and 106.
Scheme 70.
I+/Oxone catalyzed oxidative spiroetherification and spiroamination of phenols 105 and 106.

Scheme 71.
Oxone-mediated synthesis of isoxazolines 89t-ad.

Scheme 72.
Oxone-mediated synthesis of isoxazoles 92p–w.

Scheme 73.
Plausible mechanism for the catalytic cyclization of the substrate 104.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Goulart, H.A.; Araujo, D.R.; Penteado, F.; Jacob, R.G.; Perin, G.; Lenardão, E.J. Recent Advances in the Oxone-Mediated Synthesis of Heterocyclic Compounds. Molecules 2021, 26, 7523. https://doi.org/10.3390/molecules26247523
AMA Style
Goulart HA, Araujo DR, Penteado F, Jacob RG, Perin G, Lenardão EJ. Recent Advances in the Oxone-Mediated Synthesis of Heterocyclic Compounds. Molecules. 2021; 26(24):7523. https://doi.org/10.3390/molecules26247523
Chicago/Turabian StyleGoulart, Helen A., Daniela R. Araujo, Filipe Penteado, Raquel G. Jacob, Gelson Perin, and Eder J. Lenardão. 2021. "Recent Advances in the Oxone-Mediated Synthesis of Heterocyclic Compounds" Molecules 26, no. 24: 7523. https://doi.org/10.3390/molecules26247523