
Lab-In-Syringe with Bead Injection Coupled Online to High-Performance Liquid Chromatography as Versatile Tool for Determination of Nonsteroidal Anti-Inflammatory Drugs in Surface Waters



Abstract
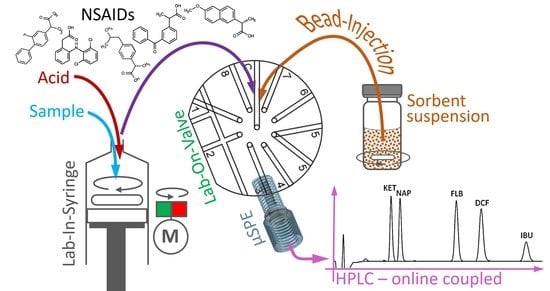
:
1. Introduction
2. Results
2.1. System Setup and Preliminary Considerations
2.2. Experiments on the Combined System Using UV Detector
2.2.1. Study of Sorbent, Loading Conditions, and Eluent Composition
2.2.2. Separation Method for Online Coupling
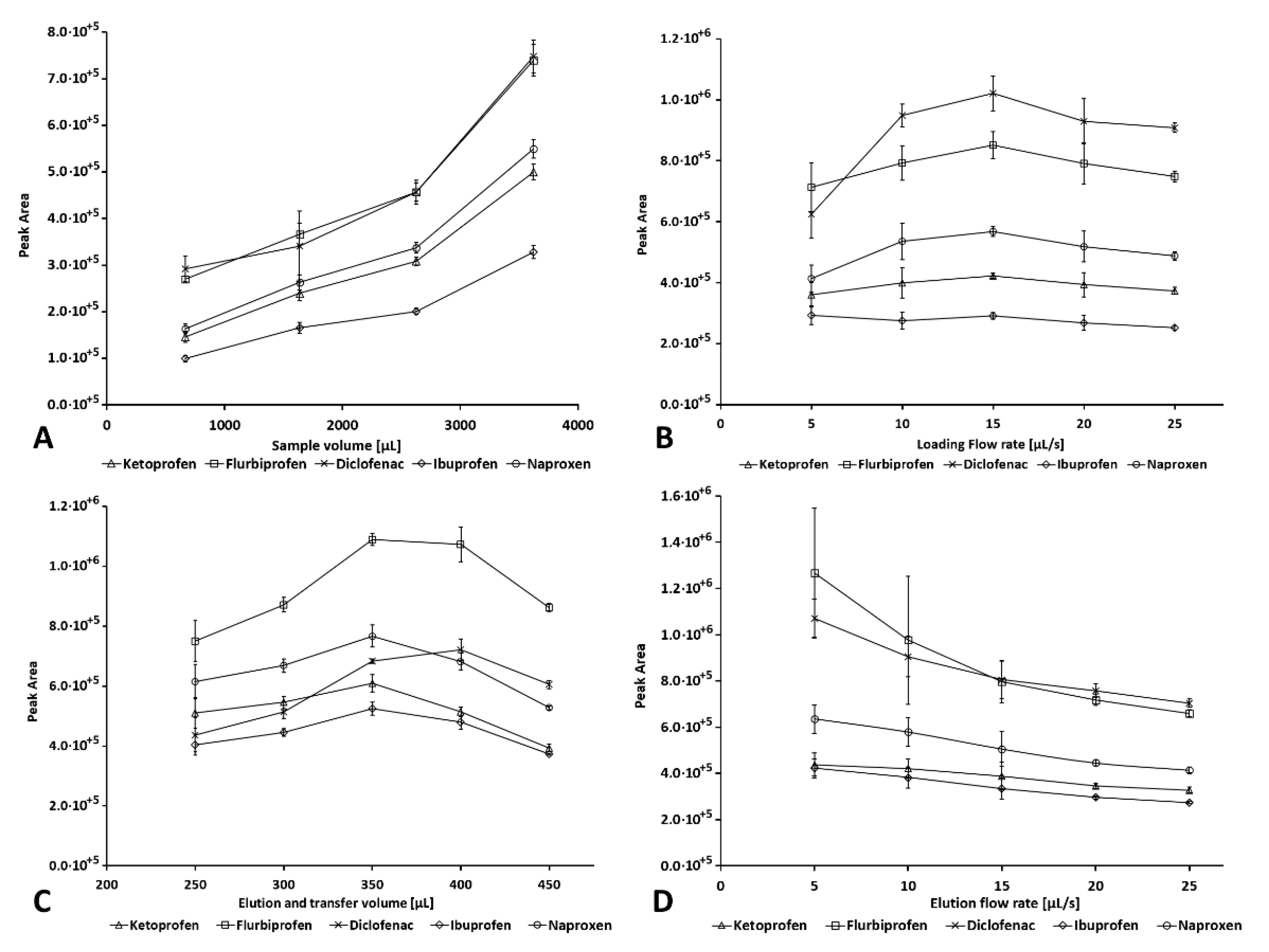
2.2.3. Sample Volume
2.2.4. Loading Flow Rate
2.2.5. Elution and Transfer Volume
2.2.6. Elution Flow Rate
2.2.7. Influence of Washing Solution
2.2.8. Loading Acidity
3. Discussion
3.1. Method Performance
3.2. Sample Measurement
4. Materials and Methods
4.1. Reagents and Solutions
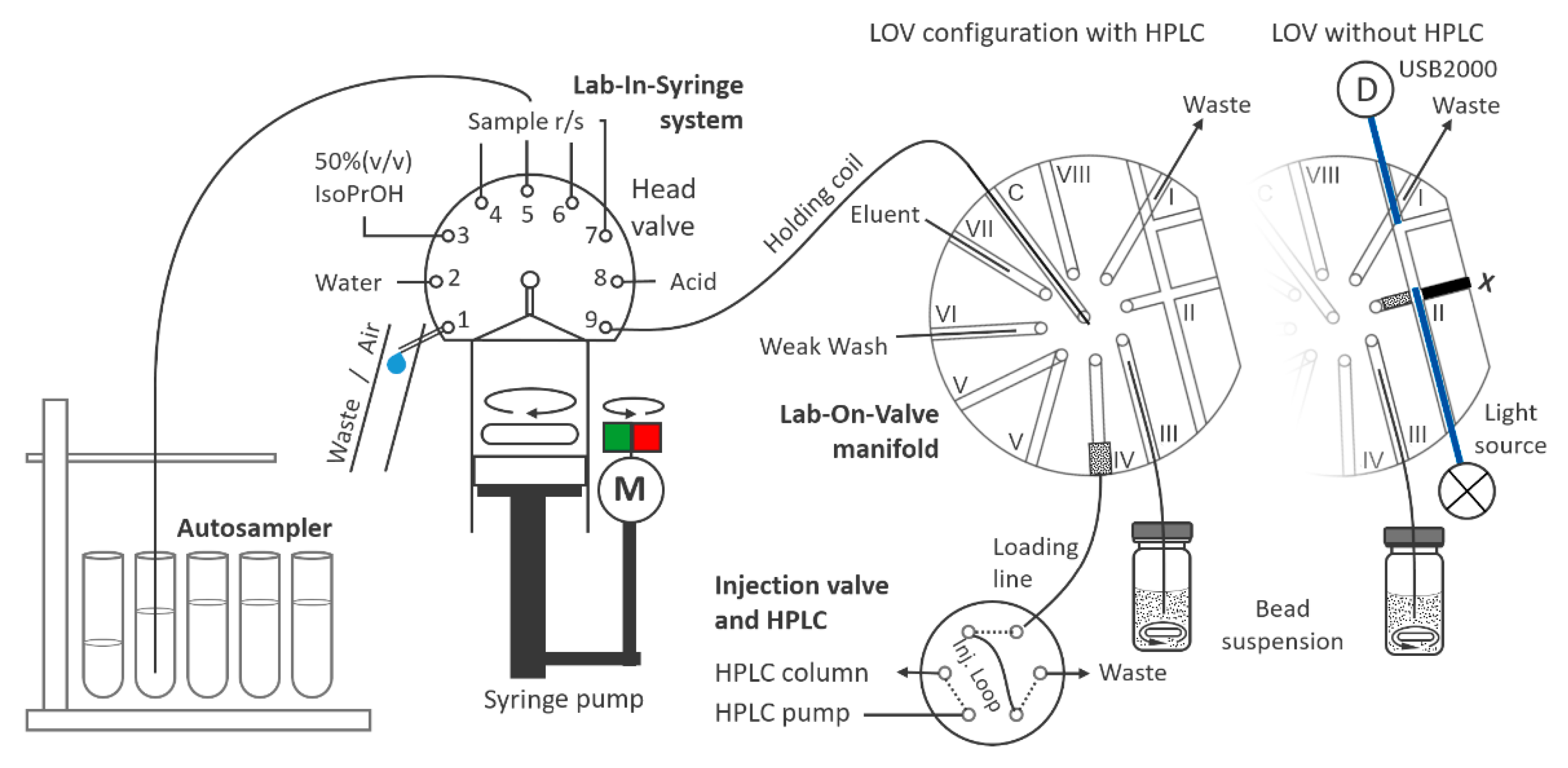
4.2. Flow System
4.3. HPLC Instrumentation
4.4. Method Operation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bacchi, S.; Palumbo, P.; Sponta, A.; Coppolino, M.F. Clinical Pharmacology of Non-Steroidal Anti-Inflammatory Drugs: A Review. Antiinflamm. Antiallergy Agents Med. Chem. 2012, 11, 52–64. [Google Scholar] [CrossRef]
- Rebecca Wong, S.Y. Role of nonsteroidal anti-inflammatory drugs (NSAIDs) in cancer prevention and cancer promotion. Adv. Pharmacol. Pharm. Sci. 2019, 2019, 3418975. [Google Scholar]
- Papich, M.G. An update on nonsteroidal anti-inflammatory drugs (NSAIDs) in small animals. Vet. Clin. N. Am. Small Anim. Pract. 2008, 38, 1243–1266. [Google Scholar] [CrossRef]
- Ramos-Payan, M.; Maspoch, S.; Llobera, A. An effective micro fluidic based liquid-phase microextraction device (m LPME) for extraction of non-steroidal anti-inflammatory drugs from biological and environmental samples. Anal. Chim. Acta 2016, 946, 56–63. [Google Scholar] [CrossRef]
- Farré, M.; Petrovic, M.; Barceló, D. Recently developed GC/MS and LC/MS methods for determining NSAIDs in water samples. Anal. Bioanal. Chem. 2007, 387, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Tixier, C.; Müller, S.R. Occurrence and fate of carbamazepine, clofibric acid, diclofenac, Ibuprofen, ketoprofen and Naproxen in Surface Waters. Environ Sci. Technol. 2003, 37, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Geng, J.; Hu, H.; Ma, H.; Gao, X.; Ren, H. Impact of selected non-steroidal anti- inflammatory pharmaceuticals on microbial community assembly and activity in sequencing batch reactors. PLoS ONE 2017, 12, e0179236. [Google Scholar] [CrossRef] [Green Version]
- Fent, K.; Weston, A.A.; Caminada, D. Ecotoxicology of human pharmaceuticals. Aquatic Toxicology 2006, 76, 122–159. [Google Scholar] [CrossRef] [PubMed]
- Vieno, N.; Sillanpää, M. Fate of diclofenac in municipal wastewater treatment plant—A review. Environ. Int. 2014, 69, 28–39. [Google Scholar] [CrossRef]
- EU. Directive 2013/39/EU of the European Parliament and of the Council. Available online: https://eurlex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2013:226:0001:0017:EN:PDF (accessed on 28 January 2021).
- Kramulov, B.; Tauchen, J.; Marsik, P.; Rezek, J.; Zidkov, M. Non-steroidal anti-inflammatory drugs in the watercourses of Elbe basin in Czech Republic. Chemosphere 2017, 171, 97–105. [Google Scholar]
- Commission Implementing Decision (EU) 2015/495 of 20 March 2015 Establishing a Watch List of Substances for Union-Wide Monitoring in the Field of Water Policy Pursuant to Directive 2008/105/EC of the European Parliament and of the Council. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=uriserv%3AOJ.L_.2015.078.01.0040.01.ENG (accessed on 22 March 2021).
- García, A.; Borrull, F.; Calull, M.; Aguilar, C. Single-drop microextraction combined in-line with capillary electrophoresis for the determination of nonsteroidal anti-inflammatory drugs in urine samples. Electrophoresis 2016, 37, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Gilart, N.; Marcé, R.M.; Fontanals, N.; Borrull, F. A rapid determination of acidic pharmaceuticals in environmental waters by molecularly imprinted solid-phase extraction coupled to tandem mass spectrometry without chromatography. Talanta 2013, 110, 196–201. [Google Scholar] [CrossRef]
- Martinez-Sena, T.; Armenta, S.; De la Guardia, M.; Esteve-Turrillas, F.A. Determination of non-steroidal anti-inflammatory drugs in water and urine using selective molecular imprinted polymer extraction and liquid chromatography. J. Pharm. Biomed. Anal. 2016, 131, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Arteaga, K.; Rodriguez, J.A.; Miranda, J.M.; Medina, J.; Barrado, E. Determination of non-steroidal anti-inflammatory drugs in wastewaters by magnetic matrix solid phase dispersion—HPLC. Talanta 2010, 80, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, V.; Carlucci, M.; Ettorre, V.; Cotellese, R.; Palumbo, P.; Fontana, A.; Siani, G.; Carlucci, G. Dispersive magnetic solid phase extraction exploiting magnetic graphene nanocomposite coupled with UHPLC-PDA for simultaneous determination of NSAIDs in human plasma and urine. J. Pharm. Biomed. Anal. 2018, 161, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Mao, X.; He, M.; Chen, B.; Hu, B. Development of novel sol-gel coatings by chemically bonded ionic liquids for stir bar sorptive extraction-application for the determination of NSAIDS in real samples. Anal. Bioanal. Chem. 2014, 406, 7261–7273. [Google Scholar] [CrossRef]
- Manzo, V.; Miró, M.; Richter, P. Programmable flow-based dynamic sorptive microextraction exploiting an octadecyl chemically modified rotating disk extraction system for the determination of acidic drugs in urine. J. Chromatogr. A 2014, 1368, 64–69. [Google Scholar] [CrossRef]
- Wang, R.; Li, W.; Chen, Z. Solid phase microextraction with poly (deep eutectic solvent) monolithic column online coupled to HPLC for determination of non- steroidal anti-inflammatory drugs. Anal. Chim. Acta 2018, 1018, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Abd Rahim, M.; Wan Ibrahim, W.A.; Ramli, Z.; Sanagi, M.M.; Aboul-Enein, H.Y. New sol-gel hybrid material in solid phase extraction combined with liquid chromatography for the determination of non-steroidal anti-inflammatory drugs in water samples. Chromatographia 2016, 79, 421–429. [Google Scholar] [CrossRef]
- Mirzajani, R.; Kardani, F.; Ramezani, Z. Preparation and characterization of magnetic metal–organic framework nanocomposite as solid-phase microextraction fibers coupled with high- performance liquid chromatography for determination of non-steroidal anti-inflammatory drugs in biological fluids and tablet formulation samples. Microchem. J. 2019, 144, 270–284. [Google Scholar]
- Jian, N.; Qian, L.; Wang, C.; Li, R.; Xu, Q.; Li, J. Novel nanofibers mat as an efficient, fast and reusable adsorbent for solid phase extraction of non-steroidal anti-inflammatory drugs in environmental water. J. Hazard. Mater. 2019, 363, 81–89. [Google Scholar] [CrossRef]
- Seidi, S.; Sanàti, S. Nickel-iron layered double hydroxide nanostructures for micro solid phase extraction of nonsteroidal anti-inflammatory drugs, followed by quantitation by HPLC-UV. Microchim. Acta 2019, 186, 297. [Google Scholar] [CrossRef]
- Rodriguez-Mozaz, S.; De Alda, M.J.L. Advantages and limitations of on-line solid phase extraction coupled to liquid chromatography—Mass spectrometry technologies versus biosensors for monitoring of emerging contaminants in water. J. Chromatogr. A 2007, 1152, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Togola, A.; Budzinski, H. Analytical development for analysis of pharmaceuticals in water samples by SPE and GC-MS. Anal. Bioanal. Chem. 2007, 388, 627–635. [Google Scholar] [CrossRef]
- Agunbiade, F.O.; Moodley, B. Occurrence and distribution pattern of acidic pharmaceuticals in surface water, wastewater, and sediment of the Msunduzi river, Kwazulu-Natal, South Africa. Environ. Toxicol. Chem. 2016, 35, 36–46. [Google Scholar] [CrossRef]
- Quintana, J.B.; Miró, M.; Estela, J.M.; Cerdà, V. Automated On-Line Renewable Solid-Phase Extraction-Liquid Chromatography Exploiting Multisyringe Flow Injection-Bead Injection Lab-on-Valve Analysis. Anal. Chem. 2006, 78, 2832–2840. [Google Scholar] [CrossRef]
- Cocovi-Solberg, D.J.; Miro, M. 3D Printing: The second dawn of lab-on-valve fluidic platforms for automatic biochemical assays. Anal. Chem. 2019, 91, 1140–1149. [Google Scholar] [CrossRef]
- Trojanowicz, M. Recent advances in flow injection analysis. Analyst 2016, 141, 2085–2139. [Google Scholar] [CrossRef] [PubMed]
- Růžička, J.; Scampavia, L. From Flow Injection to Bead Injection. Anal. Chem. 1999, 71, 257A–263A. [Google Scholar] [CrossRef] [PubMed]
- Růžička, J. Lab-on-valve: Universal microflow analyzer based on sequential and bead injection. Analyst. 2000, 125, 1053–1060. [Google Scholar] [CrossRef]
- Růžička, J.; Marshall, G.D. Sequential injection: A new concept for chemical sensors, process analysis and laboratory assays. Anal. Chim. Acta 1990, 237, 329–343. [Google Scholar] [CrossRef]
- Oliveira, H.M.; Miró, M.; Segundo, M.A.; Lima, J.L.F.C. Universal approach for mesofluidic handling of bead suspensions in lab-on-valve format. Talanta 2011, 84, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Vichapong, J.; Buraham, R.; Srijaranai, S.; Grudpan, K. Sequential injection-bead injection-lab-on- valve coupled to high-performance liquid chromatography for online renewable micro- solid-phase extraction of carbamate residues in food and environmental samples. J. Sep. Sci. 2011, 34, 1574–1581. [Google Scholar] [CrossRef]
- Oliveira, H.M.; Segundo, M.A.; Lima, J.L.F.C.; Miró, M.; Cerdà, V. Exploiting automatic on-line renewable molecularly imprinted solid-phase extraction in lab-on-valve format as front end to liquid chromatography: Application to the determination of riboflavin in foodstuffs. Anal. Bioanal. Chem. 2010, 397, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Vidigal, S.S.M.P.; Tóth, I.V.; Rangel, A.O.S.S. Exploiting the bead injection LOV approach to carry out spectrophotometric assays in wine: Application to the determination of iron. Talanta 2011, 84, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Miró, M.; Jończyk, S.; Wang, J.; Hansen, E.H. Exploiting the bead-injection approach in the integrated sequential injection lab-on-valve format using hydrophobic packing materials for on-line matrix removal and preconcentration of trace levels of cadmium in environmental and biological samples via formation of non-charged chelates prior to ETAAS detection. J. Anal. At. Spectrom. 2003, 18, 89–98. [Google Scholar]
- Horstkotte, B.; Chocholouš, P.; Solich, P. Large volume preconcentration and determination of nanomolar concentrations of iron in seawater using a renewable cellulose 8-hydroquinoline sorbent microcolumn and universal approach of post-column eluate utilization in a Lab-on-Valve system. Talanta 2016, 150, 213–223. [Google Scholar] [CrossRef]
- Quintana, B.; Boonjob, W.; Miro, M. Online Coupling of Bead Injection Lab-On-Valve Analysis to Gas Chromatography: Application to the Determination of Trace Levels of Polychlorinated Biphenyls in Solid Waste. Anal. Chem. 2009, 81, 4822–4830. [Google Scholar] [CrossRef] [PubMed]
- Grand, M.M.; Chocholouš, P.; Růžička, J.; Solich, P.; Measures, C.I. Determination of trace zinc in seawater by coupling solid phase extraction and fluorescence detection in the Lab-On-Valve format. Anal. Chim. Acta 2016, 923, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Maya, F.; Horstkotte, B.; Estela, J.M.; Cerdà, V. Lab in a syringe: Fully automated dispersive liquid-liquid microextraction with integrated spectrophotometric detection. Anal. Bioanal. Chem. 2012, 404, 909–917. [Google Scholar] [CrossRef]
- Horstkotte, B.; Solich, P. The Automation Technique Lab-In-Syringe: A Practical Guide. Molecules 2020, 25, 1612. [Google Scholar] [CrossRef] [Green Version]
- Alexovič, M.; Horstkotte, B.; Šrámková, I.; Solich, P.; Sabo, J. Automation of dispersive liquid-liquid microextraction and related techniques. Approaches based on flow, batch, flow-batch and in- syringe modes. Trends Anal. Chem. 2017, 86, 39–55. [Google Scholar] [CrossRef]
- Horstkotte, B.; Suárez, R.; Solich, P.; Cerdà, V. In-syringe-stirring: A novel approach for magnetic stirring-assisted dispersive liquid–liquid microextraction. Anal. Chim Acta 2013, 788, 52–60. [Google Scholar] [CrossRef]
- Horstkotte, B.; Miró, M.; Solich, P. Where are modern flow techniques heading to? Anal. Bioanal. Chem. 2018, 410, 6361–6370. [Google Scholar] [CrossRef]
- Maya, F.; Cabello, C.P.; Estela, J.M.; Cerda, V. Automatic In-Syringe Dispersive Microsolid Phase Extraction Using Magnetic Metal—Organic Frameworks. Anal. Chem. 2015, 87, 7545–7549. [Google Scholar] [CrossRef]
- Methods, Method Verification and Validation Volume 2 (fda.gov). Available online: https://www.fda.gov/media/73920/download (accessed on 11 August 2021).
- Cocoví-Solberg, D.J.; Miró, M. CocoSoft: Educational software for automation in the analytical chemistry laboratory. Anal. Bioanal. Chem. 2015, 407, 6227–6233. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Analytes | Ret. Time ± SD | Calibration Curve Slope (L/µg) | R2 | CF | LOD (µg L−1) | LOQ (µg L−1) | Repeatability, RSD (n = 6, 20 µg L−1) (%) | Inter-Day Precision, RSD (n = 12, 20 µg/L) (%) |
|---|---|---|---|---|---|---|---|---|
| KET | 4.59 ± 0.03 | 981.15 ± 46.6 | 0.9983 | 15 | 0.62 | 1.9 | 4.2 | 7.9 |
| NAP | 5.01 ± 0.04 | 10982 ± 16.9 | 0.9975 | 14 | 0.06 | 0.2 | 3.2 | 6.0 |
| FLB | 8.91 ± 0.06 | 1120.4 ± 52.8 | 0.9955 | 26 | 0.88 | 2.7 | 3.9 | 5.7 |
| DCF | 10.93 ± 0.02 | 827.56 ± 8.9 | 0.9941 | 32 | 1.45 | 4.4 | 7.6 | 9.2 |
| IBU | 13.08 ± 0.08 | 680.28 ± 10.3 | 0.9978 | 19 | 1.98 | 6.0 | 3.6 | 5.2 |
| Analyte | KET | NAP | FLB | DCF | IBU | |
|---|---|---|---|---|---|---|
| Sample 1 | Genuine | <LOQ (1.1) | 0.3 | <LOD | <LOD | <LOD |
| Added | 20 | 20 | 20 | 20 | 20 | |
| Found | 21.1 | 20.3 | 20 | 19.4 | 21 | |
| Recovery | 106 ± 2 | 101 ± 1 | 100 ± 1 | 96.7 ± 3 | 105 ± 1 | |
| Sample 2 | Genuine | <LOQ (1.1) | <LOD | <LOQ (0.3) | <LOD | <LOD |
| Added | 20 | 20 | 20 | 20 | 20 | |
| Found | 21.1 | 20 | 20.3 | 19.3 | 21.1 | |
| Recovery | 105 ± 2 | 100 ± 1 | 101 ± 1 | 96.4 ± 3 | 105 ± 1 | |
| Sample 3 | Genuine | <LOQ (1.3) | 0.6 | <LOD | <LOD | <LOD |
| Added | 20 | 20 | 20 | 20 | 20 | |
| Found | 21.3 | 20.6 | 19.7 | 19.2 | 21.4 | |
| Recovery | 106 ± 2 | 103 ± 1 | 98.7 ± 1 | 95.9 ± 3 | 107 ± 1 | |
| Sample 4 | Genuine | <LOQ (1.8) | 0.3 | <LOD | <LOD | <LOD |
| Added | 20 | 20 | 20 | 20 | 20 | |
| Found | 21.8 | 20.3 | 19.9 | 18.2 | 21.2 | |
| Recovery | 109 ± 2 | 101 ± 1 | 100 ± 1 | 90.9 ± 3 | 106 ± 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gemuh, C.V.; Horstkotte, B.; Solich, P. Lab-In-Syringe with Bead Injection Coupled Online to High-Performance Liquid Chromatography as Versatile Tool for Determination of Nonsteroidal Anti-Inflammatory Drugs in Surface Waters. Molecules 2021, 26, 5358. https://doi.org/10.3390/molecules26175358
Gemuh CV, Horstkotte B, Solich P. Lab-In-Syringe with Bead Injection Coupled Online to High-Performance Liquid Chromatography as Versatile Tool for Determination of Nonsteroidal Anti-Inflammatory Drugs in Surface Waters. Molecules. 2021; 26(17):5358. https://doi.org/10.3390/molecules26175358
Chicago/Turabian StyleGemuh, Celestine Vubangsi, Burkhard Horstkotte, and Petr Solich. 2021. "Lab-In-Syringe with Bead Injection Coupled Online to High-Performance Liquid Chromatography as Versatile Tool for Determination of Nonsteroidal Anti-Inflammatory Drugs in Surface Waters" Molecules 26, no. 17: 5358. https://doi.org/10.3390/molecules26175358