
Synthesis of 2-Substitued Indoles via Pd-Catalysed Cyclization in an Aqueous Micellar Medium


Dipartimento di Biotecnologie, Chimica e Farmacia, Università degli Studi di Siena, Via A. Moro 2, 53100 Siena, Italy
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(13), 3917; https://doi.org/10.3390/molecules26133917
Submission received: 17 May 2021
/
Revised: 21 June 2021
/
Accepted: 23 June 2021
/
Published: 26 June 2021
(This article belongs to the Special Issue Preparation of Heterocycles by Metal-Promoted Reactions)
Abstract
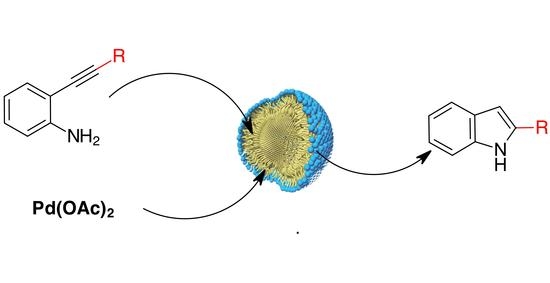
:The synthesis of 2-substituted indoles starting from the corresponding unprotected 2-alkynylanilines was made possible in 3% TPGS-750-M water using Pd(OAc)2 alone as the catalyst. The reaction was sensitive to the heating mode respect to the nature of the starting material as, in many cases, convectional heating was better than microwave dielectric heating. The MW (microwave) delivery mode had also an influence in the formation of by-products and, consequently, product yields. A tandem Sonogashira-cyclisation reaction was also accomplished using Pd(OAc)2/Xphos in the nanomicellar water environment.
1. Introduction
Amongst the plethora of methods to synthesize indoles, the cyclization reaction of 2-alkynylaniline derivatives is a significant approach to build 2-substituted- or 2,3-disubstituted-indoles [1,2,3]. One of the major benefits is the easy access to diverse 2-alkynylanilines through Sonogashira reaction from commercially available ortho-haloanilines and 1-alkynes [4,5]. Using strong bases or catalysis from transition metals are required to induce cyclization. Noble metals are generally employed [2], although catalytic (or almost stoichiometric) Cu derivatives have been employed [6,7], as other more affordable catalysts containing, for example, Hg [8], or Zn [9,10]. Metal catalysed cyclizative approaches to indoles have been recently reviewed [11,12]. Electrochemical cyclisation [13] or simple MW irradiation has been also reports as suitable technologies for cyclisation of 2-alkynyl-anilines [14].
Although the cyclization of 2-alkynylanilines has been dissected in most of its aspects, some issues are still unresolved. The need for alkynyl amines protected at N with electron-withdrawing groups (as Tf, Ts or Ns) [15,16,17], the use of ligands to stabilize the metal catalyst [14,15] or the employment of organic polar media as solvents [15,16] are some problems that should still be solved. Regarding the use of sustainable solvents, the use of copper catalysis in water have been recently reported for cyclisation of N-(2-ethynylphenyl)-sulfonamides to indoles [18,19].
Water is the environmentally benign solvent par excellence, often characterized by a different reactivity respect to organic solvents [20]. However, water is often not compatible with transition metal complexes and also low solubility of organic polar substrates hampers the success of many organic processes in aqueous media. Using surfactants appears as one of the simplest approaches to perform organic reactions in water [21,22,23]. The micellar environment enables the solubilization of organic lipophilic molecules in water behaving as a nanoreactor with a powerful influence on the reaction rate [24]. Recently a one-pot synthesis of indoles and pyrazoles have been reported under aqueous micellar catalysis [25]. The arylation of N-Boc-hydrazine was reported to occur in 3% TPGS-750-M in water in the presence of [t-BuBrettPhosPd-(allyl)]OTf. The further Fischer indole cyclisation of the arylhydrazine in the presence of ketones was carried out in the same pot under acid catalysis with good yields.
Following our interest in metal catalysed organic transformations in environmentally benign media [26,27], we became interested in studying the cyclisation of unprotected 2-alkynylaniline to 2-substituted indoles in a water/micellar environment. Starting from the unprotected aniline has the advantage to avoid the waste of reagents required for the protection and deprotection steps. The final indole could be also functionalized at the nitrogen with groups that may be not compatible with the cyclisation process.
2. Results and Discussion
Cyclisation of o-[2-(4-methoxy-3-tolyl)ethynyl]aniline (1) [28] was selected as the model reaction (Table 1). We explored different Pd catalyst/ligand combination to find the best conditions to have good conversion of the starting material into the cyclized product 2. As surfactant, we opted for TPGS-750-M (Table 1), a designed surfactant already applied in several metal-catalyzed reactions [28,29,30]. Based on literature reports on analogous reaction, we decided to investigate MW dielectric heating conditions using Pd(II) catalyst. Using Pd(PPh3)2Cl2, Pd(MeCN)2Cl2 or Pd(OAc)2 complexed with Xphos gave totally unsatisfactory results with formation of small amount of indole and starting material recovered (entries 1–5 in Table 1). Even the use of oxime-derived palladacycles [31] gave a mixture of indole 2 and starting material (entry 6). After the discovery, by chance, that the ligand had no effect on the reaction yield (entry 8), we tried to increase reaction time and temperature to improve the amount of 2 formed. The major by-products identified were the reduced forms of the starting alkyne.
However, the final improvement occurred adding 1 eq of acetic acid that gave increased yields and moderate amount of unreacted starting material (entry 10 in Table 1). A minor change of reaction temperature and time gave the complete disappearance of the starting alkynylaniline 1 providing indole 2 in 70% isolated yield after column chromatography. Attempts to increase the solubility of Pd(II) in the micelles using a more lipophilic acid (lauric acid) or a better complexing agent 4-(diphenylphosphino)benzoic acid (DPBA), had no effect in the reaction yield (entries 16 and 17 in Table 1). Analogously, a stronger acid as TFA gave lower yield of indole 2 together with a series of by-products (entry 15). Using water alone or a solution of an ionic surfactant as 3% SDS, we observed incomplete conversion of the starting material (entries 21 and 22), demonstrating that the use of the nanomicelles formed from neutral surfactant TPGS-750-M in water, was decisive for the success of the reaction.
Finally we were pleased to find that, using 5% mol of Pd(OAc)2, cyclisation occurred heating at 100 °C for 10 min, giving 72% isolated yield of indole 2 (entry 18). Attempts to further reduce the amount of Pd gave incomplete conversion even after extended heating under MWs (entry 19).
Irradiation mode seemed to have an influence in the yield of 2. Using the standard MW heating program that keeps the set temperature through magnetron on/off intervals, the crude reaction mixture obtained was characterized by several by-products and the yield of 2 was lower. Using the PowerMax™ technology, instead, using compressed gas to cool the reaction maintaining the temperature at 100 °C while the magnetron is on, gave a cleaner reaction mixture with less by-products resulting in higher isolated yield of indole 2 (entry 18). This last refined MW heating technology, however, did not prevent the formation of by-products as highlighted by the chromatographic analysis of the crude reaction mixture, probably due to the formation of hot-spot during irradiation [32]. A comparable result was observed with convectional heating of the reaction vial at 100 °C (external bath temperature) for 4 h, while longer reaction time produced a more complex mixture of by-products (entries 12 and 13 in Table 1).
Thus, we explored lower temperature for longer time, and we were pleased to observe that heating at 80 °C for 6 h gave a slightly better yield (76%), mainly due to complete conversion of the starting material and formation of less by-products. Attempts to use MW at 80 °C, on the other hand, gave incomplete conversion of aniline 1 after 6 h of heating (data not shown). In conclusion we succeeded in find conditions for cyclisation of unprotected alkynyl-aniline 1 into indole 2 in water using either MW dielectric or convectional heating with a certain preference for the convectional mode. The reaction required the presence of AcOH, probably due to limited coordination of Pd to the free amine, assistance in triple bond activation and facilitated protonolysis of C-Pd bond. This trend did not change when differently substituted o-alkynyl-anilines [33] were submitted to cyclisation to access other 2-substitued indoles (Scheme 1). With-not-coordinating residues, cyclisation occurred with acceptable yields stirring for 6 h under convectional heating (products 12–16 in Scheme 1).
When more coordinative elements were inserted in the residue, cyclisation of the free amine was more difficult. As the presence of the acid environment was indispensable for cyclisation, the use of alkynes with heteroatoms or functional groups with acid-labile protections (e.g., TBDMS of THP) was not possible. With our procedure, the only indoles with heteroatoms present at the substituents in position 2, were compounds 18–20 in Scheme 1. 2-Benzyloxymethyl-indole 18 was isolated in low yield after a careful purification from different by-products. With morpholine as substituent (20), cyclisation was even harder as the corresponding alkynylaniline seemed very unstable under heating. After several attempts, product 20 was isolated after stirring the reaction mixture for 7 days at room temperature. Although the tert-butoxycarbonyl protective group is known to be thermally labile, indole 19 was obtained under MW dielectric heating. In this case MWs were better than oil bath, as convectional heating gave largely deprotection of the starting material. An analogous behavior was observed with cyclopropyl derivative 17 that was obtained in better yields under MWs. The MW heating mode had again an influence in the reaction success. The “harder” reaction conditions, using PowerMax on, gave crudes containing larger amounts of by-products respect to the MW based procedure that maintains constant the temperature switching off the magnetron for much of the reaction time.
Finally, we tried to carry out a one pot Sonogashira/indole cyclisation, by relying on the later insight in the Sonogashira copper free mechanism. A tandem Pd/Pd cycle linked, via a multistep transmetallation process, with Pd(II) species which takes the place of Cu(I), has been proposed, suggesting that some Pd(II) was always present in the medium at the end of the reaction [34].
This hypothesis was explored starting from unprotected o-iodo-aniline 21 and alkynes 22–24 (Scheme 2). The first step was carried out in the presence of Et3N as the reaction requires an alkaline environment. When the starting aniline 21 was completely consumed, more Pd(OAc)2 was added and the vial heated at 100 °C under MWs. Following this procedure, compounds 2, and 13 were obtained in acceptable yields while cyclization with alkyne 24 did not occur and only the Sonogashira product was recovered.
3. Materials and Methods
3.1. General Information
The reactions were carried out in a previously dried MW vial or Schlenk tubes. Product purification was carried out by flash column chromatography Merck silica gel 60, 0.040–0.063 mm (230–400 mesh) or using a 55 µm, 70 Å Strata® SCX column from Phenomenex (Torrance, CA, USA) (phase: 2 g/12 mL strong cation exchange, SCX-SO3H). Merck (Darmstadt, Germany) aluminium-backed plates pre-coated with silica gel 60 (UV254) were used for analytical thin layer chromatography and were visualized by staining a solution of ninhydrin in EtOH.
1H-NMR (600 MHz) and 13C-NMR (JMOD) (150 MHz) spectra were recorded on an Avance NMR spectrometer (Bruker, Billerica, MA, USA). Deuterated chloroform, methanol and acetone were used as the solvent, and chemical shift values (δ) are reported in parts per million referred to the residual signals of this solvent (CDCl3 δ 7.26 for 1H and δ 77.16 for 13C; MeOD δ 3.31 for 1H and δ 49.00 for 13C; (CD3)2CO δ 2.05 for 1H and δ 205.87 and 30.70 for 13C). Data are represented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet or multiple resonances, bs = broad singlet), coupling constant (J) in Hertz and the integration. HPLC/MS analysis were performed with an Agilent 1260 Infinity II Preparative LC/MSD System Single Quadrupole (LC/MSD iQ, Agilent Technologies, Santa Clara, CA, USA) using an InfinityLab Poroshell 120 EC-C18 column (2.1 × 50 mm, 2.7 μm), flow 0.4 mL/min, MeCN/H2O gradient from 5/95 to 95/5 in 12 min. ES-MS analysis were performed with an Agilent 1100 series LC/MSD system, connected with a UV detector (254 nm), flow 0.4 mL/min, 95% MeOH, 5% H2O. Direct injection. ESI ionization, flow of the drying gas (N2) 9 mL/min, temperature 350 °C, atomizing pressure 40 PSI, fragmentation 70. Melting points were determined on an IA 9100 apparatus (Electrothermal, Staffordshire, UK). Elemental analyses were provided by an external supplier. Reactions carried out under MW dielectric heating were performed with a microwave oven (Discovery from CEM, Matthews, NC, USA) under monomode irradiation in a 10 mL sealed vials. The internal temperature was monitored through an internal IR sensor, and the maximal internal pressure monitored and maintained under the value of 150 psi. All reagents were used as purchased from commercial suppliers without further purification. Compounds 1 and 3–11 were prepared following literature procedures [28,35]. TPGS-750-M was used at higher concentration respect to the critical micellar concentration (CMC) [36].
3.2. Procedures
2-(4-Methoxy-3-methylphenyl)-1H-indole (2). General procedure, method A: In a Schlenk tube, a suspension of compound 1 (100 mg, 0.42 mmol) in TPGS-750-M 3 wt% in H2O (1 mL) was prepared under a nitrogen atmosphere. Acetic acid (24 µL, 0.42 mmol) and Pd(OAc)2 (5 mg, 0.02 mmol) were added sequentially. The reaction mixture was heated at 80 °C (external thermometer) in oil bath for 6 h, then diluted into EtOAc (5 mL). The organic layer was washed with H2O (2 × 5 mL) and brine (10 mL), dried over Na2SO4 and filtered. Removal of the solvent under reduce pressure gave a dark residue which was purified by silica gel flash chromatography or SCX column.
2-(4-Methoxy-3-methylphenyl)-1H-indole (2). General procedure, method B: A 10 mL MW tube was charged with a suspension of the compound 1 (100 mg, 0.42 mmol) in TPGS-750-M 3 wt% in H2O (1 mL). Acetic acid (24 µL, 0.42 mmol) and Pd(OAc)2 (5 mg, 0.02 mmol) were added sequentially under nitrogen atmosphere. The reaction mixture was submitted to microwave dielectric heating (MWs) for 10 min at 100 °C constantly cooling (ramp time 5 min; internal max pressure 250 psi; 150 W). Organic mixture was diluted into EtOAc (5 mL), the resulting phases were separated, and the organic layer was washed with H2O (2 × 5 mL), and brine (10 mL), dried over Na2SO4, filtered and the solvent was removed under vacuo. The dark residue was purified by flash chromatography on silica gel or SCX column. In alternative, the reaction mixture was submitted to microwave dielectric heating (MWs) for 3 cycles of 40 min at 80 °C (ramp time 1 min; internal max pressure 250 psi; power max 150 W) without constantly cooling.
2-(4-Methoxy-3-methylphenyl)-1H-indole (2) via Sonogashira/indole tandem cyclization reaction. A 10 mL MW tube was charged with iodoaniline (50 mg, 0.23 mmol), 4-ethynyl-1-methoxy-2-methylbenzene (50 μL, 0.34 mmol), triethylamine (80 μL, 0.58 mmol), a solution of TPGS-750-M 3 wt% in H2O (1 mL). Pd(CH3CN)2Cl2 (3 mg, 0.01 mmol) and Xphos (9 mg, 0.02 mmol) were added and the tube sealed. The mixture was submitted to microwave dielectric heating (MWs) for 30 min at 80 °C (max internal pressure 150 psi). Then Pd(OAc)2 (3 mg, 0.01 mmol) was added and the resulting mixture was heated under MW irradiations for further 10 min at 100 °C constantly cooling (PowerMax ON). The organic layer was diluted in EtOAc (5 mL) and washed with H2O (2 × 5 mL), dried over Na2SO4 and concentrated in vacuo. The residue was purified by silica gel flash chromatography (PE:EtOAc = 8:2) to afford 1 as a white solid (20 mg, 35%).
2-(4-Methoxy-3-methylphenyl)-1H-indole (2). Prepared from 2-((4-methoxy-3-methylphenyl)ethynyl)aniline in according with general procedure A, by heating at 80 °C (external thermometer) for 6 h in oil bath. Purified by silica gel flash chromatography (PE:EtOAc = 9:1). White solid; yield 76% (76 mg). Mp: 118–120 °C. MS-ESI: m/z 236 [M−H]−. 1H-NMR (600 MHz, CDCl3) δ 8.08 (s, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.40 (d, J = 6.9 Hz, 2H), 7.21 (t, J = 7.3 Hz, 1H), 7.15 (t, J = 7.3 Hz, 1H), 6.85 (dd, J = 11.8, 9.6 Hz, 2H), 6.55 (s, 1H), 3.86 (s, 3H), 2.49 (s, 3H). 13C-NMR (151 MHz, CDCl3) δ 159.29, 137.83, 137.46, 135.98, 130.28, 128.97, 125.38, 121.79, 120.35, 119.99, 116.41, 111.48, 110.66, 102.39, 55.33, 21.30. C16H15NO calcd C 80.98, H 6.37, N 5.90; O 6.74; found C 80.91, H 6.35, N 5.88.
2-([1,1′-Biphenyl]-4-yl)-1H-indole (12). Prepared from 2-([1,1′-biphenyl]-4-ylethynyl)aniline following general procedure A. Purified by SCX column eluting with MeOH. Pale yellow solid: yield 60% (60 mg). Mp: 281–283 °C. MS-ESI: m/z 268 [M−H]−. 1H-NMR (600 MHz, CDCl3) δ 8.35 (s, 1H), 7.76–7.62 (m, 6H), 7.53–7.35 (m, 5H), 7.21 (t, J = 7.6 Hz, 1H), 7.14 (t, J = 7.4 Hz, 1H), 6.89 (s, 1H). 13C-NMR (151 MHz, acetone-d6) δ 140.31, 139.78, 131.71, 128.90, 127.41, 127.30, 126.63, 125.51, 121.83, 120.20, 119.62, 111.11, 99.21. C20H15N calcd C 89.19, H 5.61, N 5.20; found C 89.24, H 5.64, N 5.23.
2-(7-Methoxynaphthalen-2-yl)-1H-indole (13). Prepared from 2-((7-methoxynaphthalen-2-yl)ethynyl)aniline following general procedure B. Purified by trituration with petroleum ether. Pale yellow solid; yield 65% (65 mg). Mp: 238–240 °C. MS-ESI: m/z 272 [M−H]−. 1H-NMR (600 MHz, acetone) δ 8.27 (s, 1H), 7.97 (dd, J = 8.5, 1.4 Hz, 1H), 7.87 (d, J = 8.5 Hz, 1H), 7.83 (d, J = 8.9, 1H), 7.58 (d, J = 7.7 Hz, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.31 (s, 1H), 7.18 (dd, J = 8.8, 2.3 Hz, 1H), 7.12 (t, J = 7.4 Hz, 1H), 7.03 (t, J = 7.3 Hz, 1H), 6.97 (s, 1H), 3.93 (s, 3H). 13C-NMR (151 MHz, acetone) δ 158.05, 138.08, 137.52, 134.20, 129.47, 129.19, 127.93, 127.43, 124.28, 122.97, 121.70, 120.14, 119.57, 119.25, 111.04, 105.92, 99.08, 54.80. C19H15NO calcd C 83.49, H 5.53; N 5.12; O 5.85; found C 83.53, H 5.50; N 5.08.
2-(Thiophen-3-yl)-1H-indole (14). Prepared from 2-(thiophen-3-ylethynyl)aniline by classical heating in oil bath at 80 °C (external thermometer) for 6 h. Purified by silica gel flash chromatography (PE:EtOAc = 9:1). White solid; yield 55% (55 mg). Mp: 210–213 °C. MS-ESI: m/z 198 [M−H]−. 1H-NMR (600 MHz, CDCl3) δ 8.27 (s, 1H), 7.62 (d, J = 7.7 Hz, 1H), 7.42 (s, 4H), 7.20 (t, J = 6.8 Hz, 1H), 7.14 (d, J = 6.8 Hz, 1H), 6.71 (s, 1H). 13C-NMR [37]. C12H9NS calcd C 72.33; H 4.55; N 7.03; S 16.09; found C 72.28; H 4.53; N 6.99.
2-Hexyl-1H-indole (15). Prepared from 2-(oct-1-yn-1-yl)aniline according to general procedure A, for 4 h. Purified by silica gel flash chromatography (PE:EtOAc = 9:1). Pale yellow oil; yield 56% (56 mg). MS-ESI: m/z 200 [M−H]−1H-NMR (600 MHz, CDCl3) δ 7.86 (s, 1H), 7.55 (d, J = 7.6 Hz, 1H), 7.31 (d, J = 7.8 Hz, 1H), 7.13 (t, J = 7.4 Hz, 1H), 7.09 (t, J = 7.4 Hz, 1H), 6.25 (s, 1H), 2.77 (t, J = 7.6 Hz, 2H), 1.75 (dd, J = 14.8, 7.3 Hz,25H), 1.49–1.32 (m, 6H), 0.93 (d, J = 6.7 Hz, 3H).13C-NMR [38]. HRMS (ESI) calcd for C14H18N [M−H]− 200.1439; found 200.1438.
2-Isopentyl-1H-indole (16). Prepared from 2-(5-methylhex-1-yn-1-yl)aniline following general procedure A. Purified by SCX column eluting with MeOH. Waxy material; yield 49% (49 mg). MS-ESI: m/z 186 [M−H]−. 1H-NMR (600 MHz, MeOD) δ 7.39 (d, J = 7.8 Hz, 1H), 7.25 (dd, J = 8.0, 0.5 Hz, 1H), 7.01–6.94 (m, 1H), 6.94-6.86 (m, 1H), 6.10 (d, J = 0.5 Hz, 1H), 2.86–2.67 (m, 2H), 1.71–1.53 (m, 3H), 0.97 (d, J = 6.2 Hz, 6H). 13C-NMR (151 MHz, MeOD) δ 140.18, 136.45, 128.90, 119.74, 118.78, 118.29, 109.94, 97.84, 38.24, 27.46, 25.70, 21.45. HRMS (ESI) calcd for C13H16N [M−H]− 186.1283; found 186.1284.
2-Cyclopropyl-1H-indole (17). Prepared from 2-(cyclopropylethynyl)aniline by heating under microwave irradiations at 80 °C for 3 cycles of 40 min, 150 W, 250 psi, PowerMax OFF. Purified by silica gel flash chromatography (PE:EtOAc = 9:1). Pale yellow solid; yield 61% (61 mg). Mp: 59–60 °C. MS-ESI: m/z 156 [M−H]−. 1H-NMR (600 MHz, CDCl3) δ 7.98 (s, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.29 (s, 1H), 7.13–7.08 (m, 2H), 6.16 (s, 1H), 1.12–0.56 (m, 5H). 13C-NMR [39] C11H11N calcd C 84.04, H 7.05, N 8.91; found C 83.97, H 7.03, N 8.88.
2-((Benzyloxy)methyl)-1H-indole (18). Prepared from 2-(3-(benzyloxy)prop-1-yn-1-yl)aniline following general procedure A. Purified by silica gel flash chromatography (PE:EtOAc = 6:4). Pale yellow oil; yield 25% (25 mg). MS-ESI: m/z 236 [M−H]−. 1H-NMR (600 MHz, MeOD) δ 7.49 (d, J = 7.8 Hz, 1H), 7.37–7.28 (m, 6H), 7.08 (t, J = 7.4 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 6.39 (s, 1H), 4.68 (s, 2H), 4.55 (s, 2H). 13C-NMR [40]. HRMS (ESI) calcd for C16H14NO [M−H]− calcd 236.1075; found 236.1075.
tert-Butyl((1H-indol-2-yl)methyl)carbamate (19). Prepared from tert-butyl (3-(2-aminophenyl)prop-2-yn-1-yl)carbamate by heating under MW irradiations at 80 °C for 3 cycles of 20 min, 150 W, 250 psi. (In according with general procedure B). Purified by silica gel flash chromatography (PE:EtOAc = 8:2). White solid; yield 28% (28 mg). Mp 108–111 °C MS-ESI: m/z 245 [M−H]−. 1H-NMR (600 MHz, MeOD) δ 7.44 (d, J = 7.8 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.04 (t, J = 7.3 Hz, 1H), 6.95 (t, J = 7.3 Hz, 1H), 6.27 (s, 1H), 4.85 (s, 2H), 1.47 (s, 9H).13C-NMR (151 MHz, MeOD) δ 120.65, 119.42, 118.65, 110.40, 98.97, 37.52, 27.37. HRMS (ESI) calcd for C14H17N2O2 [M−H]− 245.1290; found 245.1292.
4-((1H-Indol-2-yl)methyl)morpholine (20): a 25 mL bottom flask was charged with a suspension of 11 (90 mg, 0.42 mmol) in TPGS-750-M 3 wt% in H2O (1 mL); Pd(OAc)2 (10 mg, 0.04 mmol) were added sequentially under nitrogen atmosphere. The reaction mixture was stirred at room temperature for 7 days. The organic layer was diluted in CH2Cl2 (5 mL) and washed with H2O (2 × 5 mL), dried over Na2SO4 and concentrated in vacuo. The residue was purified by silica gel flash chromatography (CH2Cl2:MeOH = 85:15). Orange oil; yield 25% (23 mg). 1H-NMR (600 MHz, CDCl3) δ 8.87 (s, 1H), 7.68 (dd, J = 10.0, 8.9 Hz, 1H), 7.37 (d, J = 7.9 Hz, 1H), 7.18 (t, J = 7.4 Hz, 1H), 7.09 (t, J = 7.2 Hz, 1H), 6.41 (s, 1H), 3.78 (s, 6H), 2.59 (s, 4H). 13C-NMR [41]. The sample was contaminated with Ph3PO coming from the starting alkyne.
4. Conclusions
The TPGS-750-M/water environment was found to be a suitable medium to carry out cyclisation of N-unprotected o-alkynyl-aniline under Pd(OAc)2 catalysis. The reaction occurred without ligands but was very sensitive to the heating mode employed. Convectional heating resulted superior to MW dielectric heating as it allowed to work at lower temperature for longer times, thus reducing the number of by-products formed during the reaction. Importantly, when MWs were employed, the use of simultaneous cooling and continuous irradiation at variable power was more efficient for thermally stable substrates.
Although with low yields, we show also that a tandem Sonogashira-cyclisation reaction it is possible. Further optimization of the conditions is still required for application to larger scale processes, the procedure described here possess marked benefits in terms of atom economy and overall safety if compared with other previously reported methodologies.
Supplementary Materials
The following are available online. General experimental procedure and product characterization, 1H and 13C-NMR spectra Figures S1–S15 and HPLC spectrum Figure S16.
Author Contributions
Investigation, E.C. and M.T.; Methodology, S.S. and G.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MIUR (Rome) for financial support within the programme “Dipartimenti di Eccellenza 2018–2022. Chemessentia-Chemo (Novara, Italy) is also gratefully acknowledged for financial support to GV.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of compounds 2, 12–20 are available from authors.
References
- Herraiz-Cobo, J.; Albericio, F.; Álvarez, M. The Larock Reaction in the Synthesis of Heterocyclic Compounds. Adv. Heterocycl. Chem. 2015, 116, 1–35. [Google Scholar] [CrossRef]
- Sinha, A.K.; Equbal, D.; Uttam, M.R. Metal-catalyzed privileged 2- and 3-functionalized indole synthesis. Chem. Heterocycl. Compd. 2018, 54, 292–301. [Google Scholar] [CrossRef]
- Colella, M.; Degennaro, L.; Luisi, R. Continuous Flow Synthesis of Heterocycles: A Recent Update on the Flow Synthesis of Indoles. Molecules 2020, 25, 3242. [Google Scholar] [CrossRef]
- Plenio, H. Catalysts for the Sonogashira Coupling-The Crownless Again Shall Be King. Angew. Chem. Int. Ed. 2008, 47, 6954–6956. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C. The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry. Chem. Rev. 2007, 107, 874–922. [Google Scholar] [CrossRef] [PubMed]
- Hiroya, K.; Itoh, S.; Sakamoto, T. Mild and efficient cyclization reaction of 2-ethynylaniline derivatives to indoles in aqueous medium. Tetrahedron 2005, 61, 10958–10964. [Google Scholar] [CrossRef]
- Hiroya, K.; Itoh, S.; Sakamoto, T. Development of an Efficient Procedure for Indole Ring Synthesis from 2-Ethynylaniline Derivatives Catalyzed by Cu(II) Salts and Its Application to Natural Product Synthesis. J. Org. Chem. 2004, 69, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Sasaki, I.; Hirai, Y.; Namba, K.; Imagawa, H.; Nishizawa, M. Silaphenylmercuric Triflate Catalyzed Reactions: Synthesis of a Solid-Supported Mercuric Salt Catalyst. Angew. Chem. Int. Ed. 2009, 48, 1244–1247. [Google Scholar] [CrossRef]
- Yin, Y.; Ma, W.; Chai, Z.; Zhao, G. Et2Zn-Catalyzed Intramolecular Hydroamination of Alkynyl Sulfonamides and the Related Tandem Cyclization/Addition Reaction. J. Org. Chem. 2007, 72, 5731–5736. [Google Scholar] [CrossRef] [PubMed]
- Marsiciano, V.; Arcadi, A.; Chiarini, M.; Fabrizi, G.; Goggiamani, A.; Iazzetti, A. Synthesis of functionalised 2-3,dihydroquinolin-4(1H)-ones vs.quinoline or N-alkenylindole derivates through sequential reaction of 2-alkynylanilines with ketones. Org. Biomol. Chem. 2021, 19, 421–438. [Google Scholar] [CrossRef]
- Mancuso, R.; Dalpozzo, R. Recent Progress in the Transition Metal Catalyzed Synthesis of Indoles. Catalysts 2018, 8, 458. [Google Scholar] [CrossRef] [Green Version]
- Reddy, V.; Anand, R.V. Expedient Access to Unsymmetrical Diarlyindoliylmethanes through Palladium-Catalyzed Domino Elettrophilic Cyclization -Extended Conjugate Addition Approch. Org. Lett. 2015, 17, 3390–3393. [Google Scholar] [CrossRef]
- Arcadi, A.; Bianchi, G.; Inesi, A.; Marinelli, F.; Rossi, L. Electrochemical-mediated cyclization of 2-alkynylanilines: A clean and safe synthesis of indole derivatives. Eur. J. Org. Chem. 2008, 783–787. [Google Scholar] [CrossRef]
- Carpita, A.; Ribecai, A. Microwave-assisted synthesis of indole-derivatives via cycloisomerization of 2-alkynylanilines in water without added catalysts, acids, or bases. Tetrahedron Lett. 2009, 50, 6877–6881. [Google Scholar] [CrossRef]
- Cacchi, S.; Fabrizi, G.; Parisi, L.M. 2-Aryl and 2-Heteroaryl Indoles from 1-Alkynes and o -Iodotrifluoroacetanilide through a Domino Copper-Catalyzed Coupling−Cyclization Process. Org. Lett. 2003, 5, 3843–3846. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.Z.; Zhao, W.; Wei, H.X.; Dufour, M.; Farina, V.; Senanayake, C.H. A practical mild, one-pot, regiospecific synthesis of 2,3-disubstituted indoles via consecutive Sonogashira and Cacchi reactions. Org. Lett. 2006, 8, 3271–3274. [Google Scholar] [CrossRef]
- Li, J.; Li, C.; Ouyang, L.; Li, C.; Wu, W.; Jiang, H. N-Heterocyclic carbene palladium-catalyzed cascade annulation/alkynylation of 2-alkynylanilines with terminal alkynes. Org. Biomol. Chem. 2017, 15, 7898–7908. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, Z.; Zhang, F.; Xie, S.; Li, H.; Li, P.; Zhou, X. Palladium nanoparticles confined in the cages of MIL-101: An efficient catalyst for the one-pot indole synthesis in water. ACS Catal. 2011, 1, 1604–1612. [Google Scholar] [CrossRef]
- Song, S.; Huang, M.; Li, W.; Zhu, X.; Wan, Y. Efficient synthesis of indoles from 2-alkynylaniline derivatives in water using a recyclable copper catalyst system. Tetrahedron 2015, 71, 451–456. [Google Scholar] [CrossRef]
- Sun, H.; Xiao, L.; Li, W.; Xie, Q.; Shao, L. On Water Silver(I)-Catalyzed Cycloisomerization of Acetylenic Free Amine/Amide towards 7-Azaindole/Indoles/Isochinolone Derivates. Synthesis 2017, 49, 4845–4852. [Google Scholar] [CrossRef] [Green Version]
- Lippincott, D.J.; Landstrom, E.; Cortes-Clerget, M.; Lipshutz, B.H.; Buescher, K.; Schreiber, R.; Durano, C.; Parmentier, M.; Ye, N.; Wu, B.; et al. Surfactant Technology: With New Rules, Designing New Sequences Is Required! Org. Process Res. Dev. 2020, 24, 841–849. [Google Scholar] [CrossRef]
- Anisari, T.N.; Jasinski, J.B.; Leahy, D.K.; Handa, S. Metal–Micelle Cooperativity: Phosphine Ligand-Free Ultrasmall Palladium(II) Nanoparticles for Oxidative Mizoroki–Heck-type Couplings in Water at Room Temperature. JACS Au 2021, 1, 308–315. [Google Scholar] [CrossRef]
- Pang, H.; Hu, Y.; Gallou, F.; Lipshutz, B. Water-Sculpting of a Heterogeneous Nanoparticle Precatalyst for Mizoroki–Heck Couplings under Aqueous Micellar Catalysis Conditions. J. Am. Chem. Soc. 2021, 143, 3373–3382. [Google Scholar] [CrossRef]
- Lipshutz, B.H. Synthetic chemistry in a water world. New rules ripe for discovery. Curr. Opin. Green Sustain. Chem. 2018, 11, 1–8. [Google Scholar] [CrossRef]
- Landstrom, E.B.; Akporji, N.; Lee, N.R.; Gabriel, C.M.; Braga, F.C.; Lipshutz, B.H. One-Pot Synthesis of Indoles and Pyrazoles via Pd-Catalyzed Couplings/Cyclizations Enabled by Aqueous Micellar Catalysis. Org. Lett. 2020, 22, 6543–6546. [Google Scholar] [CrossRef] [PubMed]
- Risi, C.; Cini, E.; Petricci, E.; Saponaro, S.; Taddei, M. In water Markovnikov hydration and one-pot reductive hydroamination of terminal alkynes under Ruthenium nanoparticle catalysis. Eur. J. Inorg. Chem. 2020, 2020, 1000–1003. [Google Scholar] [CrossRef]
- Risi, C.; Calamante, M.; Cini, E.; Faltoni, V.; Petricci, E.; Rosati, F.; Taddei, M. In water alkylation of amines with alcohols through a borrowing hydrogen process catalysed by ruthenium nanoparticles. Green Chem. 2020, 22, 327–331. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Ghorai, S.; Abela, A.R.; Moser, R.; Nishikata, T.; Duplais, C.; Krasovskiy, A.; Gaston, R.D.; Gadwood, R.C. TPGS-750-M: A Second-Generation Amphiphile for Metal-Catalyzed Cross-Couplings in Water at Room Temperature. J. Org. Chem. 2011, 76, 4379–4391. [Google Scholar] [CrossRef] [Green Version]
- Andersson, M.P.; Gallou, F.; Klumphu, P.; Takale, B.S.; Lipshutz, B.H. Structure of Nanoparticles Derived from Designer Surfactant TPGS-750-M in Water, As Used in Organic Synthesis. Chem. A Eur. J. 2018, 24, 6778–6786. [Google Scholar] [CrossRef] [PubMed]
- Lipshutz, B.H.; Ghorai, S.; Cortes-Clerget, M. The Hydrophobic Effect Applied to Organic Synthesis: Recent Synthetic Chemistry “in Water”. Chem. A Eur. J. 2018, 24, 6672–6695. [Google Scholar] [CrossRef]
- Alacid, E.; Alonso, D.A.; Botella, L.; Nájera, C.; Pacheco, M.C. Oxime palladacycles revisited: Stone-stable complexes nonetheless very active catalysts. Chem. Rec. 2006, 6, 117–132. [Google Scholar] [CrossRef] [PubMed]
- Petricci, E.; Risi, C.; Ferlin, F.; Lanari, D.; Vaccaro, L. Avoiding hot-spots in Microwave-Assisted Pd/C catalysed reactions by using the biomass derived solvent γ-Valerolactone. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Feng, Y.; Li, F.; Han, J.; He, Y.; Fan, Q.H. A Synthetic Route to Chiral Benzo-Fused N-Heterocycle via Sequential Intramolecular Hydroammination and Asymmetric Hydrogenation of Anilino-Alkynes. Organometallics 2019, 38, 3979–3990. [Google Scholar] [CrossRef]
- Gazvoda, M.; Virant, M.; Pinter, B.; Košmrlj, J. Mechanism of copper-free Sonogashira reaction operates through palladium-palladium transmetallation. Nat. Commun. 2018, 9, 4814. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Song, Q. Copper-Catalyzed Radical Difluoroalkylation and Redox Annulation of Nitroalkynes for the Costruction of C2-Tetrasustituted Indolin-3-Ones. Org. Lett. 2018, 20, 393–396. [Google Scholar] [CrossRef]
- Krause, N. New surfactants for chemistry in water. Curr. Opin. Green Sustain. Chem. 2017, 7, 18–22. [Google Scholar] [CrossRef]
- Fang, Y.Q.; Lautens, M. A Highly Selective Tandem Cross-Coupling of Gem-Dihaloolefins for a Modular, Efficient, Synthesis of Highly Functionalyzed Indoles. J. Org. Chem. 2008, 73, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Maizuru, N.; Inami, T.; Kurahashi, T.; Matsubara, S. Nickel -Catalyzed Cycloaddition of Anthranilic Acid Derivates to Alkynes. Org. Lett. 2011, 13, 1206–1209. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Fu, H.; Qiao, R.; Jiang, Y.; Zhao, Y. Palladium-Free Copper -Catalyzed Sonogashira Cross-Coupling at Room Temperature. Synthesis 2008, 15, 2417–2426. [Google Scholar] [CrossRef]
- Barluenga, J.; Fenàndez, M.A.; Aznar, F.; Valdés, C. Cascade Alkenyl Amination/Heck Reaction Promoted by a Bifuctional Palladium Catalyst: A Novel One-Pot Synthesis of Indoles from o-Haloaniline and Alkenyl Halides. Chem. Eur. J. 2005, 11, 2276–2283. [Google Scholar] [CrossRef] [PubMed]
- Ambrogio, I.; Cacchi, S.; Fabrizi, G. Palladium-Catalyzed Synthesis of 2-(Aminomethyl)indoles from Ethyl 3-(o-Trifluoroacetamidophenyl)-1- propargyl Carbonate. Org. Lett. 2006, 8, 2083–2086. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Scope of the reaction. 1 80 °C, oil bath, 6 h. Yield obtained working under MW dieletric heating: (12, 35%), (14, 30%), (15, 27%), (16, 20%), (18, <10%). 2 MW (PowerMax on), see Supplementary Material (SM) 3 MW (PowerMax off), see SM. 4 Pd(OAc)2 (10%), see SM.
Scheme 1.
Scope of the reaction. 1 80 °C, oil bath, 6 h. Yield obtained working under MW dieletric heating: (12, 35%), (14, 30%), (15, 27%), (16, 20%), (18, <10%). 2 MW (PowerMax on), see Supplementary Material (SM) 3 MW (PowerMax off), see SM. 4 Pd(OAc)2 (10%), see SM.

Scheme 2.
Tandem Sonogashira-indole cyclisation.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization of reaction conditions.

| Ent. | Metal Salts/Ligand 1 | Additive | Type of Heating | Time (min)/ Temperature (T°) 2 | Yield (2) | (1) Recovered |
|---|---|---|---|---|---|---|
| 1 | Pd(PPh3)2Cl2 (10 mol%)/XPhos (10 mol%) | -- | MW | 100 °C, 30 min 3 | 0% | 80% |
| 2 | Pd(MeCN)2Cl2 (10 mol%)/XPhos (10 mol) | -- | MW | 100 °C, 30 min, then 150 °C 30 min 3 | 25% | 60% |
| 3 | Pd(OAc)2 (10 mol%)/Xphos (10 mol%) | -- | MW | 100 °C, 30 min 3 | 25% | 63% |
| 4 | Pd(OAc)2 (10 mol%)/Xphos (10 mol%) | -- | MW | 80 °C, 20 +10 min 3 | 25% | 55% |
| 5 | Pd(OAc)2 (10 mol%)/PPh3 (10 mol%) | -- | MW | 80 °C, 20 +10 min 3 | 0% | 82% |
| 6 | Pd-Oxime (10 mol) | -- | MW | 80 °C, 3 × 20 min 3 | 45% | 30% |
| 7 | Pd(OAc)2 (10 mol%) | MW | 80 °C, 3 × 20 min 3 | 35% | 40% | |
| 8 | Pd(OAc)2 (10 mol%) | -- | MW | 80 °C, 3 × 40 min 3 | 45% | 25% |
| 9 | Pd(OAc)2 (10 mol%) | -- | Oil bath | 80 °C, 16 h | 35% | 30% |
| 10 | Pd(OAc)2 (10 mol%) | AcOH (1 eq) | MW | 80 °C, 3 × 20 min 3 | 50% | 10% |
| 11 | Pd(OAc)2 (10 mol%) | AcOH (1 eq) | MW | 100 °C. 10 min | 70% | -- |
| 12 | Pd(OAc)2 (10 mol%) | AcOH (1 eq) | Oil bath | 100 °C, 4 h | 67% | -- |
| 13 | Pd(OAc)2 (10 mol%) | AcOH (1 eq) | Oil bath | 100 °C, 8 h | 30% | -- |
| 14 | Pd(OAc)2 (10 mol%) | AcOH (1 eq) | Oil bath | 80 °C, 6 h | 76% | -- |
| 15 | Pd(OAc)2 (10 mol%) | TFA (1 eq) | MW | 100 °C, 10 min | 53% | -- |
| 16 | Pd(OAc)2 (10 mol%) | C11H23COOH (15 mol%) | MW | 100 °C, 10 min | 40% | 35% |
| 17 | Pd(OAc)2 (10 mol%) | DPBA (15 mol%) | MW | 100 °C, 10 min | 45% | 30% |
| 18 | Pd(OAc)2 (5 mol%) | AcOH (1 eq)) | MW | 100 °C, 10 min 4 | 72% | -- |
| 19 | Pd(OAc)2 (2 mol%) | AcOH (1 eq) | MW | 100 °C, 10 min 5 | 43% | 39% |
| 20 | Pd(OAc)2 (10 mol) | AcOH (1 eq) | MW | 100 °C, 10 min 6 | 37% | 40% |
| 21 | Pd(OAc)2 (10 mol%) | AcOH (1 eq) | MW | 100 °C, 10 min 7 | 48% | 35% |
| 22 | Pd(OAc)2 (10 mol%) | AcOH (1 eq) | MW | 100 °C, 10 min 8 | 45% | 35% |
1 0.1 eq of catalyst and ligand, unless otherwise reported, and 1 eq of AcOH. 2 For reactions under MW dielectric heating: 1 mmol of compound 1 was mixed with 1 mL of a degassed 3% solution of TGPS-750M in H2O followed by the catalyst. The vial was heated with the program keeping the temperature through continuous irradiation and contemporary cooling (PowerMax = on). Alternatively, the vial was immersed in an oil bath at a set temperature. 3 MW irradiation to maintain the settled temperature (PowerMax = off). 4 0.05 eq of Pd(OAc)2 employed. 5 0.02 eq of Pd(OAc)2 employed. 6 Reaction done in H2O with 3% of SDS. 7 Reaction done in pure H2O. 8 Reaction done in MeCN.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Siciliano, S.; Cini, E.; Taddei, M.; Vinciarelli, G. Synthesis of 2-Substitued Indoles via Pd-Catalysed Cyclization in an Aqueous Micellar Medium. Molecules 2021, 26, 3917. https://doi.org/10.3390/molecules26133917
AMA Style
Siciliano S, Cini E, Taddei M, Vinciarelli G. Synthesis of 2-Substitued Indoles via Pd-Catalysed Cyclization in an Aqueous Micellar Medium. Molecules. 2021; 26(13):3917. https://doi.org/10.3390/molecules26133917
Chicago/Turabian StyleSiciliano, Sofia, Elena Cini, Maurizio Taddei, and Giorgia Vinciarelli. 2021. "Synthesis of 2-Substitued Indoles via Pd-Catalysed Cyclization in an Aqueous Micellar Medium" Molecules 26, no. 13: 3917. https://doi.org/10.3390/molecules26133917