
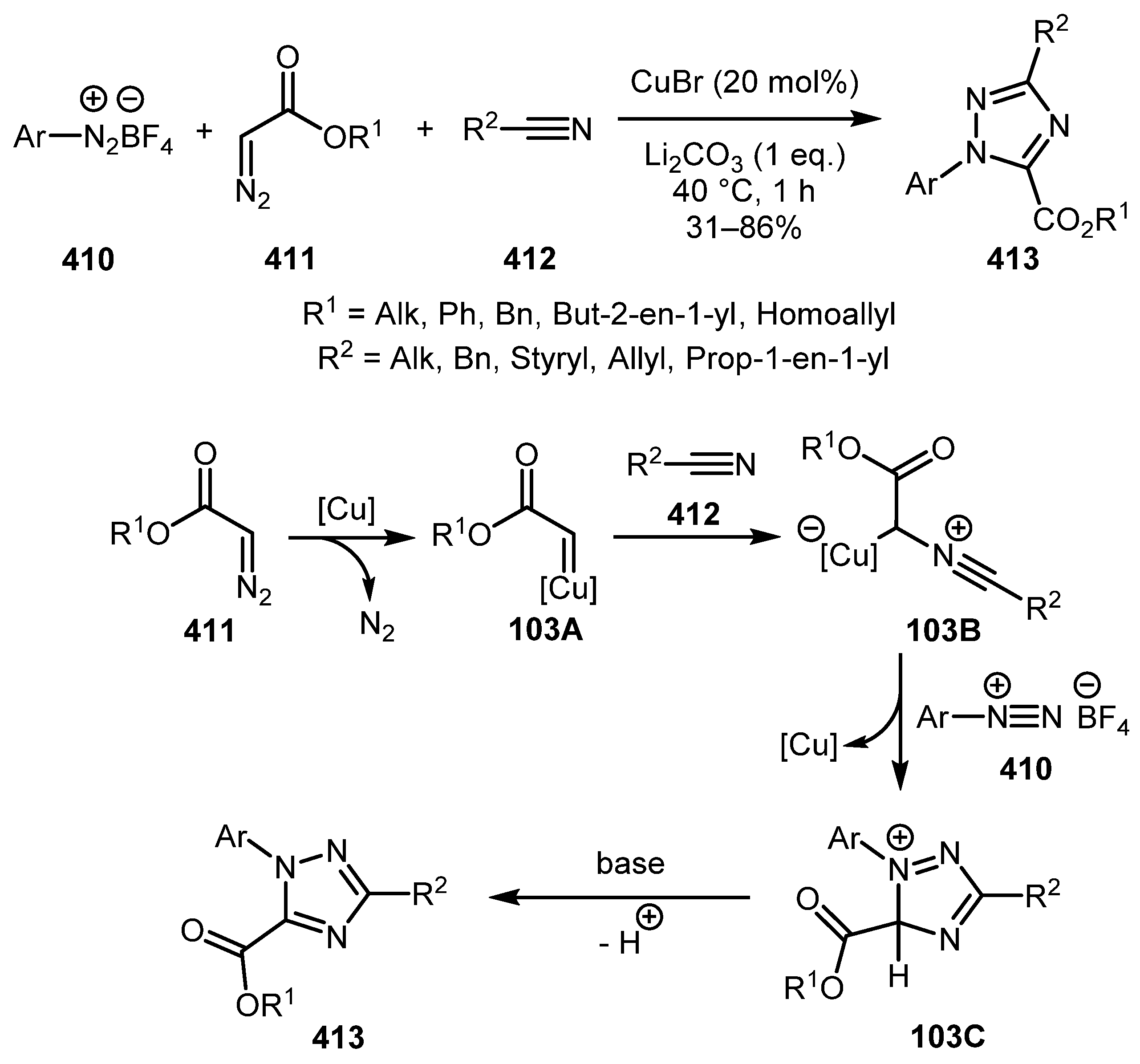
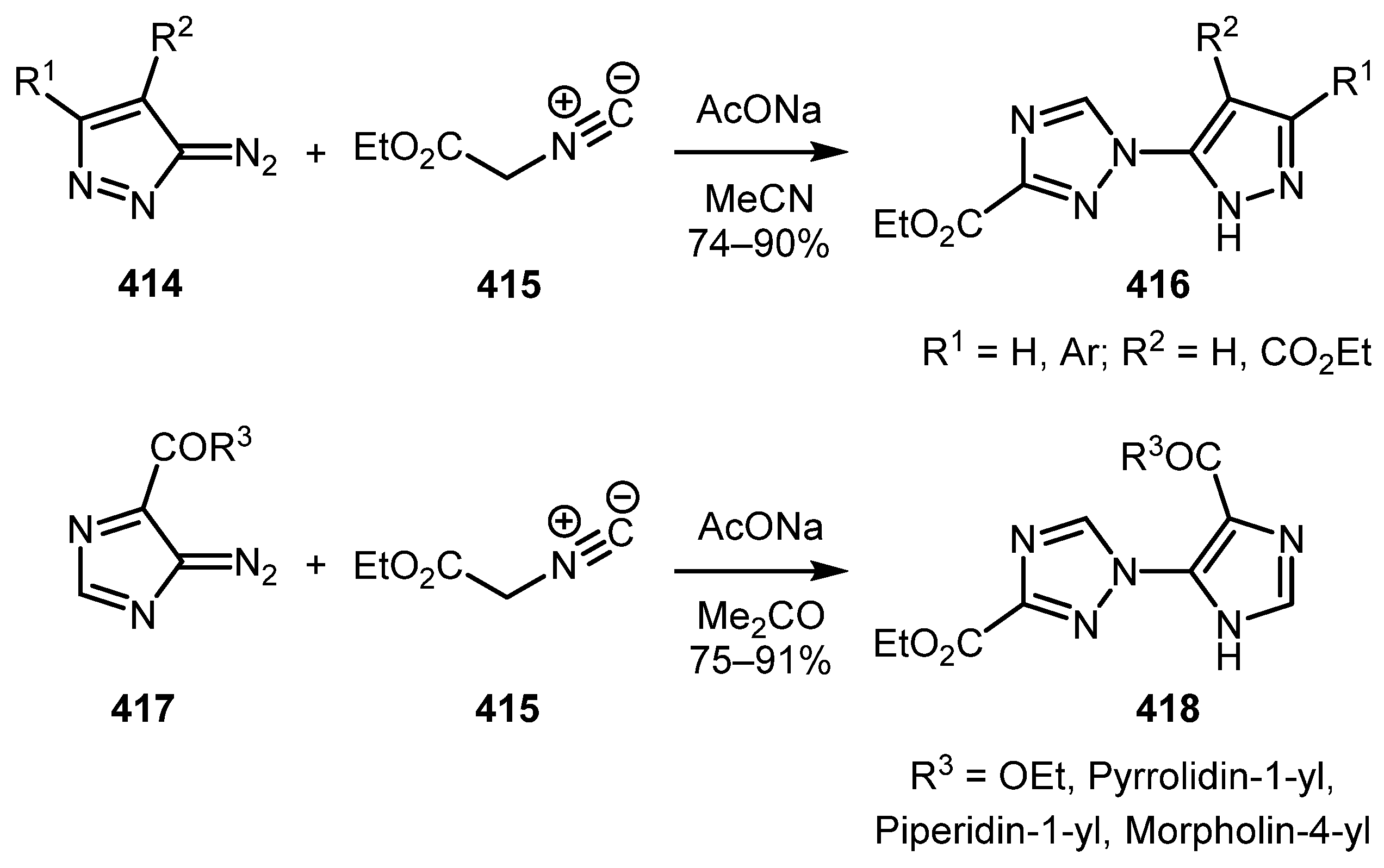
Diazocarbonyl and Related Compounds in the Synthesis of Azoles


Institute of Chemistry, St. Petersburg State University, 198504 Peterhof, Russia
*
Authors to whom correspondence should be addressed.
Molecules 2021, 26(9), 2530; https://doi.org/10.3390/molecules26092530
Submission received: 7 April 2021
/
Revised: 21 April 2021
/
Accepted: 22 April 2021
/
Published: 26 April 2021
(This article belongs to the Special Issue Diazo Chemistry)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Diazocarbonyl compounds have found numerous applications in many areas of chemistry. Among the most developed fields of diazo chemistry is the preparation of azoles from diazo compounds. This approach represents a useful alternative to more conventional methods of the synthesis of azoles. A comprehensive review on the preparation of various azoles (oxazoles, thiazoles, imidazoles, pyrazoles, triazoles, and tetrazoles) from diazocarbonyl and related compounds is presented for the first time along with discussion of advantages and disadvantages of «diazo» approaches to azoles.
1. Introduction
Diazo compounds are incredibly useful reagents in synthetic organic chemistry due to their extremely versatile and unique reactivity [1,2,3,4,5]. These compounds are able to undergo transformations with the loss of nitrogen (under catalytic, photochemical, or thermal decomposition) or with its retention as a constituent part of a reaction product. Along with diazoalkanes, EWG-substituted diazo compounds (diazo-carbonyl, -sulfonyl, -phosphonyl compounds, diazonitriles) are extensively used in organic synthesis. The latter stand out due to their higher stability, safety, and convenience in handling.
One of the highly demanded applications of diazocarbonyl compounds and other stabilized analogs is the construction of carbo- and heterocyclic systems. Recently, a range of new methods have been developed for the synthesis of various heterocycles from diazo compounds, some of which are described in reviews [6,7,8,9,10,11,12]. Versatility and efficiency of diazo compounds are most clearly manifested in the synthesis of azoles—five-membered heterocycles containing a nitrogen atom and at least one other non-carbon atom of either nitrogen, oxygen, or sulfur. Diazo reagents allow synthesis of azoles of many types with two, three, and four heteroatoms. These heterocycles have found broad application in many fields such as organic electronics, functional materials, explosives, dyes, fluorophores, and especially medicine. According to the Drug Bank, there are over 300 FDA-approved drugs containing azole moiety.
However, to our knowledge, there are only a few reviews on the synthesis of azoles from diazocarbonyl compounds and these reviews are focused on one specific azole class, namely 1,3-oxazoles [13], pyrazoles [14,15], and 1,2,3-thiadiazoles [16]. This review aims to summarize all recent developments in this area covering all types of azoles available from diazocarbonyl and related compounds (oxazoles, thiazoles, imidazoles, pyrazoles, triazoles, tetrazoles, with the exception of 1,2,3-thiadiazoles), as well as to consider earlier useful methodologies not mentioned in other reviews.
2. Azoles with Two Heteroatoms
2.1. 1,3-Oxazoles
1,3-Oxazole (in brief, oxazole) is a privileged motif for drug design. Oxazole derivatives have shown a broad range of biological activities such as anti-inflammatory, antibacterial, anticancer, antifungal, antiviral, antihyperglycemic, and so on [17,18]. Numerous oxazole medicinal agents are of natural origin. Oxazole alkaloids, including macrolides, cyclic and acyclic peptides, siderophores, and small molecules are found in marine invertebrates, bacteria, fungi, and higher plants [19]. Therefore, a vast array of methods for synthesis of 1,3-oxazoles have been developed to date [20].
Among these methodologies, the following two strategies are rather commonly employed: (i) synthesis of 1,3-oxazoles by formal [3+2] cycloaddition between metal carbenes derived from diazo carbonyl compounds and nitriles, and (ii) metal carbene insertion into the N–H bond of amides with subsequent dehydrative cyclization. These routes have been comprehensively reviewed elsewhere [13,21]. In this section, we chose to present only fairly recent examples of these strategies as well as a few rare approaches to 1,3-oxazoles.
2.1.1. Synthesis of 1,3-Oxazoles by Reaction of Diazocarbonyl Compounds and Nitriles
Since the pioneering work of Huisgen on the formal [3+2] cycloaddition between nitriles and diazo carbonyls [22], this method has become the most frequently employed diazo strategy for the synthesis of 1,3-oxazoles. The main disadvantage of this approach is the need to use an excess amount of the nitrile (often as a solvent). However, some protocols have been developed that allow reducing the excess of the nitrile down to 5-fold.
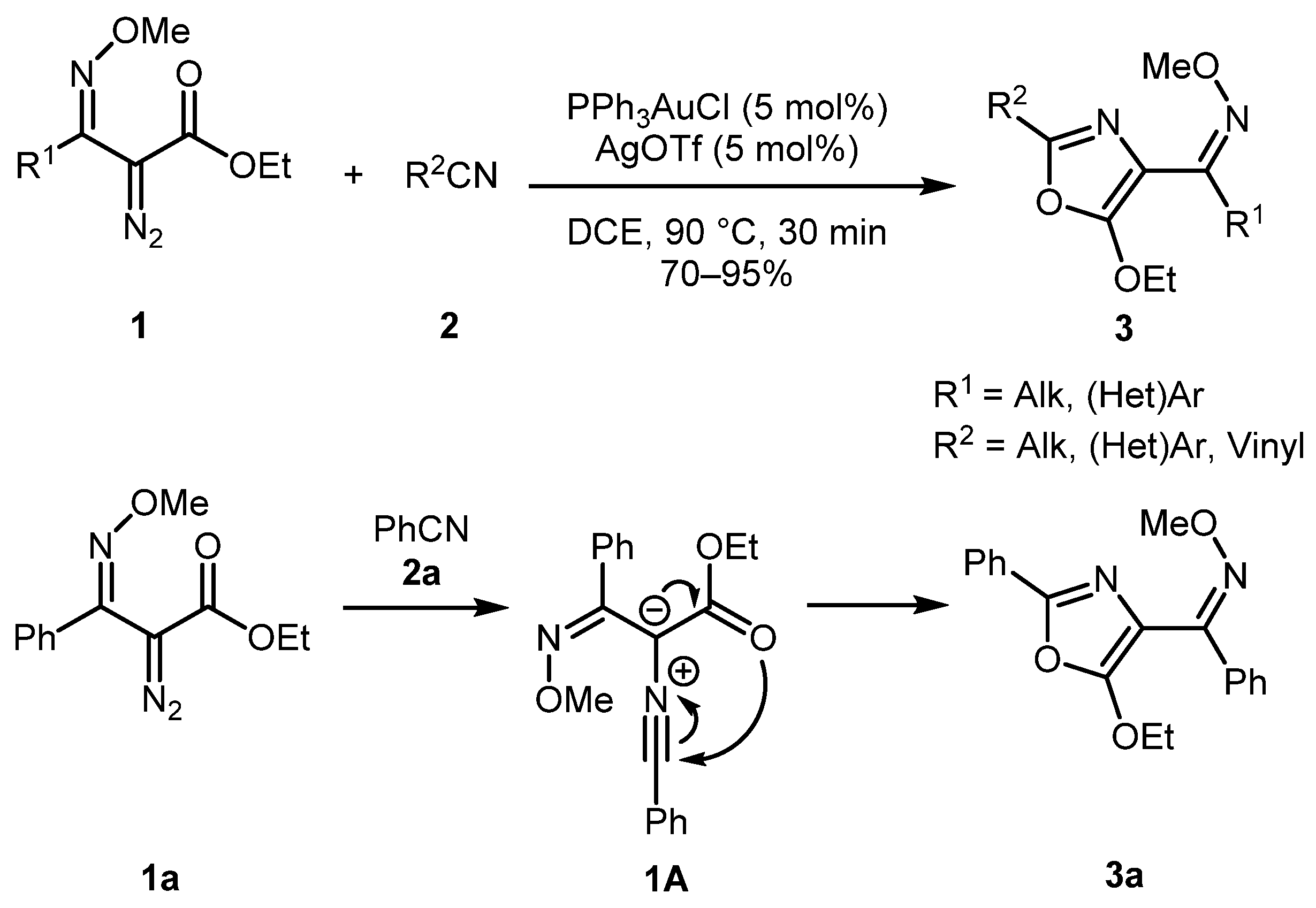
In 2016, the Park group developed an efficient method for the synthesis of highly substituted oxazoles 3 by gold-catalyzed cycloaddition reaction of α-diazo oxime ethers 1 and nitriles 2 (Scheme 1) [23]. The reaction tolerated a broad range of substituents (including alkyl, aryl, heteroaryl, and vinyl) and afforded good to excellent product yields. The formation of the oxazole ring is believed to proceed via the formation of zwitterionic intermediate 1A, which subsequently undergoes cyclization to oxazole 3. Interestingly, the cyclization occurred via the attack of the carbonyl group rather than the attack of the oxime ether group on the nitrilium electrophilic center, presumably due to the Z-configuration of the C=N double bond (Scheme 1).
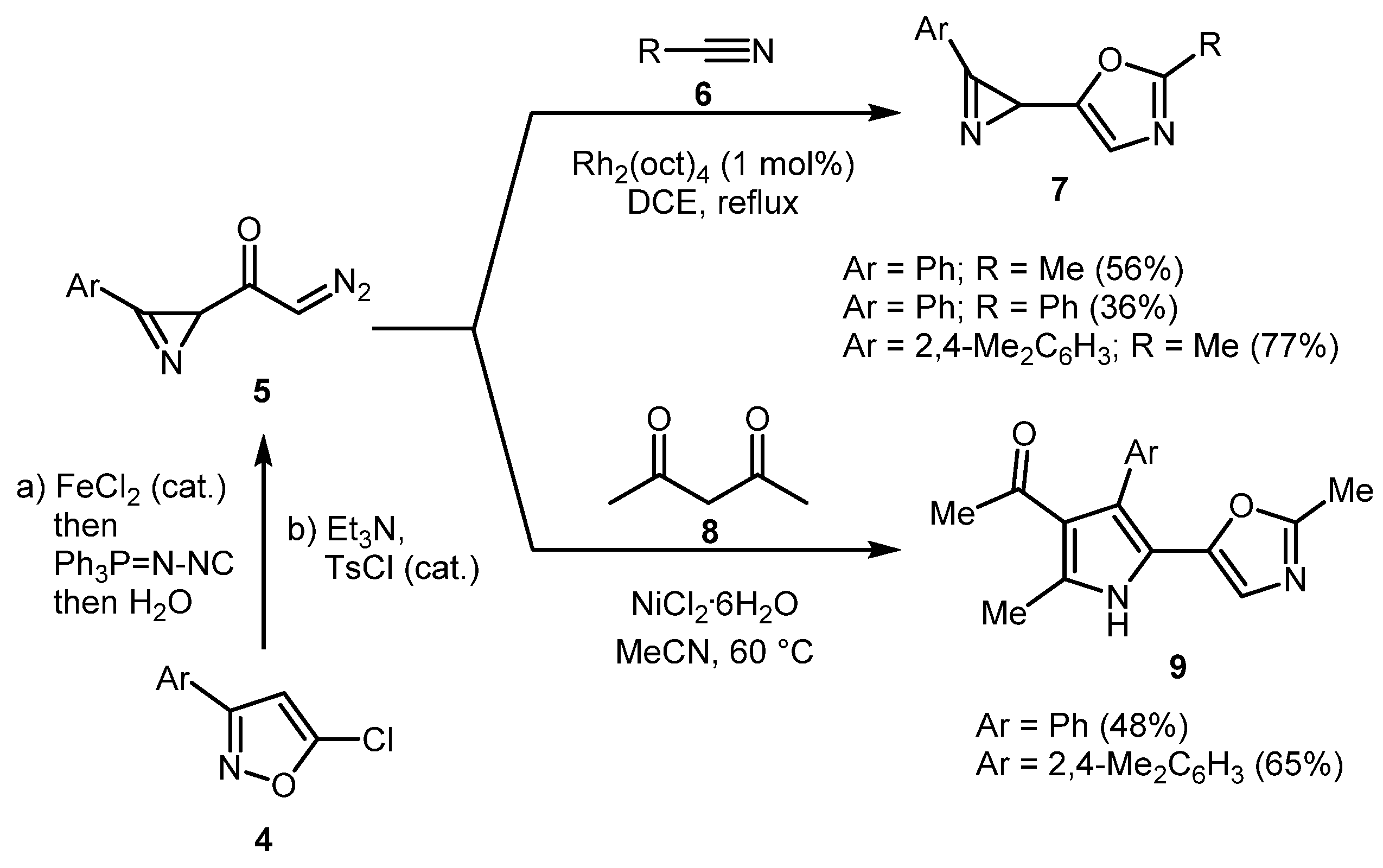
In the synthetic exploration of 2-diazoacetyl-2H-azirines 5, which are available from 5-chloroisoxazoles 4, Khlebnikov and co-workers demonstrated the utility of 5 for the preparation of oxazoles [24]. Rhodium-catalyzed reaction with nitriles 6 afforded oxazoles 7 with preservation of azirine functionality. On the other hand, NiCl2-catalyzed reaction with acetylacetone 8 in acetonitrile provided pyrrolyl-substituted oxazoles 9 (Scheme 2).
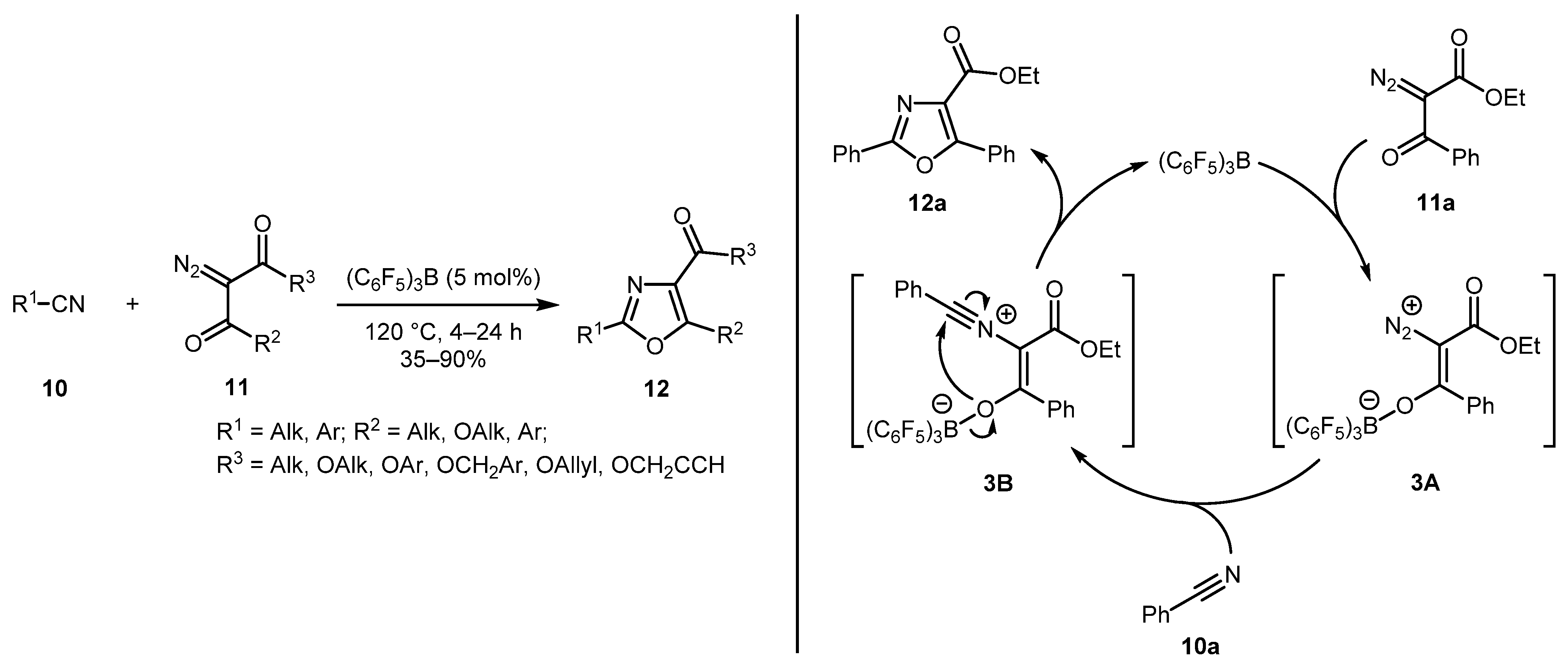
An unusual tris(pentafluorophenyl)borane catalyst was employed for the reaction of nitriles 10 and diazo carbonyls 11 by Kumaraswamy and Gangadhar [25]. This metal-free protocol was found compatible with many functional groups including alkyl, aryl, alkyl(aryl)oxy, and allowed using a lower (5-fold) excess of the nitrile partners. A plausible reaction mechanism implies initial activation of diazocarbonyl compound 11 by the catalyst generating alkenediazonium salt 3A, which is attacked by nitrile 10 with extrusion of N2 molecule to give intermediate 3B. The latter undergoes cyclization to form oxazole product 12 (Scheme 3).
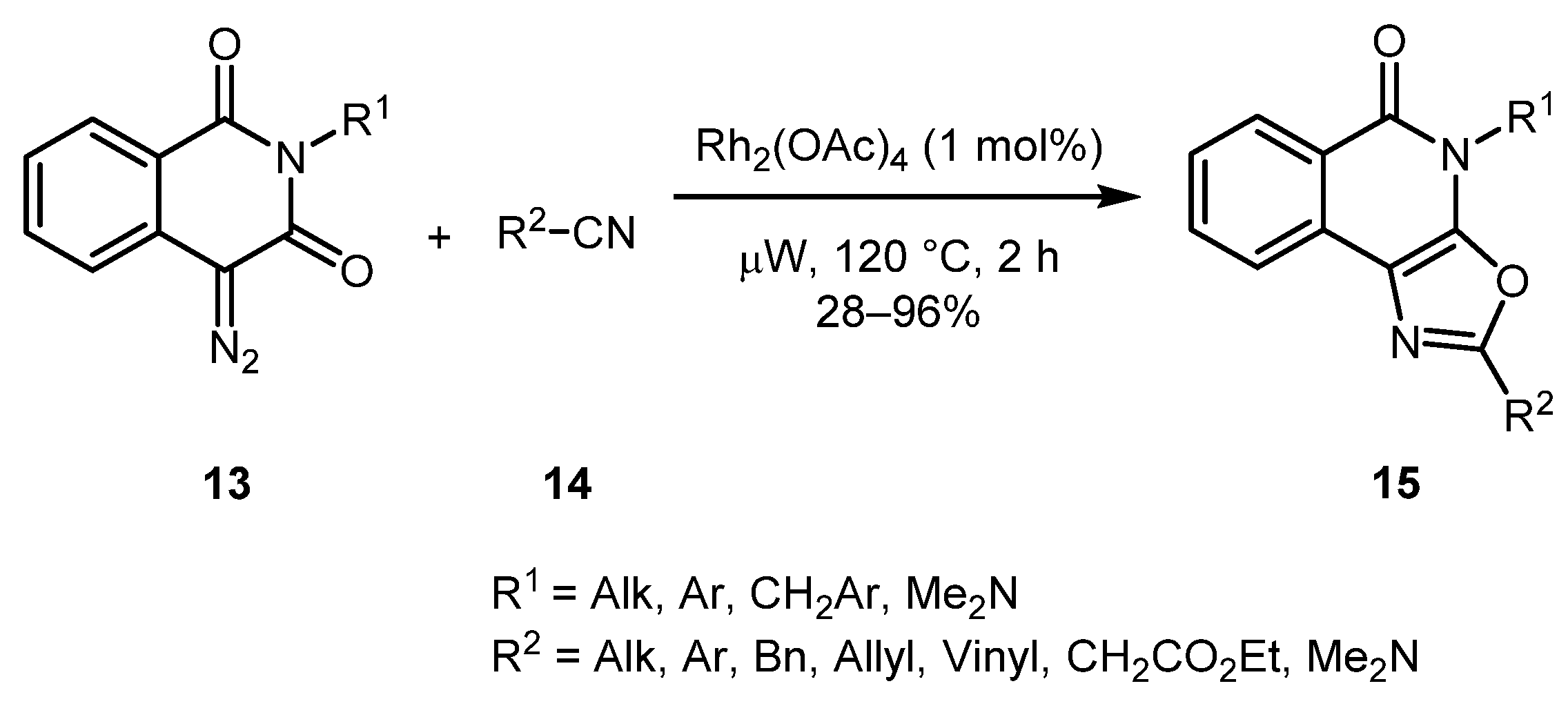
Diazo homophthalimides 13 were the first heterocycles containing an α-diazocarbonyl moiety, which were involved in a [3+2] cycloaddition with nitriles 14 to deliver oxazole-fused adducts 15 as reported by the Krasavin group [26]. The reaction displayed a generally high functional group tolerance giving low yields only in case of olefinic, benzylic, and sterically hindered nitriles. Diazo homophthalimides 13 are easily available via diazo transfer to the corresponding homophthalimides (Scheme 4).
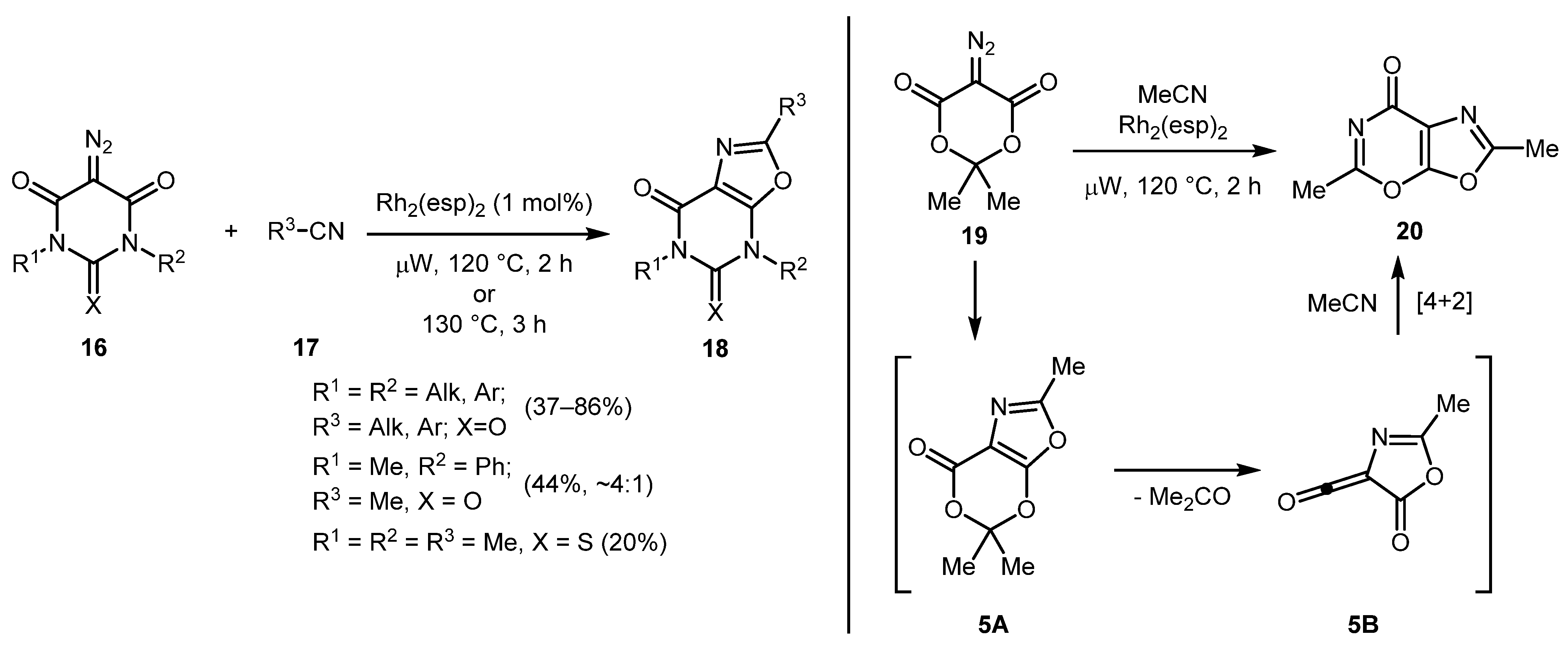
Further exploring the synthesis of fused oxazoles from diazo containing heterocycles, the same group reported a [3+2] cycloaddition reaction of diazobarbituric acid derivatives 16 (available from barbituric acids via a novel sulfonyl-azide-free (SAFE) diazo transfer protocol [27]) with nitriles 17 [28]. Diversely substituted oxazolo[5,4-d]pyrimidine-5,7(4H,6H)-diones 18 were obtained in generally moderate to good yields. However, mono-N-substituted diazobarbituric acid failed to give the desired product. Non-symmetric diazobarbituric acid gave mixture of regioisomers. Notably, the attempt of performing this reaction with diazo Meldrum’s acid 19 led to the formation of oxazolo[4,5-e][1,3]oxazinone 20 via acyl ketene intermediate 5B, which was involved int [4+2] cycloaddition with another molecule of acetonitrile (Scheme 5).
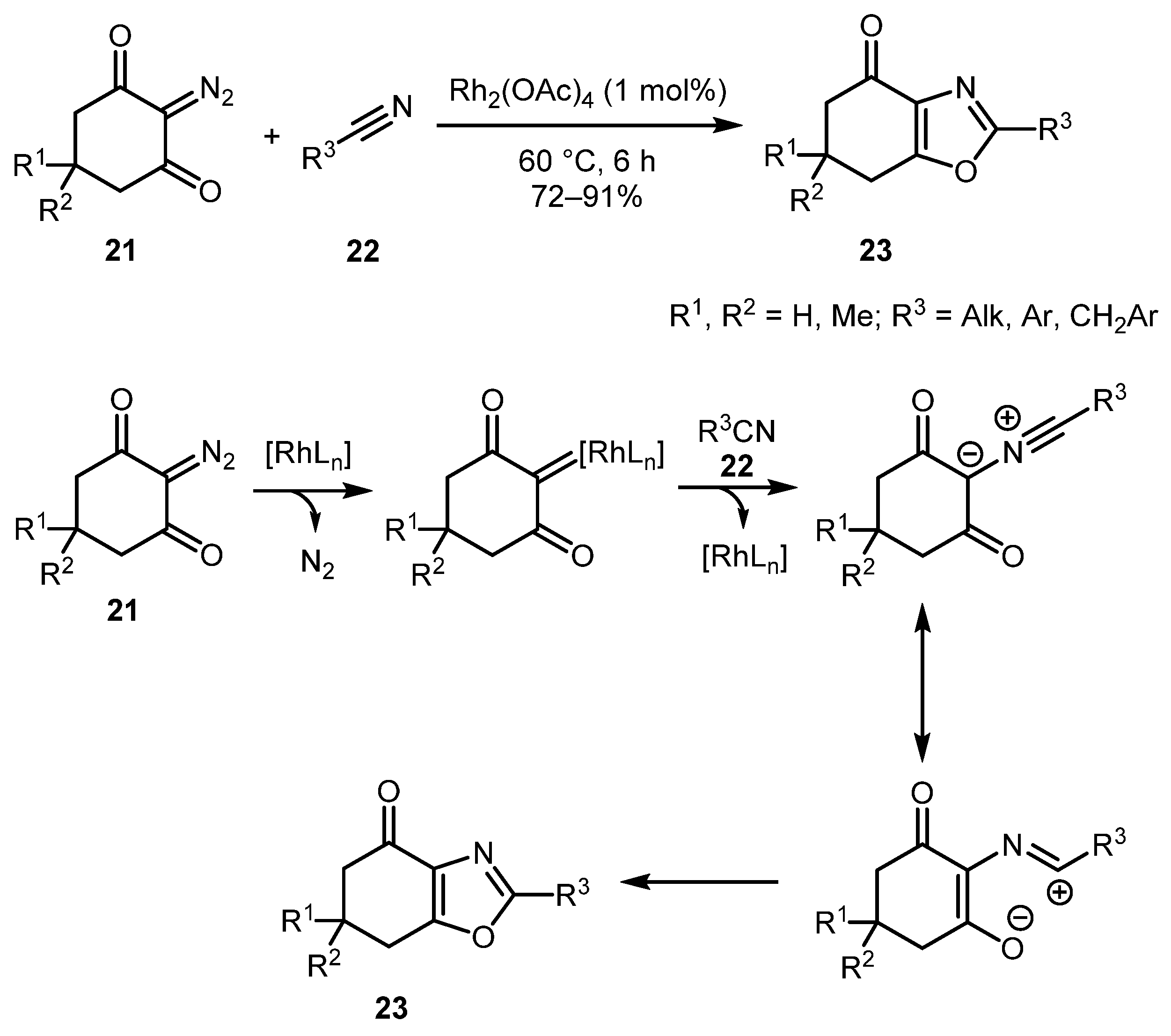
Alicyclic diazocarbonyl compounds have also been employed in oxazole synthesis. Thus, in 2018, Fan and co-workers described the synthesis of 6,7-dihydrobenzo[d]oxazol-4(5H)-ones 23 by reacting cyclic diazo diketones 21 with nitriles 22 [29]. The best yields were obtained with non-substituted diazo substrates. Analogously to the example discussed above, diazo Meldrum’s acid did not provide the desired oxazole. From the mechanistic standpoint, the reaction likely proceeds via the attack of the nitrile on the rhodium carbene, generated from diazo species, following steps similar to the above-mentioned examples (Scheme 6).
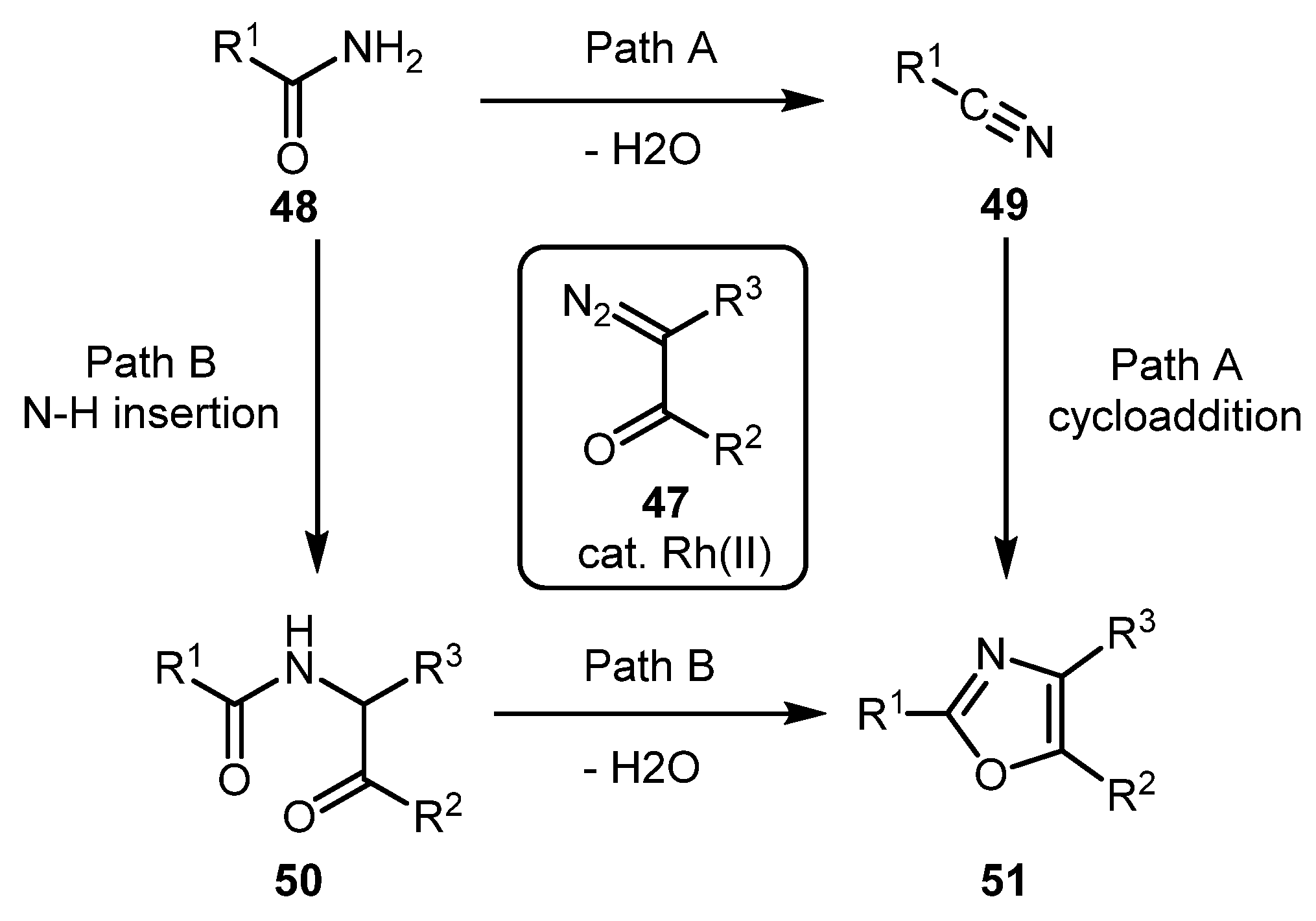
2.1.2. Synthesis of 1,3-Oxazoles by Reaction of Diazocarbonyl Compounds with Amides
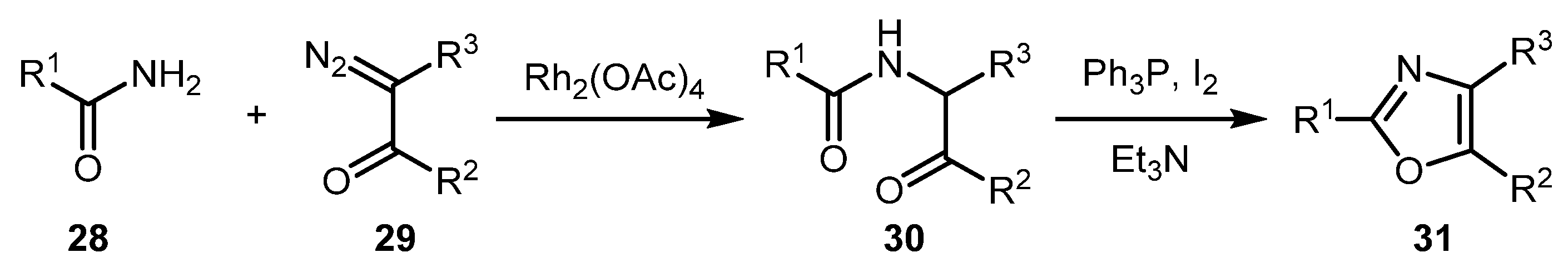
The currently well-known approach to oxazoles 31 from amides 28 and diazo compounds 29 was first described by Moody and co-workers in 1996 (Scheme 8) [34,35]. It was developed as an alternative to the cycloaddition of nitriles, which gave unsatisfactory results when applied to N-protected α-aminonitriles derived by dehydration of α-amino acid amides. The new approach involves Rh carbene NH-insertion into the amide molecule followed by cyclodehydration via the so-called Wipf protocol, i.e., using a mixture of Ph3P, I2 and Et3N (Scheme 8).
Notably, several methodologies have been developed that allow the synthesis of oxazoles regioisomeric to the ones obtained by the Moody approach. The key step in these alternative methods is the formation of a carbonyl ylide via amide oxygen atom attack on metal carbene rather than an NH-insertion as the first step of the synthetic sequence (vide infra). Another attractive feature contrasting these protocols to the Moody oxazole synthesis is that the oxazole ring is constructed in one step.
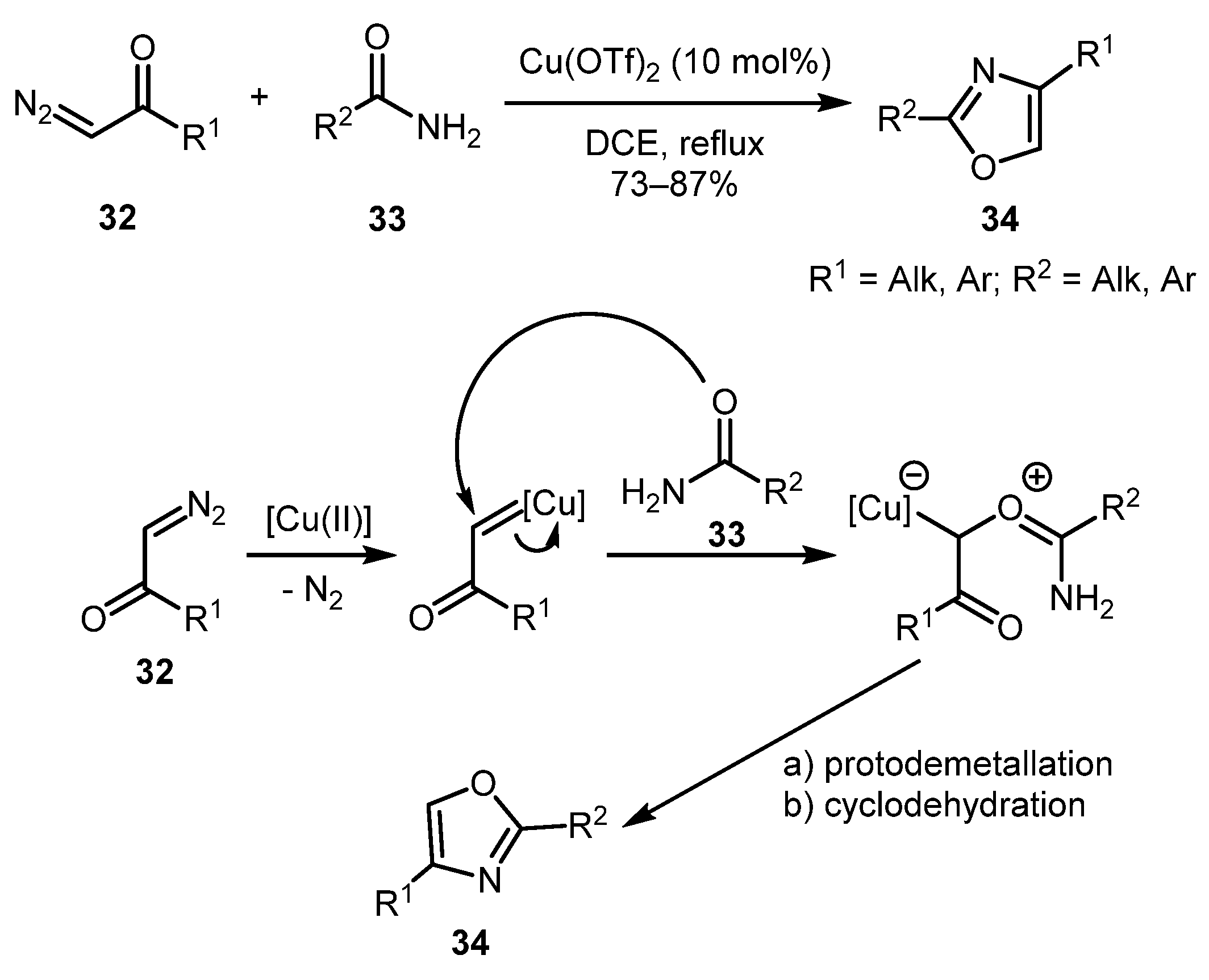
Cu(OTf)2-catalyzed synthesis of 2,4-substituted oxazoles 34 from terminal diazocarbonyl compounds 32 and amides 33 was developed by the Subba Reddy group [36]. This method works well for both aliphatic and aromatic substrates providing good product yields. The reaction was unsuccessful only when both substrates were aliphatic. The following mechanism was invoked. Initially, copper carbene intermediate is formed on interaction of 32 with the Cu-catalyst. Rather unexpectedly, the subsequent attack of the amide on the Cu-carbene occurs through the carbonyl group, giving corresponding ylide in preference to the attack by the nitrogen atom, which would result in an NH-insertion product. Subsequent protodemetallation and cyclodehydration gives the observed oxazole 34 (Scheme 9).
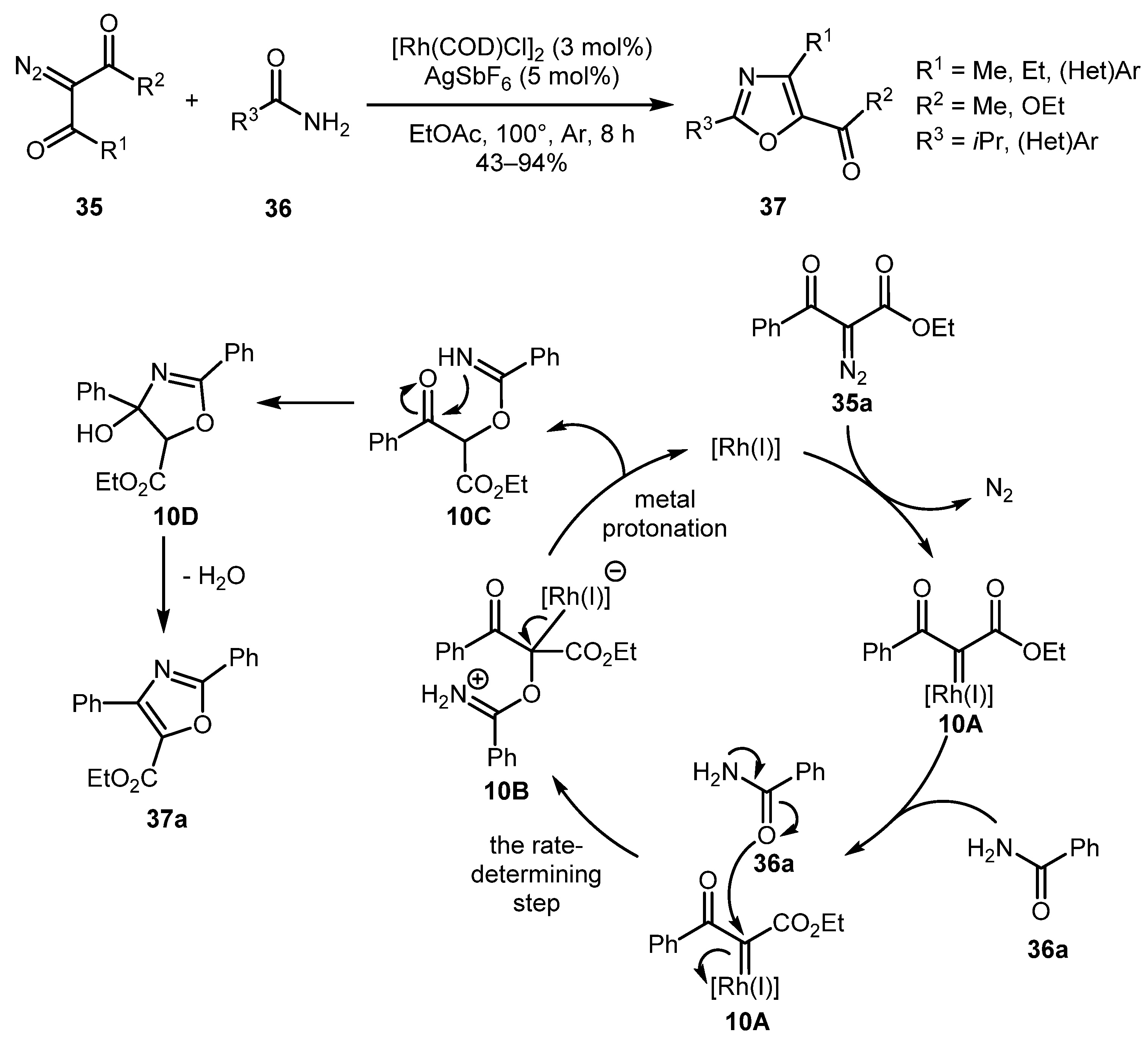
In 2018, Chen and co-workers reported the involvement of diazo dicarbonyl compounds 35 in a similar reaction under Rh(I) catalysis, which afforded fully substituted oxazoles 37 [37]. In this case, however, the yields were significantly lower for aliphatic substrates in comparison to the previous example. From the mechanistic perspective, rhodium(I) carbene complex 10A likely reacted with the carbonyl oxygen atom of amide 36a to form intermediate 10B, which underwent metal protonation (with regeneration of the catalyst) to afford β-ketoester 10C. The latter entered the cyclization/dehydration sequence, thus producing trisubstituted oxazole 37a. Preliminary mechanistic studies revealed that the attack on Rh(I) carbenes was likely the rate-determining step (Scheme 10).
Thus, by using either of these methods of the Moody protocol, both oxazole regioisomers can be accessed starting from the same set of substrates. The reasons for such a switch in regiospecificity are not entirely clear. Presumably, it can be explained by the change in electrophilicity of the intermediate metal carbene either upon changing metal, upon changing ligands, or both [38].
2.1.3. Other Approaches to the Synthesis of 1,3-Oxazoles
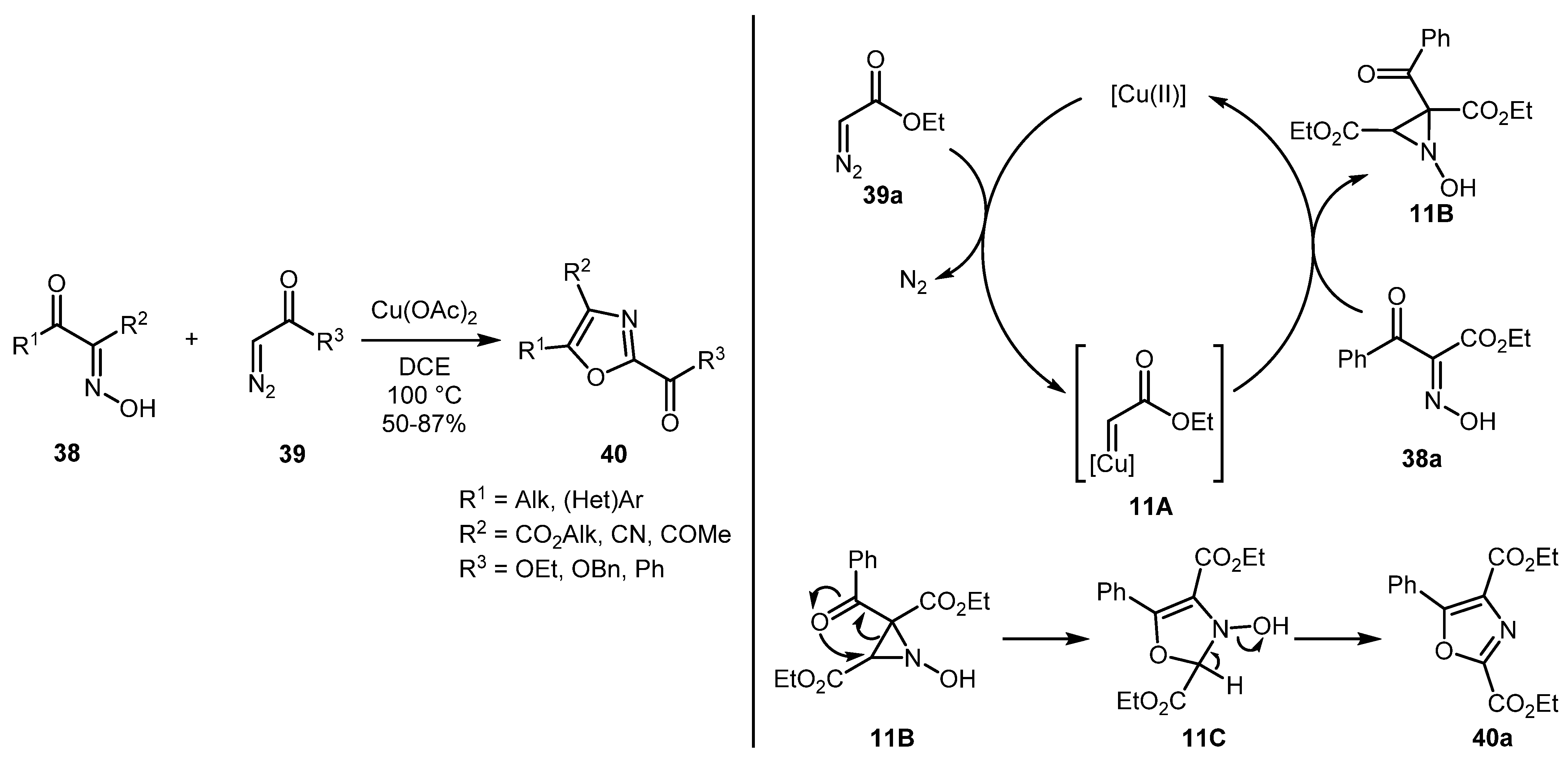
The reaction of α-ketooximes 38 with terminal α-diazo carbonyl compounds 39 afforded polysubstituted oxazoles 40 in moderate to good yields as reported in 2018 by Swamy and co-workers [39]. The reaction tolerates many substituents, including alkyl, (hetero)aryl, as well as ester, ketone, and nitrile functional groups. The following reaction mechanism was proposed by the authors. The reaction is believed to be initiated by the interaction between copper carbene 11A and ketooxime 38a, which leads to aziridine 11B. The latter undergoes an intramolecular rearrangement to form intermediate 11C. Subsequent dehydration of 11C leads to oxazole 40a (Scheme 11).
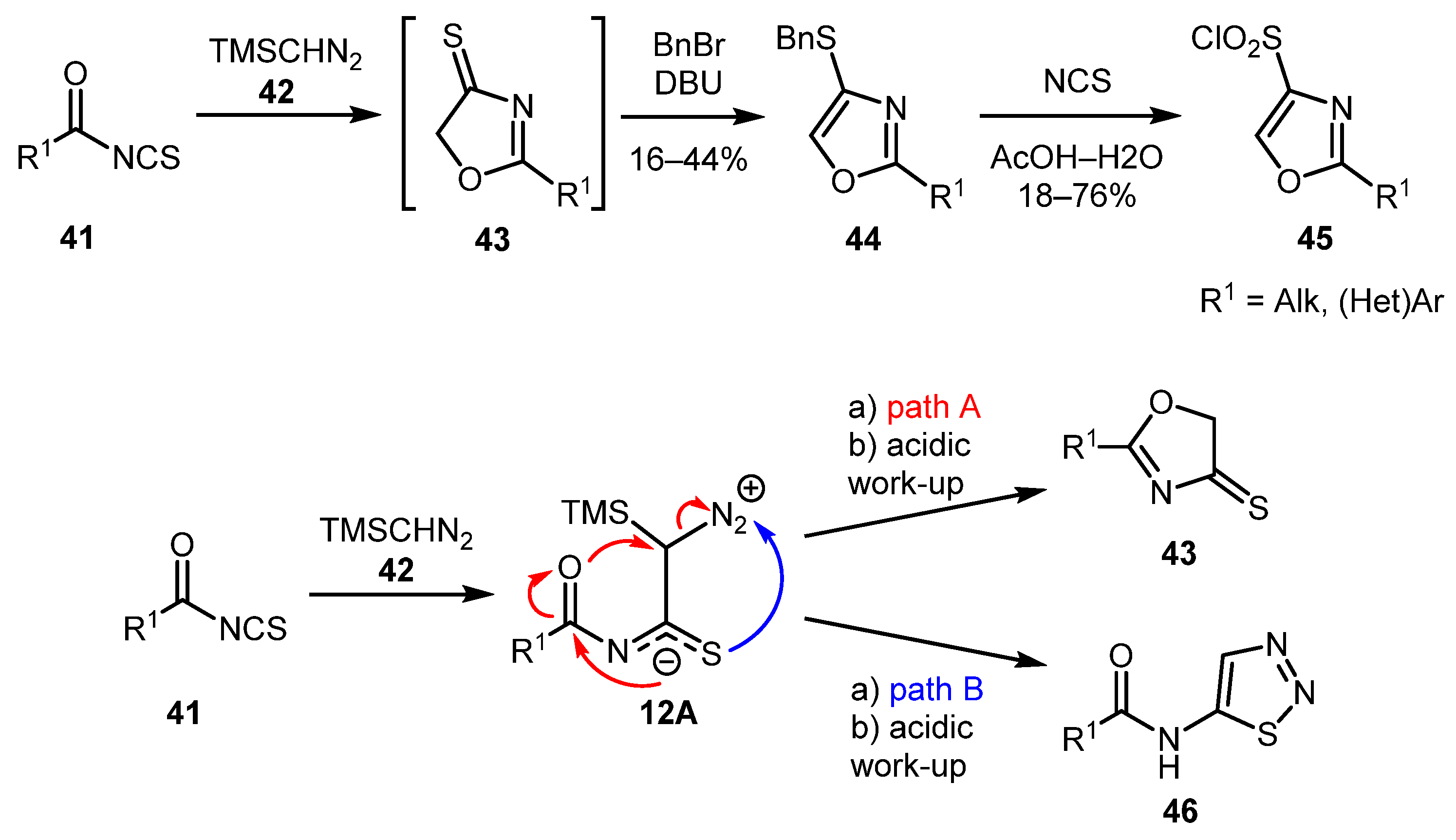
In 2015, Trujillo and co-workers synthesized a series of novel 4-(benzylthio)oxazoles 44 by reacting acyl isothiocyanates 41 with trimethylsilyl diazomethane (TMSCHN2) 42 with subsequent treatment of the oxazole thiones 43 with BnBr-DBU [40]. Isolated in moderate yields, compounds 44 were then converted to oxazole sulfonyl chlorides 45 in treatment with N-chlorosuccinimide (NCS). BnS-derivatives 44 were formed along with thiadiazoles 46, which is explained by two possible reaction pathways. In path A, initially formed intermediate 12A underwent cyclization via the attack of the carbonyl group on the carbon atom adjacent to N2+, while in path B, intermediate 12A undergoes cyclization via the attack of the thiocarbonyl group on the N2+ moiety (Scheme 12).
2.1.4. Total Synthesis of 1,3-oxazole Containing Natural Products
Both nitrile cycloaddition (Scheme 13, path A) and amide NH-insertion/cyclization (Scheme 13, path B) diazo strategies have found application in total synthesis of oxazole-containing natural products. The former strategy works best with simple alkyl and aryl nitriles 49 or in those cases when 5-unsubstituted oxazole is the target. For fully decorated oxazoles, the latter strategy is often preferred. The choice of a particular synthetic approach can also be dictated by the availability of the requisite nitrile 49 or amide 48. However, both routes are complementary and can be considered interchangeable as was shown by Moody and co-workers [34]. Hence, this subsection deals with selected works by the Moody group that have not yet been reviewed.
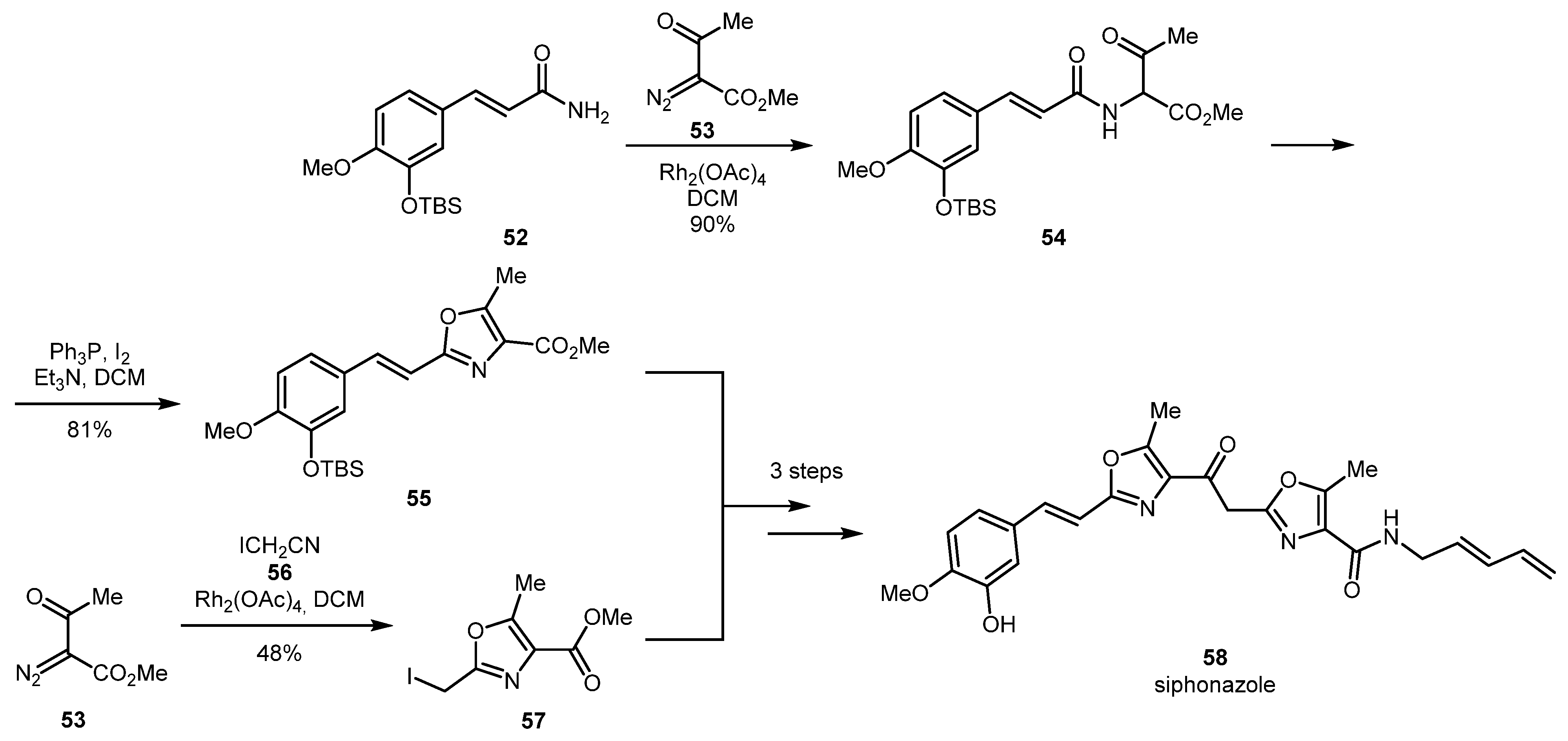
Total synthesis of siphonazole 58, an unusual metabolite from Herpetosiphon species [41], involves the construction of the oxazole intermediate containing a bulky styrene substituent (55) via an NH-insertion reaction of TBS-protected 3-hydroxy-4-methoxycinnamamide 52 and methyl 2-diazo-3-oxobutanoate 53 followed by the Wipf cyclodehydration. The other oxazole portion of the siphonazole molecule (57) was obtained by a [3+2] cycloaddition of 53 with iodoacetonitrile 56. From oxazoles 55 and 57, the natural product was assembled in three steps (Scheme 14).
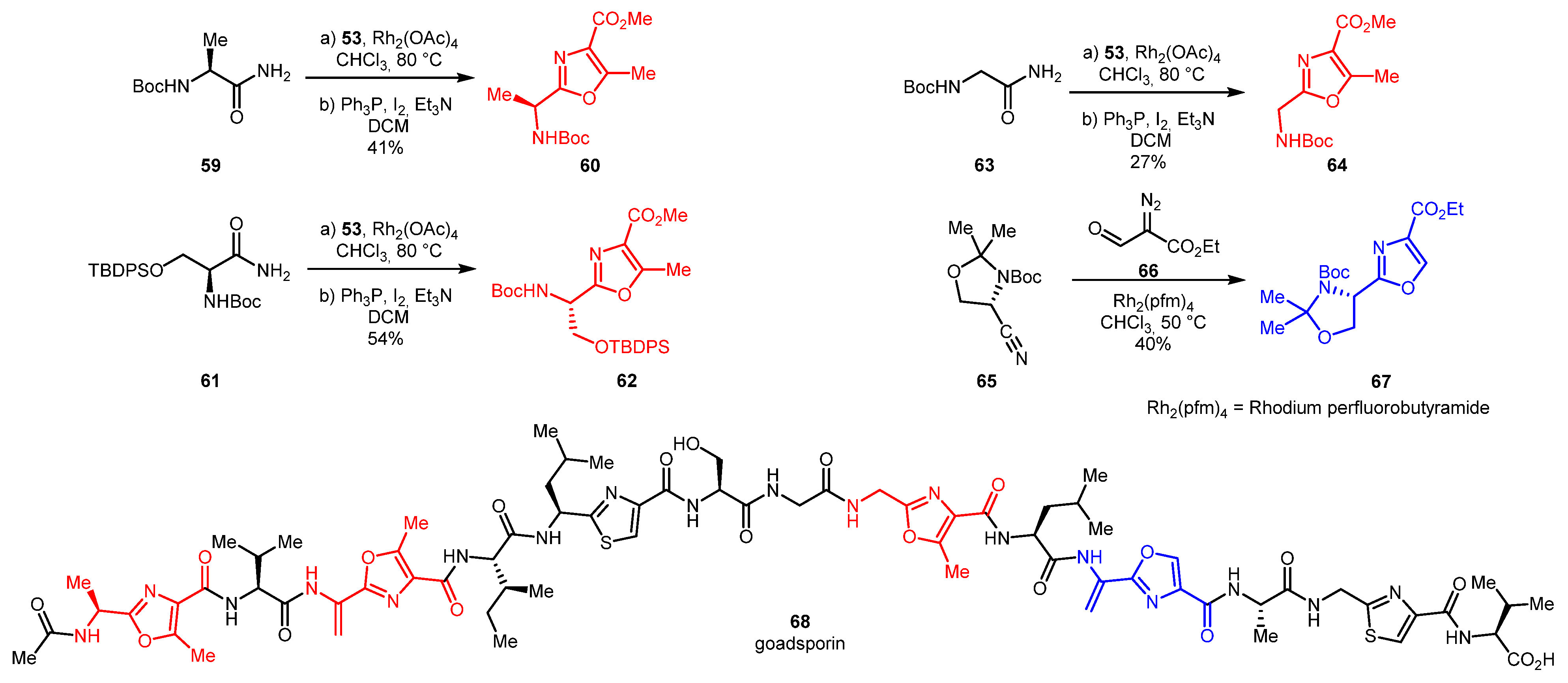
A more complex target polyazole peptide antibiotic goadsporin 68 was synthesized in 2017 [42]. It consists of four oxazole and two thiazole rings, linked through a number of amino acid residues. 5-Substituted oxazoles 60, 62, 64 were synthesized from the corresponding amino acids amides 59, 61, 63 using NH-insertion/cyclization strategy, whilst 5-unsubstituted oxazole 67 was synthesized by [3+2] cycloaddition strategy from L-serine-derived nitrile 65 and ethyl 2-diazo-3-oxopropanoate 66 (Scheme 15).
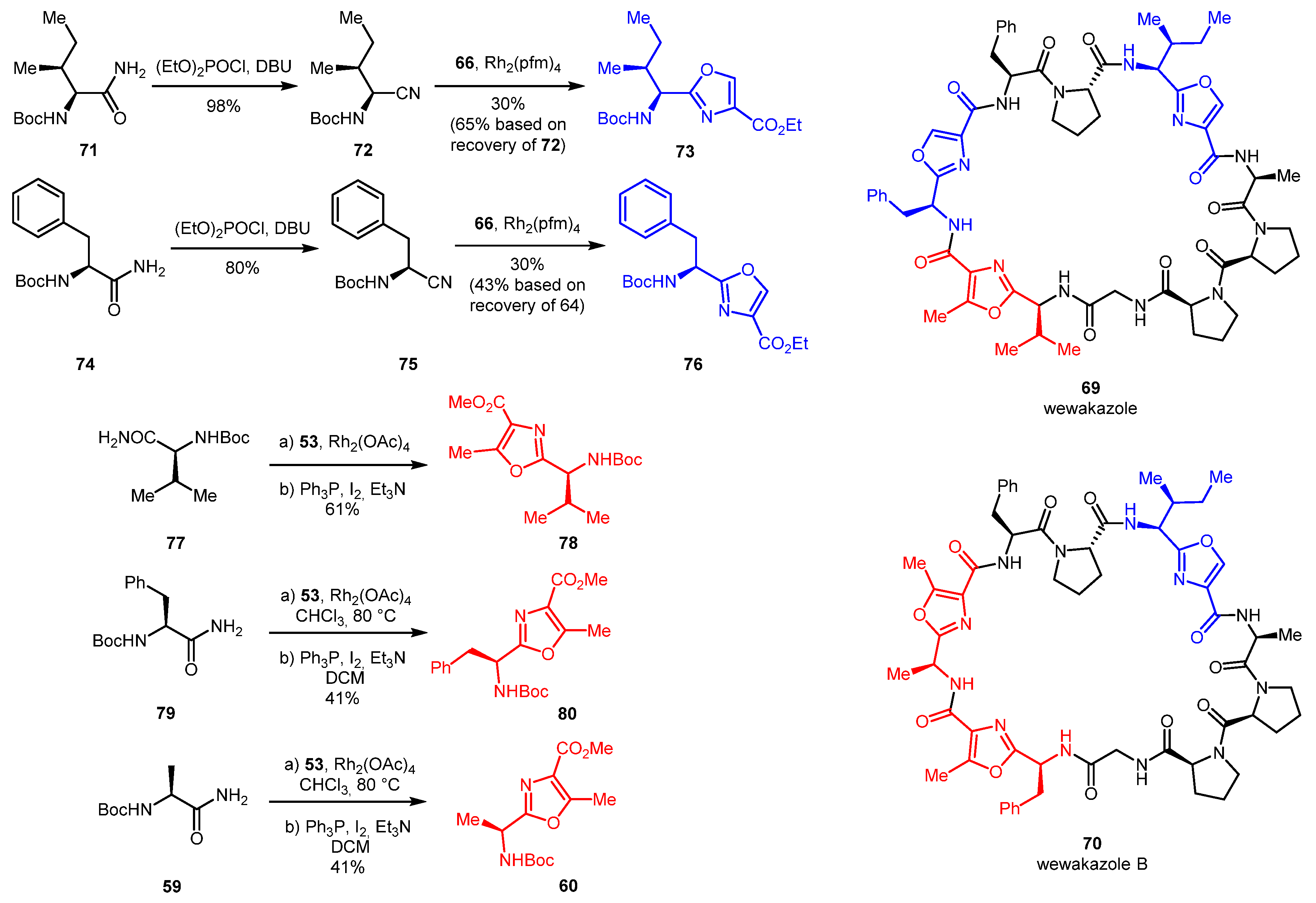
Similarly, with a goal of synthesizing cyclic dodecapeptides wewakazole 69 and wewakazole B 70 [43], 2,4-disubstituted oxazoles 73 and 76 were accessed via the nitrile cycloaddition strategy while 2,4,5-trisubstituted oxazoles 60, 78, and 80 were prepared by the NH-insertion/cyclization strategy. Nitriles 72 and 75 were obtained by dehydration of amino acid amides 71 and 74, respectively (Scheme 16).
2.2. 1,2-Oxazoles (Isoxazoles)
1,2-Oxazole ring plays an important role in organic synthesis. This heterocycle serves as a masked equivalent of a 1,3-dicarbonyl compound, which can be useful in total synthesis of natural compounds [44]. Moreover, other heterocycles are available from isoxazoles, such as 1,3-oxazoles, azirines [45], pyridines, and pyrroles [46]. Biological profile of isoxazoles is somewhat similar to 1,3-oxazoles [47]. Classic routes to isoxazoles include condensation of hydroxylamine with 1,3-dicarbonyl compounds and 1,3-dipolar cycloaddition of nitrile oxides to alkynes, although many other methods have been developed [46,48]. As for diazo methodologies for the synthesis of isoxazoles, information remains scarce. Only a few reports can be found in the literature to date.
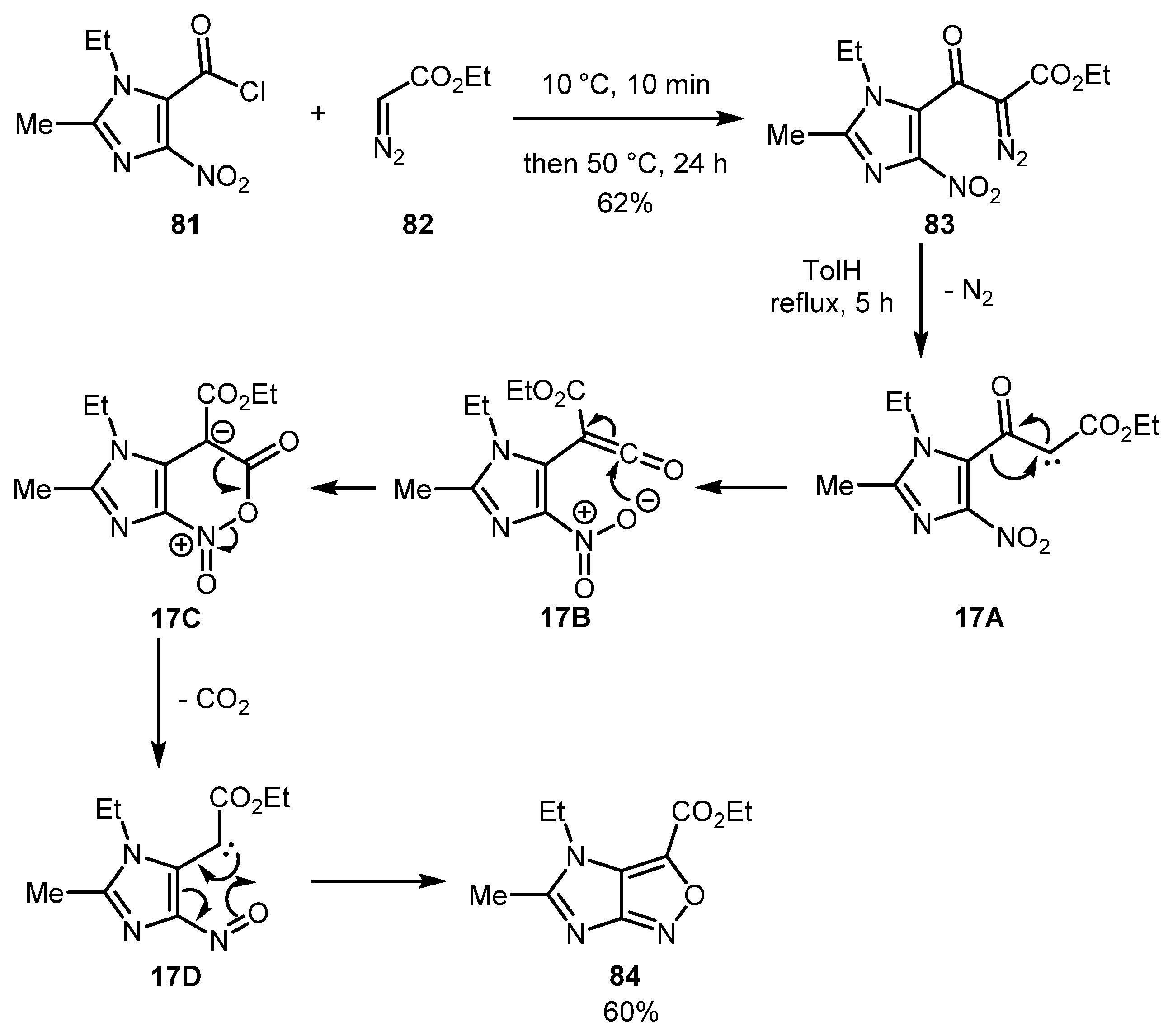
Duffy and co-workers described the preparation of unusual 4H-imidazo[4,5-c]isoxazole 84 from diazo compound 83 which, in turn, was obtained via the coupling of acid chloride 81 with ethyl diazoacetate 82 [49]. On heating, 83 underwent the Wolff rearrangement to give ketene 17B, which cyclized to form zwitterionic intermediate 17C. The latter eliminated a CO2 molecule, thereby producing ortho-nitroso carbene intermediate 17D. The electrocyclization that followed afforded imidazoisoxazole 84 (Scheme 17).
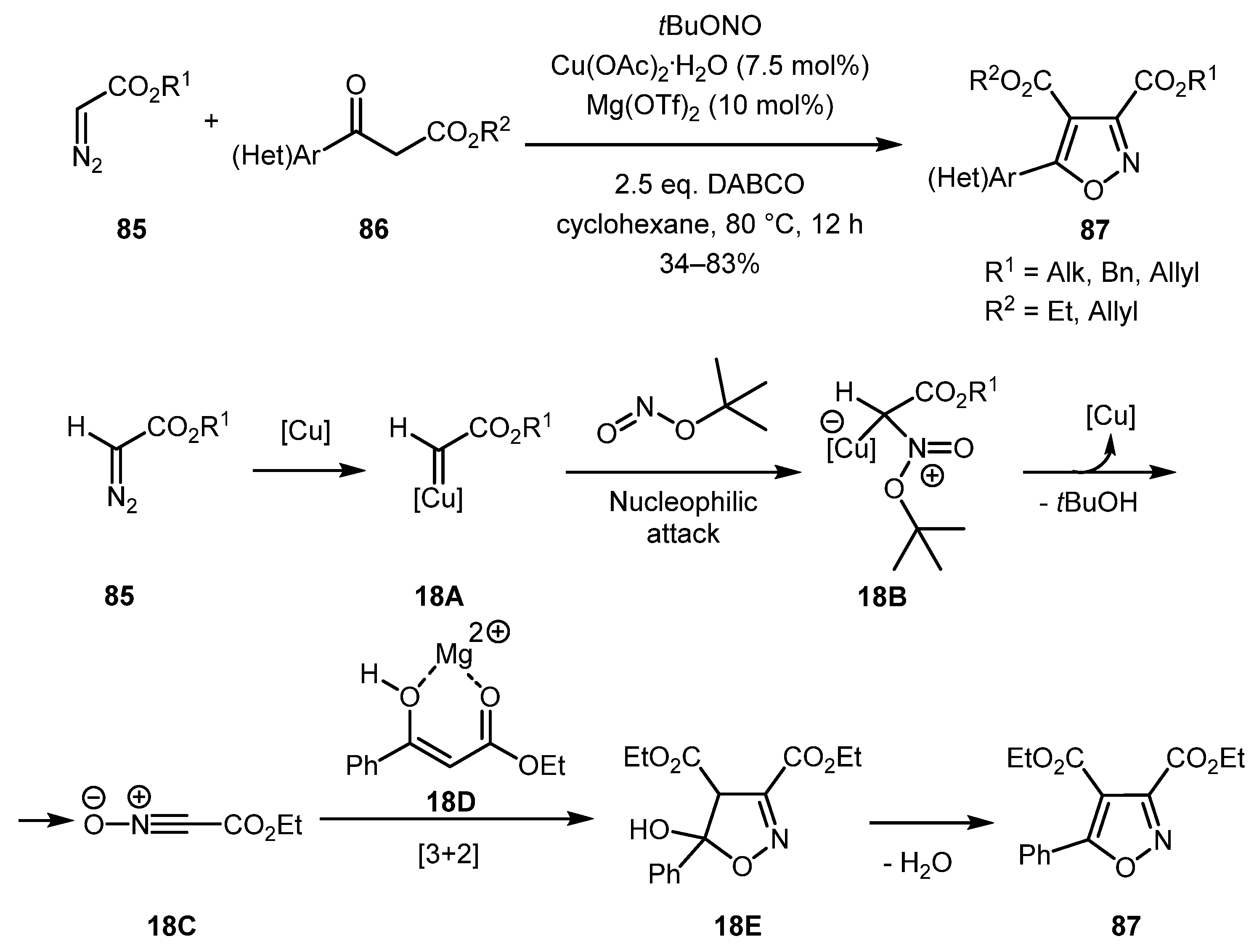
In a 2018 work by the Wan group, diazo esters 85 were used for in situ preparation of nitrile oxides, which underwent highly regioselective [3+2] cycloaddition with enols generated from β-ketoesters 86 [50]. The reaction tolerated a broad range of substituents in the aromatic ring and furnished 3,4,5-substituted isoxazoles 87 in moderate to good yields. Based on preliminary studies, authors proposed the following mechanism for the reaction (shown for (Het)Ar = Ph). Presumably, copper carbene 18A generated from 85 was attacked by tert-butyl nitrite to form intermediate 18B. The latter decomposed to nitrile oxide 18C, which, in turn, underwent a [3+2] cycloaddition with magnesium enolate 18D to produce intermediate 18E. Isoxazole 87 was likely produced via the dehydration of hydroxyisoxazoline 18E (Scheme 18).
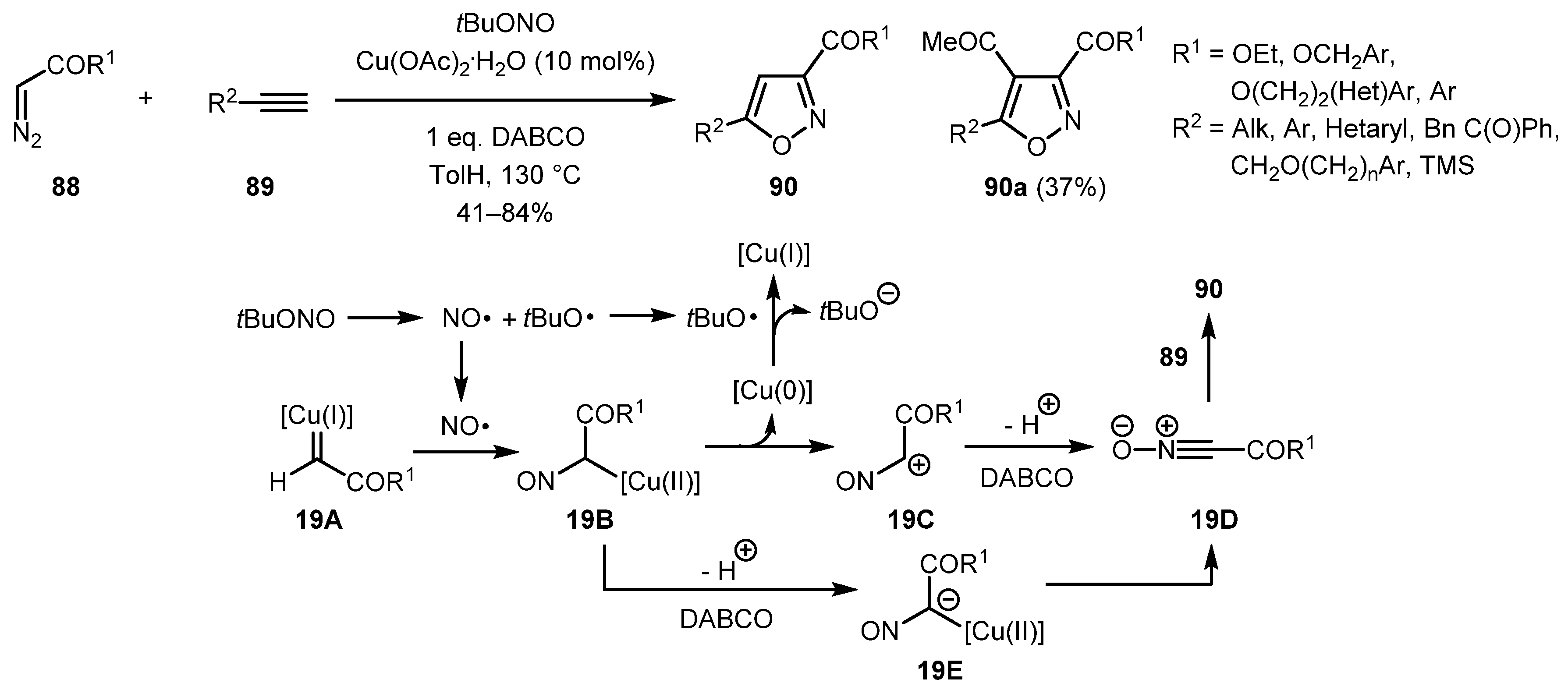
Alkynes 89 can also be effective replacements for β-ketoesters in this reaction as demonstrated by Wang and co-workers [51]. The reaction proceeded in a highly regioselective manner even with internal alkynes (isoxazoles 90a), albeit in lower yield. The authors proposed the following radical mechanism for the reaction. Nitroso radical generated by homolytic cleavage of tert-butyl nitrite along with tert-butoxy radical is thought to react with Cu(I) carbene intermediate 19A to give organocopper intermediate 19B. Heterolytic bond cleavage of 19B with elimination of copper(0) followed by deprotonation could give nitrile oxide 19D, which could then undergo a [3+2] cycloaddition to alkyne 89 to produce desired isoxazole 90. In the course of the reaction, Cu(0) is oxidized to Cu(I) by the tBuO radical, which completes the catalytic cycle. Alternatively, deprotonation of intermediate 19B could occur with subsequent heterolytic C-Cu bond cleavage to give the nitrile oxide intermediate 19D with the same subsequent events (Scheme 19).
2.3. 1,3-Thiazoles
1,3-Thiazole-based materials are now extensively studied for application in organic electronics due to their suitable electronic, optical, and spatial properties. 1,3-Thiazoles have been employed for the preparation of organic field-effect transistors (OFETs), organic photovoltaic cells (OPVs), and organic light-emitting diodes (OLEDs) [52]. Moreover, the thiazole ring has been utilized in the design of medicinal agents for the treatment of viral and bacterial infections, diabetes, Alzheimer’s disease, neglected protozoan diseases, and more [53]. Standard approaches to the synthesis of 1,3-thiazoles, such as thionation of α-acylaminoketones (the Gabriel synthesis) and condensation of α-halocarbonyl compounds with thioamides (the Hantzsch synthesis) are frequently employed [54,55].
The diazo approaches to thiazoles discussed in this subsection of the review are somewhat similar to these classic methods: (i) reaction of α-diazocarbonyl compounds (acting as the α-halocarbonyl compounds equivalent) with thioamides or thioureas; (ii) α-carbonyl carbene NH insertion into thioamides or thiourea with subsequent thionation; and (iii) less conventional reactions.
2.3.1. Synthesis of 1,3-Thiazoles from Diazocarbonyl Compounds and Thioamides or Thioureas
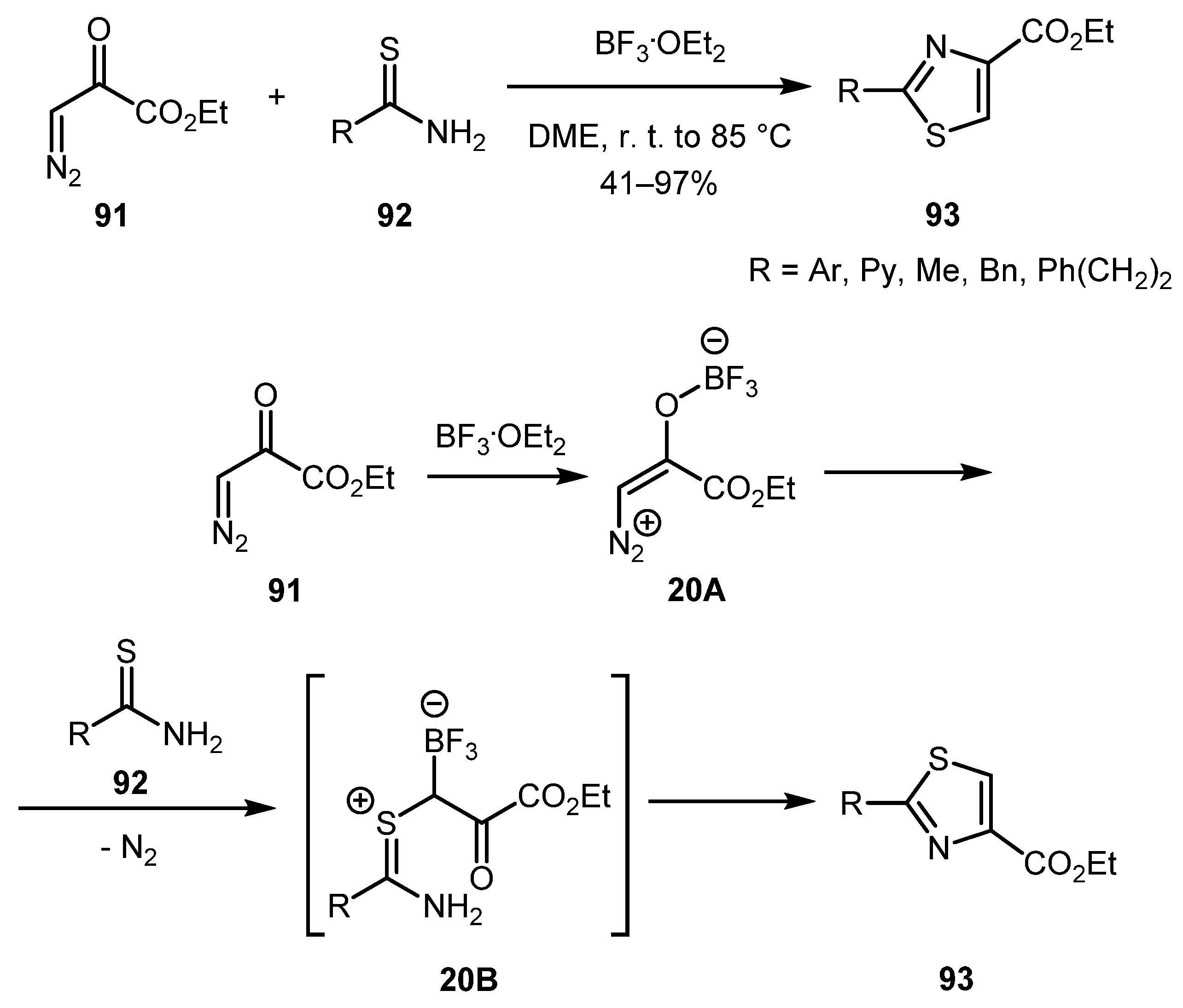
In 1995, Kim and co-workers developed a method for the synthesis of 2,4-disubstituted thiazoles 93 by BF3∙OEt2-promoted reaction of ethyl diazopyruvate 91 with thioamides 92 [56,57]. The best yields were obtained with aromatic thioamides, while for aliphatic and heteroaromatic thioamides, lower product yield were obtained. The following mechanism was proposed as plausible: The initially formed boron enolate 20A could be attacked by thioamide 92 to produce thiocarbonyl ylide 20B. The thiazole product (93) could arise as the result of the cyclodehydration of the intermediate 20B (Scheme 20).
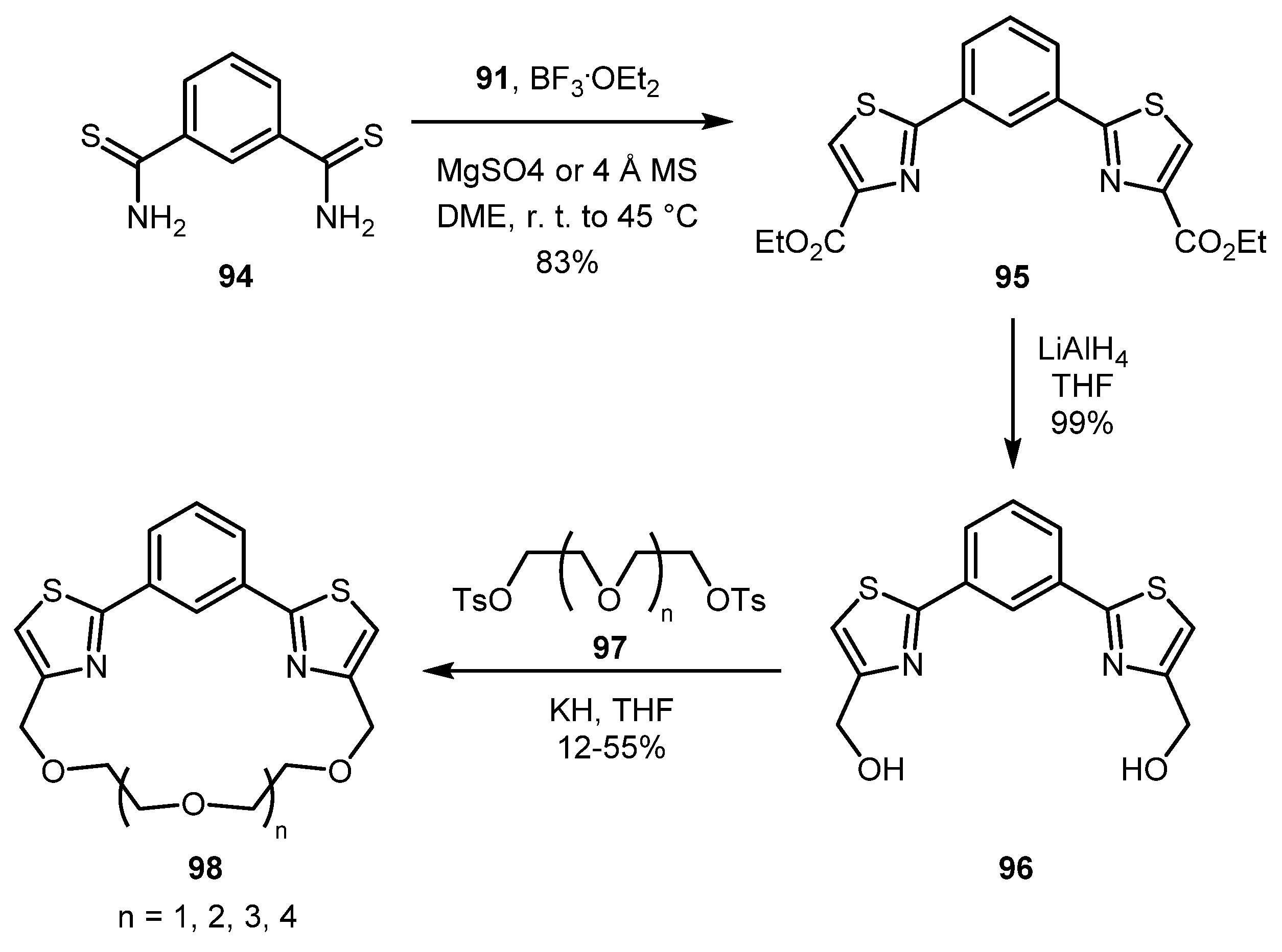
Later, the same group demonstrated the applicability of this procedure to bisthioamides, namely benzene-1,3-bis(carbothioamide) 94 [58]. Thus, bisthiazolylbenzene 95 was obtained in 83% yield. In this case, the use of MgSO4 or molecular sieves as desiccants was essential to avoid the formation of by-products. Compound 95 was used for preparation of unusual thiazole-containing crown ethers 98 (Scheme 21).
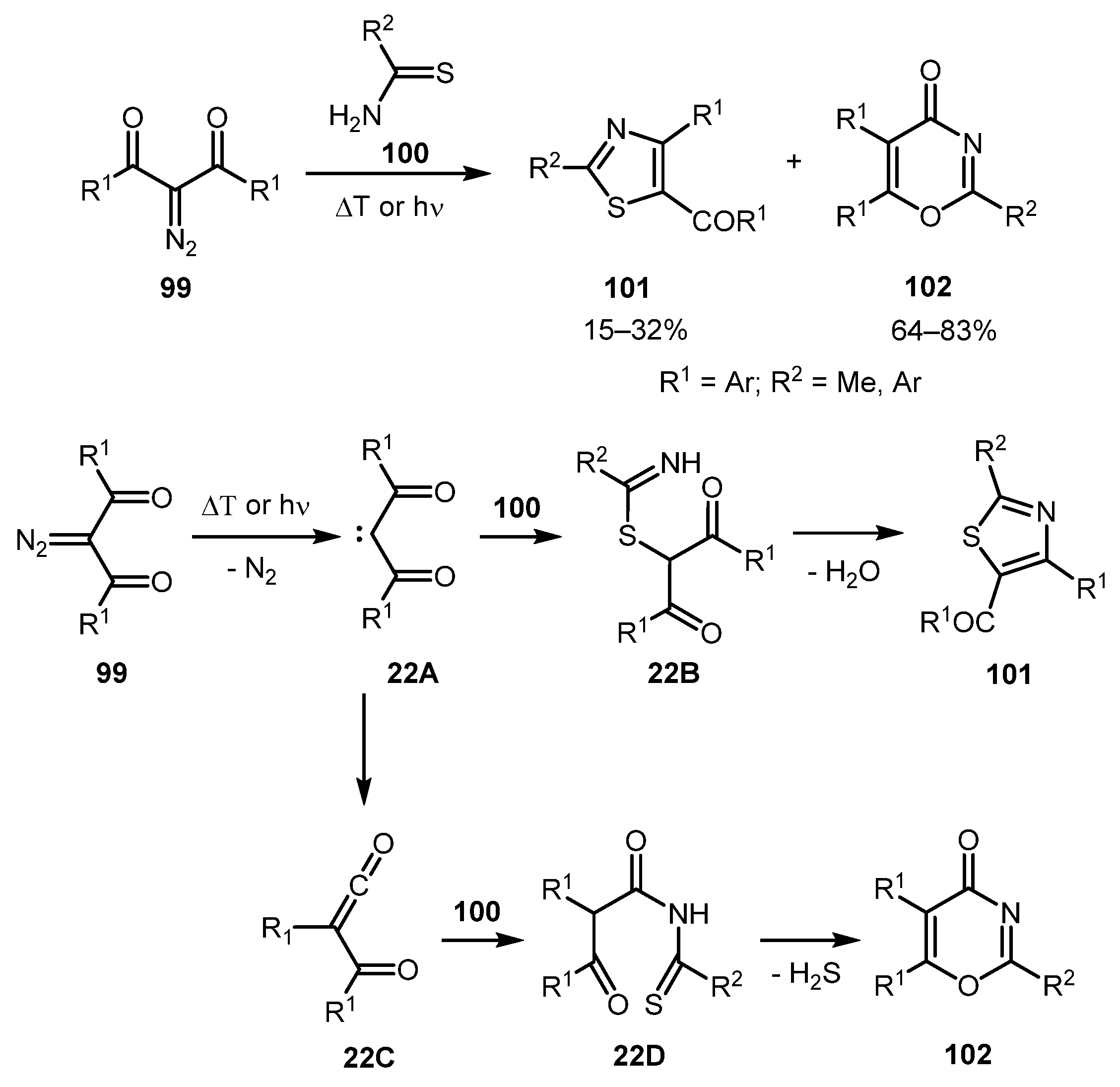
In 1990, Capuano and co-workers observed the formation of 2,4,5-substituted thiazoles 101 upon thermolysis or irradiation of 2-diazo-1,3-diketones 99 in the presence of thioamides 100 [59]. Under thermolysis conditions, thiazoles 101 were formed as a mixture with 4H-1,3-oxazin-4-ones 102 (with a predominance of the latter), while on irradiation, thiazoles were the only products. Unfortunately, the yields were only modest in both cases. The reaction presumably proceeded via ketocarbene 22A, which can react with thiourea to produce intermediate 22B. Subsequent cyclodehydration can afford thiazole 101. Alternatively, 22A can undergo the Wolff rearrangement with the formation of α-oxoketene intermediate 22C, which could be attacked by the NH moiety of thioamide 100 to give intermediate 22D. The latter could cyclize with the loss of H2S and afford 4H-1,3-oxazin-4-one 102 (Scheme 22).
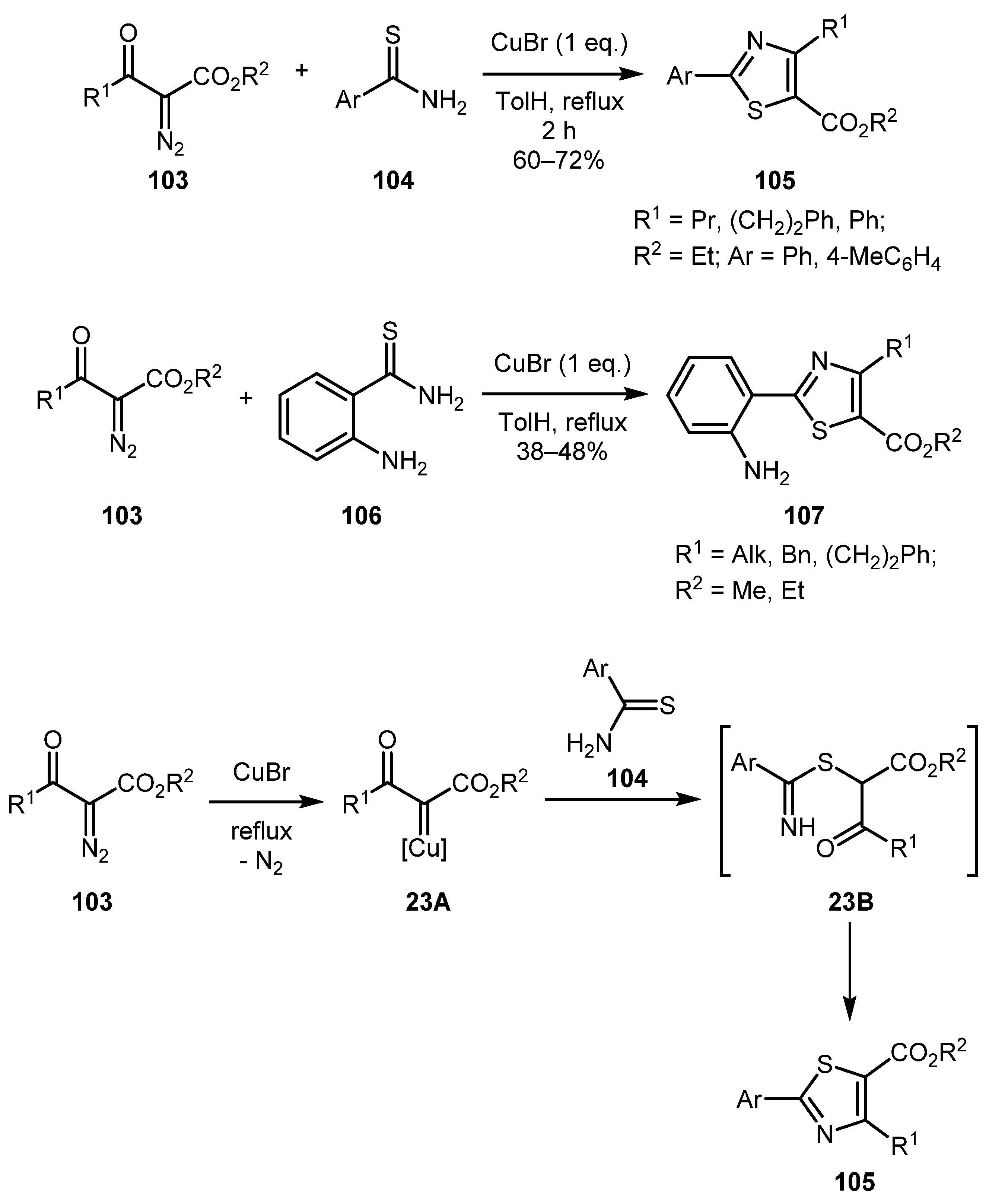
The Villalgordo group employed Cu(I) bromide for this reaction to selectively prepare thiazoles 105 in good yields [60]. Further extending the scope of this methodology, functionalized 2-aminobenzothioamide 106 was used to obtain thiazoles 107, albeit in lower yields. The use of aromatic thioamides proved to be crucial for this reaction since aliphatic ones failed to give the desired products. The tentative mechanism involves the reaction of copper carbene 23A (generated from α-diazo-β-ketoester 103 on action of CuBr) with the thioamide 104 to give an intermediate 23B whose cyclodehydration produced thiazole 105 (Scheme 23).
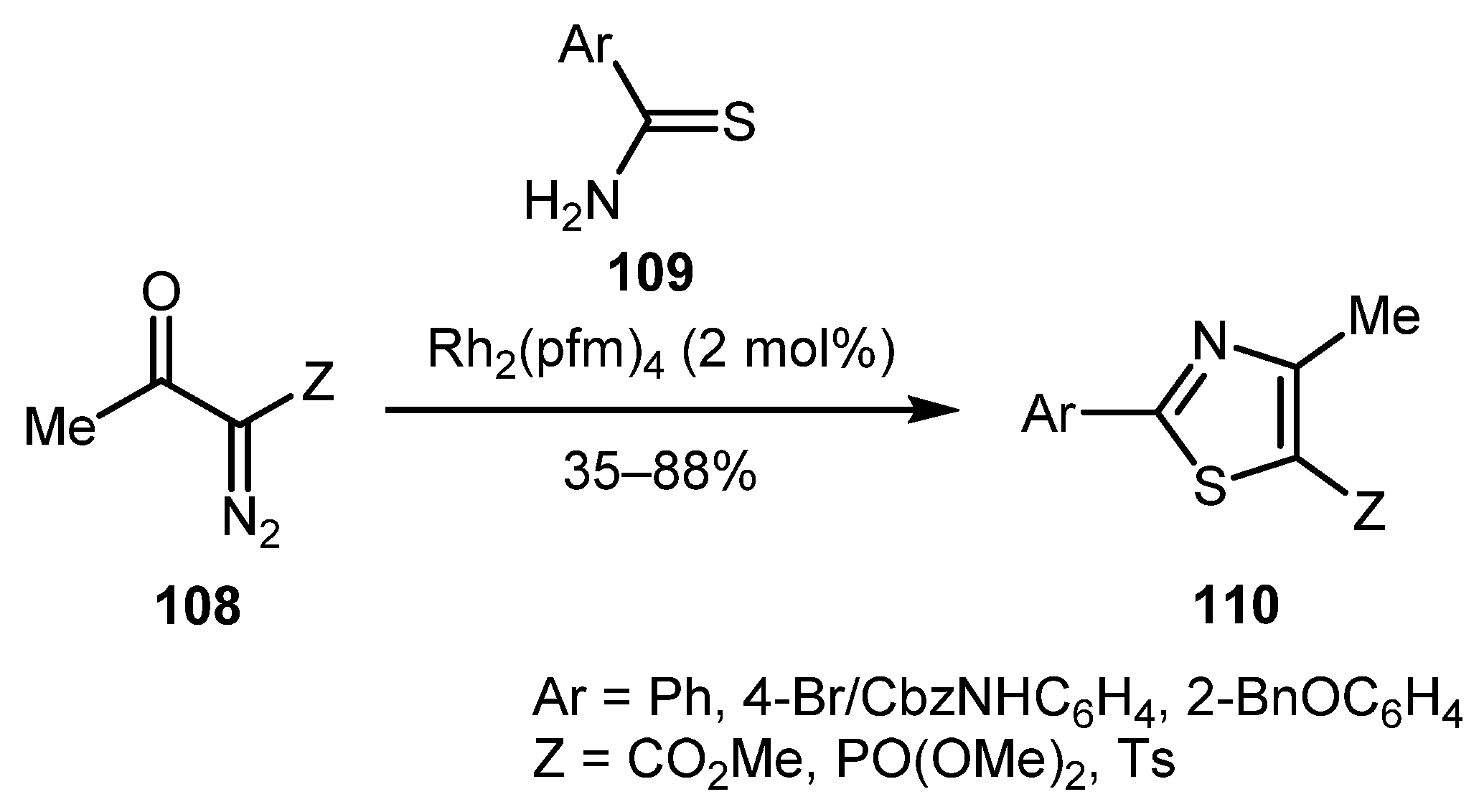
Rhodium catalysis was also successfully employed in the synthesis of thiazoles. In 2010, the Moody group reported the reaction of diversely substituted 1-diazopropan-2-ones 108 with aromatic thioamides 109 catalyzed by rhodium perfluorobutyramide (Rh2(pfm)4), which afforded fully substituted thiazoles 110 in generally good yields (Scheme 24) [38]. The mechanism is similar to that shown in Scheme 23. Other Rh(I) catalysts, namely by [Rh(COD)Cl]2, were also found suitable for promoting this reaction [37].
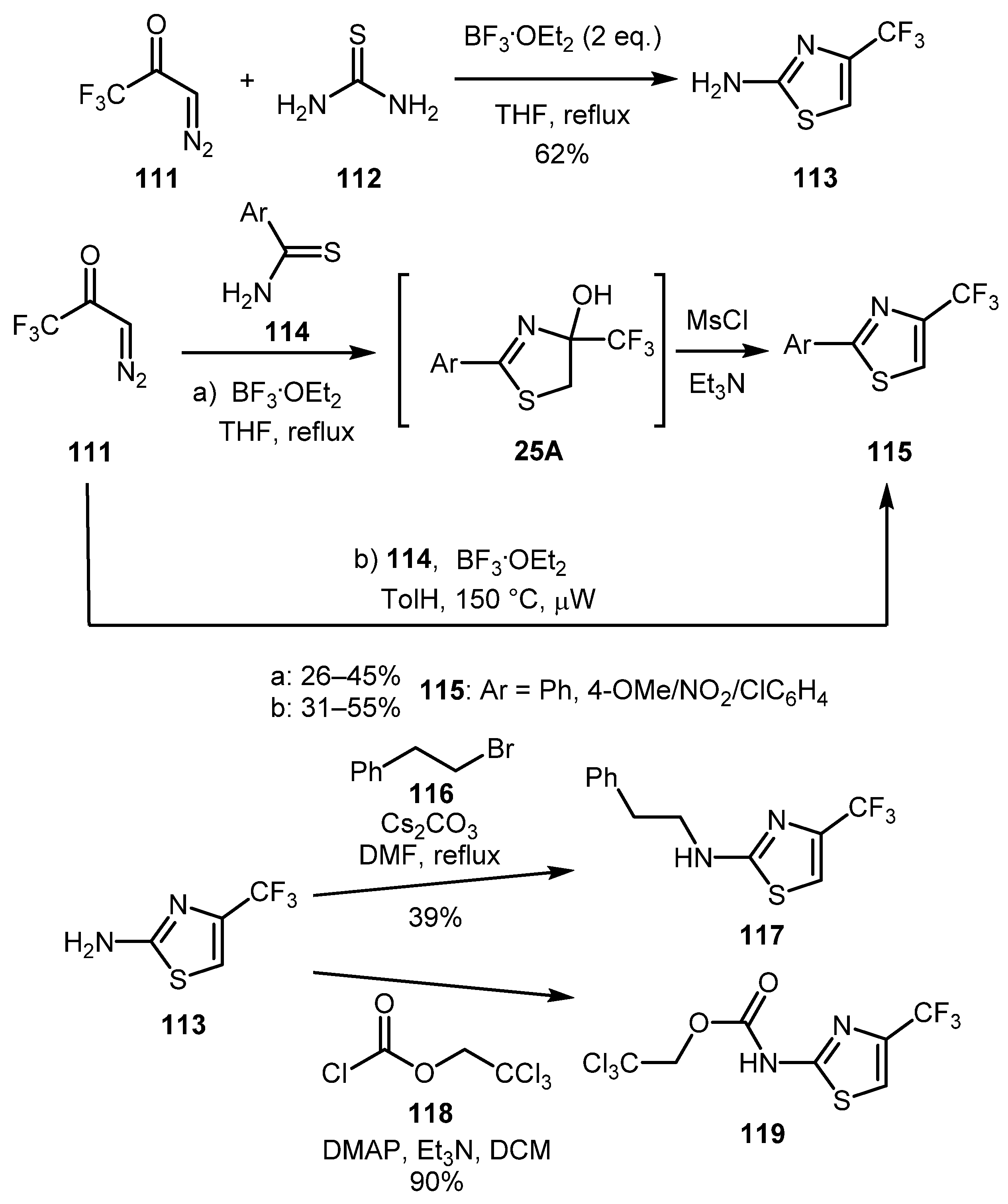
Trifluoromethyl-substituted thiazoles 113 and 115 were synthesized by Obijalska and co-workers via the BF3·OEt2-promoted condensation of 3-diazo-1,1,1-trifluoropropan-2-one 111 and thiourea 112 or benzothioamides 114 (Scheme 25) [61]. The reaction proceeded smoothly with thiourea, but when thioamides were used, dehydration under MsCl/Et3N conditions was additionally needed. To overcome this drawback, heating at 150 °C under the microwave irradiation was alternatively performed, which also gave slightly higher yields of the desired 2-arylthiazoles 115. As in some of the previous examples, the aliphatic thioamides failed to give any thiazole products. 2-Aminothiazole 113 was also furtherly derivatized to obtain CF3-analogs of immunomodulatory drug candidate Fanetizole (117) and anti-inflammatory drug Lotifazole (119) (Scheme 25).
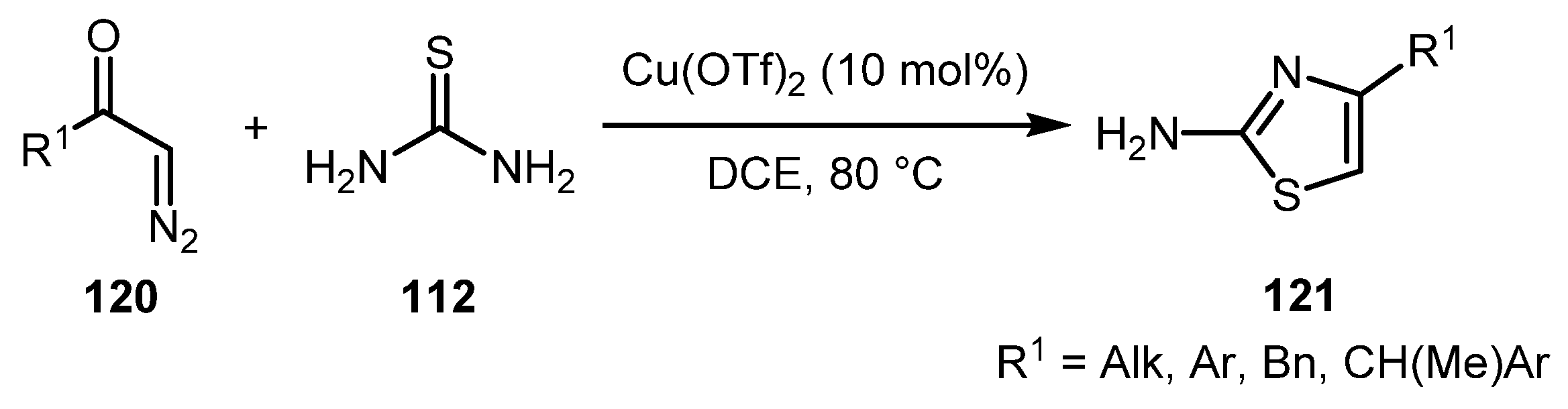
The Narsaiah group reported another example of the coupling of diazo compounds 120 with thiourea 112 under Cu(OTf)2 catalysis [62]. This protocol worked well for a variety of aliphatic, donor- and acceptor-substituted aromatic, and benzylic diazoketones, affording 2-aminothiazoles 121 in excellent yields. A catalyst-free version of this reaction using PEG-400 as a solvent was also reported (Scheme 26) [63].
2.3.2. Synthesis of 1,3-Thiazoles Through the Reaction of Diazocarbonyl Compounds and Amides followed by Cyclization
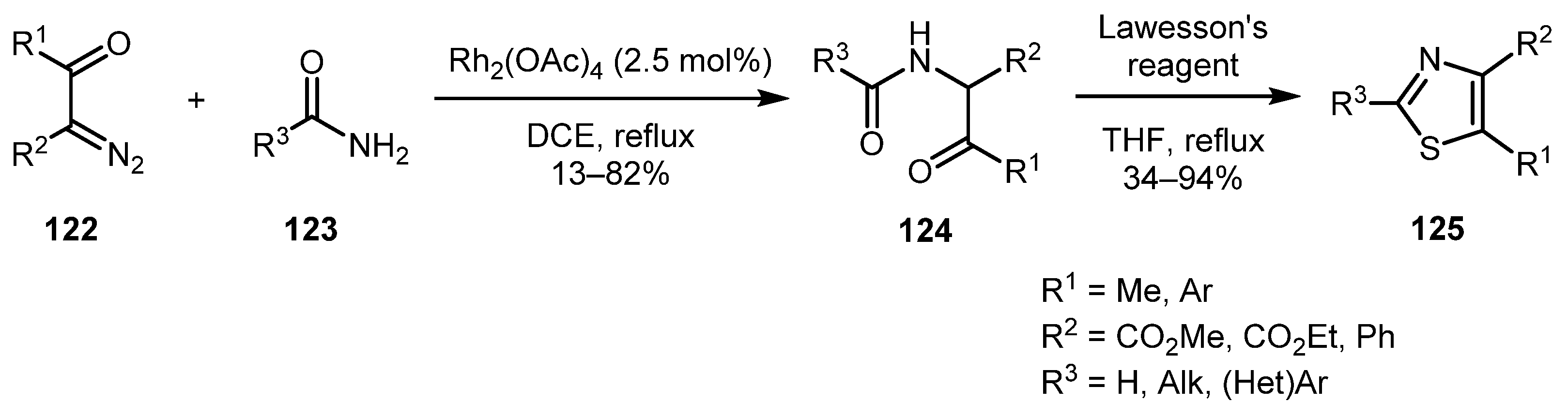
In 2004, the Moody group demonstrated the generality of their approach towards 1,3-oxazoles via NH-insertion/cyclodehydration cascade (Section 2.1.2, Scheme 8) for synthesis of other 1,3-azoles, such as thiazole and imidazole (Section 2.4.1,
Scheme 35) [64]. Thus, fully substituted thiazoles 125 were obtained in varying yields via the treatment of α-(acylamino)ketones 124 with Lawesson’s reagent in refluxing THF. The approach demonstrated high functional group tolerance in the amide moiety, also working well even with aliphatic amides (Scheme 27).
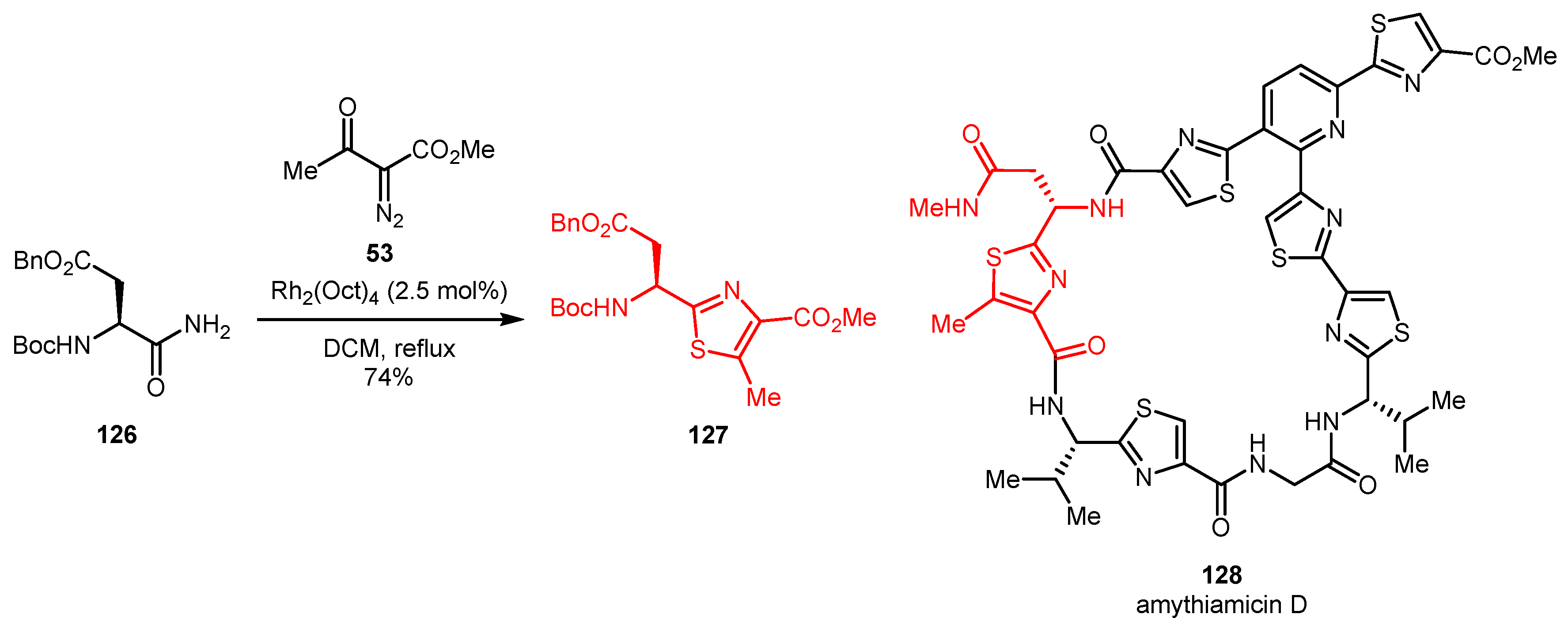
This approach has found application in the total synthesis of natural products. One of the six thiazoles (127) required for the construction of thiopeptide antibiotic amythiamicin D (128) was prepared using this methodology from aspartic acid amide derivative 126 and methyl 2-diazo-3-oxobutanoate 53 (Scheme 28) [65,66]. This route was also involved in total synthesis of other thiazole-containing antibiotics, such as GE2270A and GE2270T [67].
2.3.3. Other Approaches to 1,3-Thiazoles Starting from Diazo Compounds
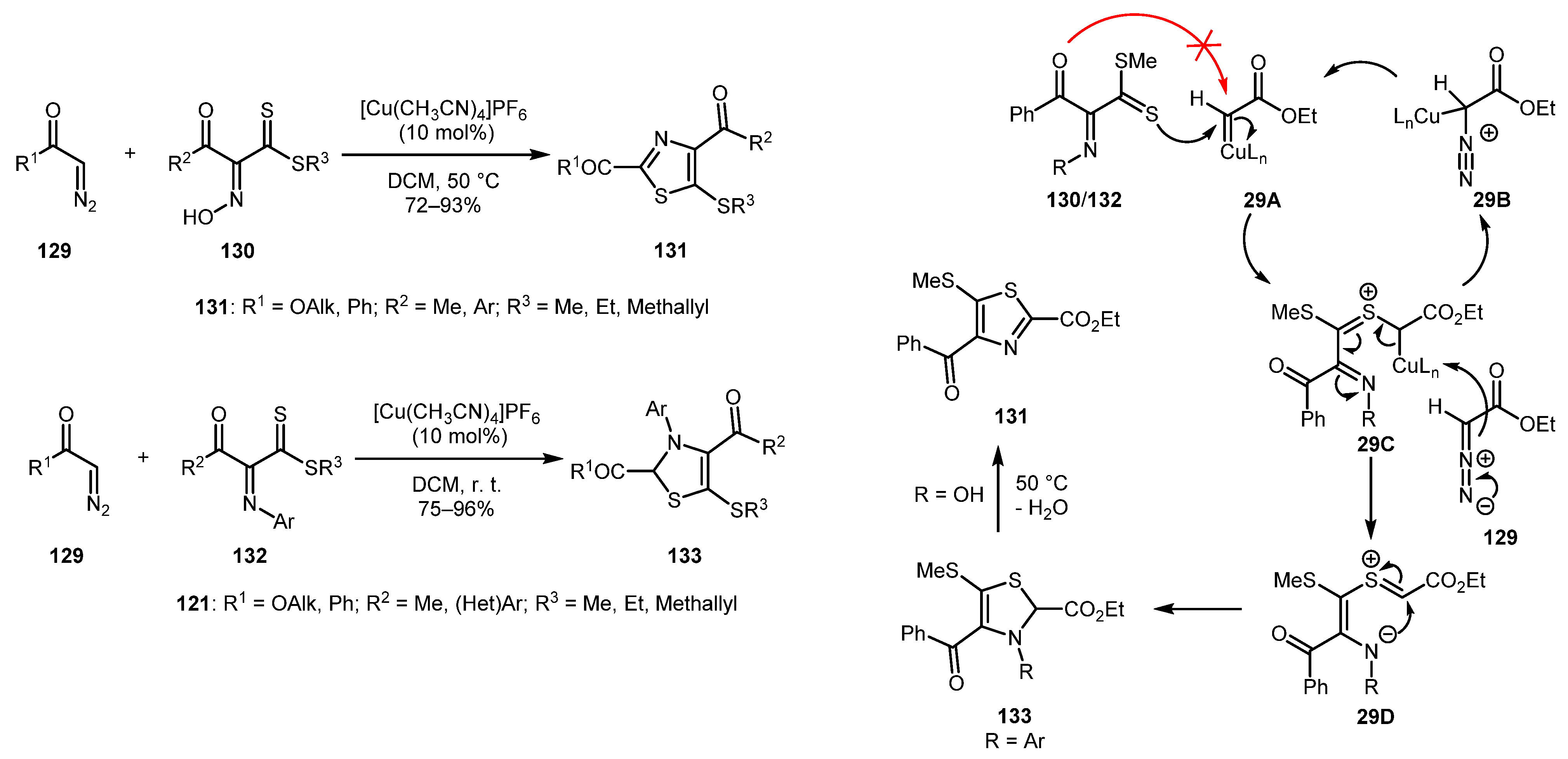
An interesting procedure for the chemoselective synthesis of 2,4,5-substituted thiazoles from diazo carbonyls 129 and α-(N-hydroxy/aryl)imino-β-oxodithioesters 130/132 was reported in 2017 by Srivastava and co-workers [68]. The [Cu(CH3CN)4]PF6-catalyzed reaction gives access to both 5-mercaptothiazoles 131 and 5-mercapto-2,3-dihydrothiazoles 133 in good to excellent yields. Mechanistically, the initially formed Cu-carbene 29A was chemoselectively attacked by thiocarbonyl sulfur of α-imino-β-oxodithioester 130/132 to give thiocarbonyl ylide 29C, which, in turn, reacted with diazocarbonyl compound 129 to afford intermediate 29D, thereby regenerating copper carbene species 29A. Intermediate 29D underwent intramolecular cyclization producing 2,3-dihydrothiazole 133. If N-hydroxy-substrate 130 was used, 133 was converted to aromatic thiazole 131 with a loss of water molecule on moderate heating (Scheme 29).
2.4. Imidazoles
Imidazole derivatives are widely used in medicinal chemistry. Agents based on imidazole moiety possess anticancer, antifungal, antibacterial, antiviral, anti-inflammatory, and other biological activities [69]. Another area of application of imidazoles is in organic light-emitting diodes (OLEDs) and semiconductors [70]. To date, many methods have been developed for the synthesis of imidazoles, including the classical Debus-Radziszewski imidazole synthesis and reaction of α-halo carbonyl compounds with amidines [71,72]. However, syntheses based on diazocarbonyl compounds have not yet become widespread.
There are two main approaches to the preparation of imidazoles using diazocarbonyl compounds: Metal catalyzed insertion into the NH bond of ureas followed by cyclization with formation of 2-imidazolones (which can be easily converted to substituted imidazoles), and NH-insertion into amides followed by cyclization in the presence of amine or an ammonia source. In addition, there are also some more exotic syntheses making use of isocyanides, imines, and nitriles.
2.4.1. Synthesis of Imidazoles by Reaction of Diazocarbonyl Compounds with Amides and Ureas
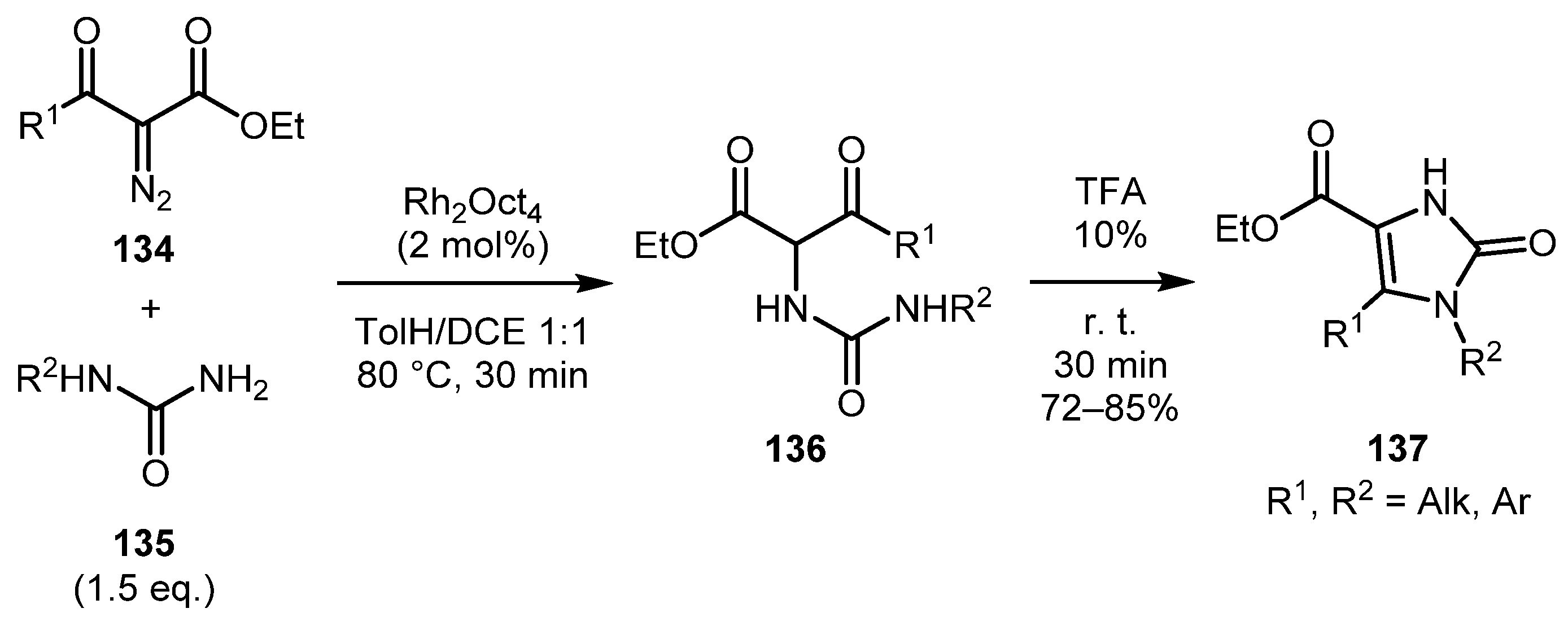
In 2003, the Janda group reported the one-pot procedure for the synthesis of 2-imidazolones 137 by rhodium(II)-catalyzed NH-insertion involving α-diazo-β-ketoesters 134 and primary ureas 135 followed by TFA-mediated cyclodehydration of insertion products 136 (Scheme 30) [73]. The reaction resulted in good yields with a full chemoselectivity.
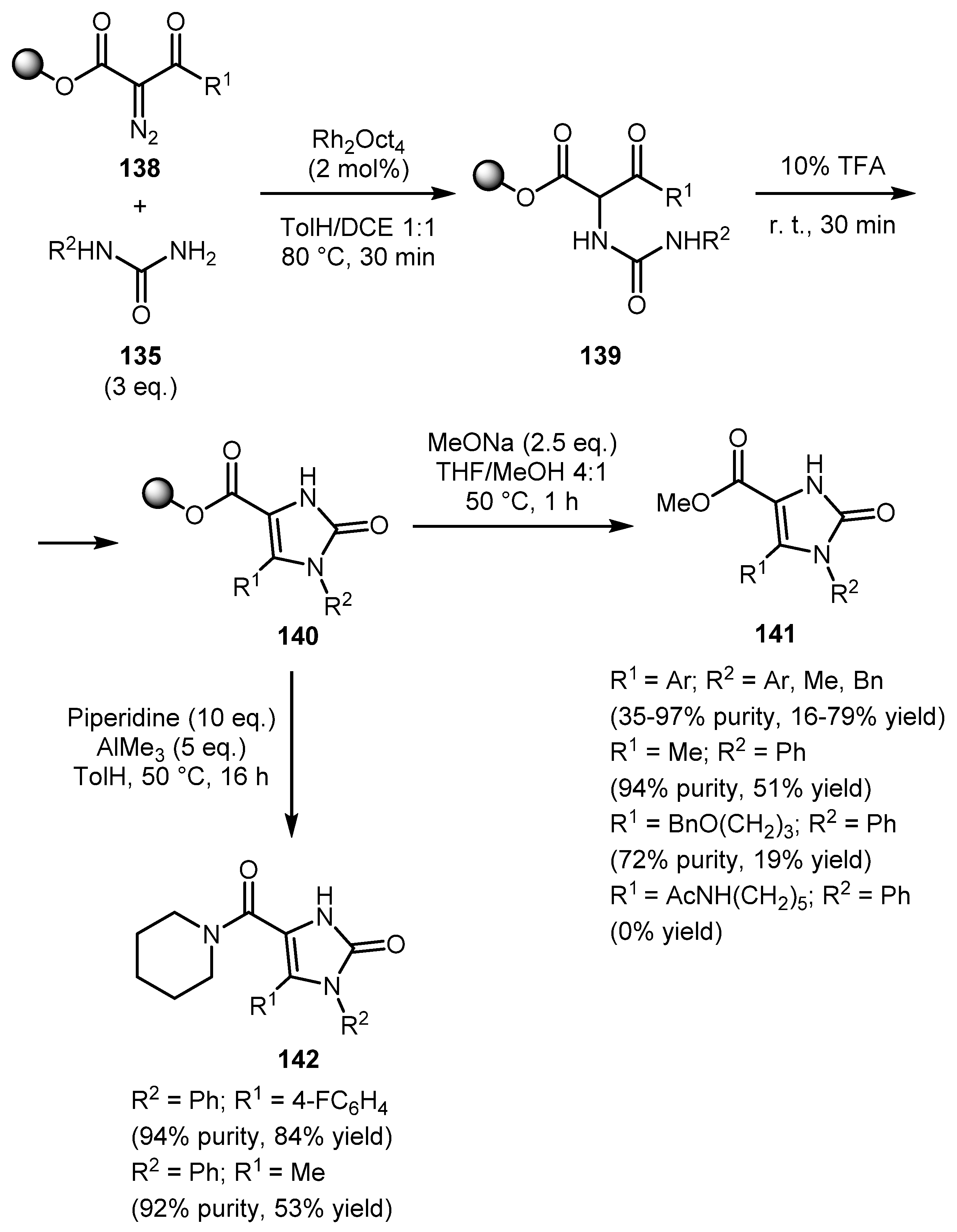
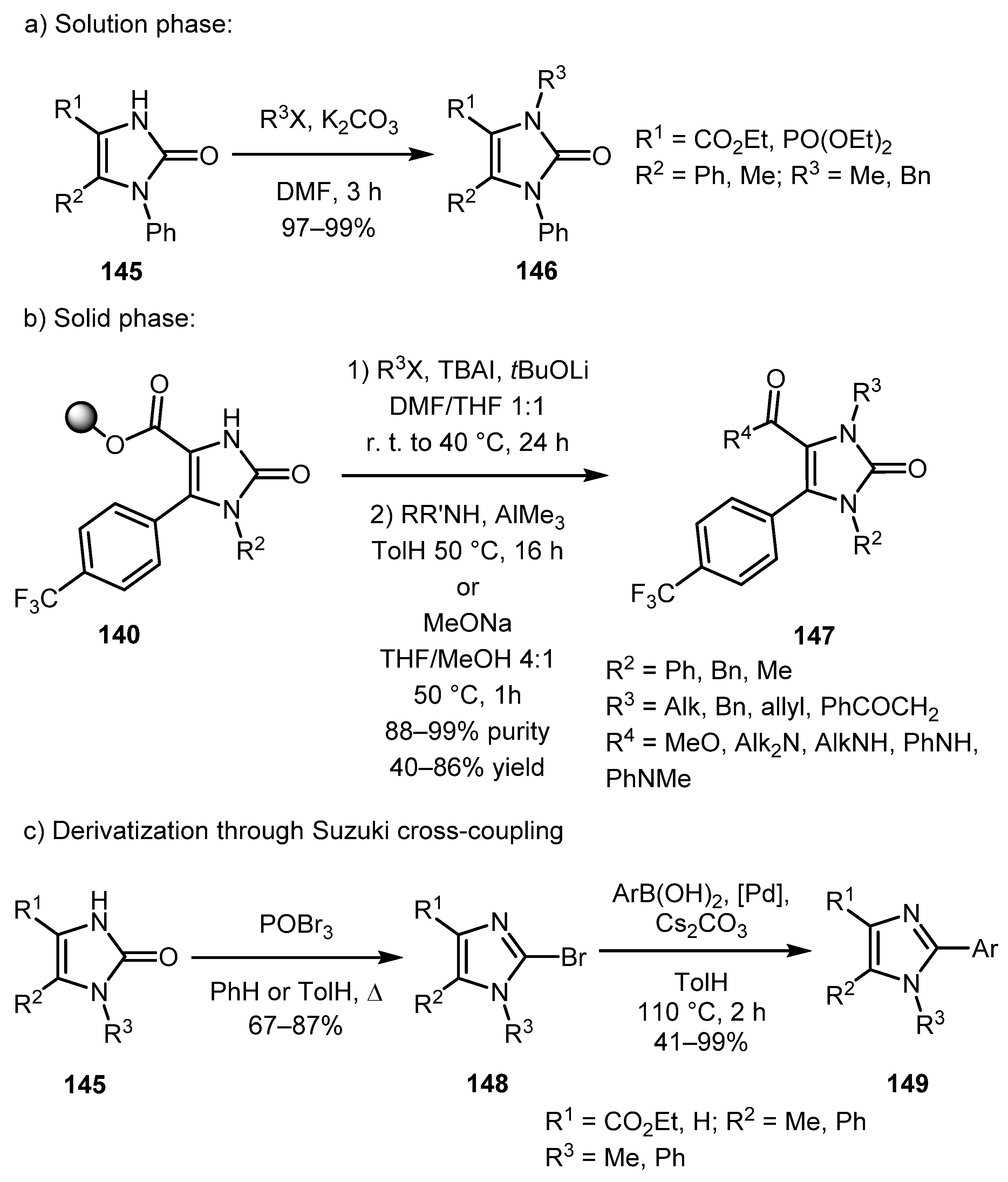
Encouraged by the successful results obtained by performing these syntheses in solution, the authors investigated this strategy on solid phase using hydroxypentyl-JandaJel polymer-bound α-diazo-β-ketoesters 138 (Scheme 31). Imidazolones 140 were then cleaved from the resin either by transesterification to give esters 141 or by the amidation reaction to give more diversely substituted amides 142 in fair yields and high purity. The only limitation was the unallowable use of diazo substrates bearing long alkyl R1 substituents. In these cases, the yield was poor, or the product could not be isolated.
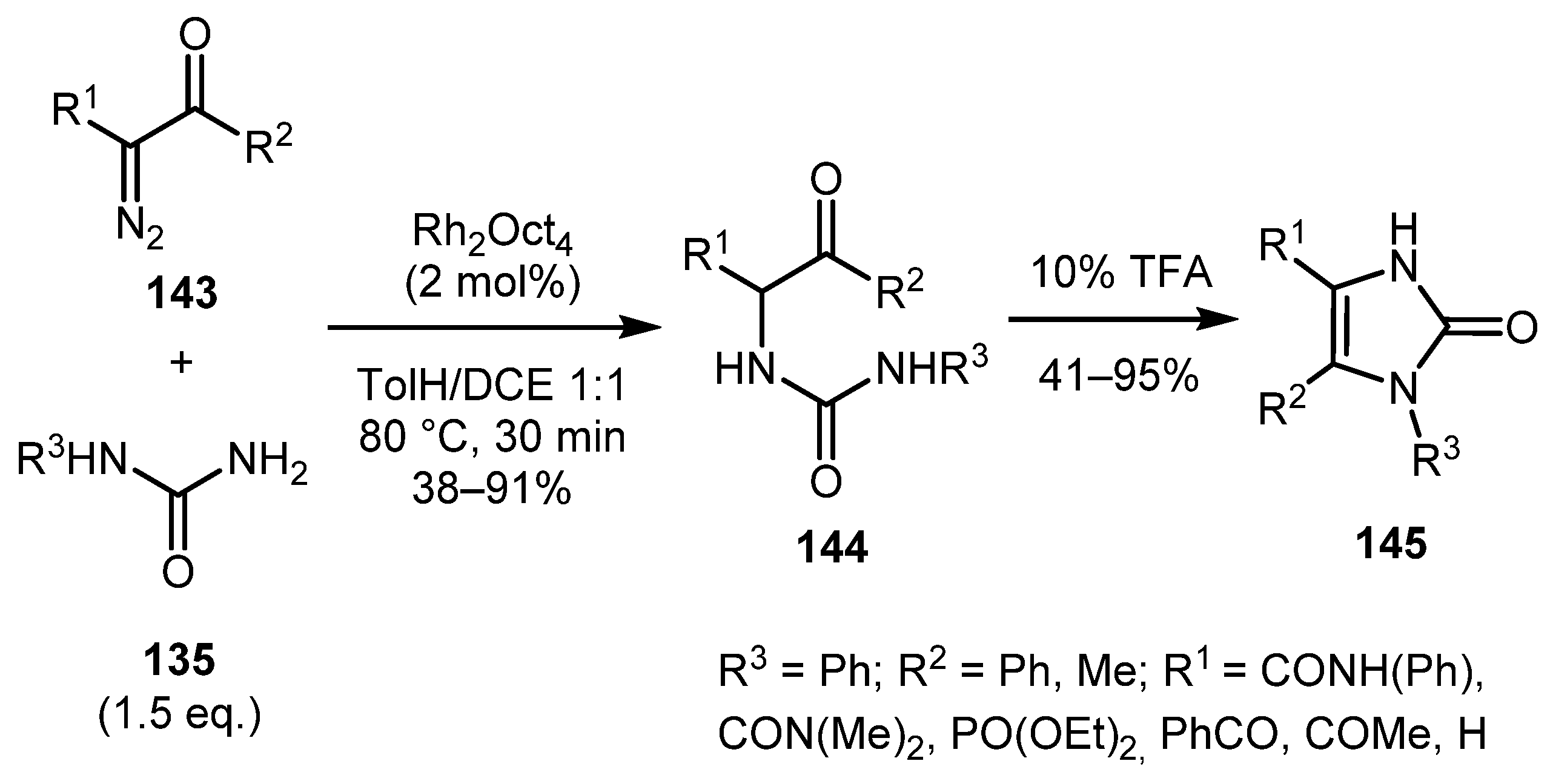
In a continuation of this topic, a work on the expansion of the range of suitable diazo carbonyls and further functionalization of imidazolones by the same authors was published in 2004 (Scheme 32) [74]. α-Diazo-β-ketophosphonates, α-diazo-1,3-diketones, α-diazo-β-ketoamides, diazomalonic ester, ethyl diazoacetate, amd ethyl 2-diazo-2-(diethoxyphosphoryl)acetate have been successfully involved in the reaction. It should be noted that some of the intermediates 144 could be isolated. The resulting imidazolones 145 could be alkylated both in solution (Scheme 33a) and on solid phase (Scheme 33b). In the second case, both transesterification and amidation can be used to cleave the product from the polymer resin.
Another aspect of this work was the conversion of imidazolones 145 to 2-bromoimidazoles 148 by the treatment with POBr3 in refluxing benzene or toluene. Compounds 148 can be further derivatized via the Suzuki cross-coupling reaction (Scheme 33c).
Thus, in this series of works, the authors have shown the applicability of this approach for the synthesis of libraries of various imidazolone and imidazole derivatives, also in an array mode.
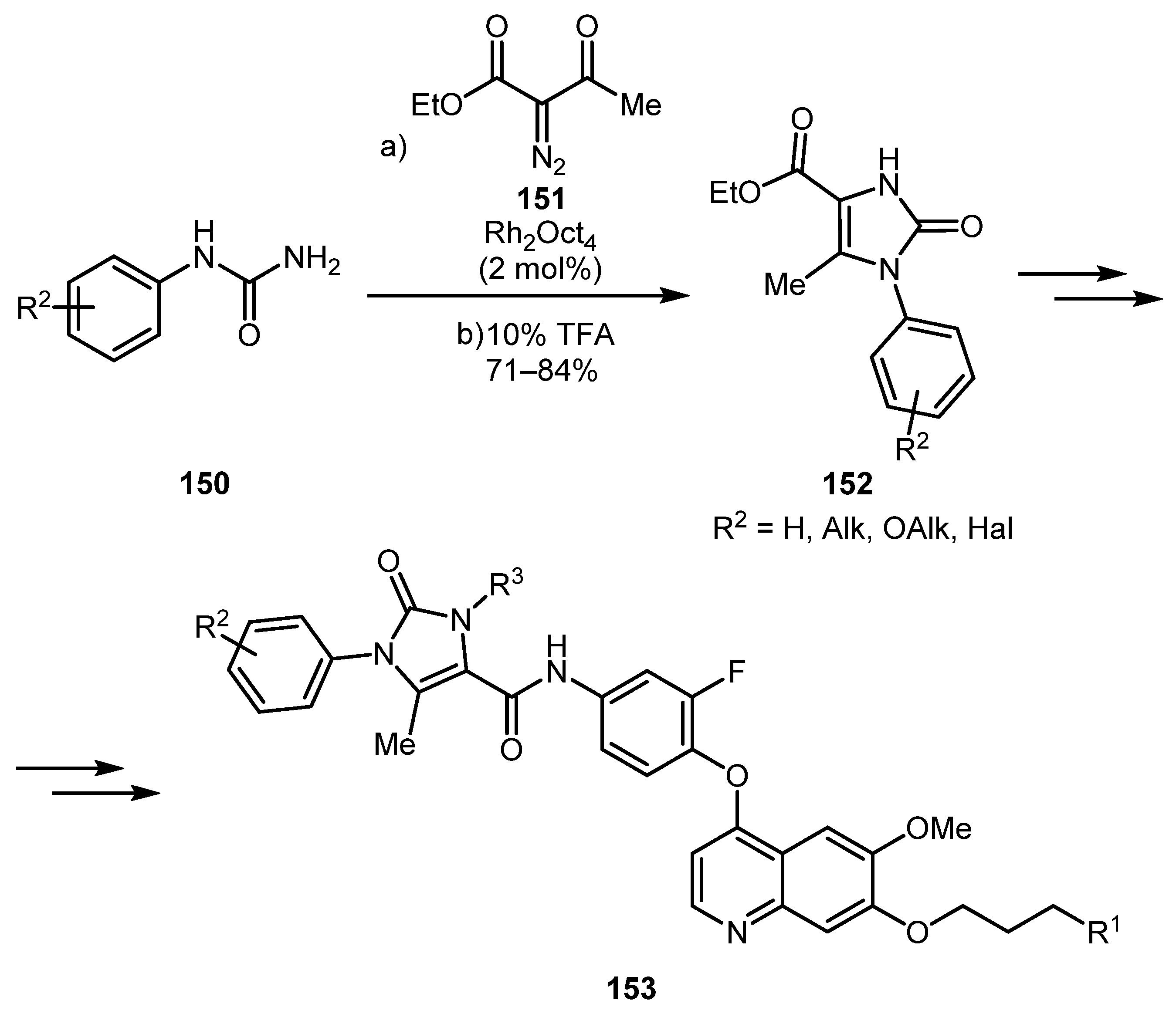
The above method has found application in medicinal chemistry [75,76]. In 2015, Gong and co-workers obtained a number of imidazolones 152, which were then used in the synthesis of a series of potential c-Met kinase inhibitors 153 (Scheme 34) [75].
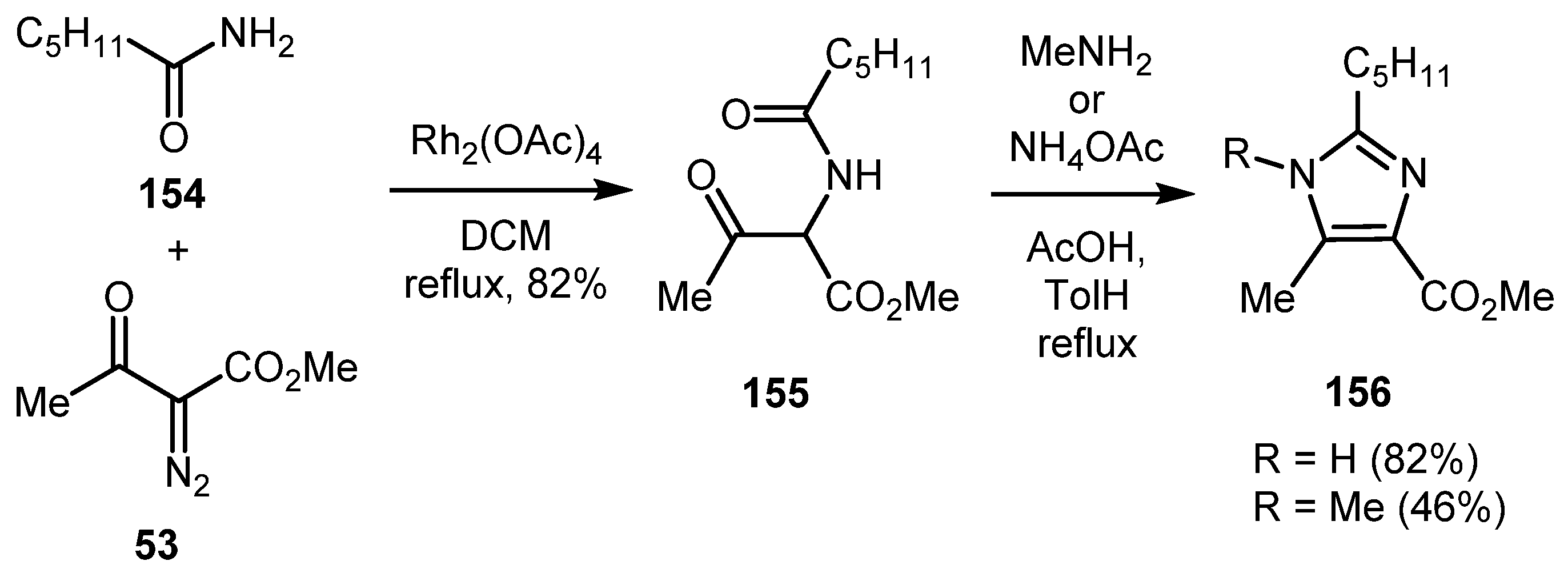
In 2004, the aforementioned Moody’s strategy for the synthesis of 1,3-oxazoles based on the NH-insertion of rhodium carbene intermediates generated from α-diazocarbonyl compounds into primary amides (Section 2.1.2, Scheme 8) was extended to the synthesis of 1,3-thiazoles (Section 2.3.2, Scheme 27) and imidazoles [64]. Thus, imidazoles 156 were obtained by treatment of intermediate 155 with a primary amine or ammonium acetate in the presence of acetic acid under reflux conditions (Scheme 35).
2.4.2. Other Syntheses of Imidazoles from Diazo Compounds
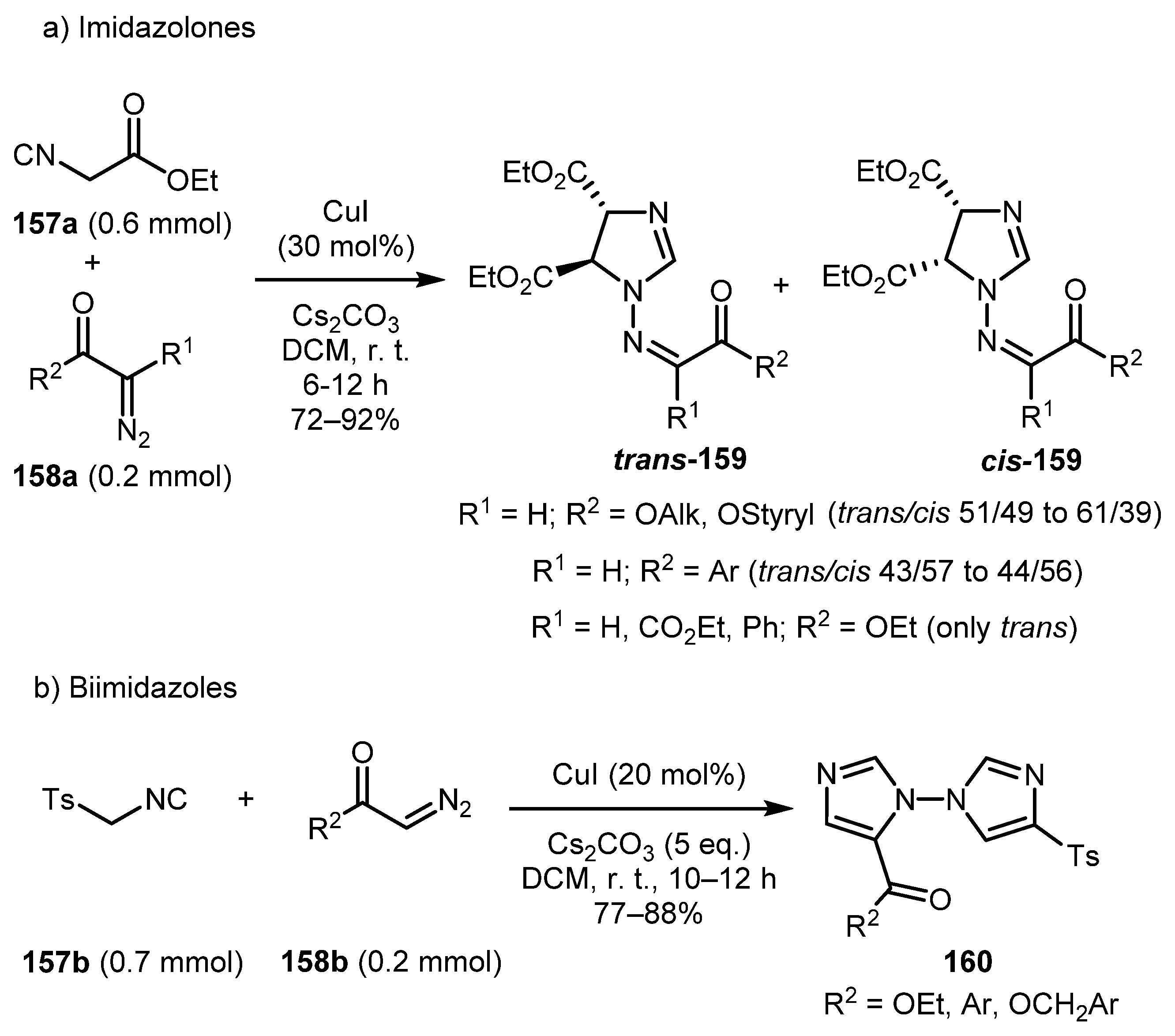
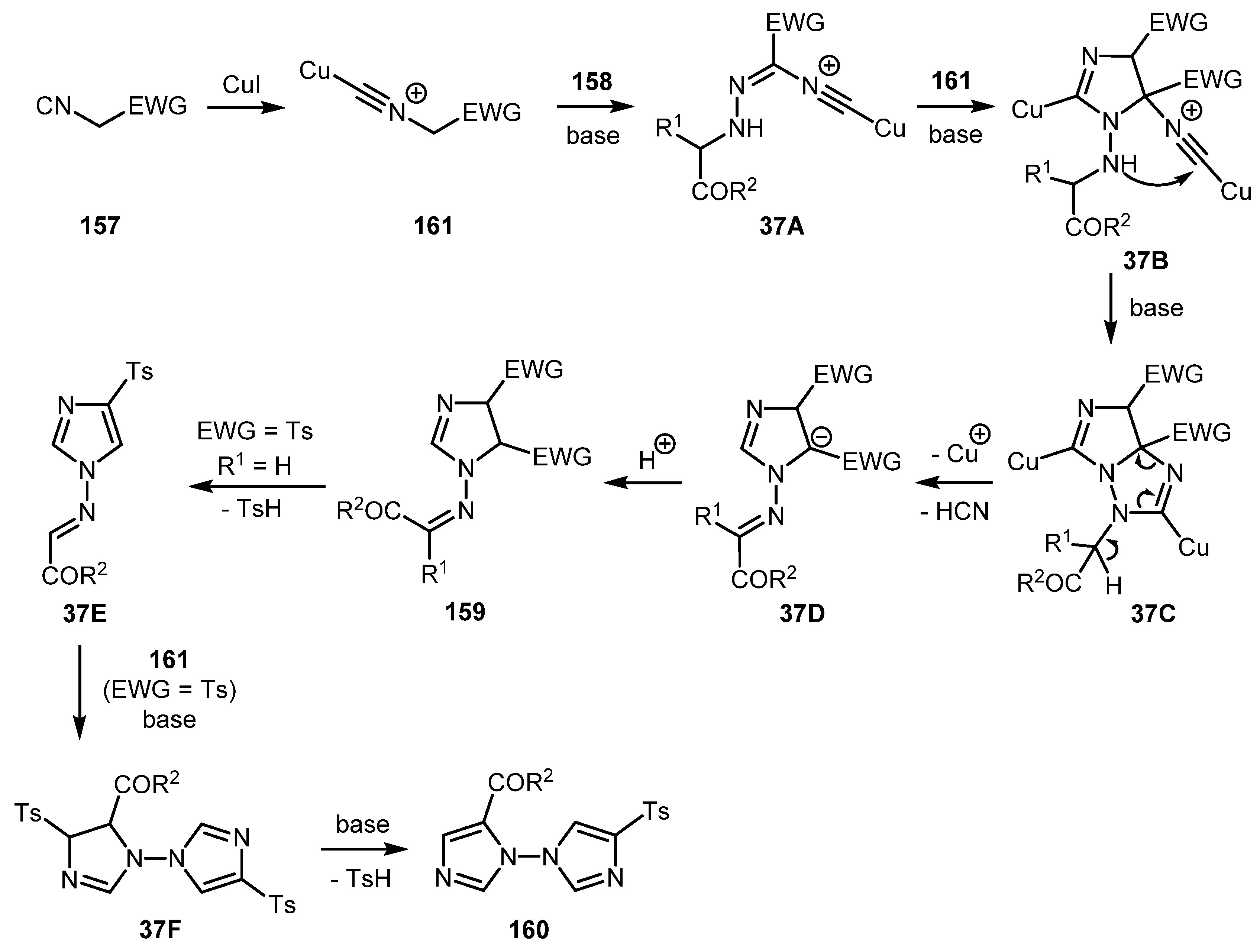
In 2017, Zhao and co-workers devised an effective copper-catalyzed cascade cyclization reaction of isocyanides 157 with α-diazocarbonyls 158 [77]. This protocol furnishes imidazolines 159 (Scheme 36, a) and biimidazoles 160 (Scheme 36b) in good yields under very mild conditions. Imidazolines 159 were obtained as a mixture of diastereomers using ethyl isocyanoacetate 157a. Stereochemistry of the product depends on the R2 substituent: Bulky hydroxyalkyl R2 substituents gave the mixture with predominance of the trans-isomer whereas aryl substituents afforded the product mixture with predominance of the cis-isomer. Only in the case of the ethoxy group was the trans-isomer obtained exclusively. Biimidazoles 160 were obtained when tosyl methyl isocyanide (TosMIC) 157b was used as the isocyanide component.
A plausible reaction mechanism is presented in Scheme 37. Initially, the copper isocyanide complex 161 is formed in which the acidity of the α-H atom is significantly enhanced compared to the non-complexed 157. Subsequently, nucleophilic attack of 161 onto the terminal nitrogen atom of α-diazocarbonyl compound 158 occurs under basic conditions to give intermediate 37A. Intermediate 37B, (generated by the formal [3+2] cycloaddition reaction of 37A with complex 161) then undergoes intramolecular cyclization to give the bicyclic intermediate 37C. Imidazoline 159 is formed from intermediate 37C via ring-opening followed by HCN elimination and protonation. When TosMIC was used, imidazoline 159 suffered base-mediated elimination of sulfinic acid to produce the intermediate 37E, which then underwent a formal [3+2] cycloaddition with the third equivalent of complex 161. The following elimination of sulfinic acid from the intermediate 37F gave the biimidazole 160.
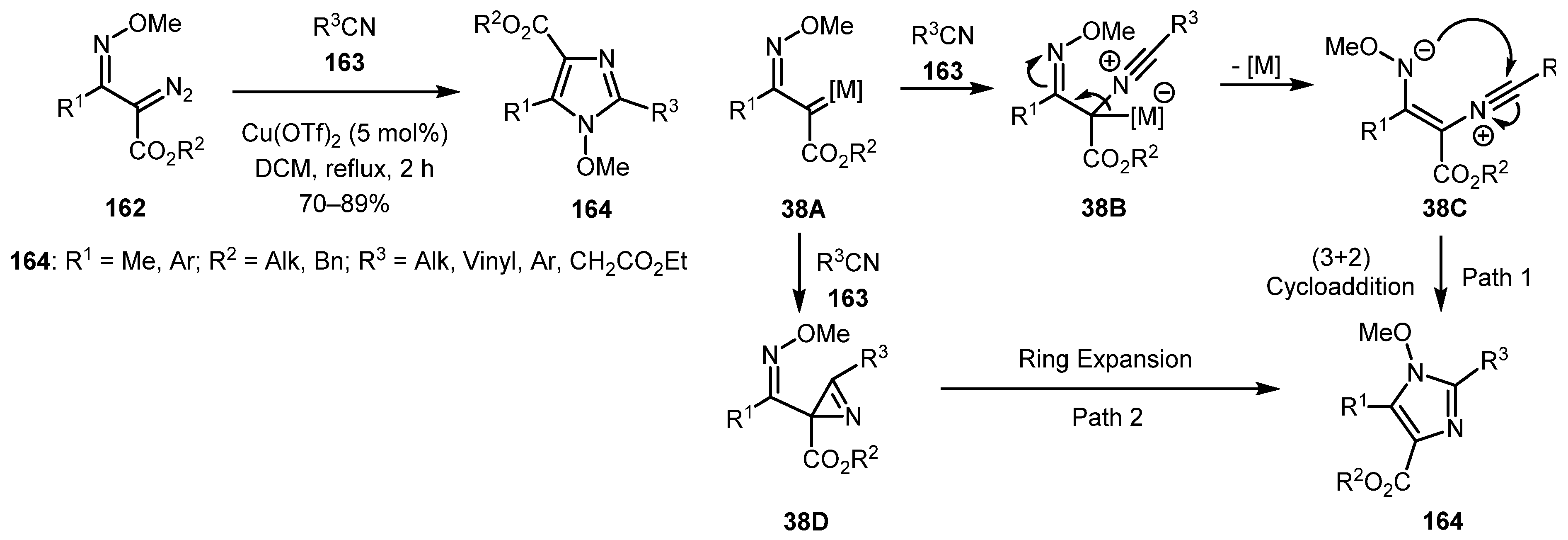
In 2016, the reaction of α-diazo oxime ethers 162 (available from β-ketoester via one-pot oxime formation with subsequent diazo transfer reaction [78]) with nitriles 163 in presence of copper triflate (Cu(OTf)2) catalyst was described by Kuruba and co-workers [79]. The developed procedure provided fully decorated N-methoxyimidazoles 164 and worked well with a broad range of nitriles, both aliphatic and aromatic. Mechanistically, two possible reaction pathways were proposed. In both cases, the reaction begins with the formation of metal carbene 38A from diazo compound 162. In Path 1, carbene 38A is attacked by nitrile 163 to form adduct 38B, which affords ylide 38C after the release of the catalyst. Ylide 38C then undergoes cyclization giving imidazole 164. Path 2 leads to the formation of azirine 38D with subsequent ring expansion. However, no azirine intermediates have been detected or isolated, so it was concluded that path 1 was favored over path 2 (Scheme 38).
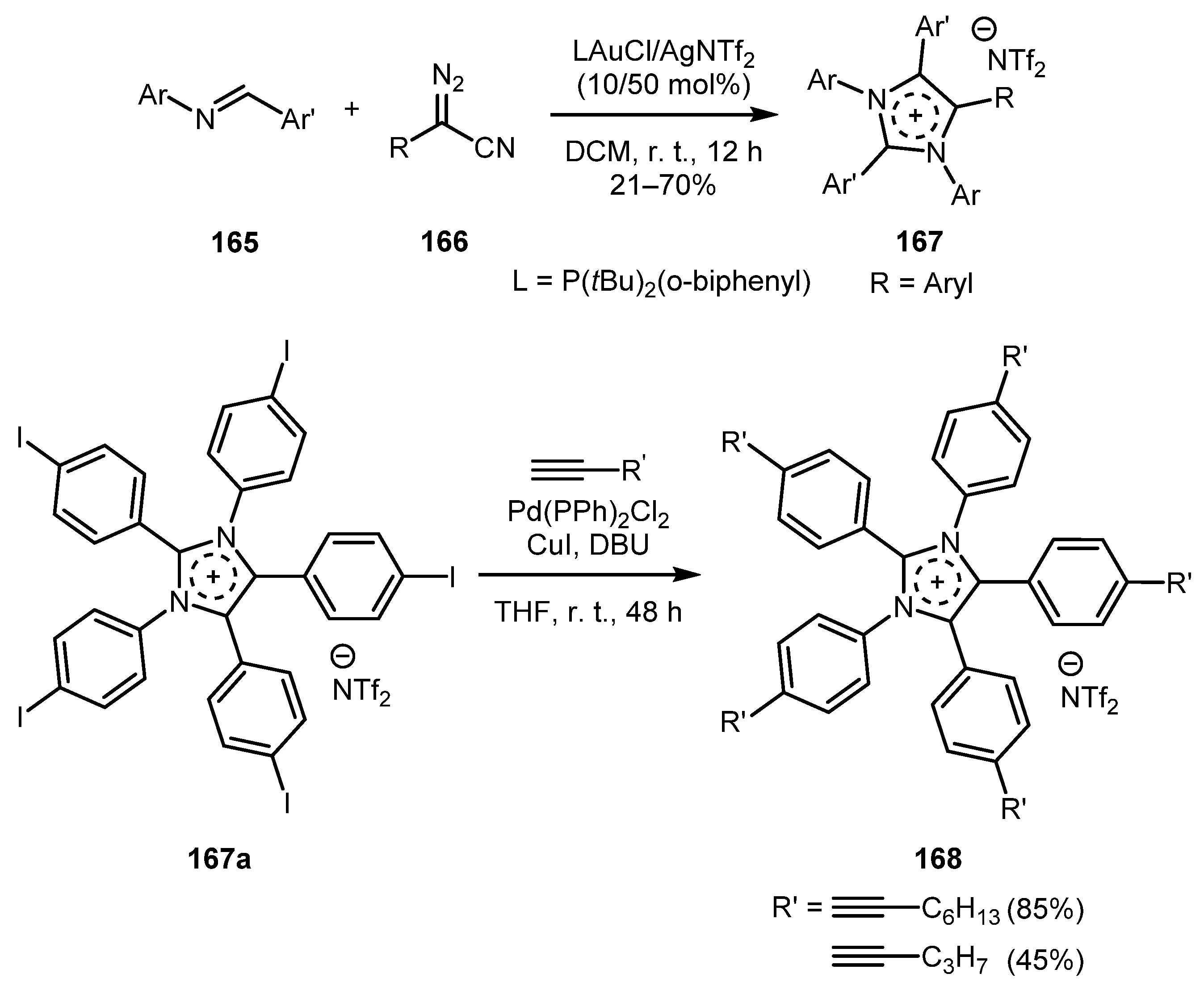
Imidazolium salts are currently being intensively studied as electrolytes, metal ligands, ionic liquids, and liquid crystals [80,81,82]. However, only a few studies have been devoted to the synthesis of polyarylated imidazolium salts. Thus, development of new methods for the synthesis of polyarylated imidazolium salts would be a timely undertaking. To achieve this goal, the Liu group developed a gold-catalyzed [2+2+1] annulation reaction involving two equivalents of imine 165 and one equivalent of aryl diazoacetonitrile 166 [83]. Introducing electron-rich aryl substituents in the imine component resulted in higher yields of imidazolium salts 167 compared to the imines bearing electron-deficient substituents. The same effects were observed from the substituents in the diazo nitrile aryl ring. Moreover, these imidazolium salts can be further modified by the Sonogashira reaction to afford polyalkynyl-substituted derivatives 168 (Scheme 39).
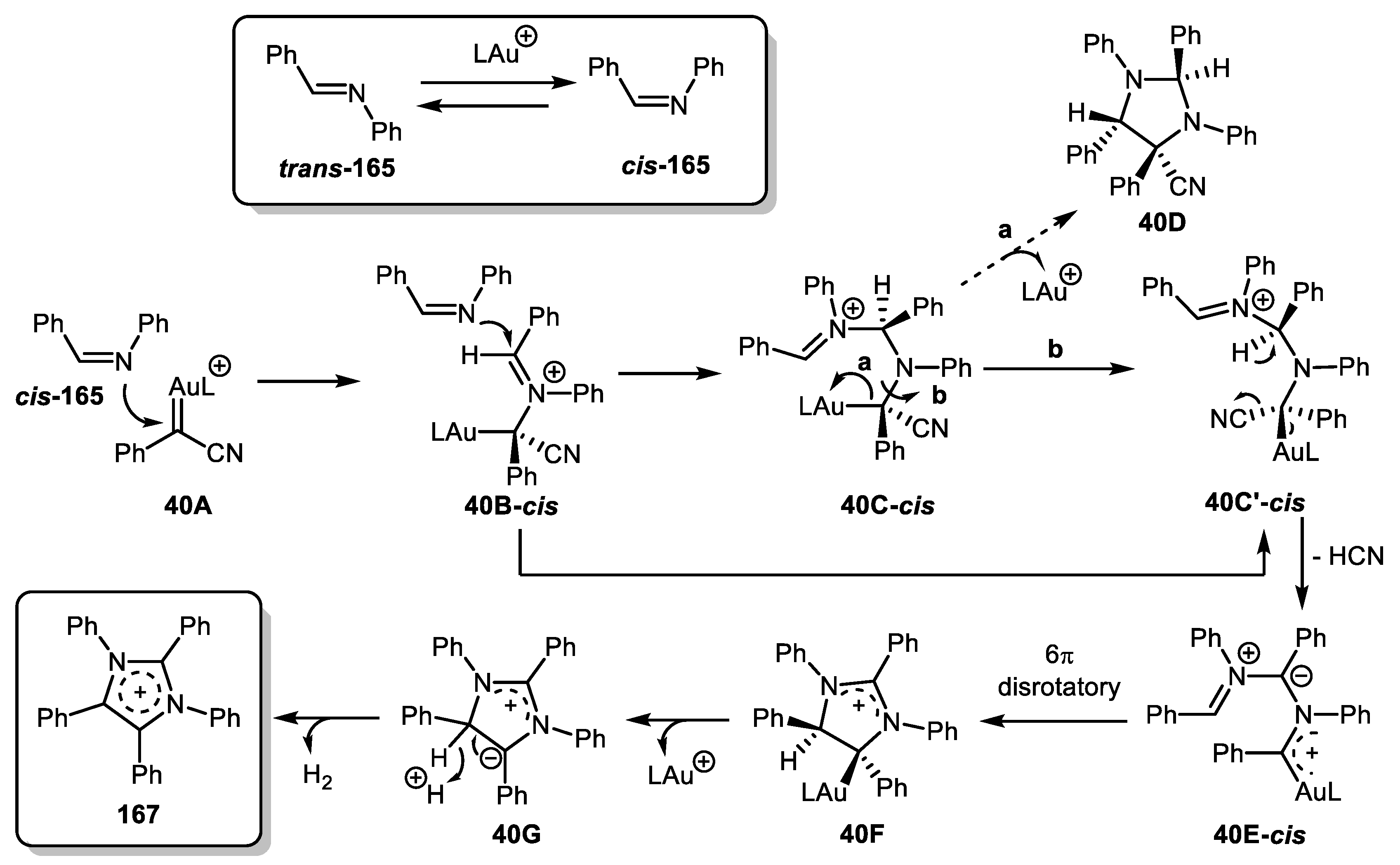
Based on DFT-calculations, the following mechanism was proposed. Imine cis-165 attacks gold carbene 40A to produce iminium intermediate 40B which is, in turn, attacked by another imine molecule to give intermediate 40C (or its conformer 40C’). The latter can evolve along two possible pathways. Path a involves the formation of imidazolidine 40D. However, this path is unlikely to occur according to DFT-calculations. In path b, 40C’ loses HCN to form a stable gold carbene 40E, which then undergoes 6π-electrocyclization in disrotatory mode. 40F is affected by a neighboring cation to release LAu+. Zwitterion 40G is subsequently oxidized to desired compound 167. It is worth mentioning that in this case, aryl diazoacetonitriles can serve as arylmethine fragment source (Scheme 40).
2.4.3. Fused Imidazoles from Diazo Compounds
The most common fused imidazole derivative being synthesized from diazo compounds is imidazo[1,2-a]pyridine. The classic approach to this heterocyclic ring is the condensation of 2-aminopyridines and α-haloketones or their synthetic equivalents [84]. The imidazo[1,2-a]pyridine-based drugs have found application in therapy of musculoskeletal, gastrointestinal, CNS disorders, cardiovascular, and infectious diseases [85]. Little information is available on the synthesis of other fused imidazole systems from diazo compounds.
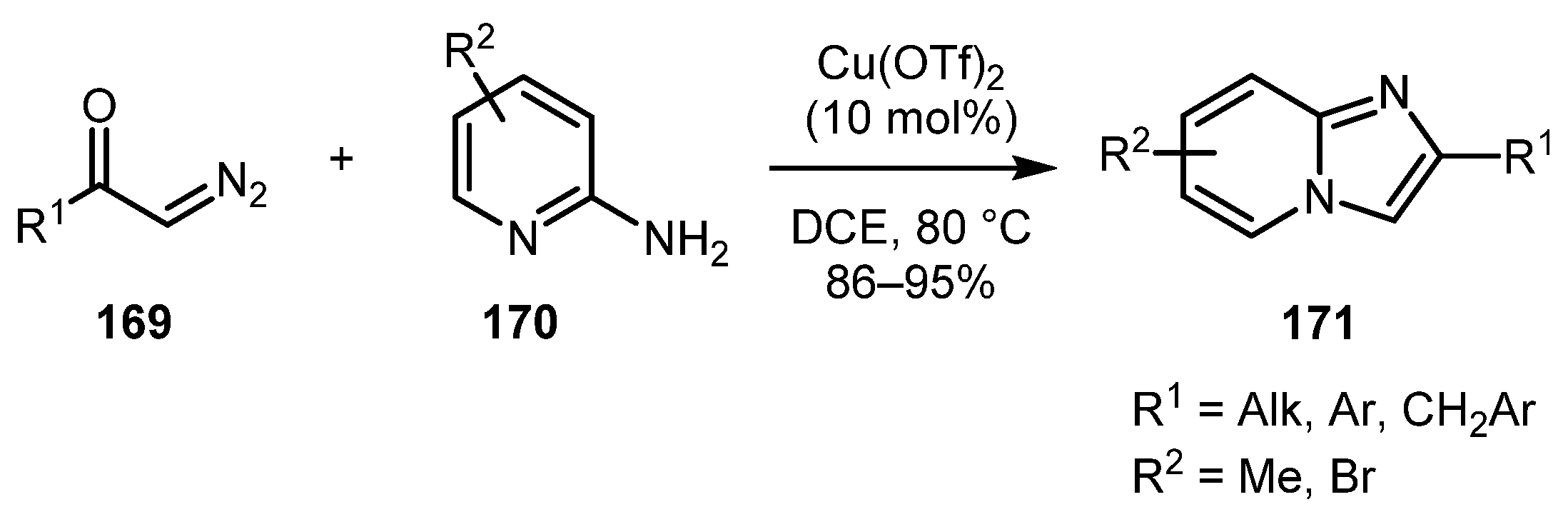
The first example of coupling of α-diazoketones 169 with 2-aminopyridines 170 was reported in 2007 by Yadav and co-workers [86]. The Cu(OTf)2-catalyzed reaction proceeded regioselectively and afforded imidazo[1,2-a]pyridines 171 in high yields. Both aromatic and aliphatic diazo ketones ere suitable partners for this reaction (Scheme 41). Later, in 2014, this method was employed towards the synthesis of glucagon-like peptide-1 receptor agonists [87].
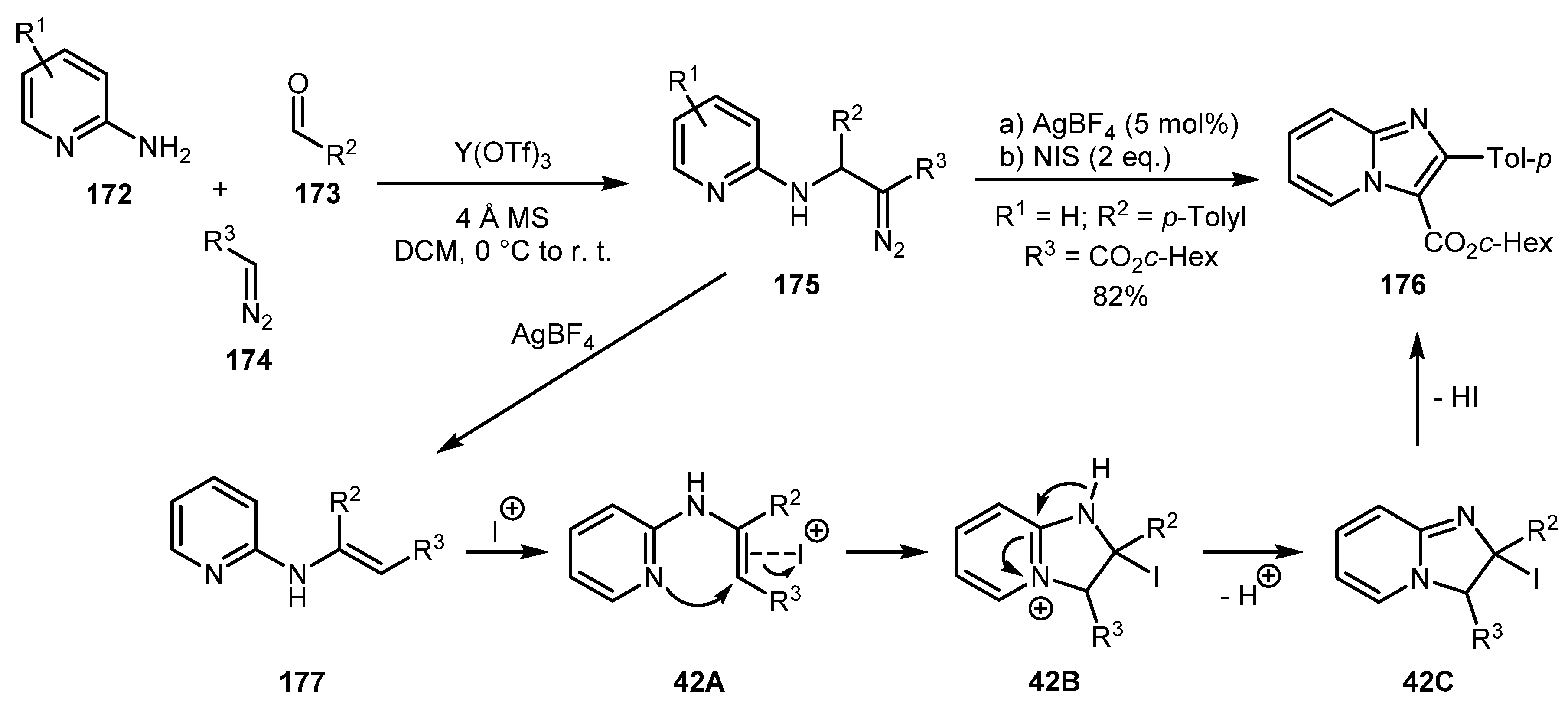
In 2013, the Gevorgyan group in their work on three-component coupling reaction of 2-aminopyridines 172, aldehydes 173, and diazo compounds 174, demonstrated that the product of this reaction 175 can be transformed to imidazo[1,2-a]pyridine 176 [88]. The reaction proceeds via intermediate 177 (formed via a AgBF4-catalyzed 1,2-hydride shift) followed by I+-mediated cyclization, tautomerization, and elimination of HI (Scheme 42).
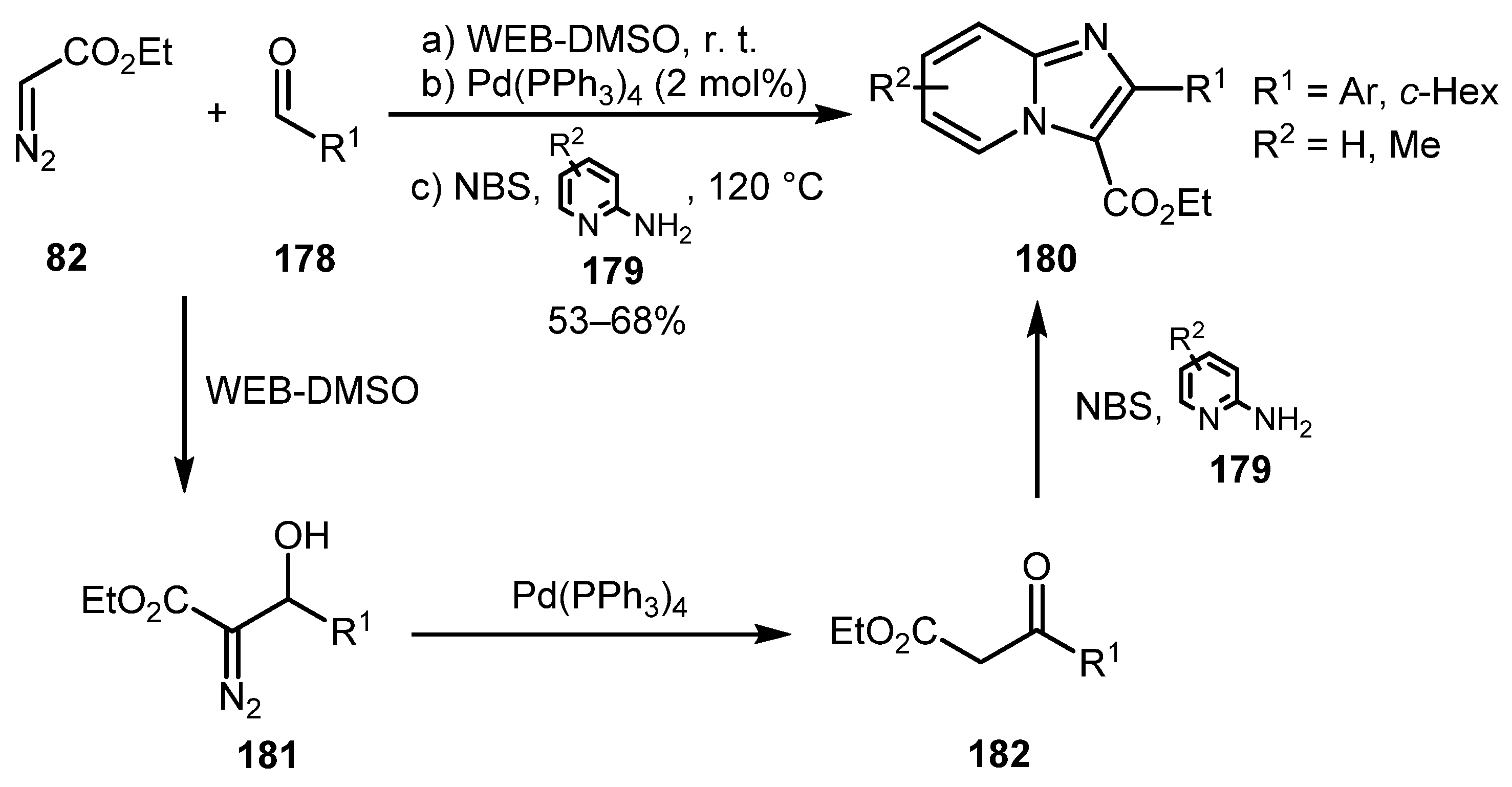
An interesting example of using a banana ash extract and dimethylsulfoxide (WEB-DMSO) as solvent cum reagent system for three-step one-pot synthesis of imidazo[1,2-a]pyridines 180 was reported by Dutta and co-authors in 2019 [89]. Initially, a WEB-DMSO-promoted aldol condensation of ethyl diazoacetate 82 and aldehyde 178 occurred to give diazo alcohol 181, which was converted to β-ketoester 182 by a Pd-catalyzed 1,2-H shift. Treatment of 182 with NBS and aminopyridine 179 on the third step afforded the corresponding imidazo[1,2-a]pyridine 180 (Scheme 43).
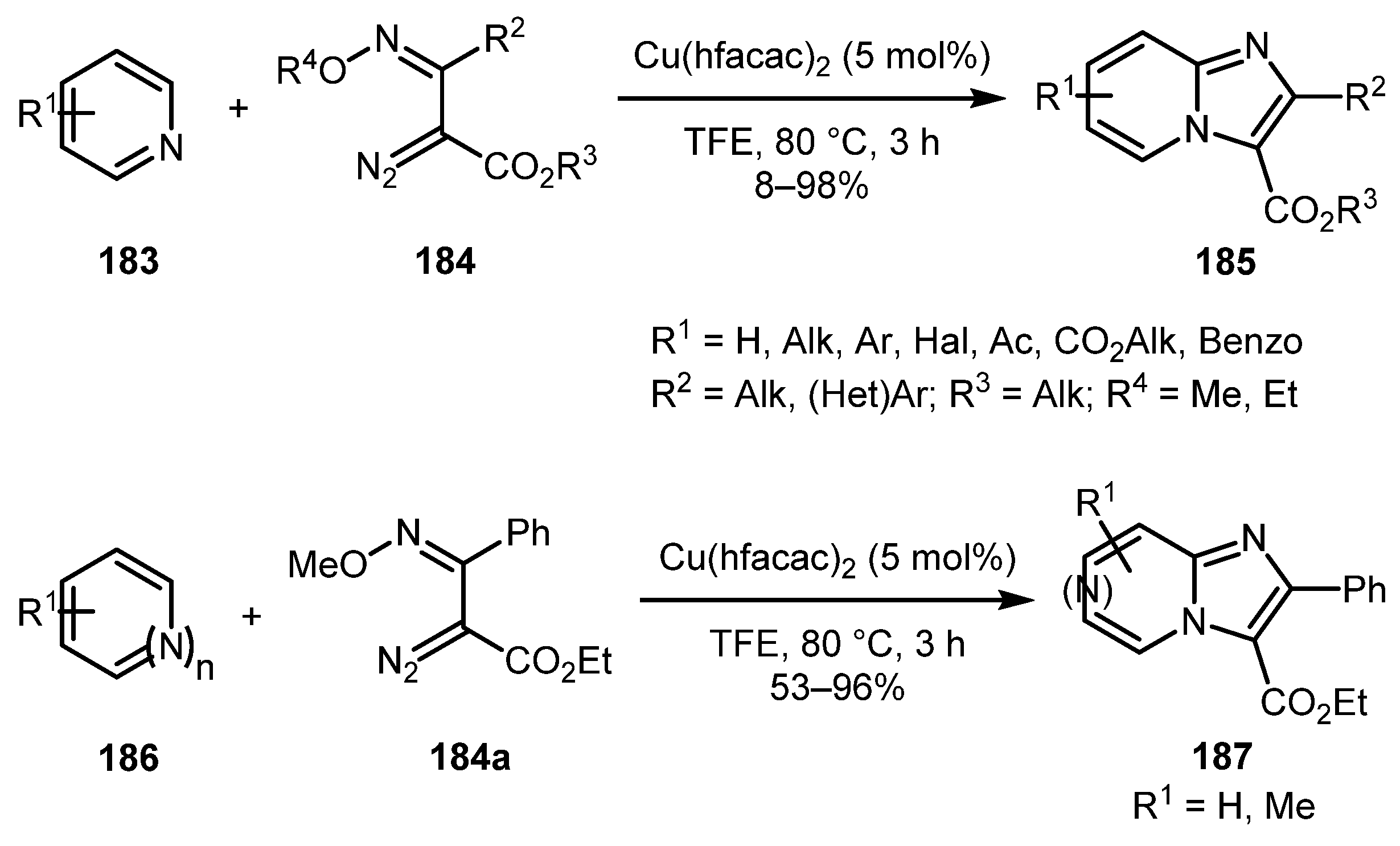
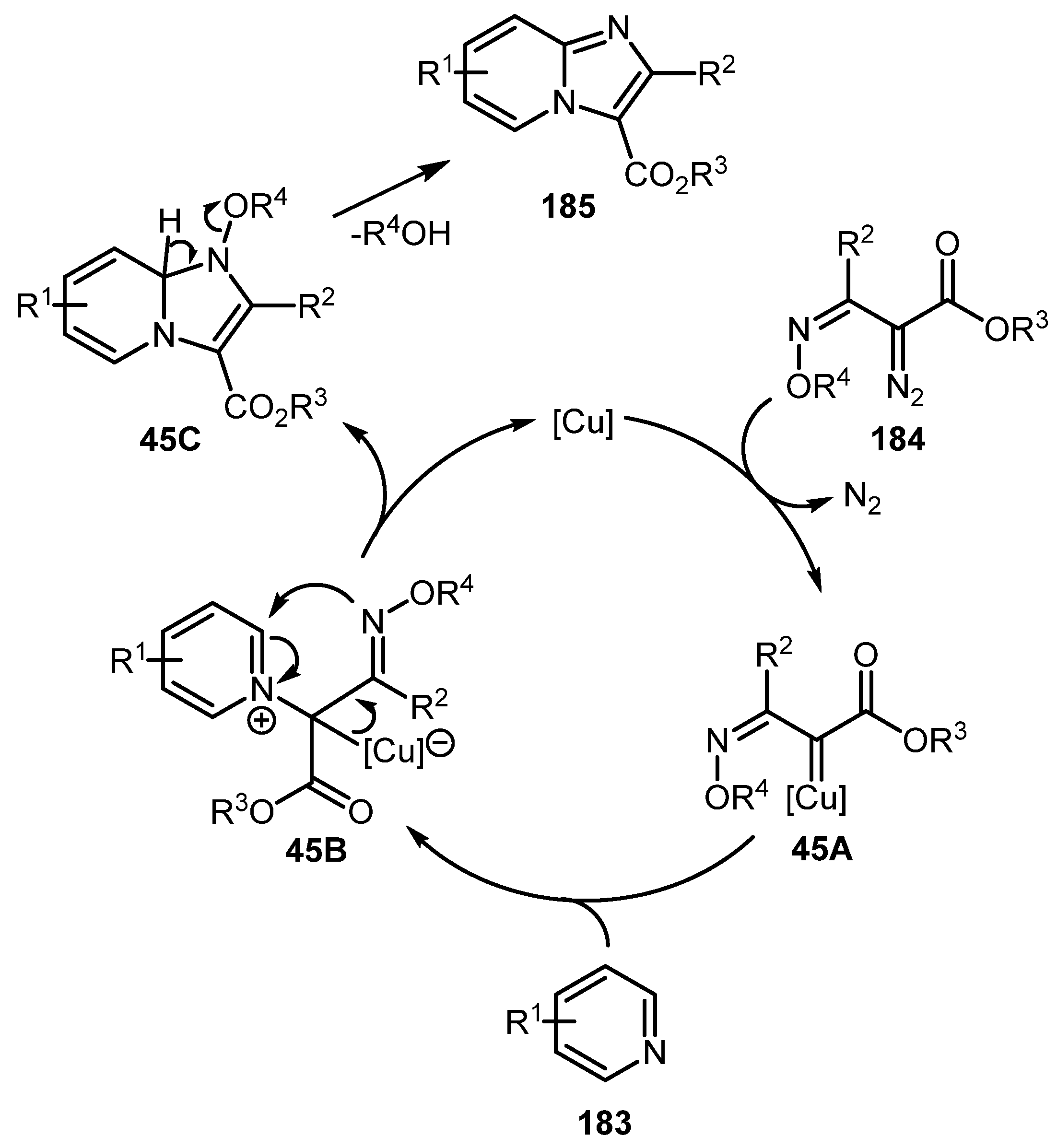
A work by the Lee group shows that imidazopyridines can be synthesized not only from aminopyridines, but also directly from pyridines 183 by a formal aza-[3+2] cycloaddition reaction with α-diazooxime ethers 184 under copper catalysis conditions [90]. The reaction works well for a broad range of substituents and also allows for preparation of other N-heterocyclic compounds such as imidazopyridazines, imidazopyrimidines, and imidazopyrazines 187 (Scheme 44). A proposed reaction mechanism begins with conversion of α-diazooxime ether 184 to Cu-carbene 45A, which is attacked by the pyridine nitrogen atom to form zwitterionic intermediate 45B. Then, 45B undergoes cyclization with regeneration of copper catalyst to give 45C, which, after elimination of alcohol, affords imidazo[1,2-a]pyridine 185 (Scheme 45).
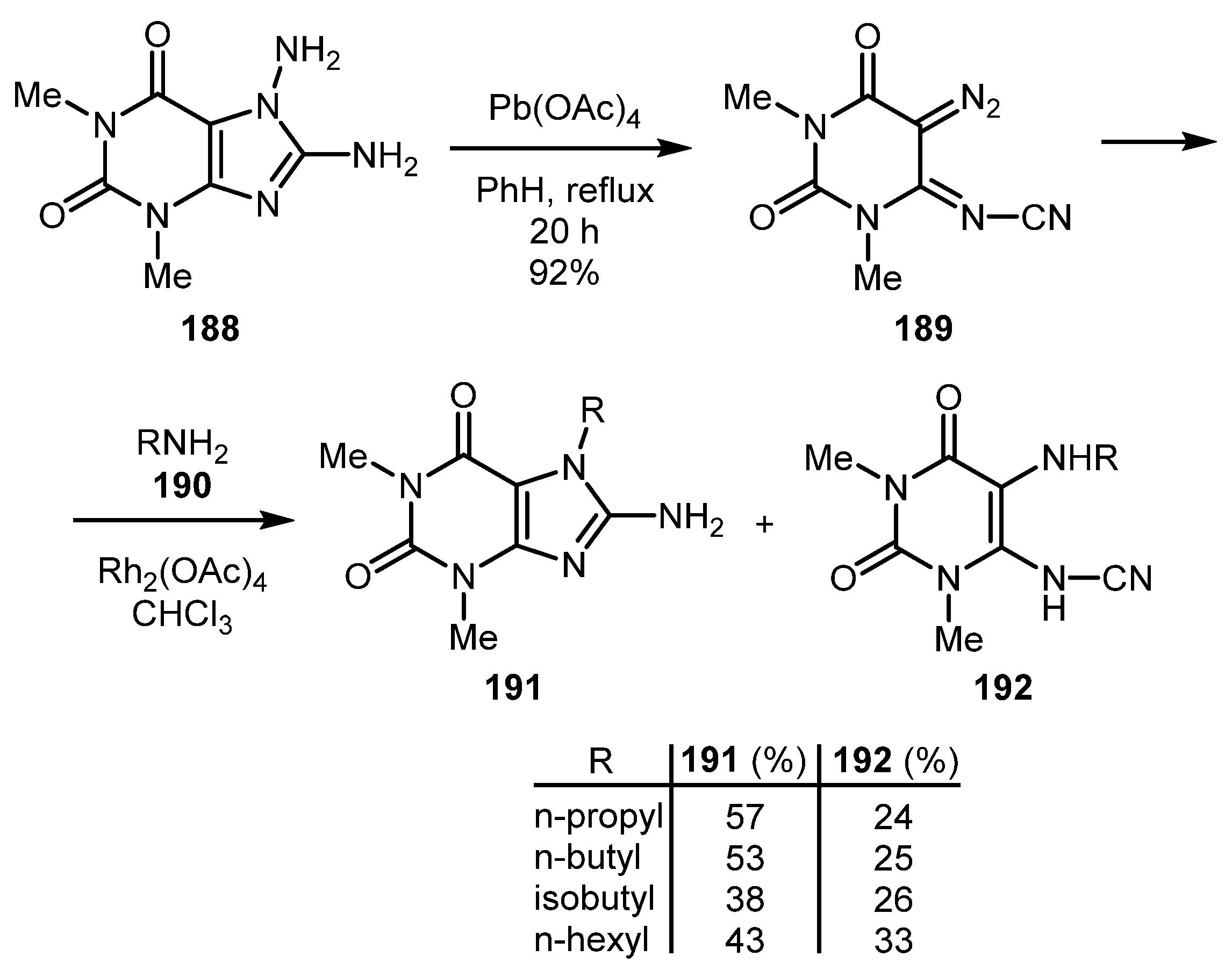
A rare example of another fused imidazole heterocycle prepared from the diazo substrate was presented in the work by Ueda and co-workers [91]. Theophyllines 191 were obtained along with cyanamides 192 from the diazobarbituric acid derivative 189 by treatment with amine 190 in the presence of Rh2(OAc)4. The imino diazo compound 189 was synthesized by oxidation of 7,8-diaminotheophyline 188 with Pb(OAc)4 (Scheme 46). Unfortunately, the method is characterized by low selectivity.
2.5. Pyrazoles (1H, 3H and 4H) and Indazoles
Pyrazole and indazole are common units in a number of synthetic products, such as tartrazine (food coloring agent), dipyrone (antipyretic and analgesic agent), sildenafil (drug used for the treatment of erectile disfunction), rimonabant (drug used to treat obesity), celecoxib (selective COX-2 inhibitors and non-steroidal anti-inflammatory drug, NSAID), fipronil (insecticide), pazopanib (tyrosine kinase inhibitor), and benzydamide (locally acting NSAID). Other biological activities of pyrazole- and indazole-containing compounds include antimicrobial, antihyperglycemic, pesticidal, leishmanicidal, antichagasic, anti-HIV, anti-depressant, and antiarrhythmic [92,93,94,95]. Among methods for the synthesis of pyrazoles diazo strategies are widely used. One commonly known approach is the 1,3-dipolar cycloaddition between diazo compounds and alkynes, or alkenes with the leaving group [96,97], including deacylative variant [14,15]. Another route is an intramolecular cyclization of vinyldiazo compounds [98]. Moreover, to date, a number of other methods involving diazo compounds have been developed.
2.5.1. 1H-Pyrazoles
Synthesis of 1H-Pyrazoles by 1,5-Electrocyclization of Vinyldiazo Compounds
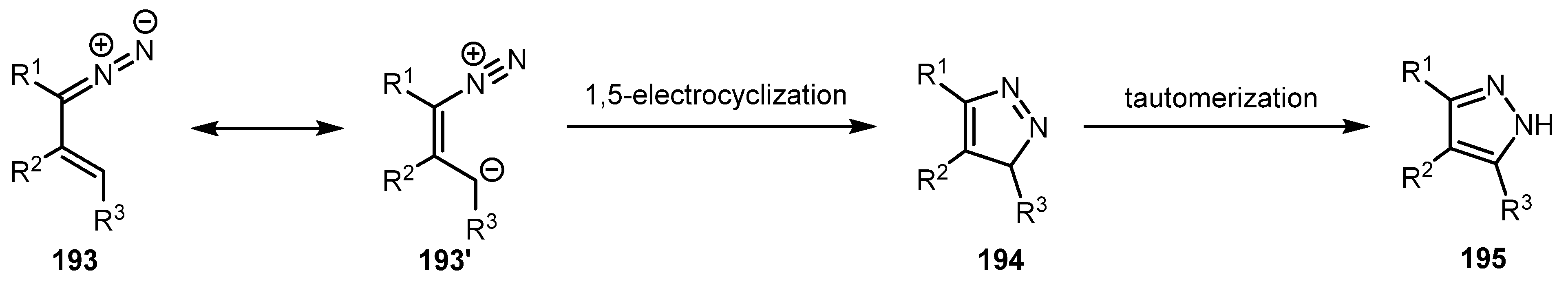
A mechanism of this transformation is depicted in Scheme 47. It involves 1,5-electrocyclization of the vinyldiazo compound 193 with subsequent tautomerization to yield 1H-pyrazole 195 [99].
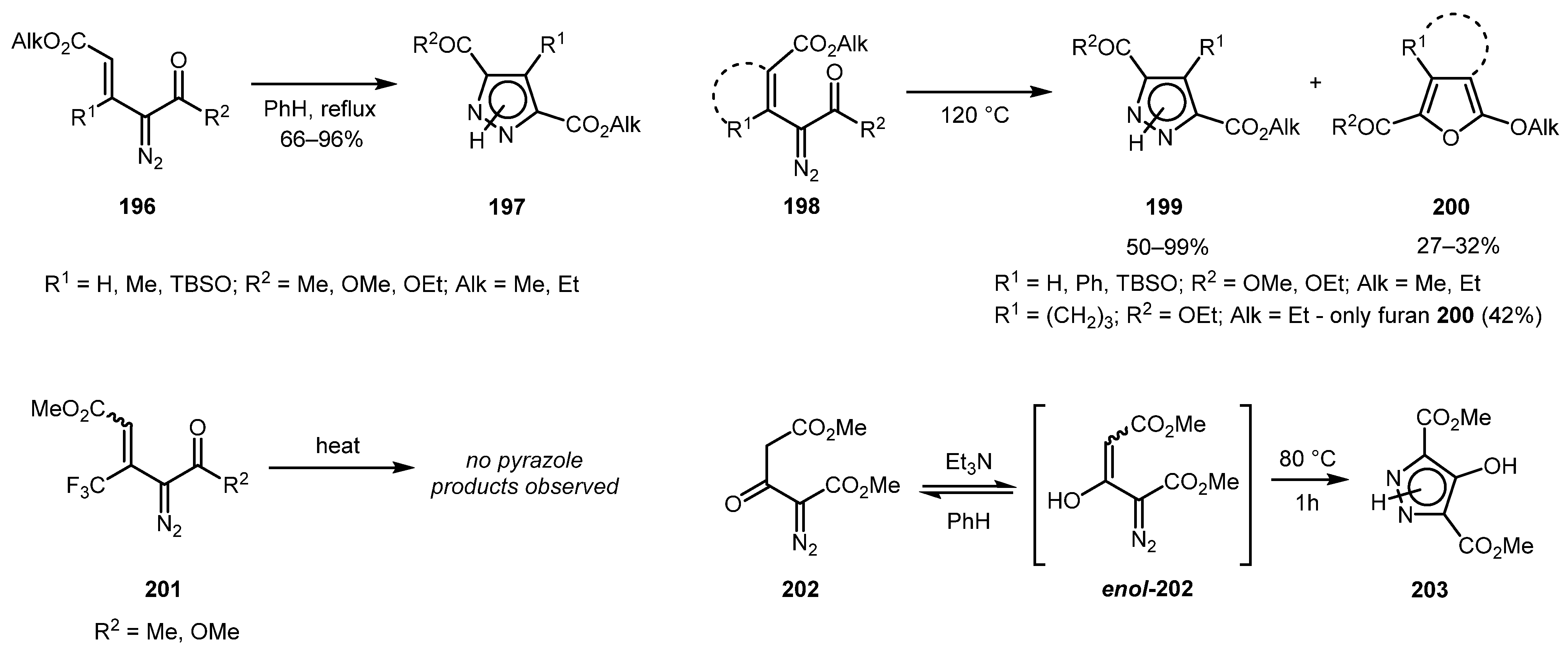
In the works by the Nikolaev group, it was shown that configuration of double bond in vinyldiazo compounds significantly affects their reactivity in this reaction (Scheme 48) [100,101]. trans-Vinyl diazo compounds 196 readily underwent electrocyclization under moderate temperature (80 °C), while their cis-counterparts 198 produced pyrazoles 199 only at elevated temperatures (up to 120 °C) or decomposed with formation of furans 200. Moreover, the authors demonstrated that fluoroalkyl-substituted vinyldiazo carbonyls 201 (both trans and cis) failed to give any pyrazole products, furnishing only carbene-derived products on heating. Interestingly, the diazo enol enol-202 formed in situ can also undergo this transformation, affording 4-hydroxypyrazole 203.
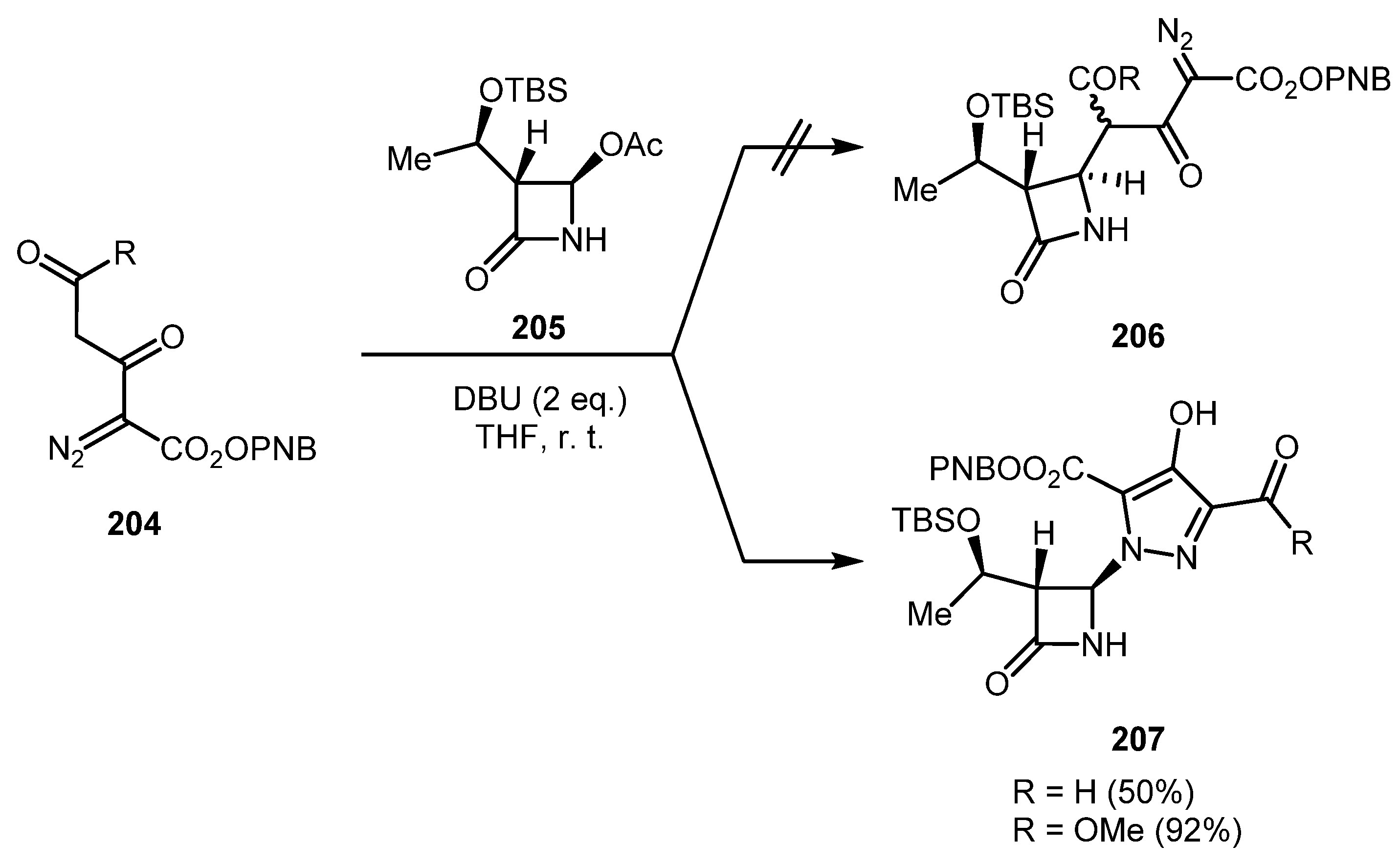
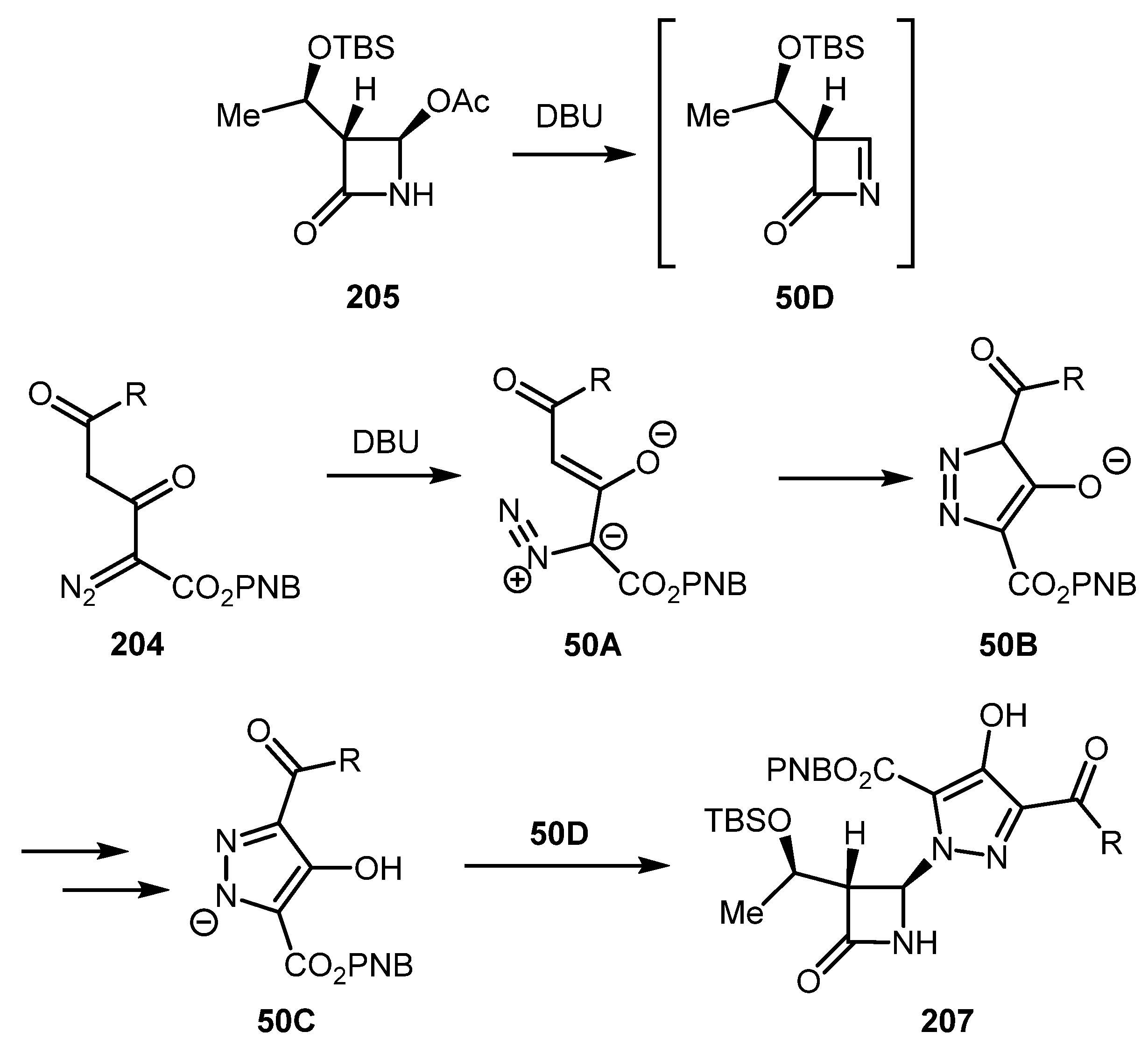
An attempt to prepare carbapenem precursor 206 by treatment of diazo carbonyls 204 with DBU and β-lactam 205 unexpectedly led to the formation of hydroxypyrazoles 207 (Scheme 49) [102]. From the mechanism standpoint, the 1,5-electrocyclization of enolate 50A likely gave pyrazolium anion 50C, which reacted with imine 50D, which was, in turn, formed by DBU-promoted elimination of acetic acid from β-lactam 205 (Scheme 50). The direct regiospecific synthesis of N-arylpyrazoles by the co-catalyzed reaction of vinyl diazo carbonyls and aryl diazonium salts has also been reported [103].
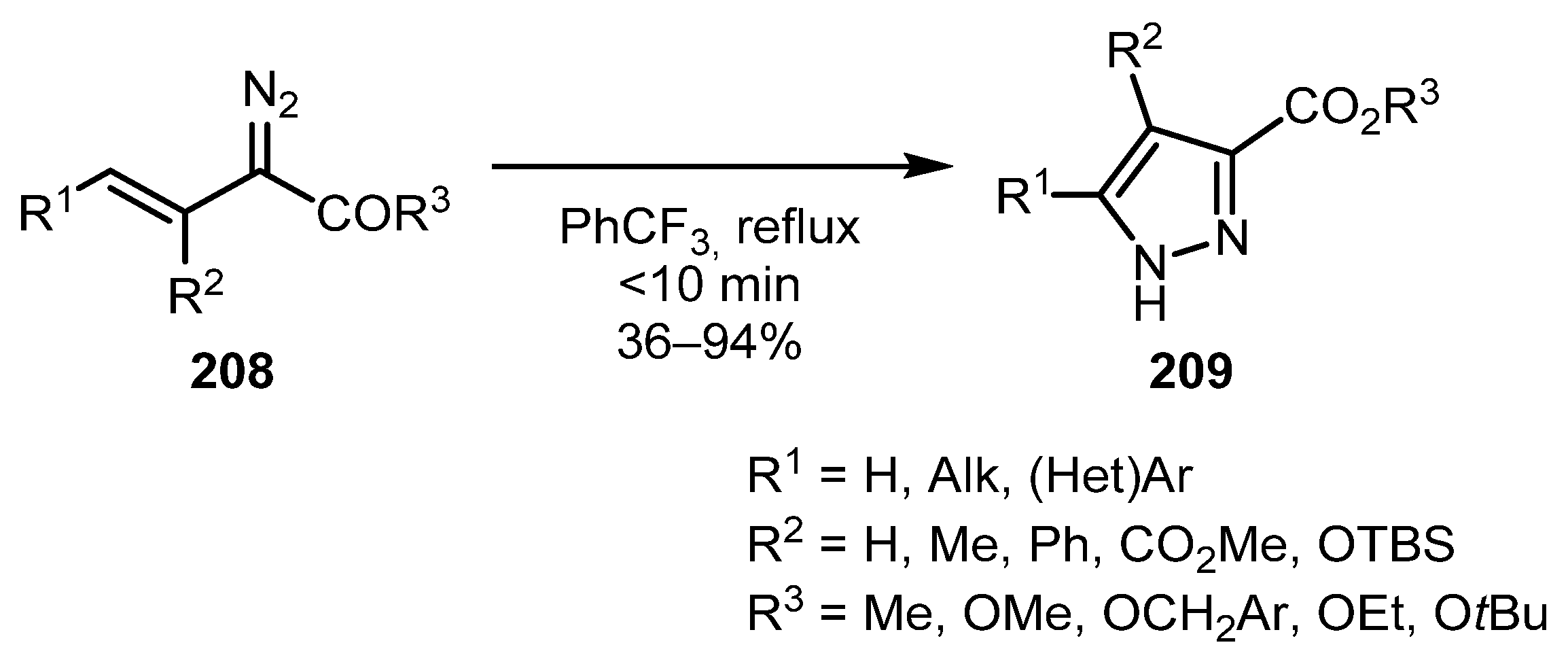
The synthesis of mono-, di-, and tri-substituted pyrazoles 209 from vinyldiazo compounds 208 was recently reported by Dikermann and co-workers (Scheme 51) [104]. The reaction proceeds smoothly on heating in trifluoromethylbenzene affording desired products in yields of up to 94%. The synthesis of 5-arylsubstituted pyrazoles by the one-pot diazo transfer-cyclization approach has also been reported [105]. Separate examples of cyclization of vinyldiazo compounds to pyrazoles are presented in a number of works by other authors [106,107,108,109,110,111].
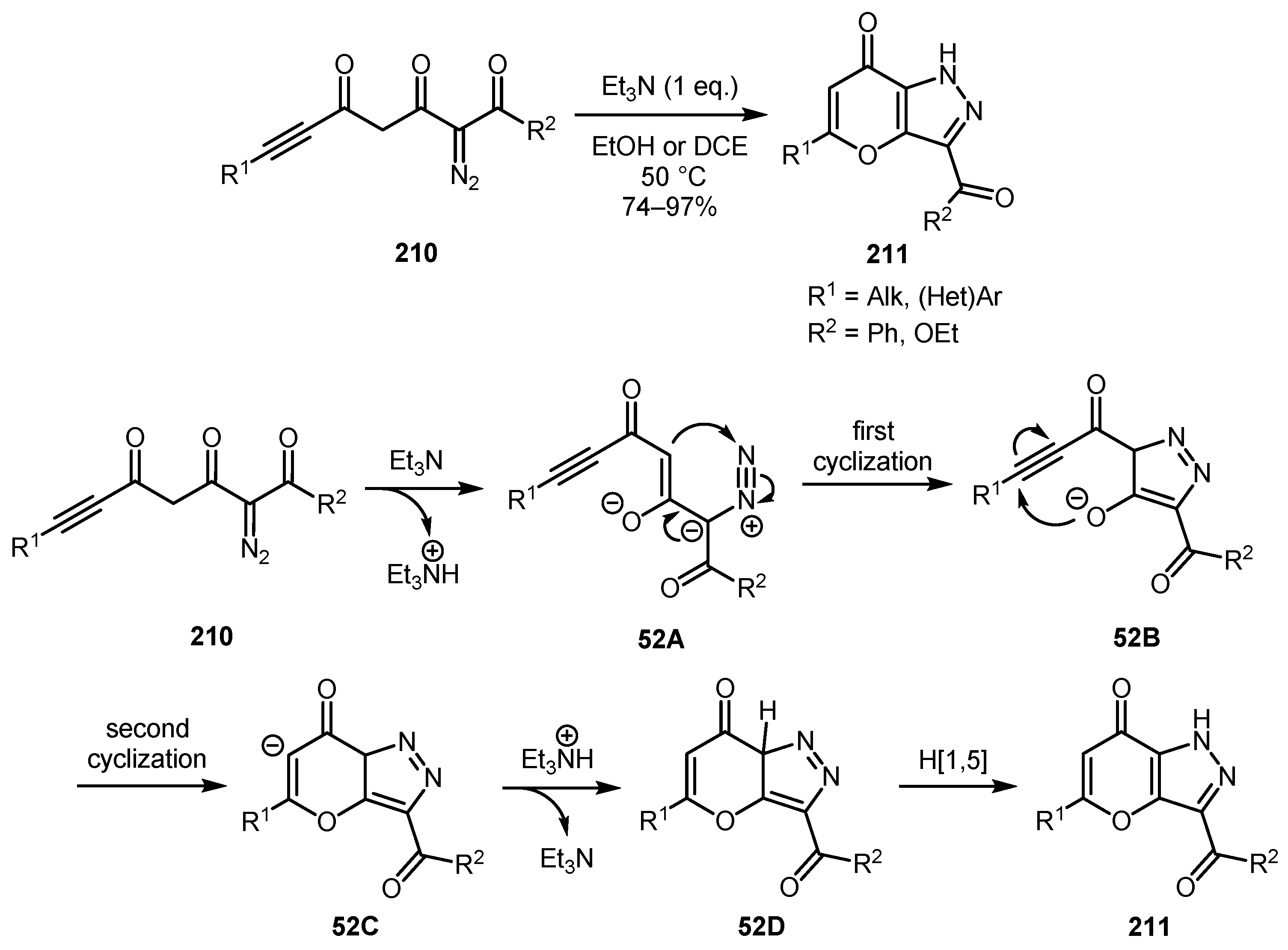
A cascade electrocyclization-Michael addition process for the synthesis of pyrano[3,2-c]pyrazol-7(1H)-ones 211 from 2-diazo-3,5-dioxo-6-ynoates (ynones) 210 was developed by Deng and co-workers [112]. The reaction tolerates a broad range of substituents giving generally excellent yields of annulated pyrazoles. Apparently, the reaction begins with the formation of enolate 52A, which undergoes a 6-π electrocyclic ring closure to produce 3H-pyrazole intermediate 52B. The following Michael addition of enolate oxygen to triple bond provides carbanion 52C. Subsequent protonation and [1,5]-H shift affords the desired product 211 (Scheme 52).
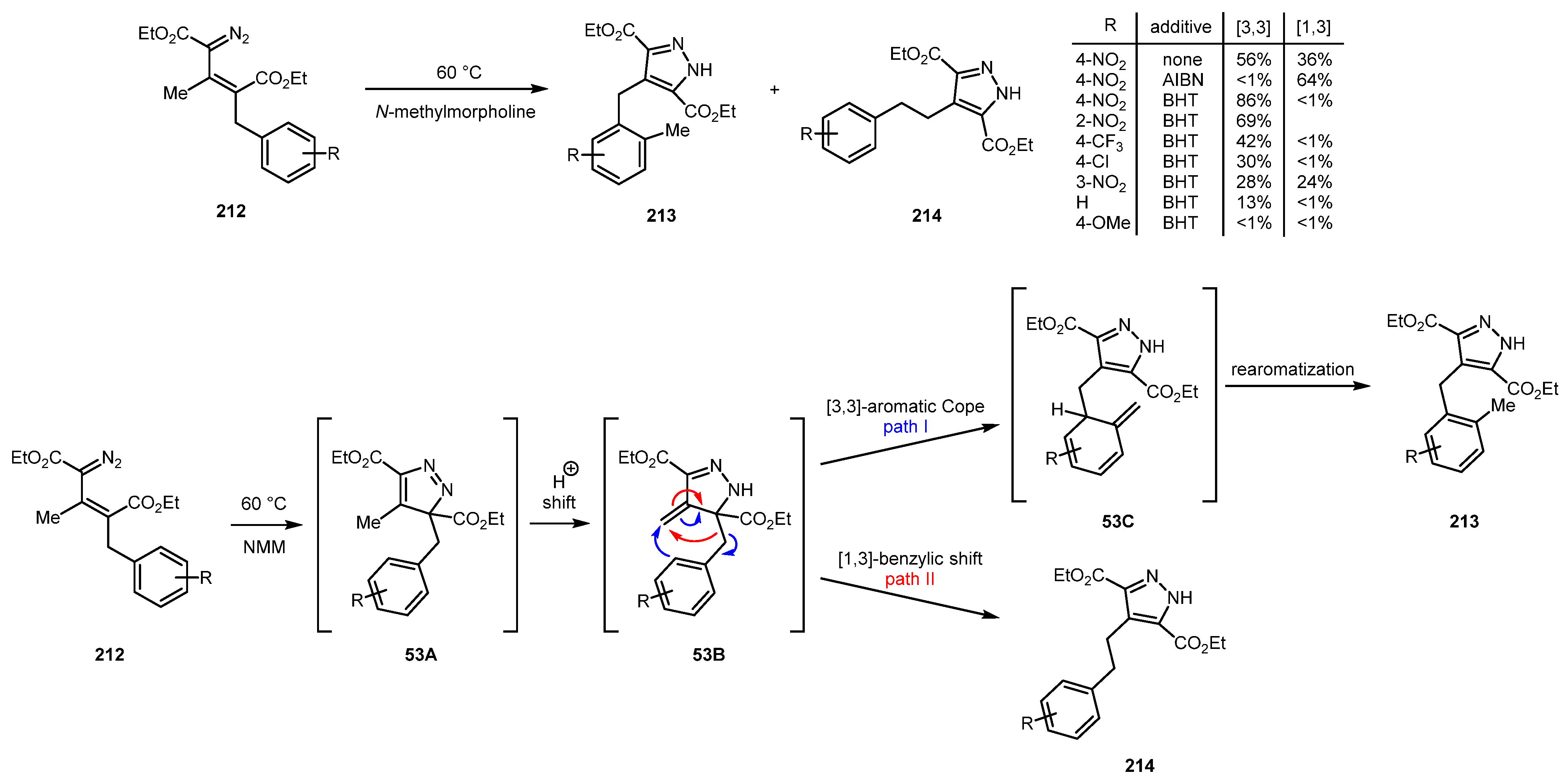
Electrocyclization of vinyl diazo compounds 212 can trigger subsequent rearrangements, such as the Cope rearrangement and 1,3-sigmatropic shift as was discovered by Babinski and co-workers [113,114]. The initially formed 3H-pyrazole 53A (although it can undergo the van Alphen-Hüttel rearrangement to become aromatic) in this case undergoes proton shift to give intermediate 53B. The latter has two pathways to transform itself to aromatic pyrazole. In path I, a rare [3,3]-aromatic Cope rearrangement can occur, followed by re-aromatization to afford pyrazole 213. In path II, intermediate 53B can suffer a 1,3-alkyl shift to give pyrazole 214 (Scheme 53) [114]. Path II presents a radical reaction, so it can be facilitated by addition of radical initiator (such as AIBN), whereas path I is favored in presence of radical inhibitor (such as BHT). Although the scope of this tunable reaction is limited to benzyls with electron-withdrawing groups in positions 2 and 4, it gives access to two types of fully substituted pyrazoles 213 and 214.
Miscellaneous Methods for Pyrazole Ring Construction from Vinyl Diazo Compounds
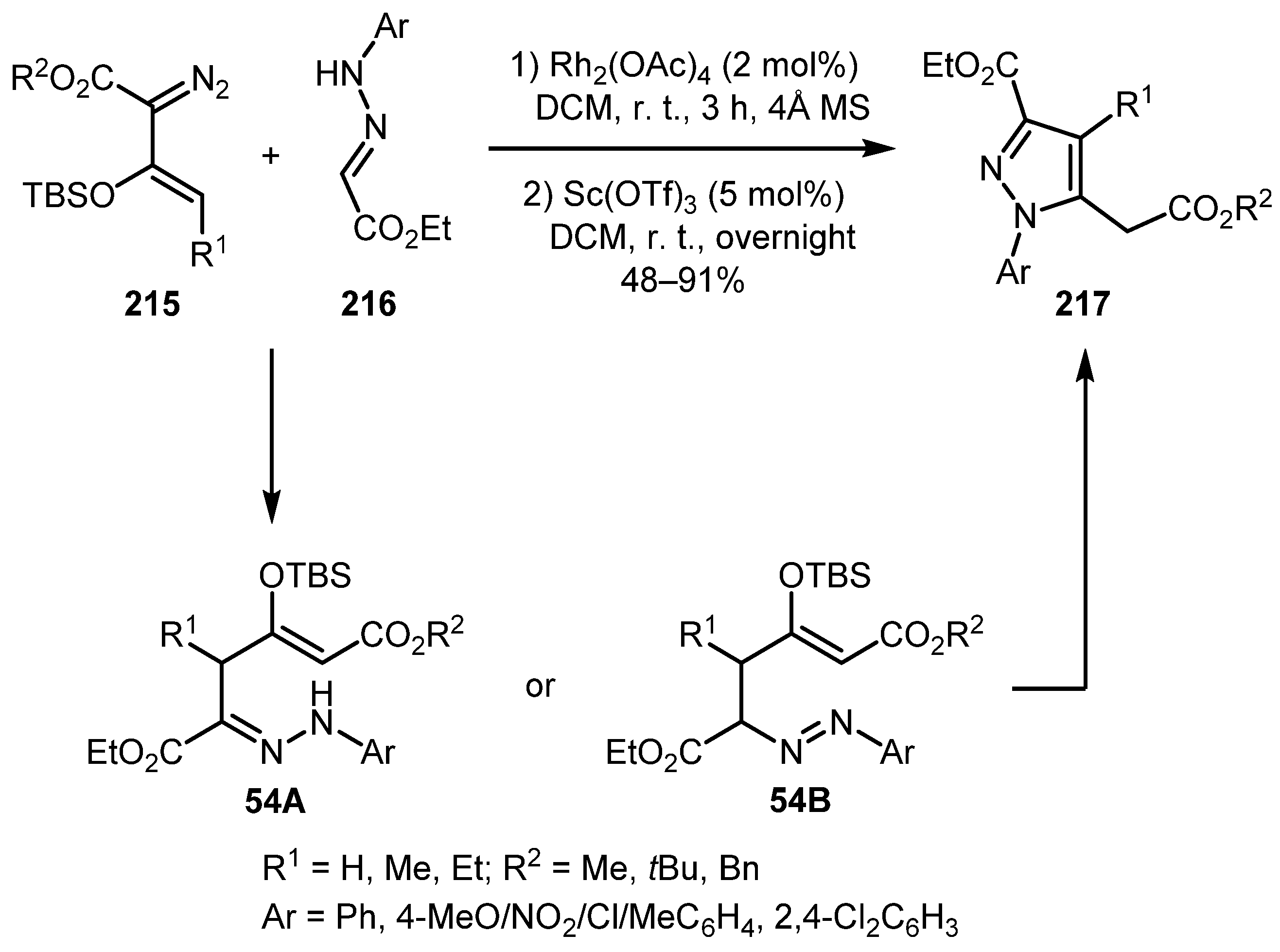
In 2013, the Doyle group devised a regiospecific one-pot, two-step method for the synthesis of pyrazoles 217 via the rhodium-catalyzed vinylogous addition of donor-acceptor hydrazones 216 to enol diazo acetates 215 followed by Lewis acid catalyzed cyclization (Scheme 54) [115]. Numerous substituents were tolerated, although in the case of substituents other than hydrogen in the vinylogous position (R1), the yields were lower.
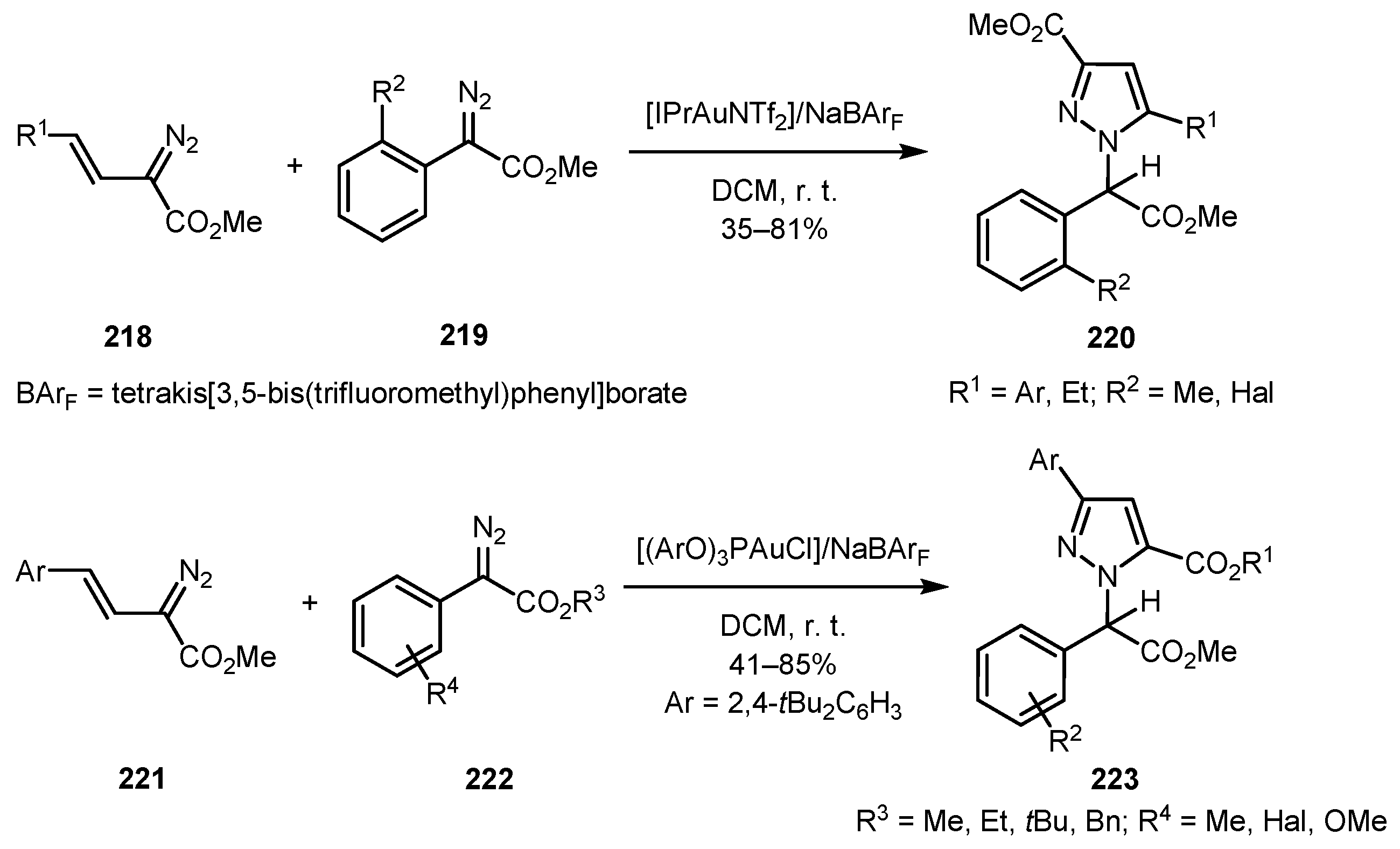
An unprecedented chemo- and regioselective gold-catalyzed cross-coupling of vinyl diazo compounds 218/221 and aryl diazo acetates 219/222 was described by Xu and co-workers [116]. The ligand-controlled reaction gave pyrazoles 220 and 223 in a position-switchable mode. If [IPrAuNTf2] was used as the catalyst, the products were pyrazoles 220 with the N-substituent adjacent to the aryl (or ethyl) group, while with [(ArO)3PAuCl] as the catalyst, the regioselectivity was reversed and the products were pyrazoles 223. For the first mode of the reaction, the presence of an ortho-substituent in the aryl moiety of aryl diazo acetates 219 was essential, whereas the second mode tolerated substituents in all positions of the phenyl ring of 222 (Scheme 55).
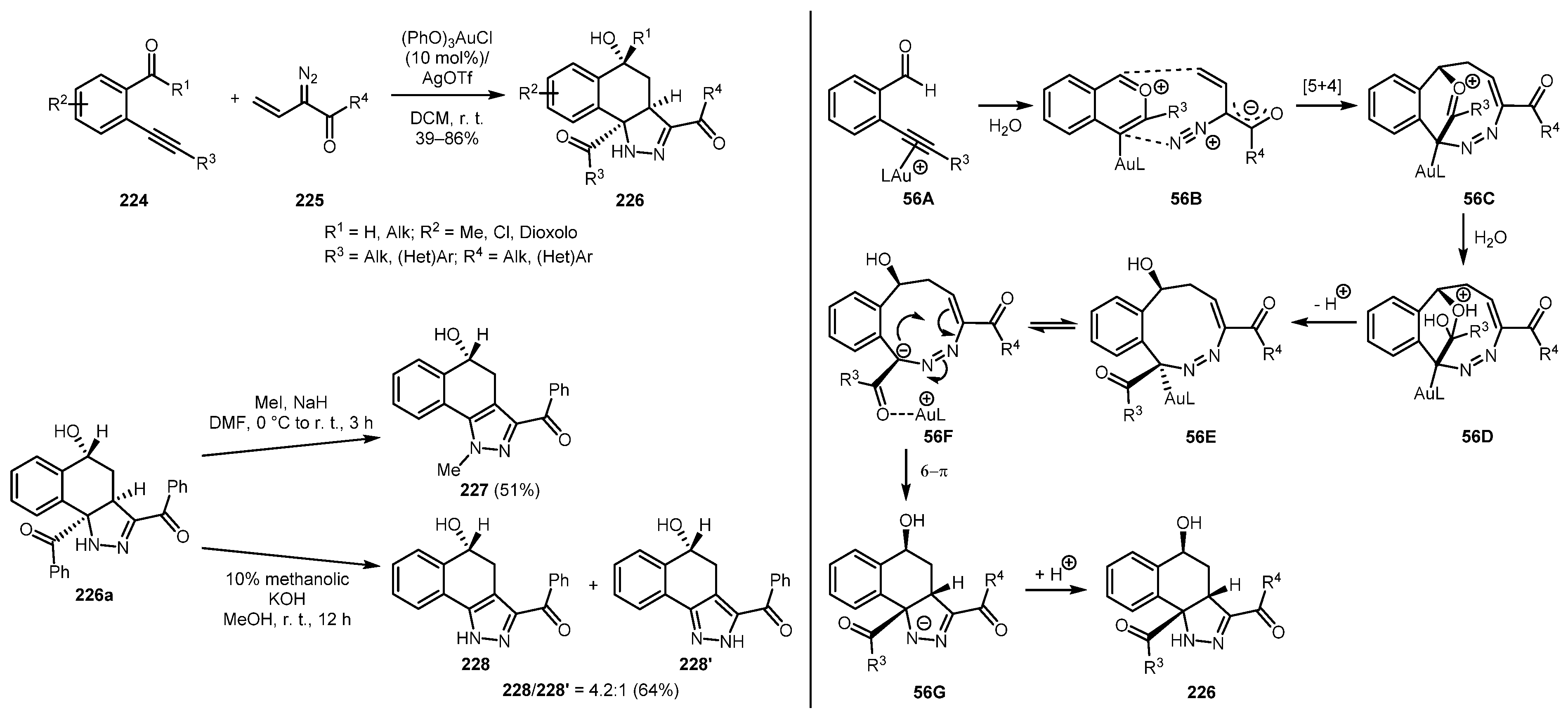
Another example of gold-catalyzed reaction of vinyl diazo compounds for the synthesis of pyrazole derivatives was described in 2019 by Raj and Liu [117]. The procedure involving [5+4]-annulation between 2-alkynyl-1-carbonylbenzenes 224 and vinyl diazo ketones 225 afforded 4,5-dihydro-benzo[g]indazoles 226 in generally good yields and with high diastereoselectivity. These initial products can be further converted to aromatic pyrazoles 227 and 228/228’. The possible reaction mechanism likely involves the gold-catalyzed formation of benzopyrilium cation 56B followed by its [5+4]-cycloaddition with vinyldiazo ketone 225 to yield intermediate 56C. The latter can be hydrolyzed to give intermediate 56E, which then undergoes a disrotatory 6π-electrocyclization with subsequent diastereoselective protonation leading to the observed pyrazoline 226 (Scheme 56).
Synthesis of 1H-Pyrazoles from Diazo Compounds and Alkynes
In this section, only selected examples are considered that have not been reviewed previously.
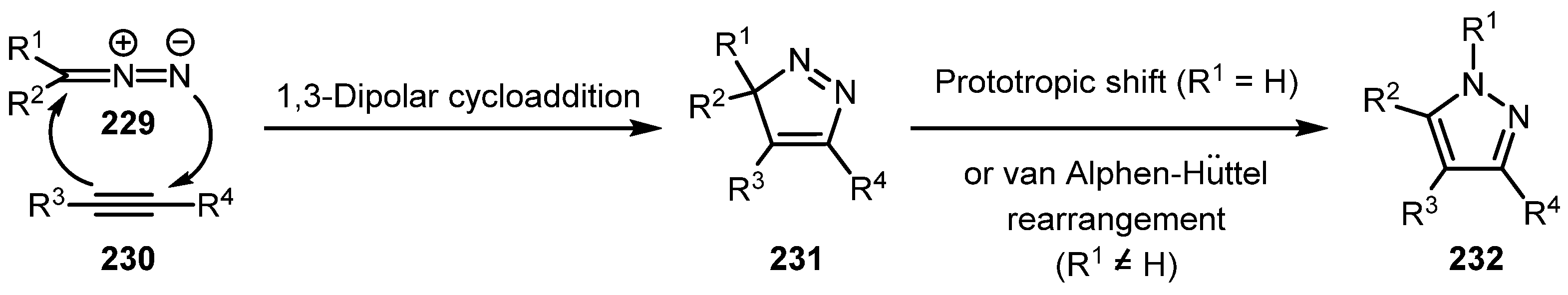
In the classic 1,3-dipolar cycloaddition of diazo compound 229 to alkyne 230, 3H-pyrazole 231 is initially formed and, in some cases, can be isolated (Section 2.5.2). However, it usually undergoes a prototropic shift or the van Alphen–Hüttel [118,119] rearrangement to produce 1H-pyrazole 232 (Scheme 57) [120].
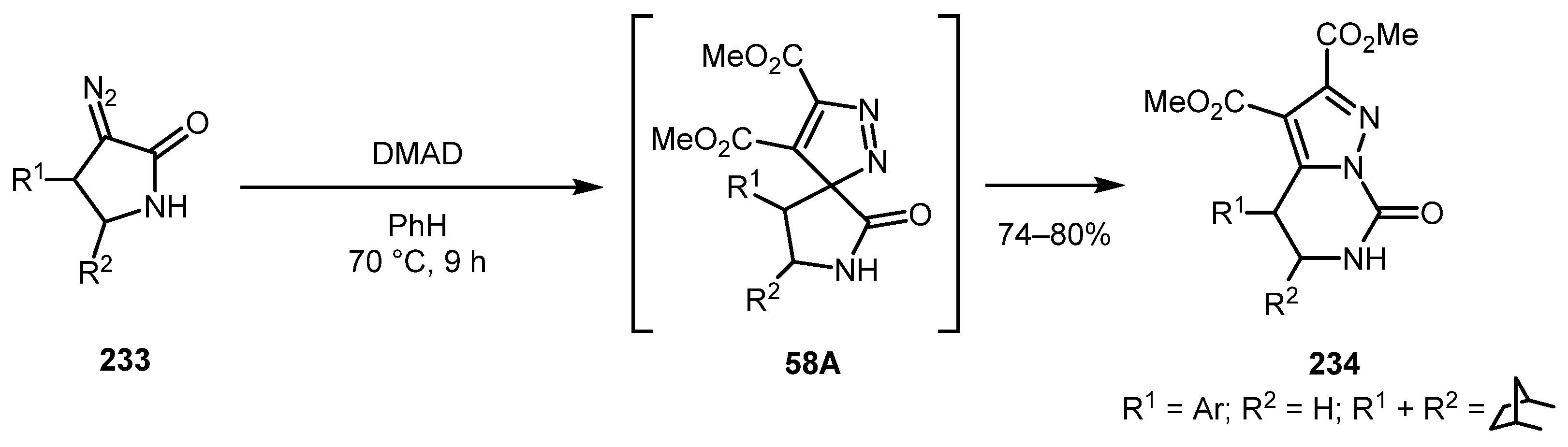
In 2015, Sultanova and co-authors reported an efficient synthesis of 7-oxo-4,5,6,7-tetrahydropyrazolo[1,5-c]pyrimidines 234 by reaction of 3-diazopyrrolidones 233 with dimethyl acetylenedicarboxylate (DMAD) [121]. The reaction is believed to proceed through the formation of a spirocyclic 3H-pyrazole intermediate 58A via 1,3-dipolar cycloaddition with subsequent ring expansion via the van Alphen–Hüttel rearrangement (Scheme 58).
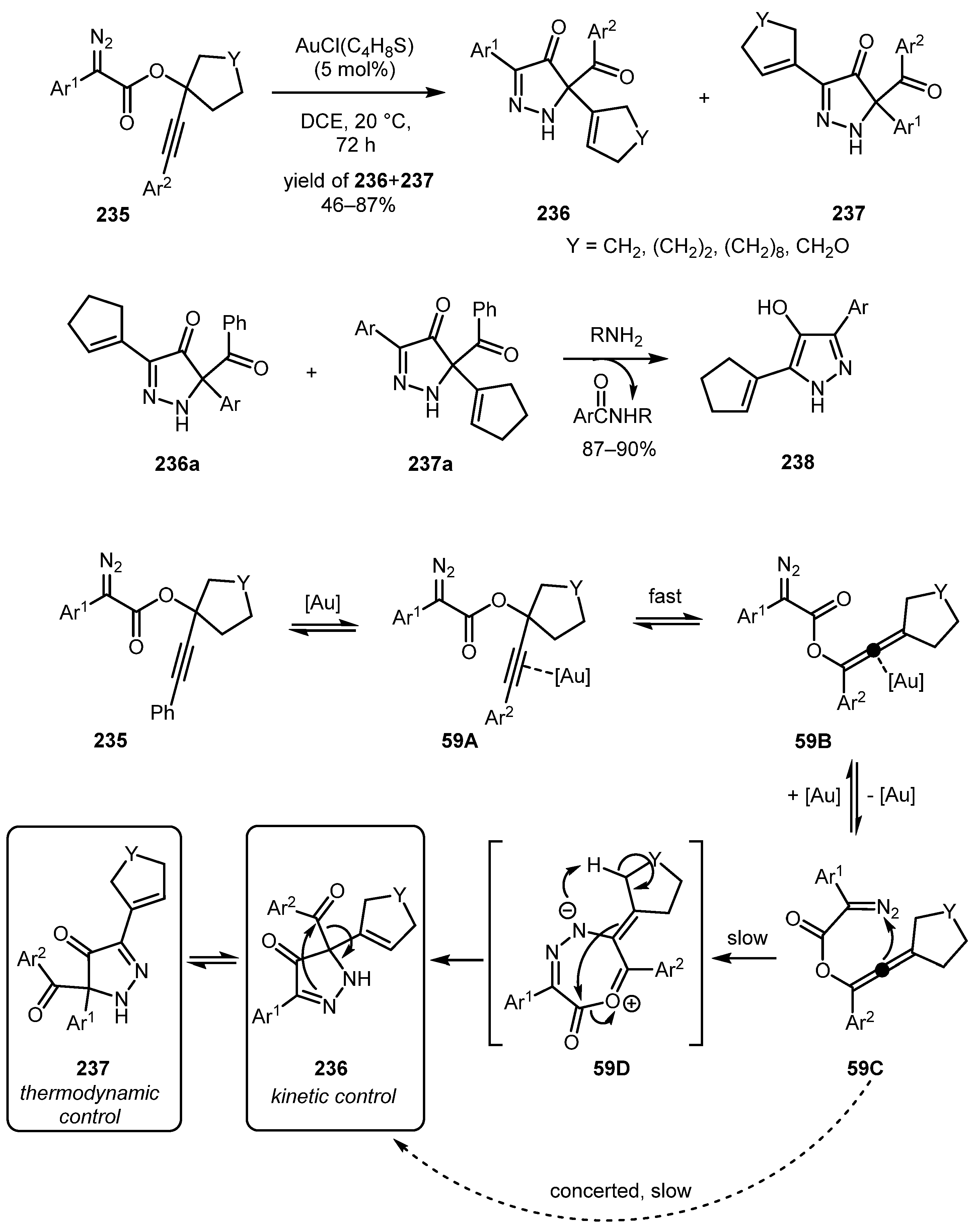
An interesting rearrangement of aryl propargyl α-aryl diazo acetates 235 to 1,5-dihydro-4H-pyrazol-4-ones 236 and 237 was reported by the Doyle group in 2016 [122]. The gold-catalyzed 1,3-acyloxy rearrangement of 235 likely gives allene species 59A, which may exist in equilibrium with 235. Intermediate 59A undergoes rearrangement to produce dihydropyrazolone 236 directly in a concerted fashion or stepwise through intermediate 59D. Under the reaction conditions, compound 236 is in equilibrium with 237, via 1,3-acyl migration. At room temperature, the ratio of 236 to 237 is generally about 3:1. It is worth mentioning that these dihydropyrazolones can act as acyl transfer reagent, and when reacted with amine, afforded 4-hydroxypyrazoles 238 (Scheme 59).
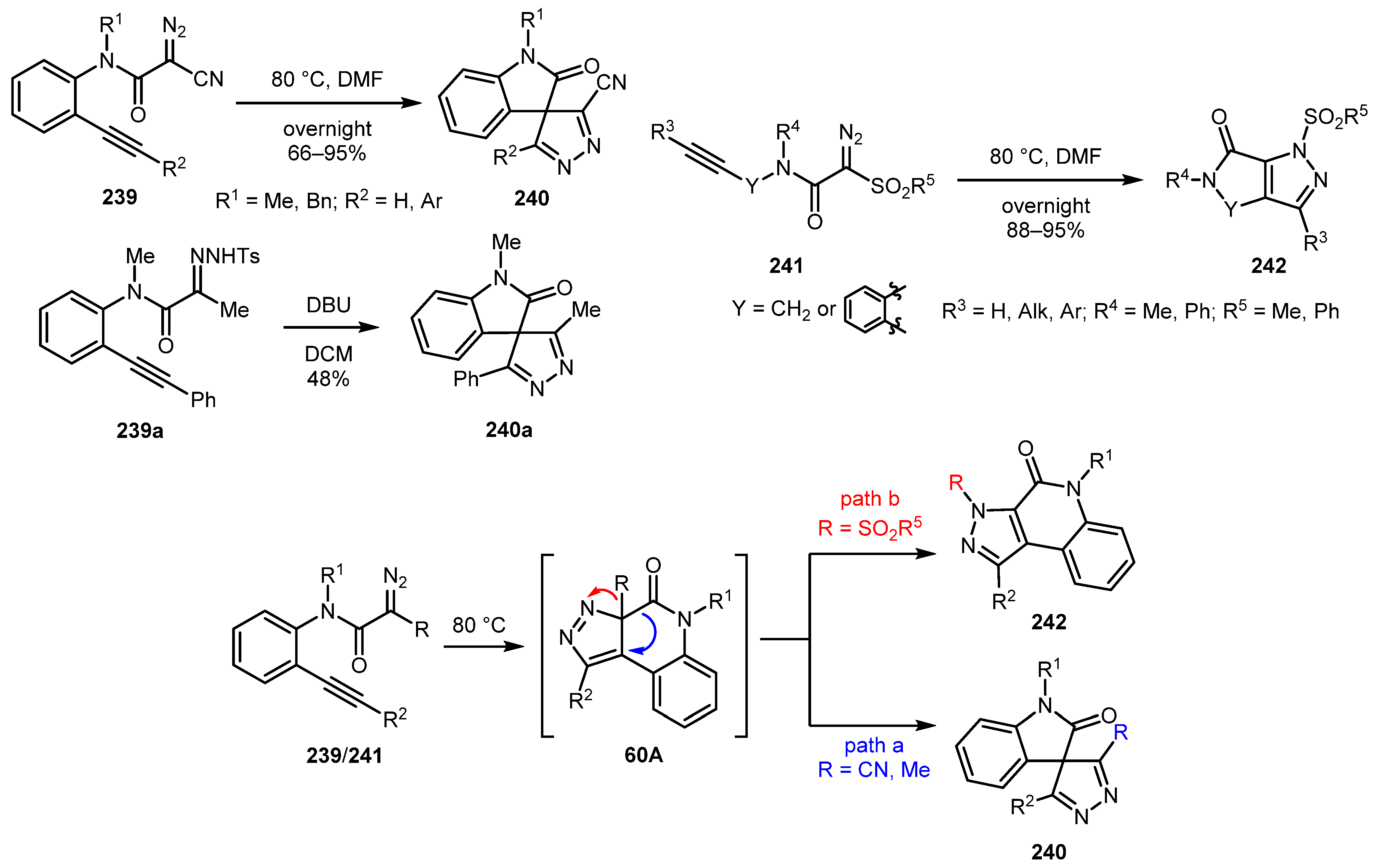
A rare example of synthesis of 4H-pyrazoles was reported in 2018 by Zhang and co-workers (Scheme 60) [123]. An intramolecular 1,3-dipolar cycloaddition of alkyne-tethered α-cyano diazoamides 239 with subsequent 1,5-carbonyl migration provided spiro-4H-pyrazole-oxindoles 240 in good to excellent yields (Scheme 60, path a). The reaction worked well even with terminal alkyne. α-Methyl diazoamide 239a also gave the corresponding 4H-pyrazole 240a, albeit in diminished yield. In addition, α-sulfonyl diazo compounds 241 were employed in the reaction but in this case, the product was 1H-pyrazole 242 resulting from 1,5-sulfonyl migration to the N-atom in the 3H-pyrazole intermediate 60A (Scheme 60, path b).
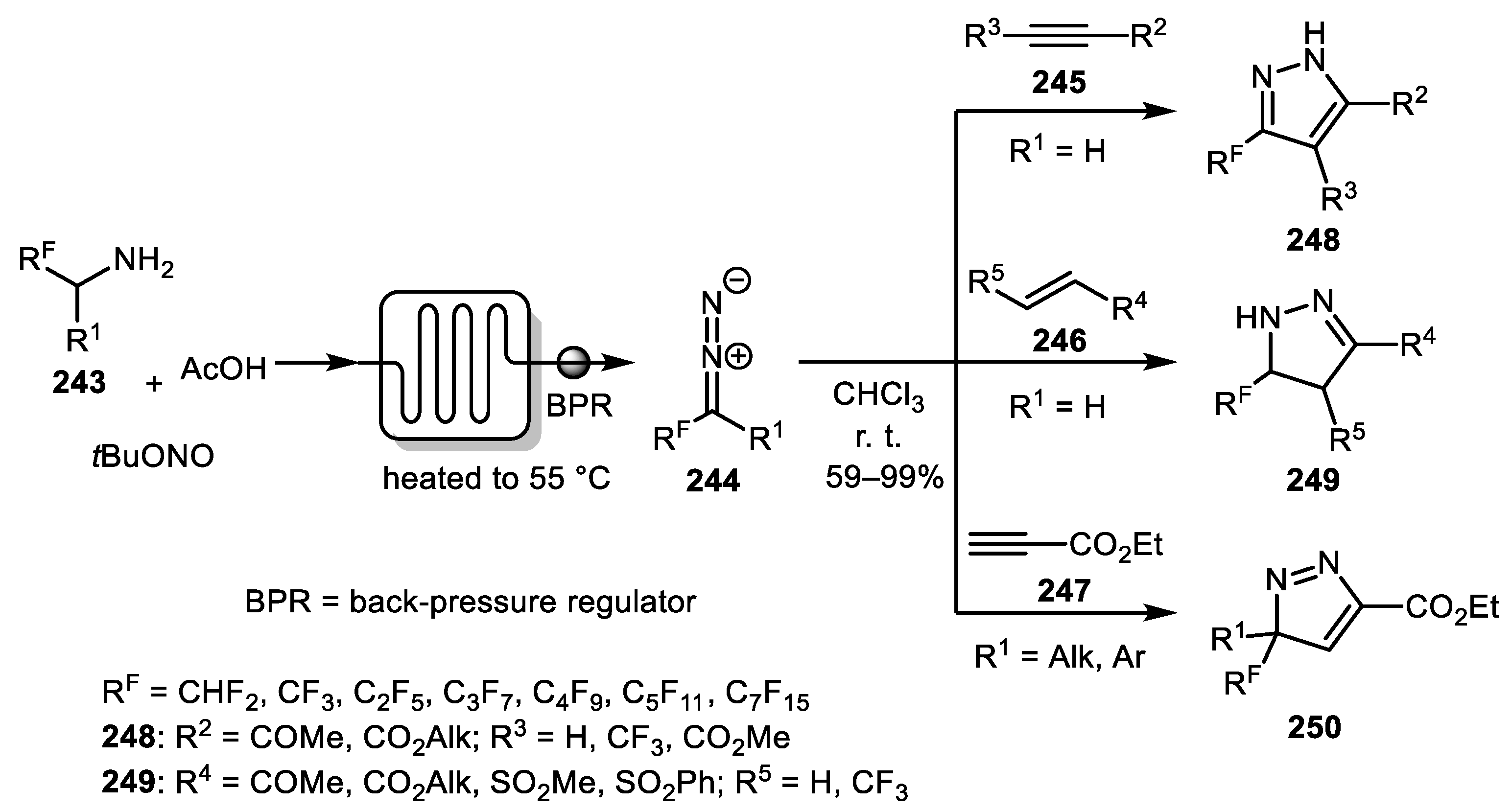
A continuous-flow synthesis of fluoroalkyl-substituted diazomethanes 244 from the corresponding amines 243 and their application in [3+2]-cycloaddition reactions with various alkynes and alkenes was described by Mertens and co-workers (Scheme 61) [124]. The method proved to be efficient for the preparation of fluoroalkyl-substituted 1H-pyrazoles 248, pyrazolines 249, and 3H-pyrazoles 250 with absolute regioselectivity in generally good to excellent yields. An approach for the preparation of mono-, bis-, and tris(trifluoromethyl)-substituted pyrazoles from ethyl 2-diazo-3,3,3-trifluoropropanoate has also been reported by the Reissig group [125].
2.5.2. 3H-Pyrazoles
Relatively stable 3H-pyrazoles, containing two substituents in position 3, are commonly prepared from disubstituted diazo compounds. Although, they are still prone to rearrange to more stable products via, for instance, the van Alphen–Hüttel rearrangement or denitrogenative transformations [126].
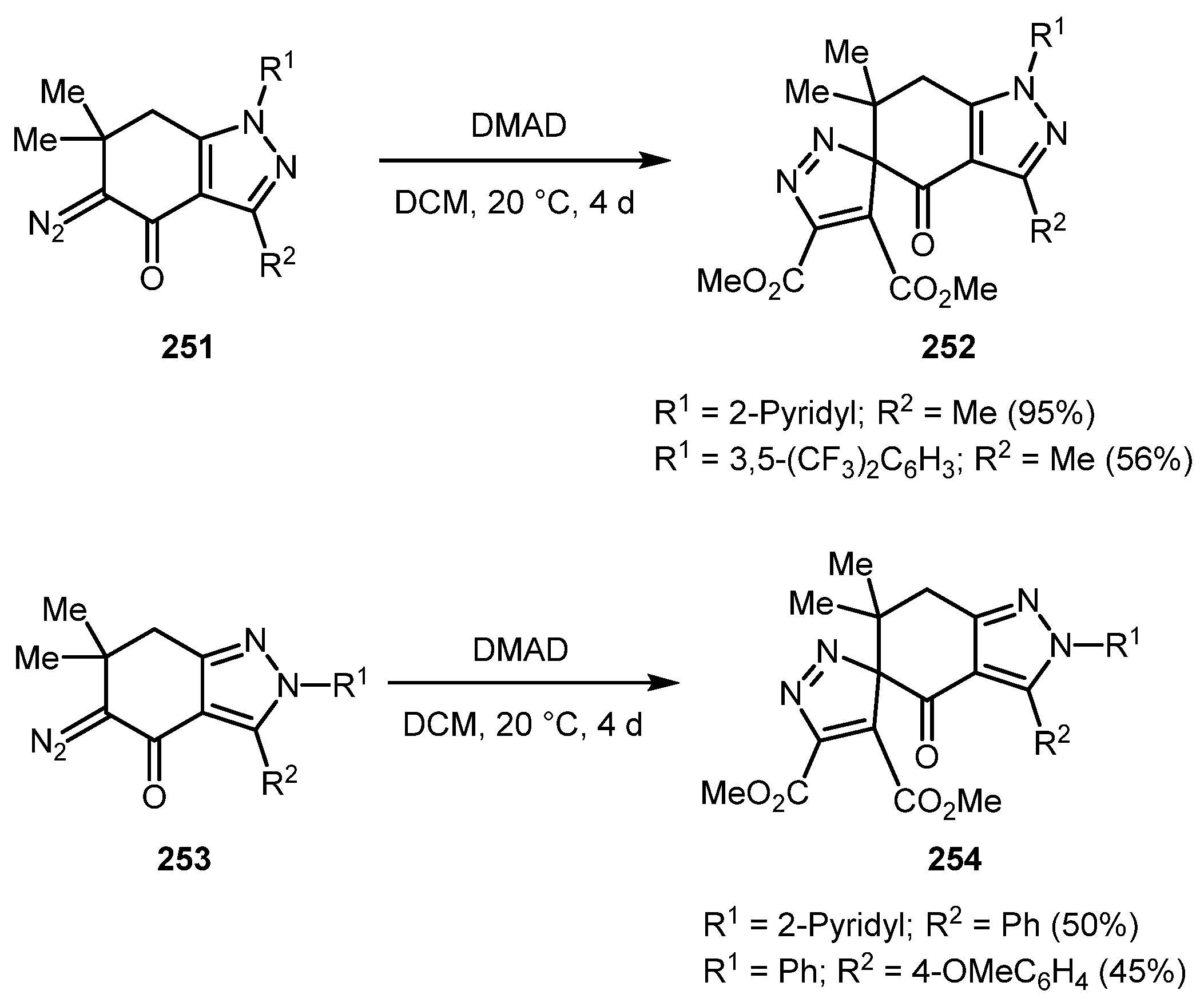
In 2007, Strakova and co-workers reported the synthesis of spiro-3H-pyrazoles adducts 252 and 254 by [3+2] cycloaddition of DMAD to diazo compounds 251 and 253, respectively (Scheme 62) [127]. The reaction proceeded smoothly at room temperature; however, the reaction took four days to go to completion.
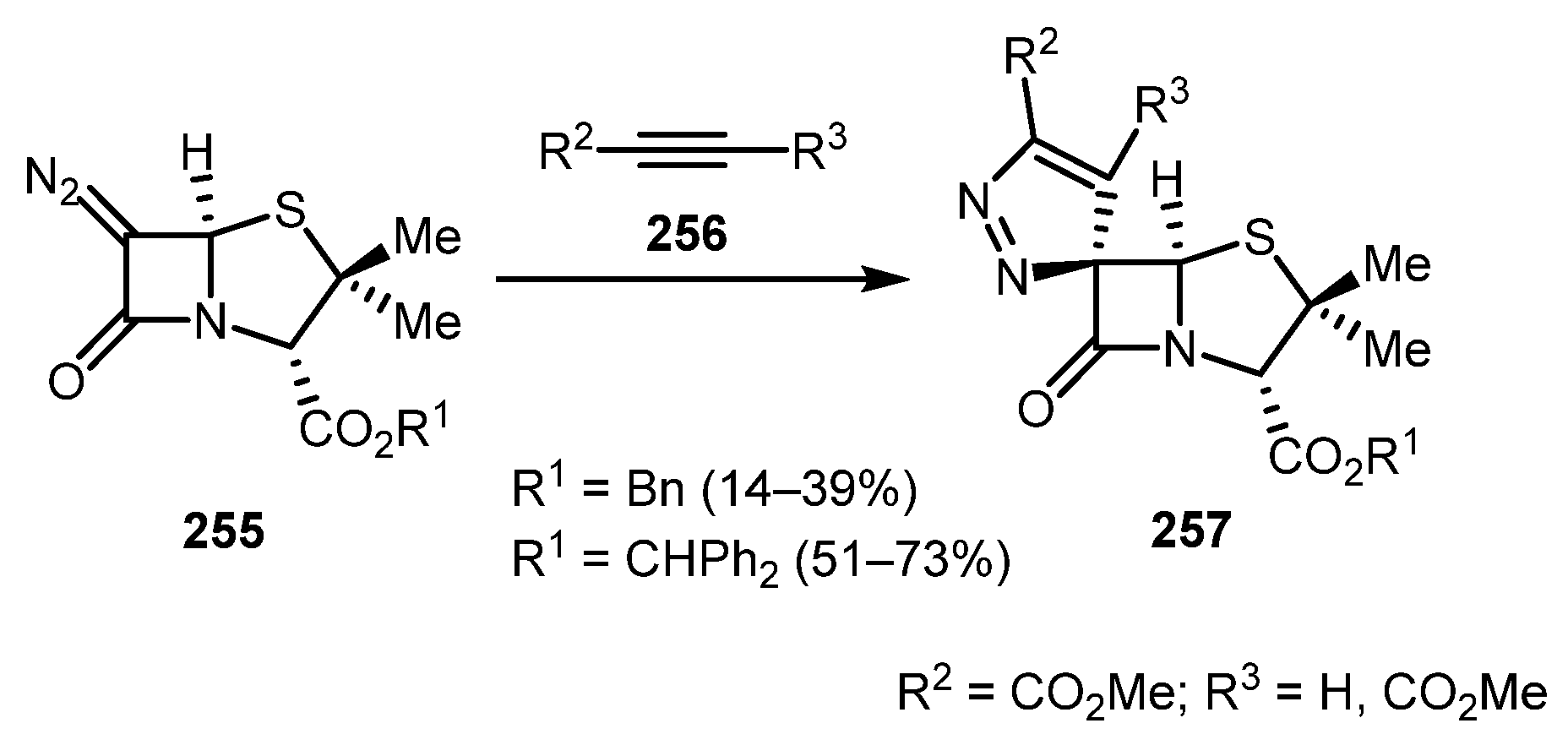
A regio- and stereoselective synthesis of spiro-3H-pyrazole-penicillanates 257 was achieved by Santos and co-workers [128]. Benzhydryl esters provided better yields than their benzyl counterparts. The observed stereoselectivity likely resulted from the alkyne 256 approaching diazo β-lactam 255 from the less sterically hindered α-side (Scheme 63).
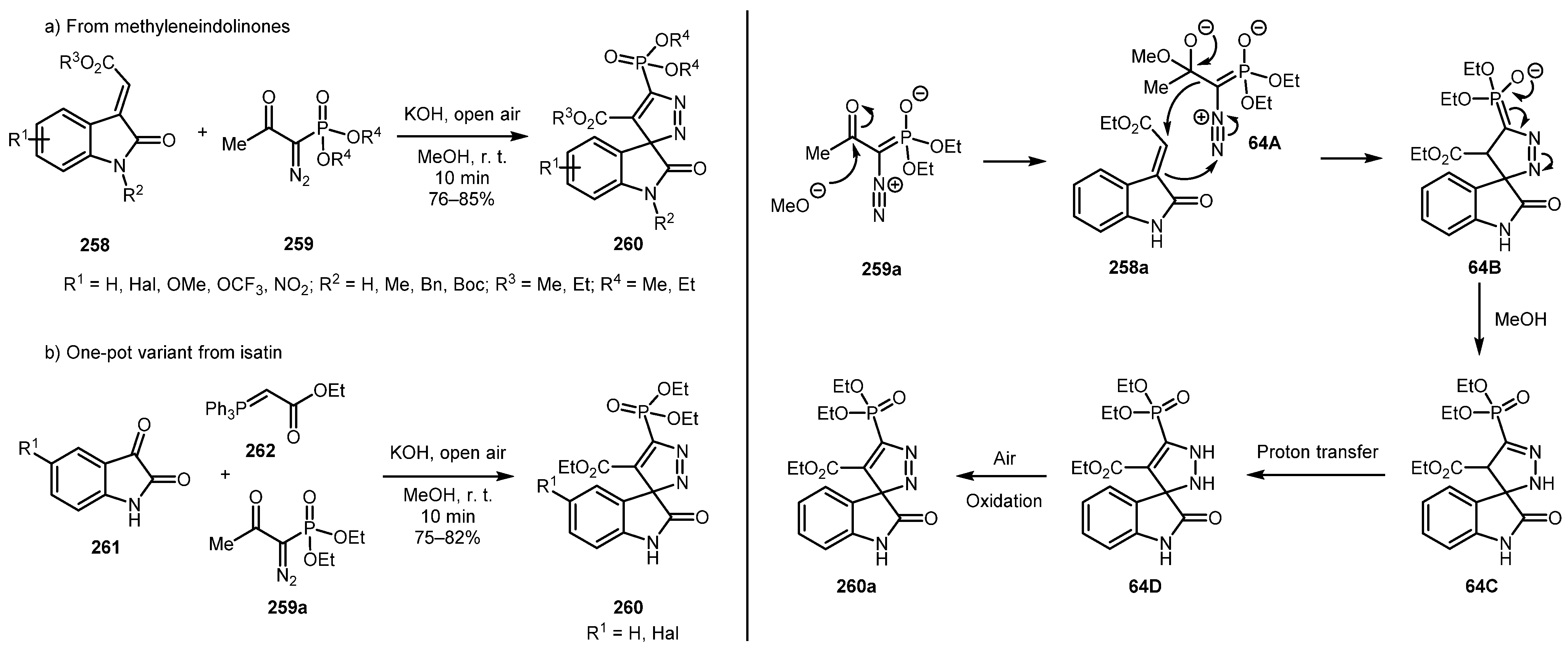
In 2015, Shelke and Suryavanshi described a method for the synthesis of 3,3′-spiro-phosphonylpyrazole-oxindoles 260 from methylidene indolinones 258 and the Bestmann-Ohira reagent (BOR) 259 (Scheme 64a) [129]. The regioselective reaction tolerated a broad range of substituents and afforded good yields of the desired products. Moreover, a one-pot procedure with generation of indolinones 258 in situ by the Wittig reaction of readily available isatins 261 and phosphonium ylides 262 was developed (Scheme 64b). The mechanism of the reaction likely involves the deacylation of BOR 259a by the methoxide ion with subsequent 1,3-dipolar cycloaddition of the diazo-phosphonate anion (arising from 64A) to methylidene indolinone 258a to form intermediate 64B. Subsequent protonation of 64B and proton transfer produces intermediate 64D, which is oxidized by air to afford the final compound 260a.
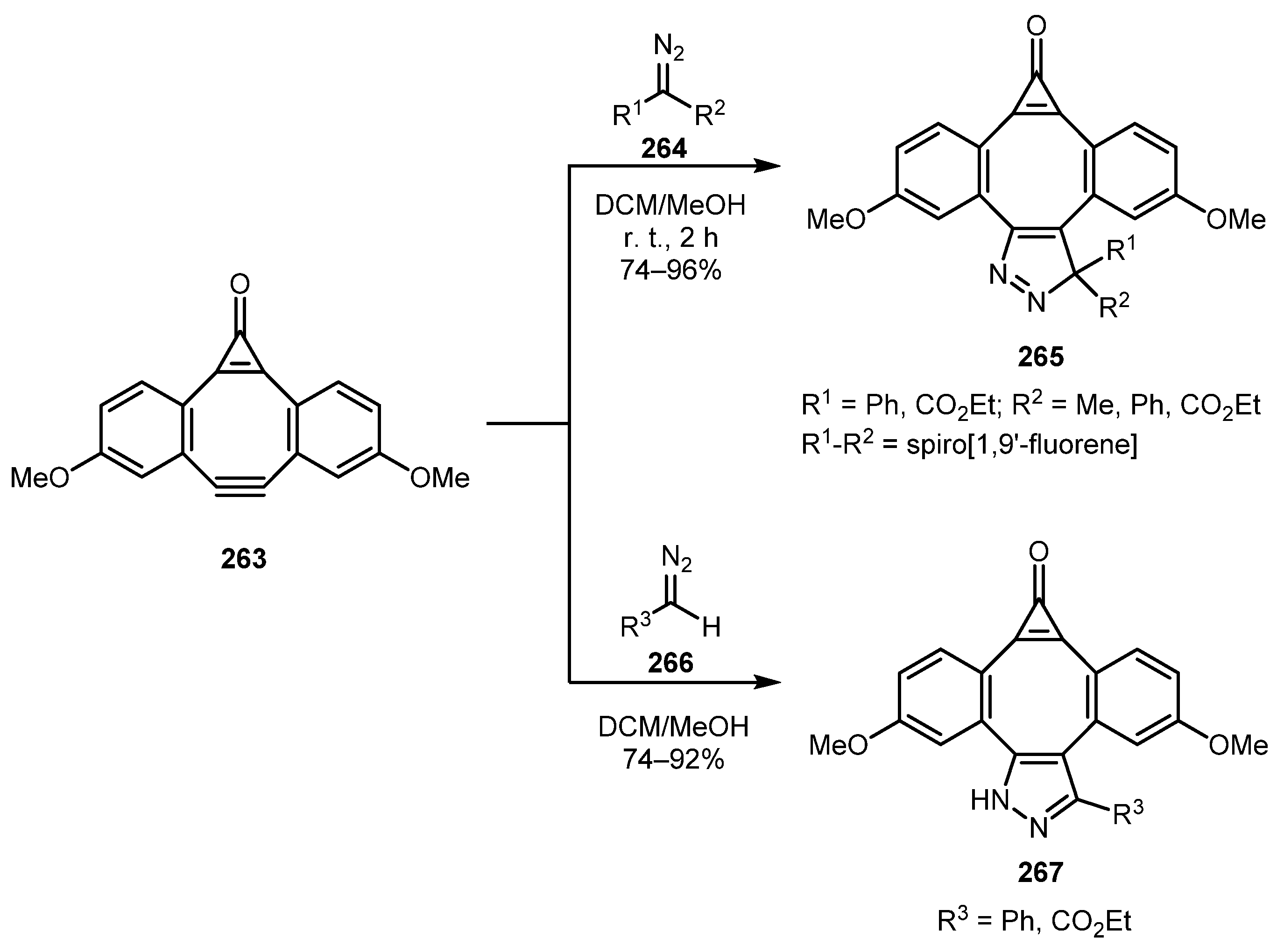
In a study devoted to the exploration of fluorescence turn-on cycloadditions of dibenzocyclooctyne derivative 263 with various 1,3-dipoles, including di- and mono-substituted diazo compounds 264 and 266, Friscourt and co-authors prepared 3H-pyrazole and 1H-pyrazole cycloadducts 265 and 267 [130]. The reaction is believed to be strain-promoted, which resulted in a short reaction time (2 h), even at room temperature. 3H-Pyrazole adducts 265 showed low quantum yields of the fluorescence whereas 1H-pyrazole adducts 267 exhibited a 160-fold fluorescence enhancement over the starting compound 263 (Scheme 65).
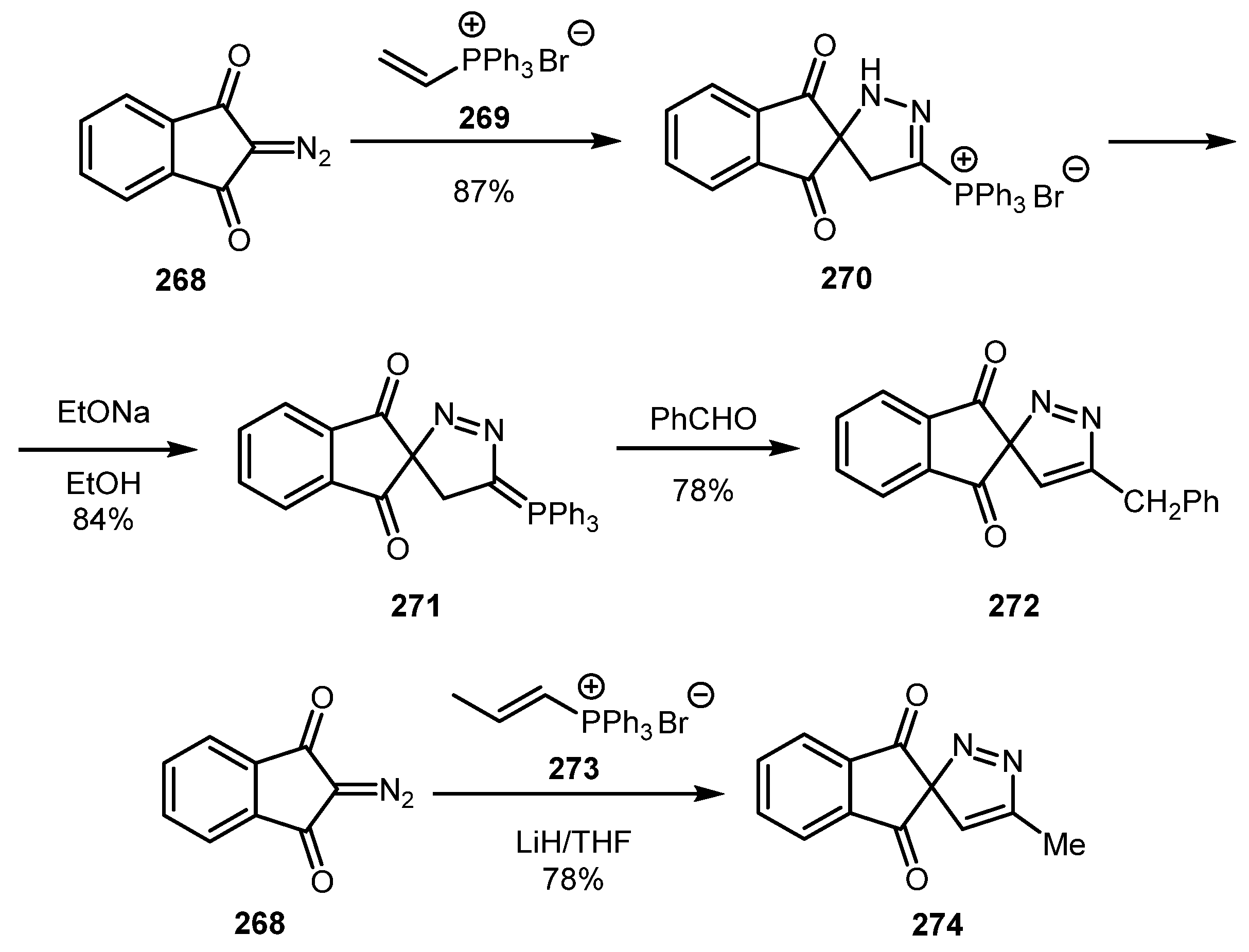
An unusual three-step approach to 5-substituted spiro-3H-pyrazoles was reported in 2008 by the Khidre group [131]. Diazo indan-1,3-dione 268 reacted with triphenyl(vinyl)phosphonium bromide 269 to give pyrazoline 270, which was then converted to phosphonium ylide 271 upon treatment with EtONa. Reaction of 271 with benzaldehyde afforded spiro-3H-pyrazole derivative 272. On the other hand, the involvement of triphenyl(prop-1-en-1-yl)phosphonium bromide (273) into the reaction allowed for the direct synthesis of 5-methyl-spiro-3H-pyrazole 274 (Scheme 66).
2.5.3. Indazoles
A vast majority of methods for the preparation of indazoles from diazo compounds make use of a 1,3-dipolar cycloaddition reaction with an aryne species generated in situ. Among aryne precursors, o-silylaryl triflates are the most commonly used.
1H-Indazoles
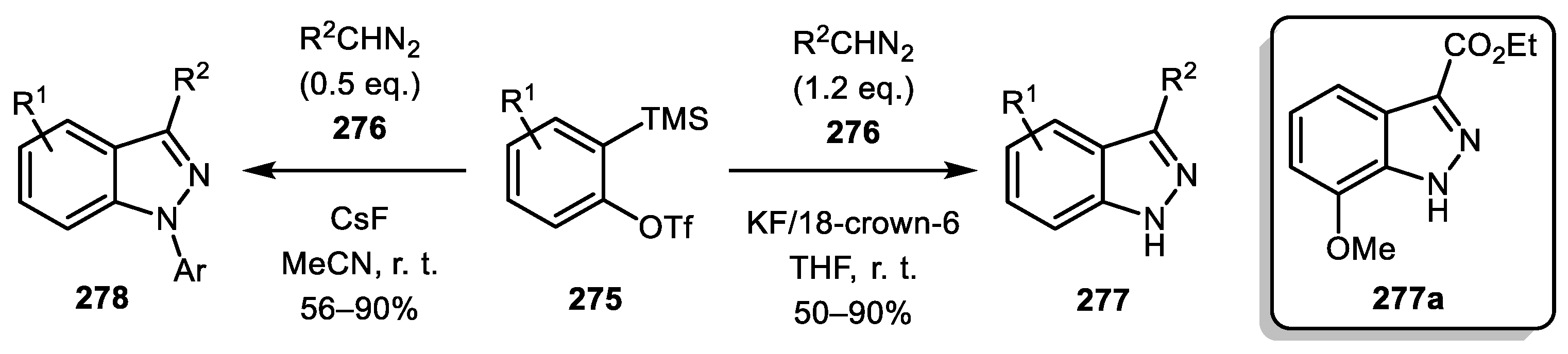
When a terminal diazo compound reacts with aryne, 3H-indazole is initially formed. Subsequently, it can undergo a prototropic isomerization to give 1H-indazole. In 2007, Jin and Yamamoto described a method for the synthesis of 1H-indazoles 277 and 278 by such an approach [132]. Using 1.2 equivalents of the diazo reagent 276 and KF/18-crown-6 in THF system provided N-unsubstituted indazoles 277 while using 0.5 equivalents of the diazo reagent 276 with CsF in acetonitrile led to N-arylated products 278. As for asymmetrical silylaryl triflates, the complete regioselectivity was achieved only in the case of methoxy-substituted one (7-methoxy isomer 277a was formed exclusively) whereas in other cases, a 1:1 mixture of regioisomers was obtained (Scheme 67).
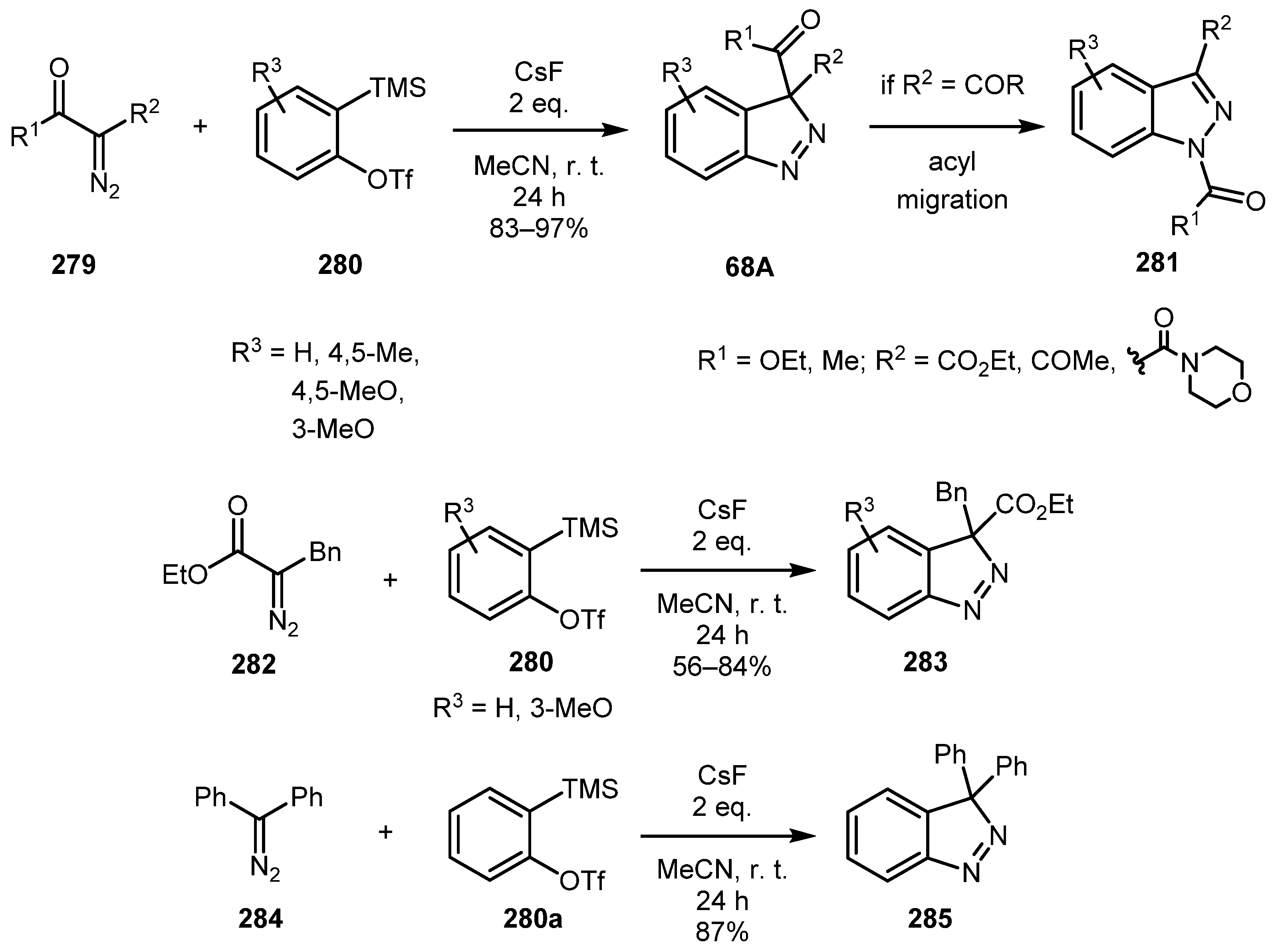
When an internal diazo compound reacted with a benzyne intermediate, 3H-indazole intermediate 68A is formed. For diazodicarbonyl compounds 279, intermediate 68A was prone to undergo acyl migration to give 1H-indazoles 281 as was shown by the Larock group [133]. On the other hand, benzyl-substituted diazo ester 282 and diazo diphenylmethane 284 gave stable 3H-indazoles 283 and 285 exclusively (Scheme 68).
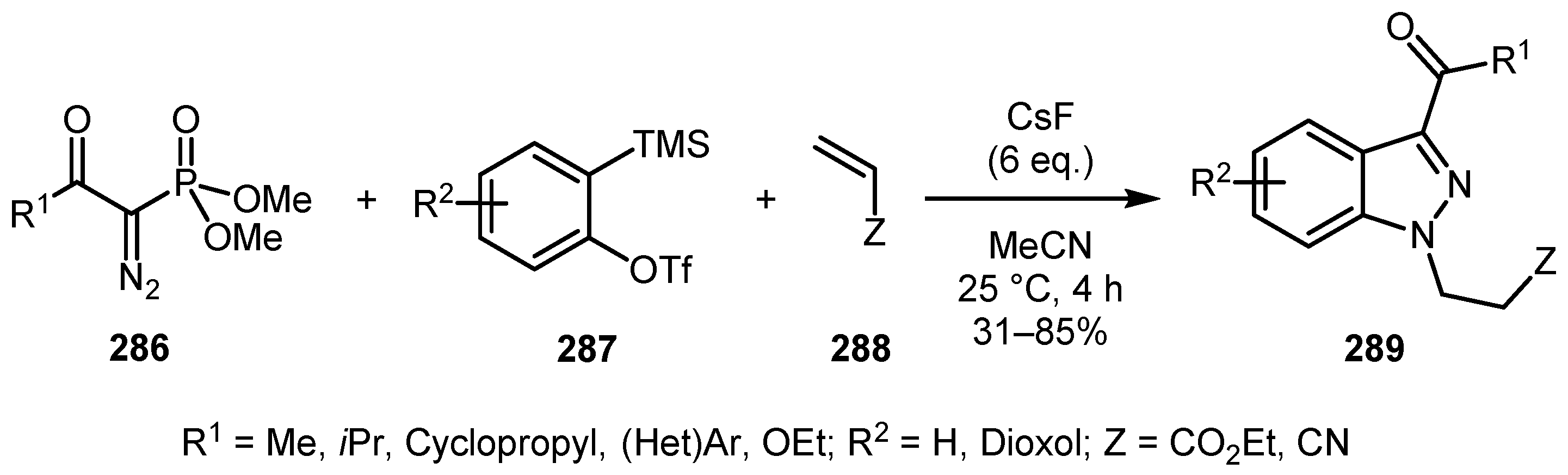
An efficient protocol for the synthesis of N-substituted indazoles from the Bestmann–Ohira reagent (BOR) analogs 286, silylaryl triflates 287, and Michael acceptors 288 was developed by Phatake and co-workers [134]. A role of fluoride anion in this reaction was not only in the generation of the aryne intermediate, but also in the dephosphonylation of BOR. The cascade [3+2] cycloaddition of the acyl diazomethane anion to aryne following the aza-Michael addition process furnished 1,3-substituted indazoles 289 in generally high yields under very mild reaction conditions (Scheme 69).
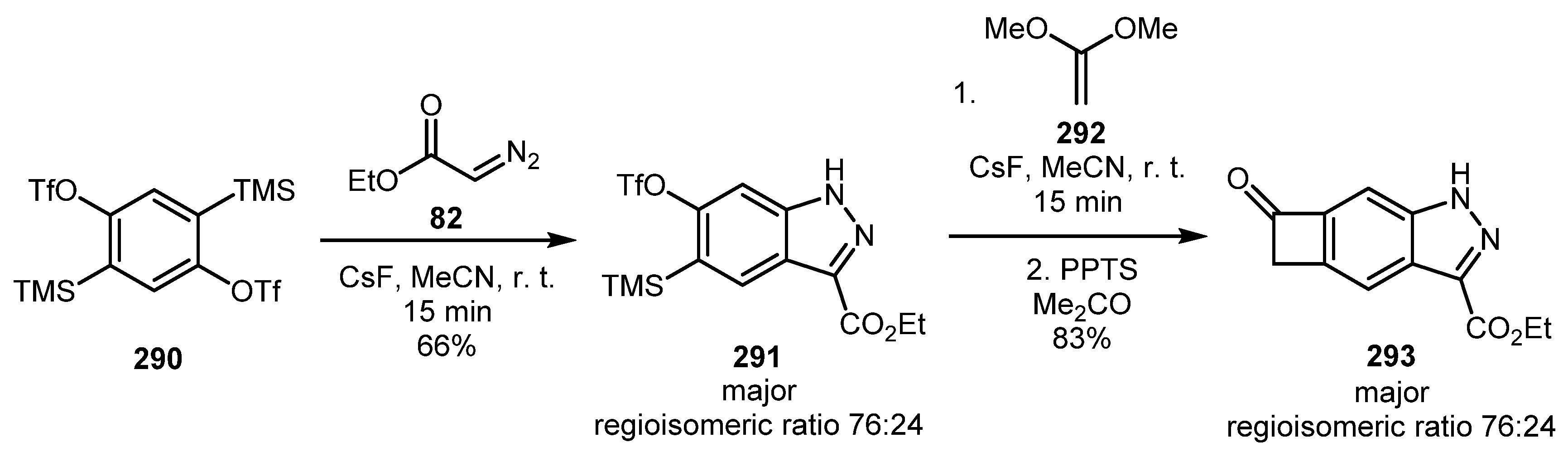
In 2016, Ikawa and co-workers demonstrated that 2,4-bis(trimethylsilyl)-1,3-bis(trifluoromethanesulfonyloxy)benzene 290 could act as a 1,4-benzdiyne equivalent [135]. Moreover, aryne “triple bonds” could be generated in a sequential manner. Using this methodology, an exotic indazole derivative 293 was prepared in a two-step procedure with good overall yield (Scheme 70).
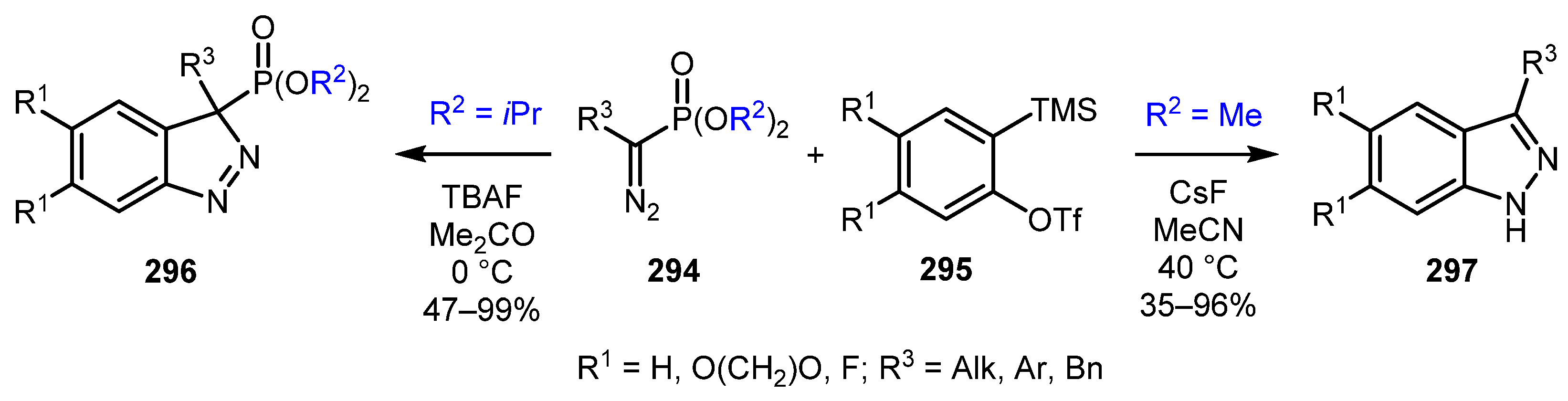
A tunable approach for the synthesis of 1H- and 3H-indazoles was reported by Chen and co-authors [136]. The product distribution depended on the structure of the phosphoryl group in α-substituted-α-diazophosphonates 294. Bulky diisopropyl phosponates provided stable 3H-indazoles 296, whereas less sterically hindered dimethyl phosphonates gave 1H-indazoles 297 (Scheme 71).
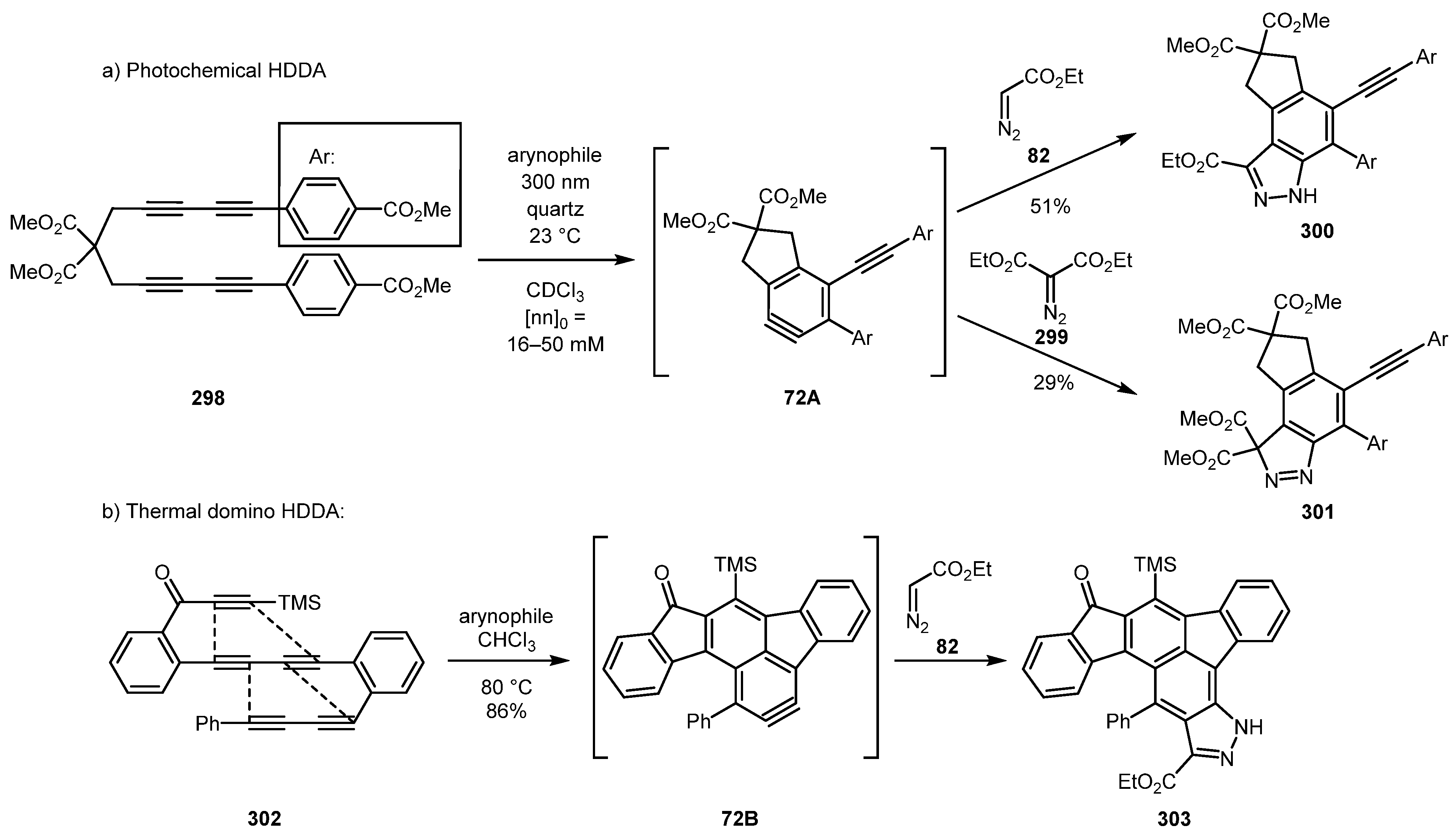
The ‘hexadehydro Diels-Alder’ (HDDA) reaction was employed in the synthesis of indazoles. In 2017, the Hoye group—in their work devoted to investigation of photochemical variation of this reaction—demonstrated that a highly functionalized 1H- and 3H-indazoles 300 and 301 can be constructed from a tetraalkyne derivative 298 and corresponding diazo compounds (Scheme 72a) [137]. While developing this methodology further, this group showed that an efficient domino HDDA reaction of polyalkynes (such as 302) could be used for the synthesis of diverse polyaromatic systems, including indazole 303 (Scheme 72b) [138].
Other aryne precursors, including heteroaromatic ones, have also been reported to-date [139,140,141,142,143,144,145]. The aryne cycloaddition approach also found its application in the synthesis of biologically active indazoles [146].
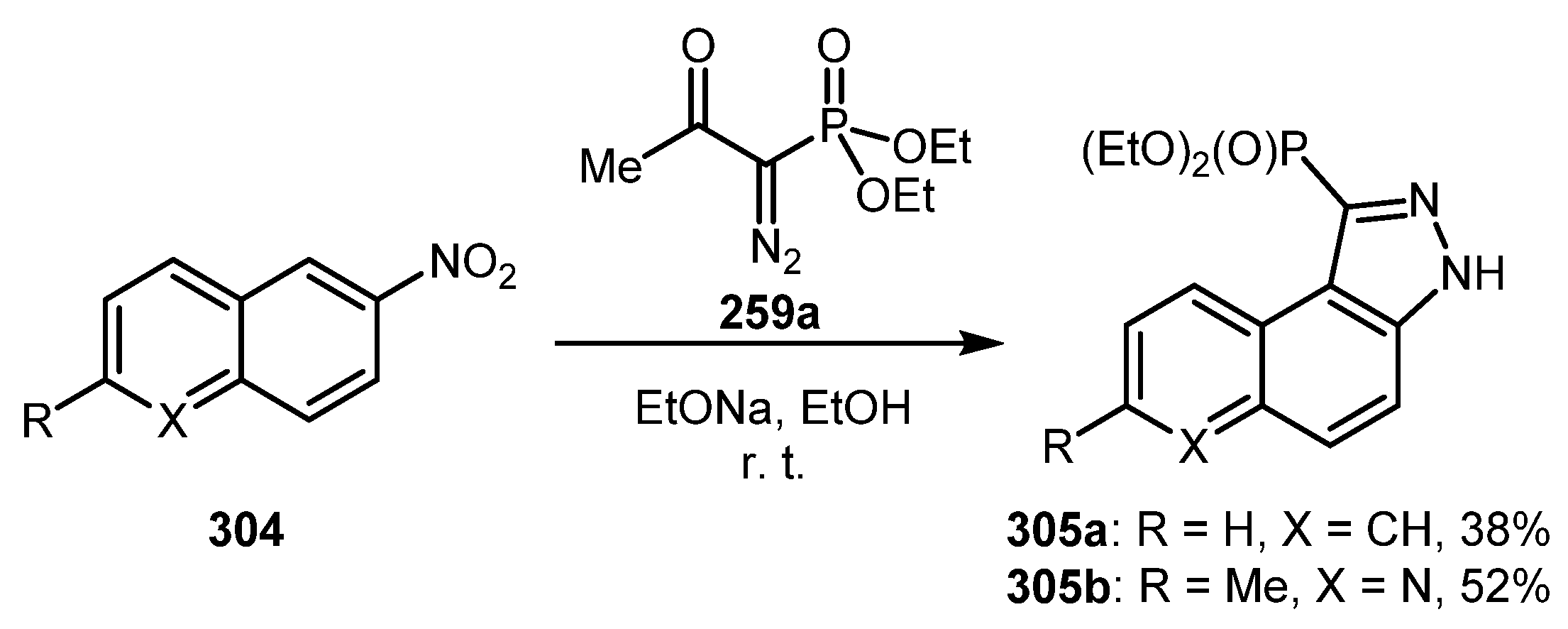
As for other methodologies not involving arynes, Muruganantham and Namboothiri reported a reaction of BOR 259a with nitroalkenes, including nitroethylene moiety as a part of aromatic or heteroaromatic ring (such as 304) [147]. The process consisting of [3+2] cycloaddition followed by HNO2 elimination provides benzo- or pyrido-fused phosphonylindazoles 305 in moderate yields (Scheme 73). Unfortunately, the reaction failed to give any indazole products with simple nitrobenzene derivatives.
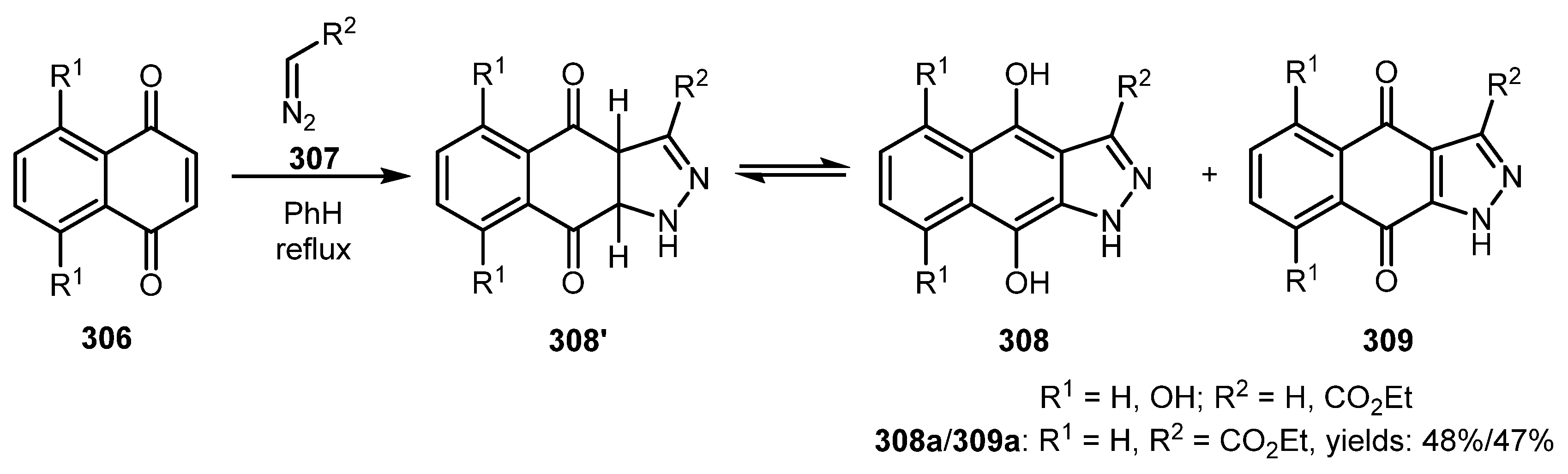
A 1,3-dipolar cycloaddition reaction of naphthoquinones 306 and diazo compounds 307 was used for the synthesis of naphthohydroquinone-derived indazoles 308 in a work by Tandon and co-workers [148]. Biological evaluation of obtained compounds showed that 308a exhibited antifungal and antibacterial activities. The drawback of this approach is that indazoles 308 were obtained as a mixture with (1,4)-naphthoquinono-[3,2-c]-1H-pyrazoles 309 (Scheme 74).
3H-Indazoles and 2H-Indazoles
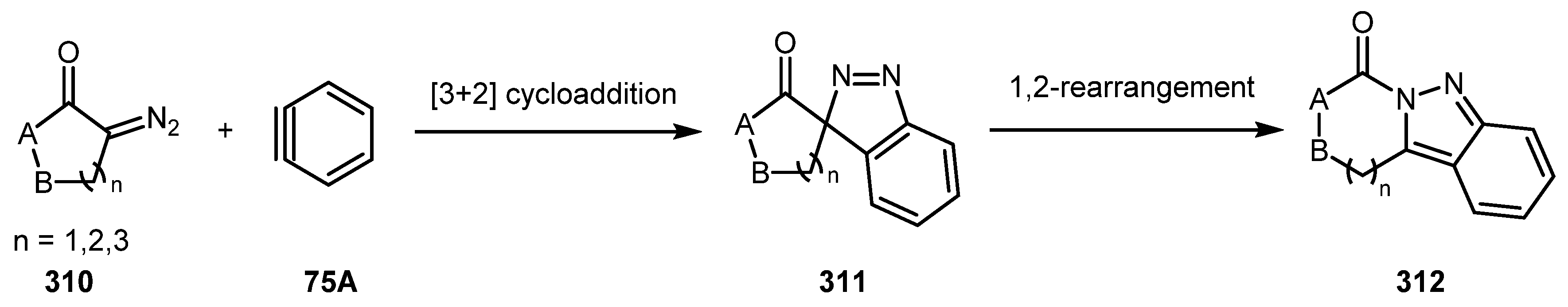
When a 5-,6- or 7-membered cyclic diazocarbonyl compound 310 reacted with an aryne intermediate 75A, the migration of a carbonyl moiety of initially formed spiro-3H-indazole 311 is restricted to 1,2-rearrangement since 1,3-migration is impossible in this case. Such a rearrangement typically gives 2H-indazole 312 (Scheme 75) [149]. Although these spiro-3H-indazoles 311 can be quite stable, some methods have been reported for their conversion to 2H-indazoles 312.
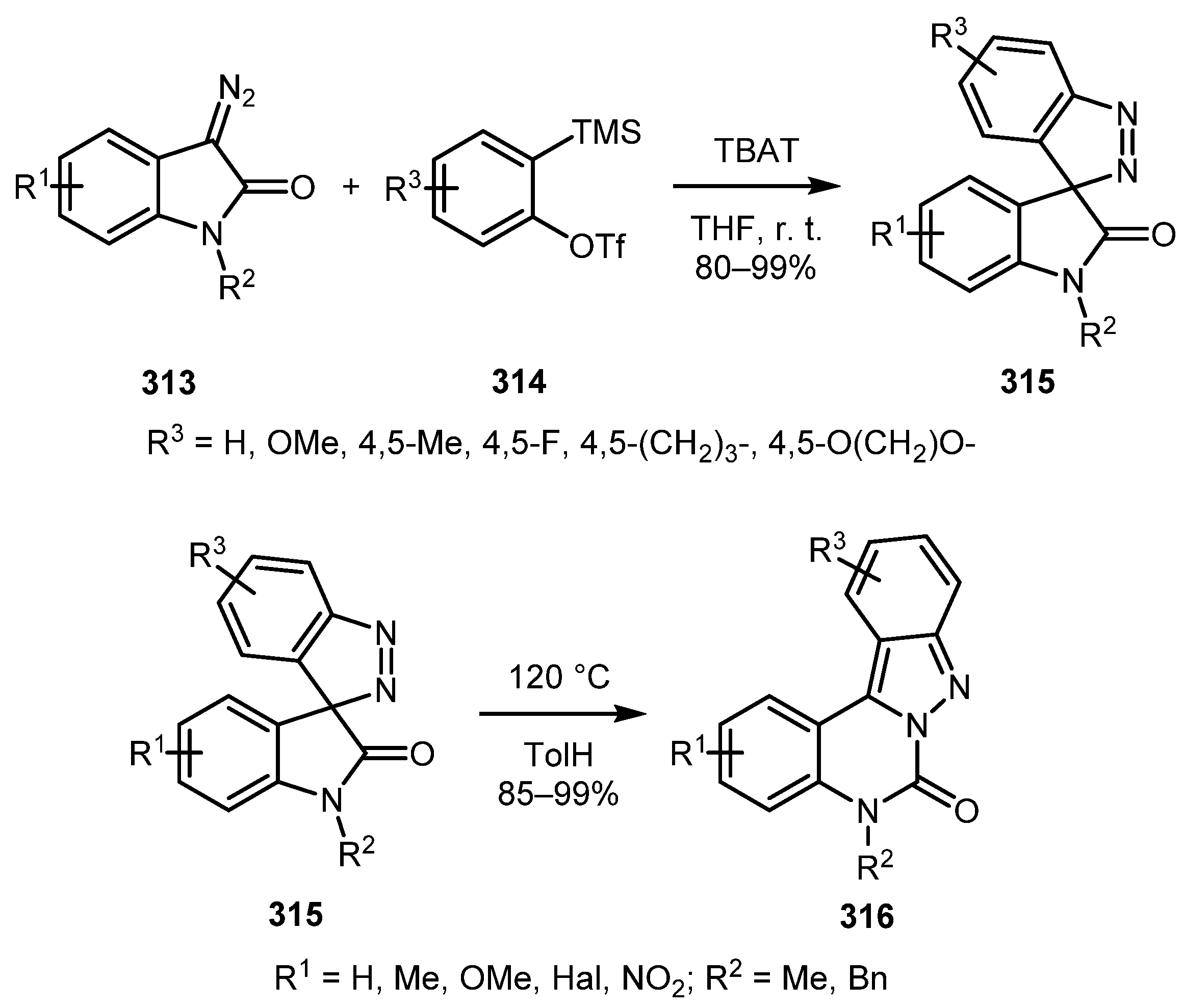
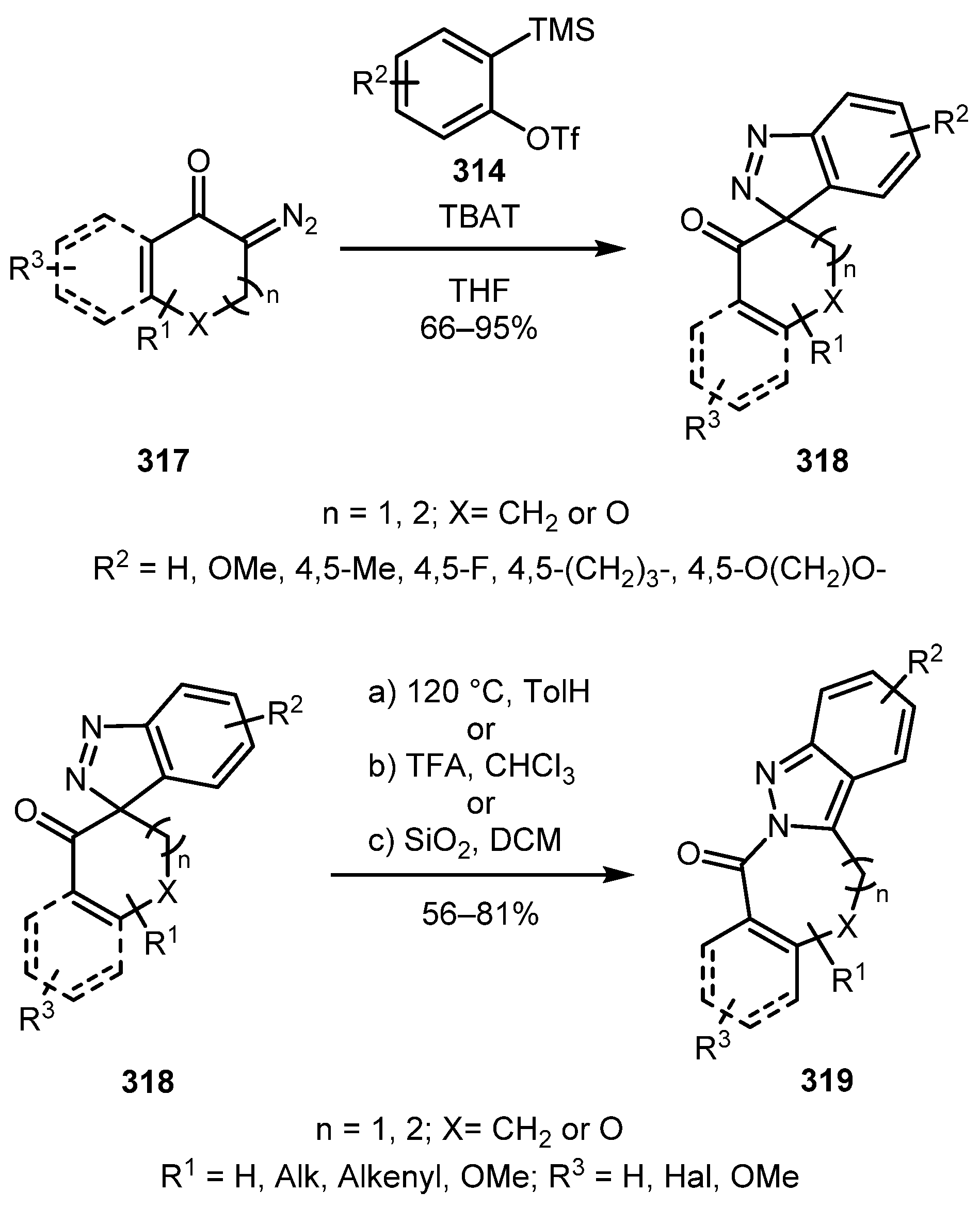
In 2017, the Zhai group devised a protocol for the preparation of stable spiro-3H-indazoles by the cycloaddition of 3-diazoindolin-2-ones 313 to arynes generated from silylaryl triflates 314 [150]. The reaction tolerated electron-withdrawing and electron-donating groups in both indolinone and silylaryl triflate moieties providing spiro[indazole-3,3′-indolin]-2′-ones 315 in good to excellent yields. Under thermal conditions, these products underwent rearrangement to produce fused 2H-indazoles 316 (Scheme 76). The syntheses of various other spiro-3H-indazoles from 3-diazoindolin-2-one derivatives have also been reported in other works [151,152].
This methodology was further extended to carbocyclic diazo compounds 317 [153]. In this case, the rearrangement into 2H-indazoles 319 could be carried out not only under thermal but also under acidic conditions, even with weakly acidic SiO2 (Scheme 77). The reaction worked well with 6-membered cyclic diazo compounds, but in the case of 7-membered ones, pure 3H-indazole could not be obtained. Fortunately, performing the two-step reaction in a one-pot fashion allowed to prepare 2H-indazole fused with the seven-membered ring in a moderate yield. Noteworthily, the five-membered cyclic diazo compound failed to give any indazole products.
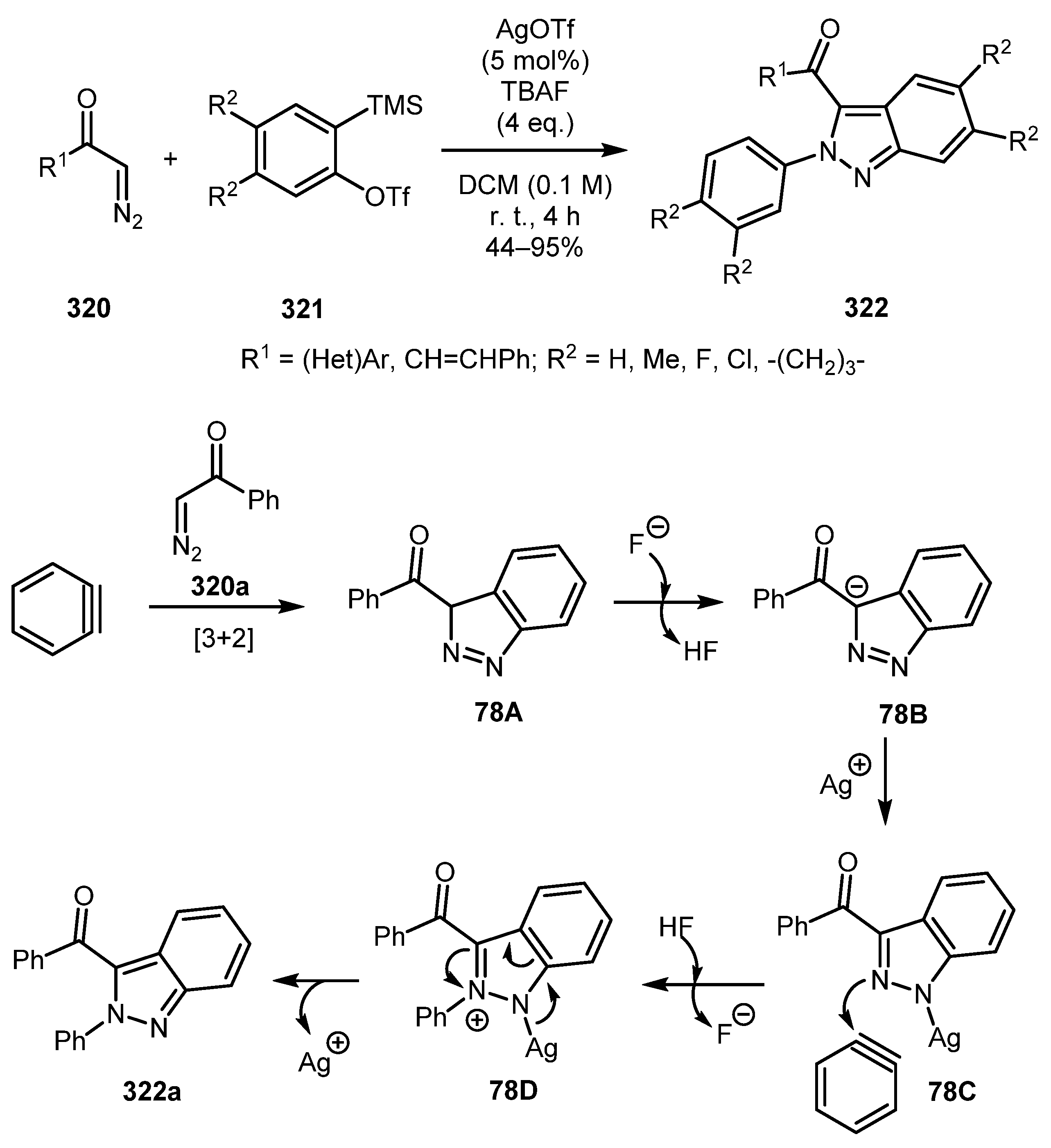
In 2012, Wang and Liu found that cationic Ag(I) species can dramatically affect the chemoselectivity of the reaction of terminal diazo carbonyls 320 with an excess of arynes [154]. Thus, in the presence of AgOTf, 2-aryl-2H-indazoles 322. This result is in contrast to 1-aryl-1H-indazoles 278 which were obtained by Jin and Yamamoto in absence of any catalyst (vide supra, Scheme 67). The authors postulated the following mechanism for this reaction. Intermediate 78A generated by initial cycloaddition is deprotonated by fluoride to give a carbonyl-stabilized anion 78B, which is, in turn, trapped by Ag(I) cation to form (1H-indazol-1-yl)silver 78C. The reaction of intermediate 78C with benzyne is likely to produce the distinct arylation product 78D. Subsequent elimination of silver with tautomerization furnishes the final 2H-indazole product 322a (Scheme 78).
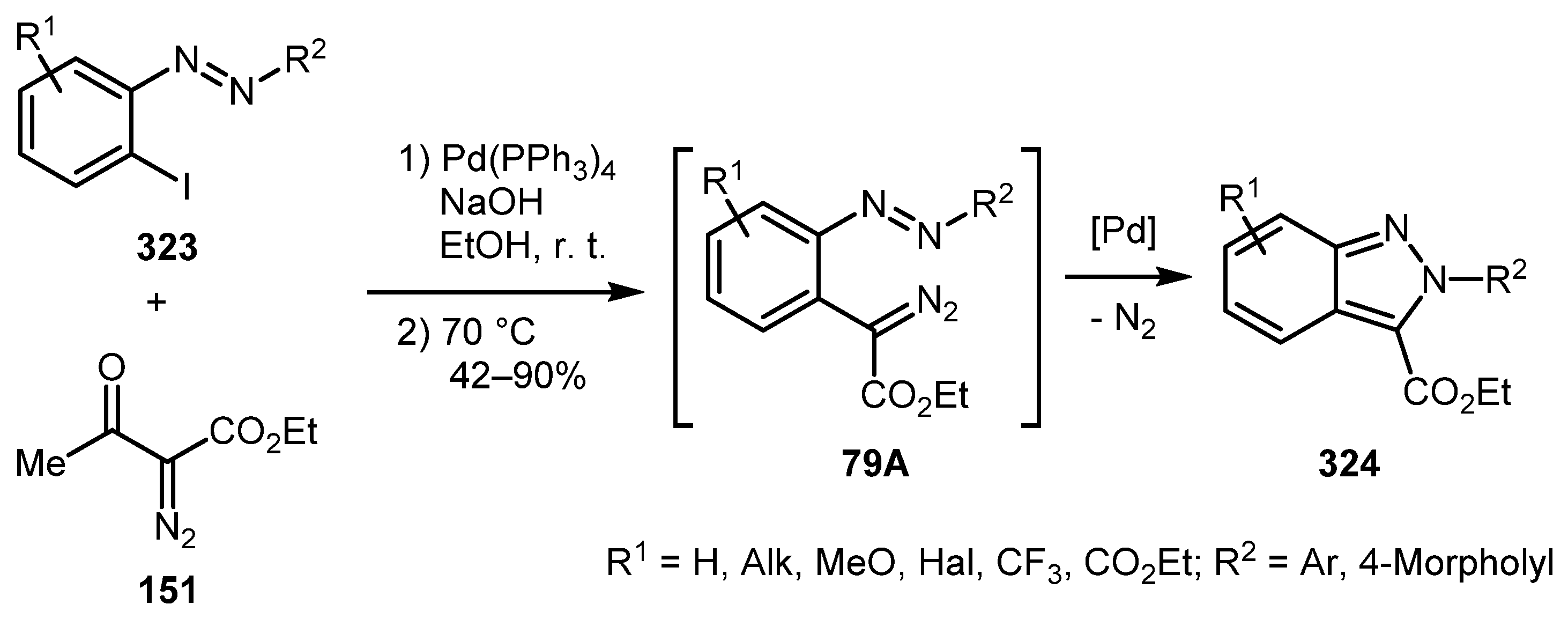
Not only «aryne» approaches have been employed for the synthesis of 2H-indazoles. In a study by the Lee group, 2H-indazoles were prepared from 2-iodoazoarenes/2-iodoaryltriazenes 323 and α-acetyldiazoacetate 151 by a one-pot tandem palladium-catalyzed deacylative cross-coupling/denitrogenative cyclization process [155]. The method was applicable to a broad range of substituents in the aryl moiety of 323 and afforded 2-substituted 2H-indazoles 324 in generally good yields (Scheme 79). Moreover, this protocol provided entry into indazoles with different substituents in the two aryl rings, which is not achievable when using «aryne» methodologies.
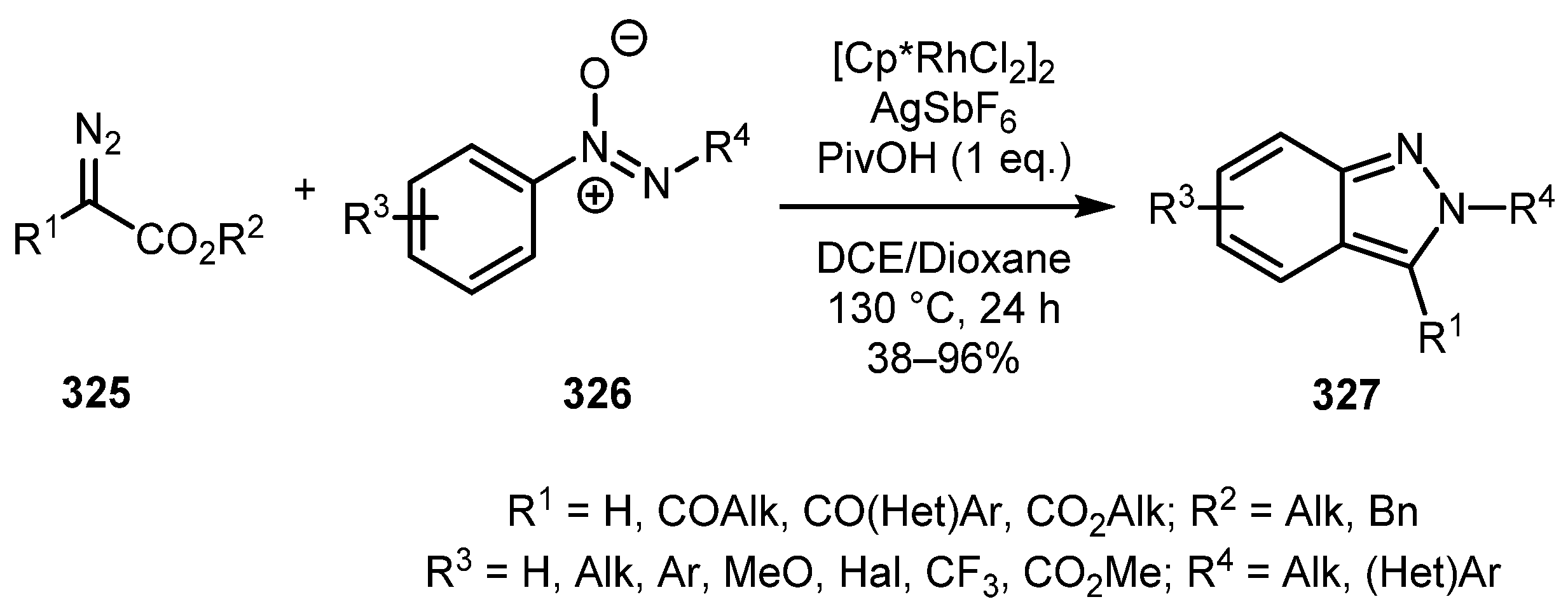
Azoxy compounds can be used for the synthesis of 2H-indazoles, as was shown by the You group [156]. The Rh(III)-catalyzed C-H-alkylation/intramolecular decarboxylative cyclization reaction of diazo esters 325 and aryldiazene oxides 326 provided 3-acyl-2H-indazoles 327 in a highly regioselective manner (Scheme 80). As in the previous case, both symmetrical and non-symmetrical diaryldiazene oxides were found suitable for the reaction. In addition, the protocol is also compatible with 2-alkyl-1-aryldiazene oxides giving N-alkyl-2H-indazoles in acceptable yields.
3. Azoles with Three Heteroatoms
3.1. 1,2,3-Thiadiazoles
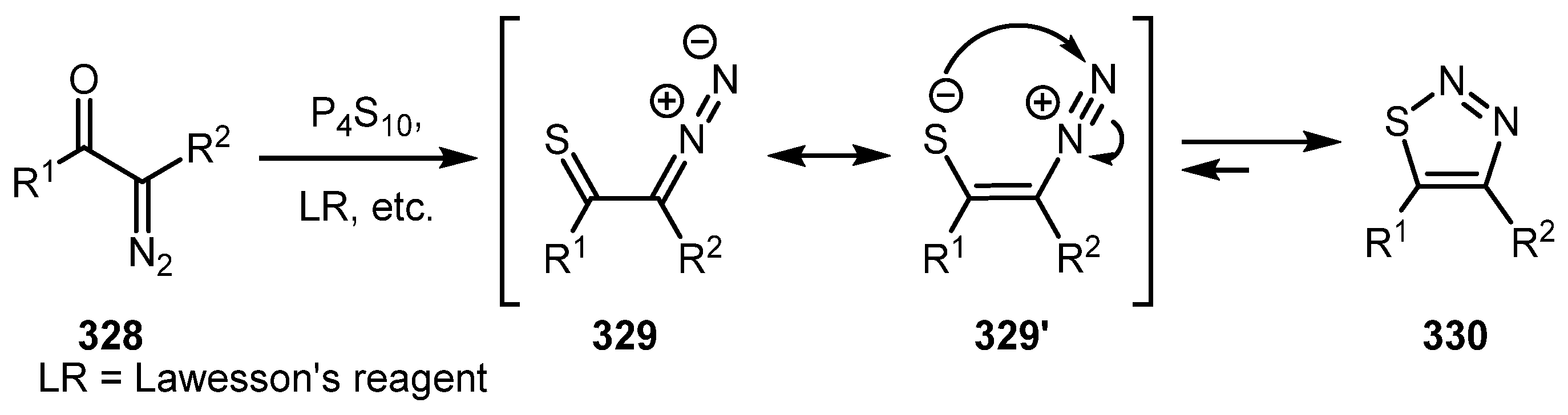
A general approach to the synthesis of 1,2,3-thiadiazoles from diazo compounds is based on the reaction of diazocarbonyl compound 328 with a thionation reagent. The initially formed diazothiocarbonyl compound 329 is usually unstable and undergoes 1,5-electrocyclization to form 1,2,3-thiadiazole 330 (Scheme 81). This topic has been recently reviewed by Shafran and co-authors [16], therefore we chose not cover it in the present review.
3.2. 1,2,3-Triazoles
Since the revolutionary discovery of Copper-Azide-Alkyne Cycloaddition (CuAAC) by Meldal [157] and Fokin and Sharpless [158], this reaction has become the most widely used method for the preparation of 1,2,3-triazoles. Due to the great versatility and simplicity of this approach, it found application in many fields of chemical science, such as bioorthogonal reactions [159], design of functional coatings [160], preparation of organic dyes and fluorophores [161], etc. It has also become a handy tool in medicinal chemistry and drug design for the preparation of triazole-containing bioactive molecules possessing anticancer, antimicrobial, anti-tubercular, antiviral, antidiabetic, antimalarial, anti-leishmanial, and neuroprotective activities [162].
Standard CuAAC is suitable for terminal alkynes and allows the synthesis of 1,4-disubstituted 1,2,3-triazoles, but the regioselectivity can be reversed by using a ruthenium catalyst for synthesis of 1,5-disubstituted triazoles. Additionally, RuAAC also works well with internal alkynes providing fully substituted triazoles, although in this case, regioselectivity cannot always be reliably controlled [163]. In contrast with this method, a number of azide-free methodologies for regiospecific synthesis of variously substituted 1,2,3-triazoles have been developed [164,165], including the «diazo» approach, specifically, the Wolff cyclocondensation. Moreover, for the latter reaction, common Lewis acids are used and often no transition metal catalyst is needed.
3.2.1. 1,2,3-Triazoles via Cyclization of Imino Diazo Compounds
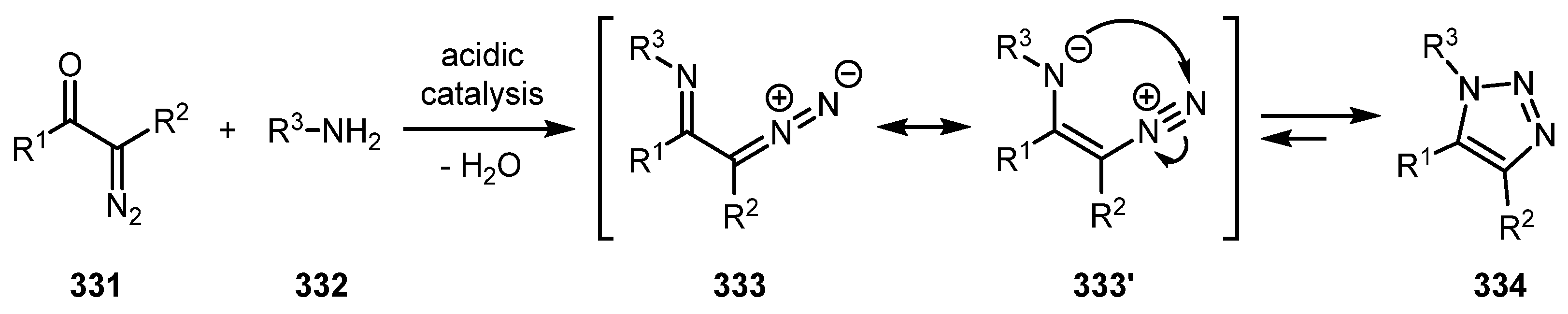
The Wolff cyclocondensation is a reaction of diazocarbonyl compound with amine leading to 1,2,3-triazoles [166]. The mechanism of this reaction is depicted in Scheme 82. The condensation of amine 332 with diazo compound 331 carbonyl group, usually occurring under acidic catalysis, gives diazo imine intermediate 333, which is in equilibrium with 1,2,3-triazole 334. When the R group in the imine moiety is not electron-withdrawing, this equilibrium is completely shifted towards triazole. The disadvantages of this approach in comparison with CuAAC include its questionable bioorthogonality and often harsh reaction conditions.
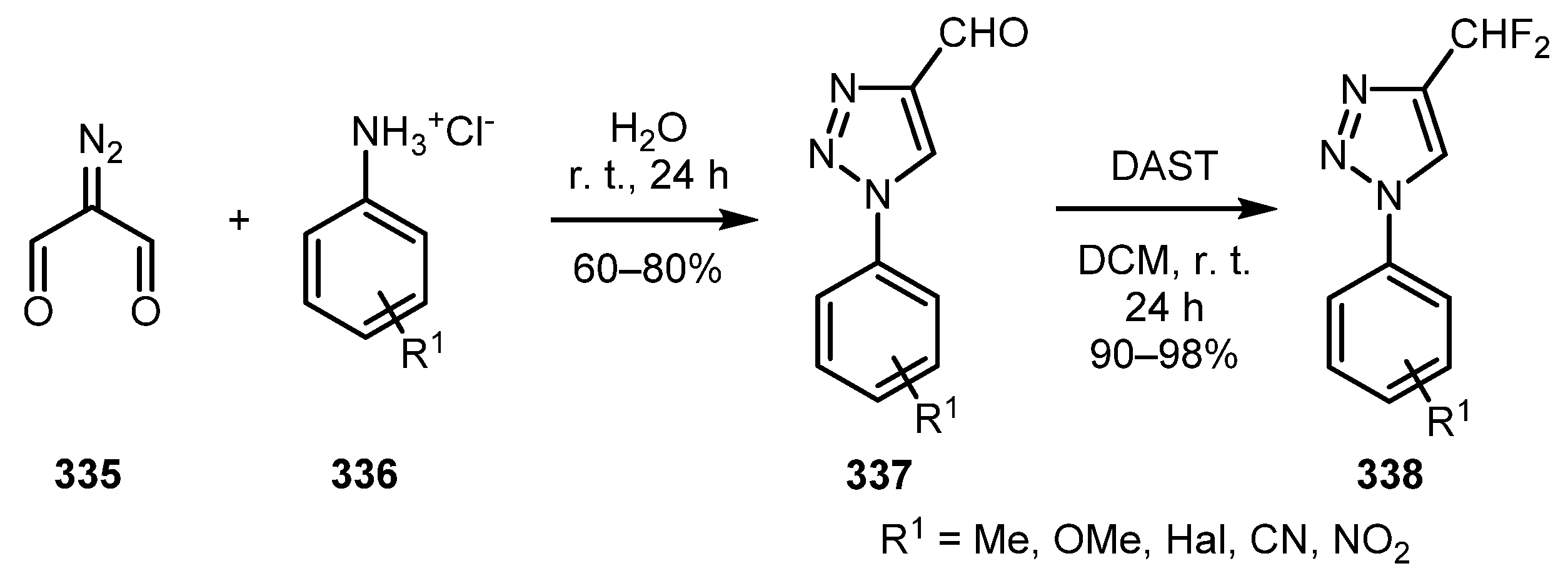
In a study devoted to the synthesis of potential anti-tubercular agents, Costa and co-workers employed a reaction of diazo malonaldehyde 335 with aniline hydrochlorides 336 conducted in water at room temperature to afford 1,2,3-triazoles with a 1,4-substitution pattern 337 (Scheme 83) [167]. It should be noted that amine hydrochlorides themselves provide an acidic medium, which facilitates the generation of intermediate imine species. Such 1-aryl-1,2,3-triazole-4-carbaldehydes 337 can be further converted into difluoromethyl-substituted triazoles 338 in excellent yields simply on treatment with diethylaminosulfur trifluoride (DAST).
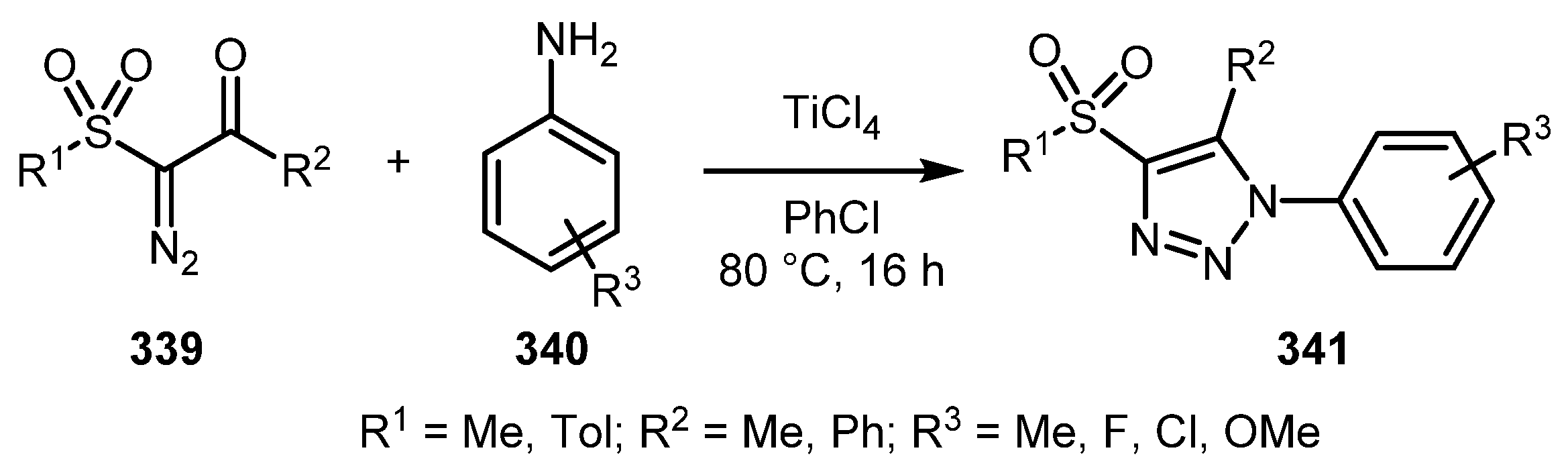
In contrast to diazo aldehydes, the reaction of amines with α-diazoketones usually requires the use of elevated temperature and the addition of Lewis acids in stoichiometric amounts [168]. α-Diazo-β-oxosulfones 339 can also be involved in this reaction, as was shown by the Krasavin group [169]. Heating at 80 °C in chlorobenzene in the presence of 1.5 equivalents of TiCl4 turned out to be the best conditions for this transformation, although the desired 4-sulfonyl-1,2,3-triazoles 341 were obtained in moderate yields (Scheme 84).
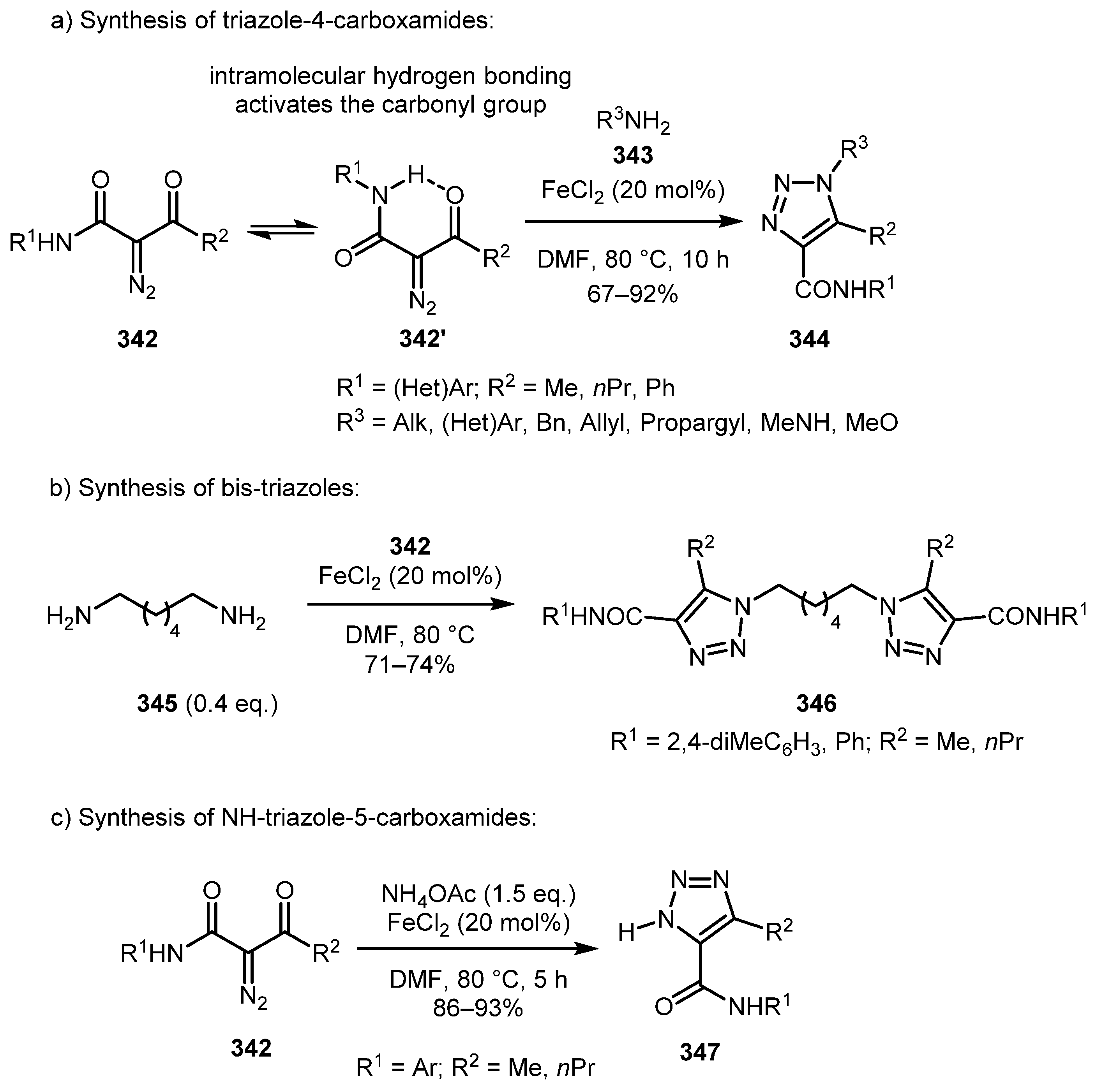
An interesting effect of intramolecular hydrogen bonding activation of carbonyl group in α-diazo-β-oxoamides 342 was described in 2012 by Wang and co-workers (Scheme 85) [170]. This effect allowed the use of catalytic amounts (20 mol.%) of Lewis acid, affording diversely substituted triazole-4-carboxamides 344 in good yields (Scheme 85a). The method works well with both aliphatic and aromatic amines, as well as diamines such as 345 (Scheme 85b), substituted hydrazine, hydroxylamine, and even ammonium acetate (Scheme 85c). In the latter case, 1H-triazole-5-carboxamides 347 were obtained. This methodology was later used for the preparation of some triazole-containing polymers [171].
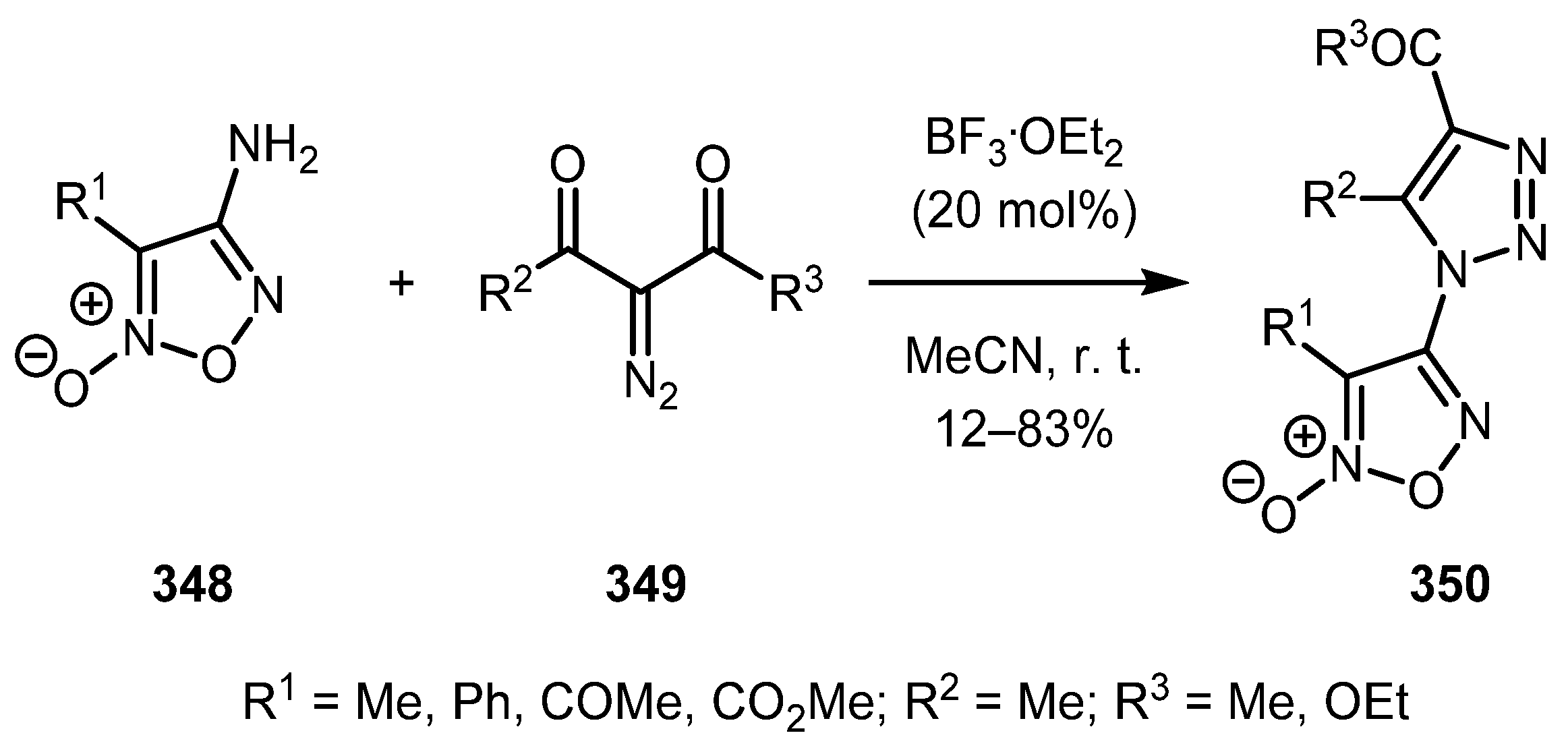
Even poorly nucleophilic amines, such as aminofuroxans 348, can be employed for this reaction, as was shown by Fershtat and co-authors [172]. The use of 20 mol.% of boron trifluoride etherate at room temperature proved to be the optimal conditions for this substrate, although the desired triazoles 350 were obtained in low to moderate yields. It is worth mentioning that in this case, α-diazodiketones gave better yields than α-diazo-β-ketoesters (Scheme 86).
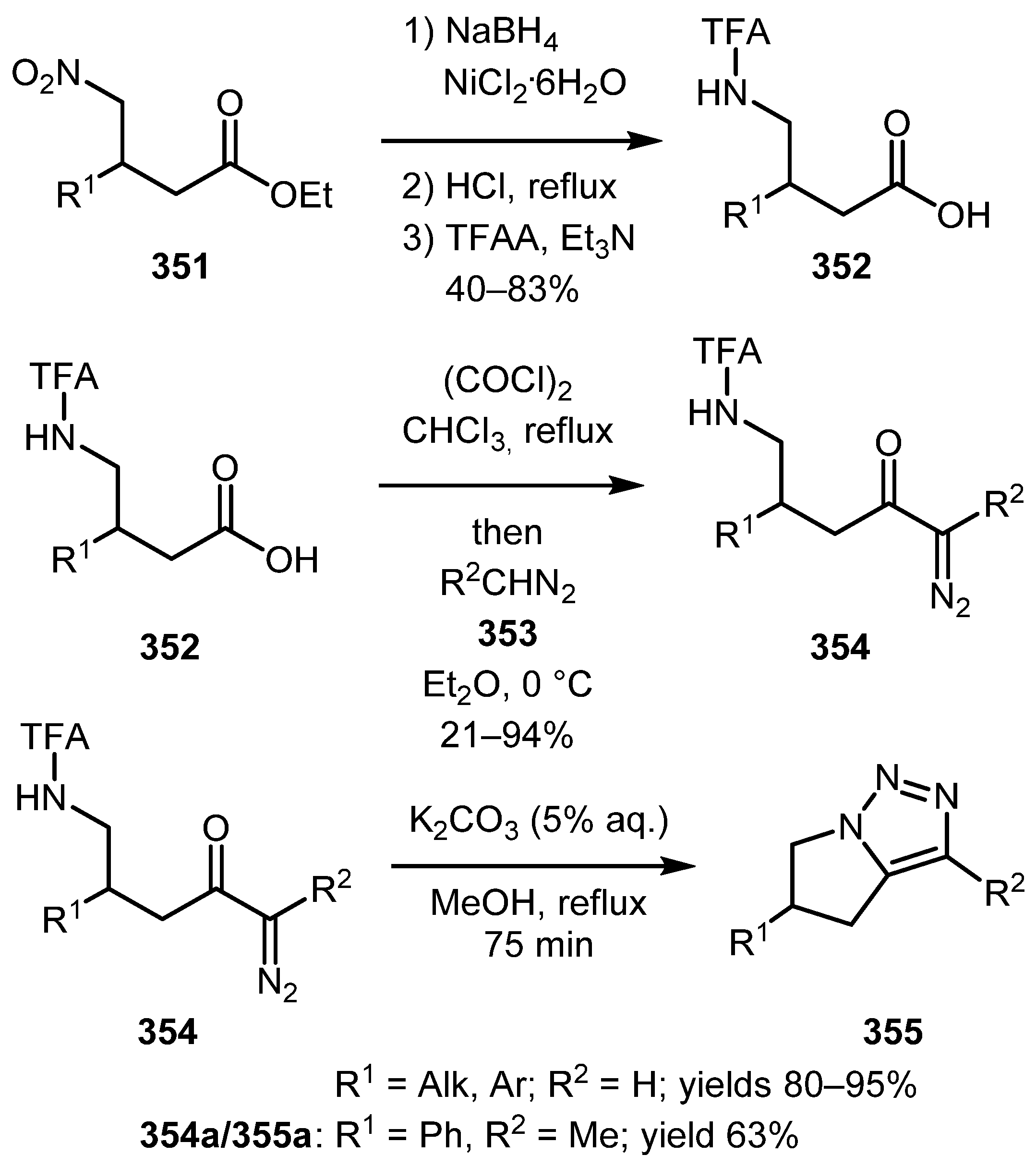
An intramolecular variant of this reaction usually requires milder conditions and often no use of a Lewis acid catalyst. In 2019, Santiago and Burtoloso reported the synthesis of fused bicyclic 1,2,3-triazoles 355 from γ-N-protected amino diazo ketones 354 [173]. The starting diazo compounds were prepared in four steps from γ-nitro esters 351. The cyclization step was performed by gently refluxing the diazo ketones 354 in a methanolic aqueous solution of potassium carbonate. The yields on the final step were good to excellent, the only diminished yield was observed with α-methyl-α-diazoketone 354a (Scheme 87).
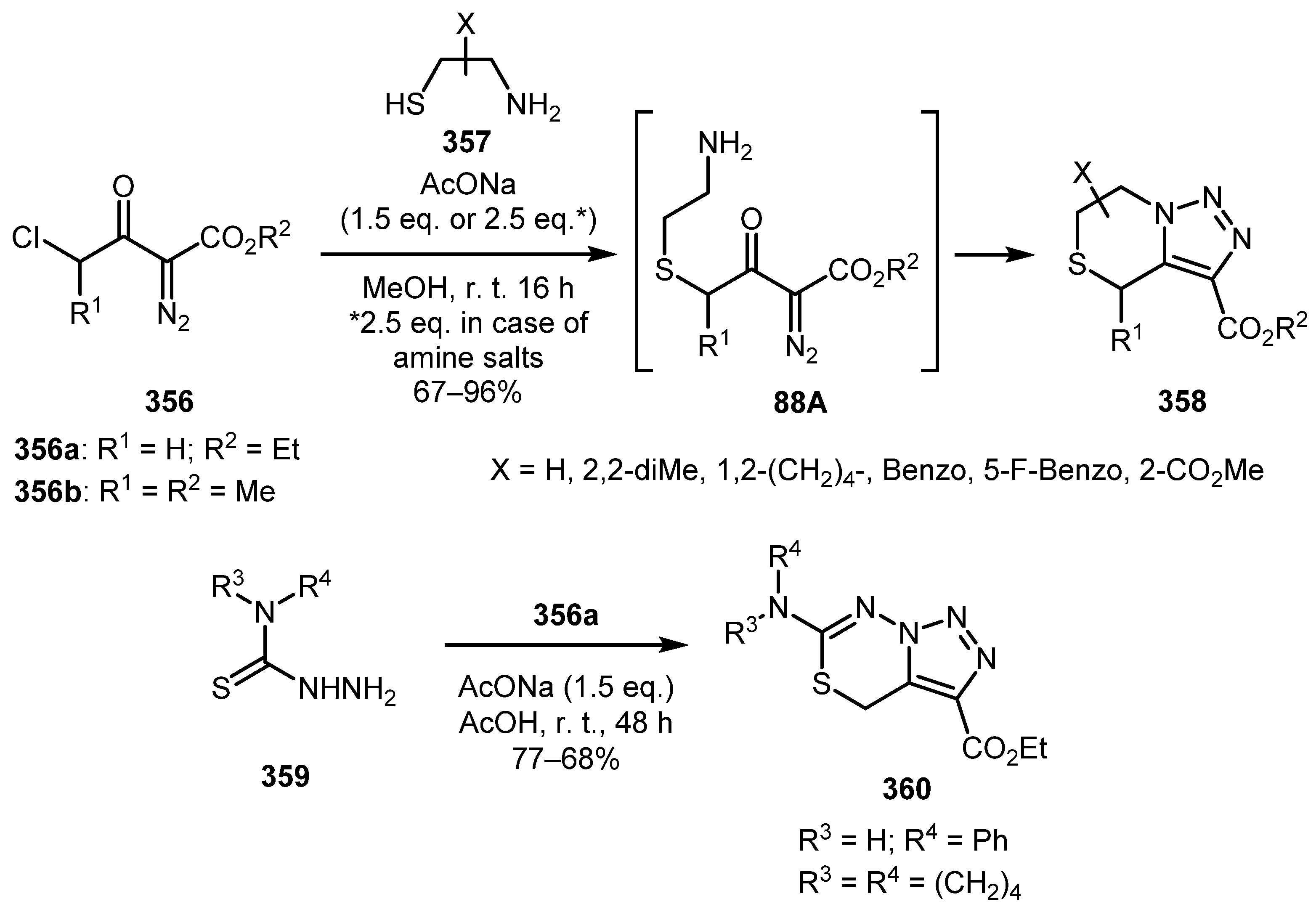
Combining the halo, keto, and diazo functions in α-diazo-β-keto-γ-haloesters 356 has a potential for constructing fused triazole systems. This potential was realized in a recent work by Krasavin and co-workers who reacted these diazo compounds with aminothiols 357 (Scheme 88) [174]. Initially, halogen is substituted by a more nucleophilic thiol group so that the subsequent imine formation becomes an intramolecular reaction and requires rather mild conditions to proceed. Both aliphatic and aromatic aminothiols worked well, providing 6,7-dihydrotriazolothiazines 358 in good to excellent yields. Attempts to perform this reaction with thiosemicarbazides 359 only resulted in the formation of substitution product, which, however, underwent rapid cyclization into triazole 360 in pure acetic acid (Scheme 88).
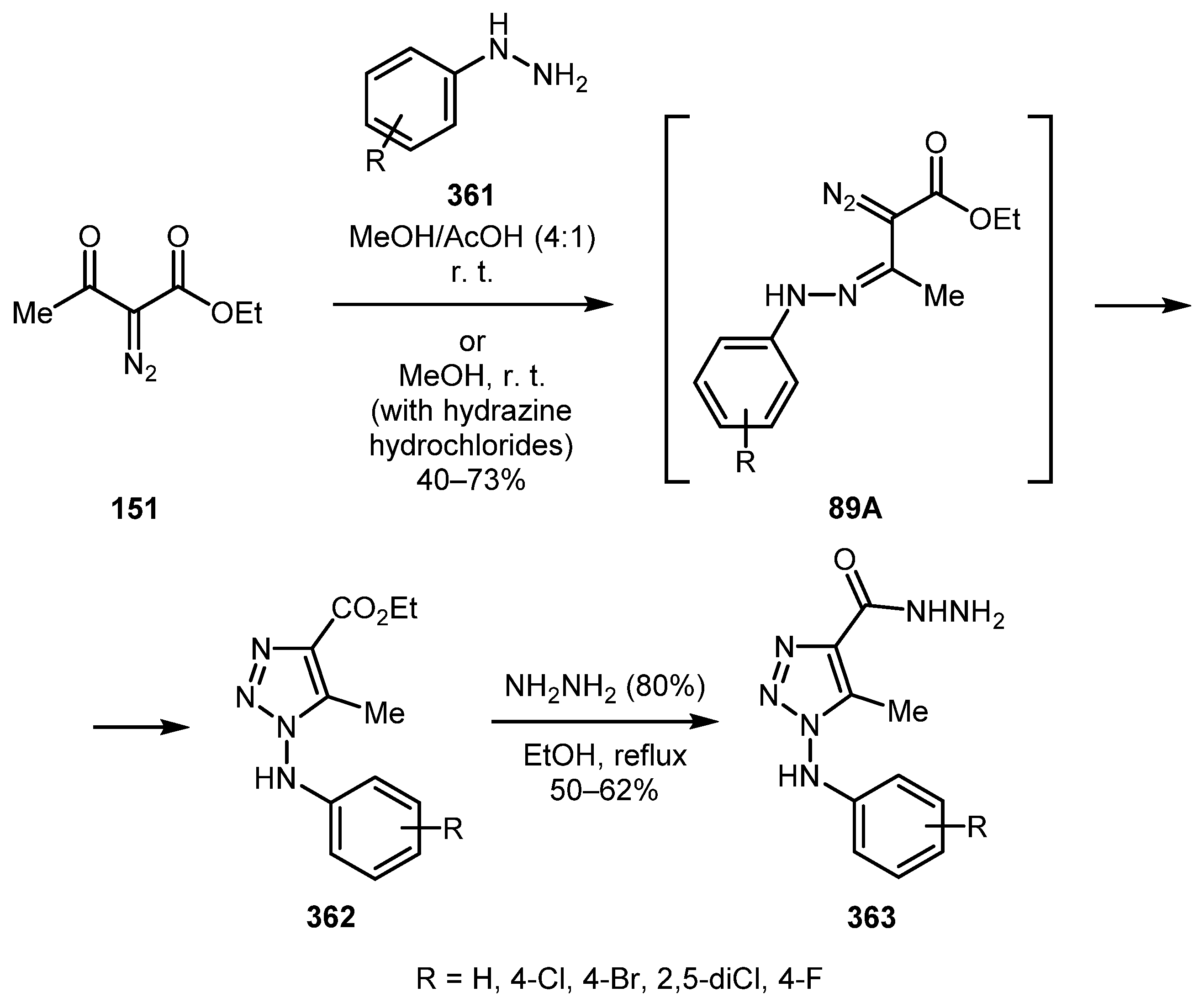
The reaction of diazo ketone 151 with substituted hydrazines 361 occurs under mild conditions and acetic acid as acidic promoter is usually sufficient for formation of hydrazone intermediates 89A [175]. Jordão and co-workers used this approach in their work on the synthesis and antiviral testing of N-amino-1,2,3-triazoles 362 and their hydrazides 363 against the Cantagalo virus (Scheme 89) [176]. The best yield was achieved with unsubstituted phenylhydrazine. The yield decreased as electron-withdrawing substituents were introduced. These triazoles were further modified in order to obtain potential anti-HSV-1 [177] and anti-candidiasis [178] agents.
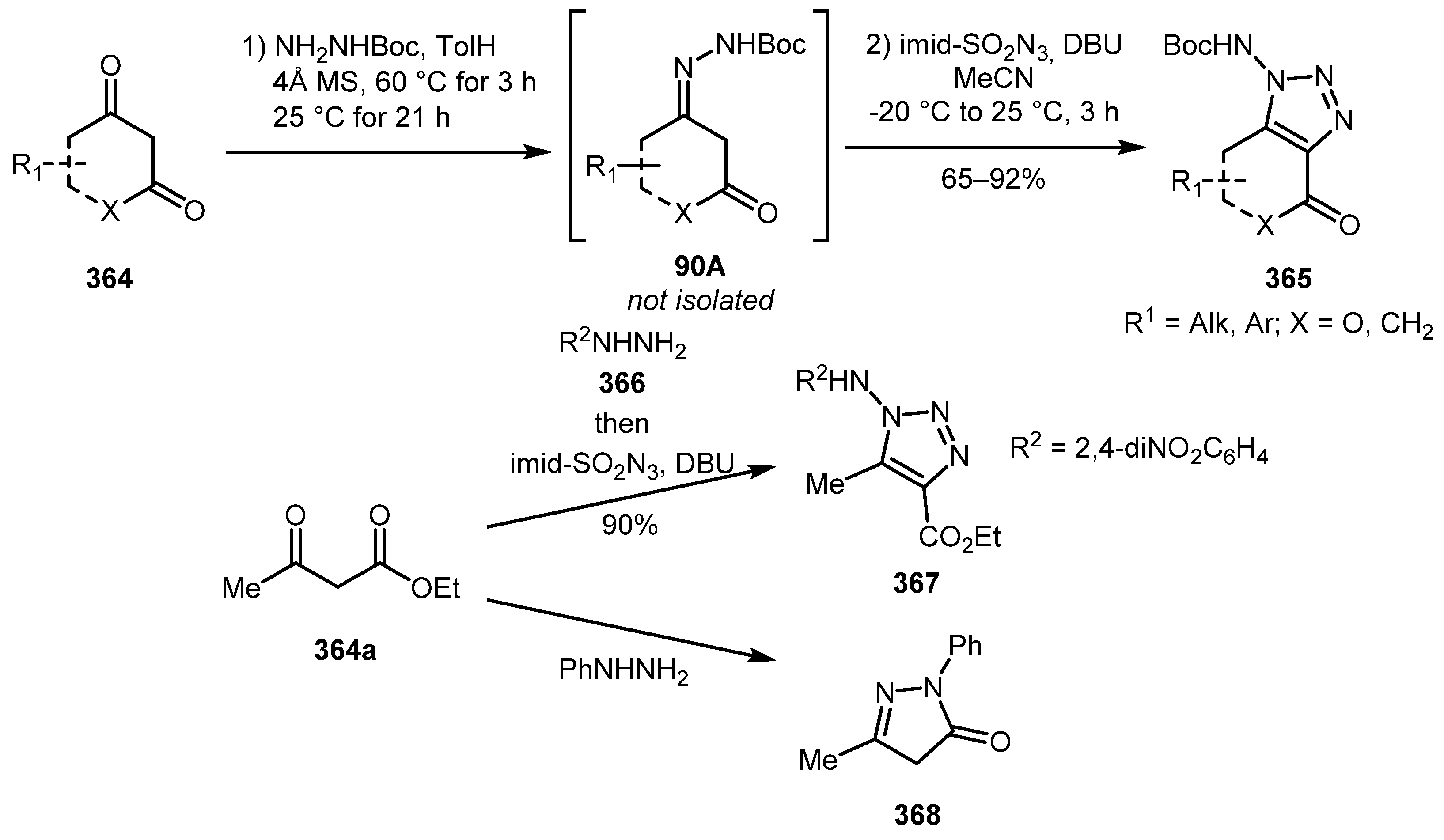
The steps sequence in a «diazo» approach to 1,2,3-triazoles can be reversed so that imine is formed first and then undergoes a diazo transfer reaction. In 2016, the Emmanuvel group developed a one-pot procedure for the synthesis of N-aminotriazoles 365 from β-ketocarbonyl compounds 364 and tert-butyl carbazate [179]. The reaction also worked well with 2,4-dinitrophenylhydrazine 366. However, when phenylhydrazine was involved into this process, pyrazolone 368 formation was observed instead of the desired 1,2,3-triazole (Scheme 90). In other work by this group, authors were able to isolate the intermediate diazo hydrazones using different diazo transfer conditions [180]. A deacylative variation of diazo transfer on enaminones for the synthesis of 1,5-disubstituted triazoles have also been reported to date [181].
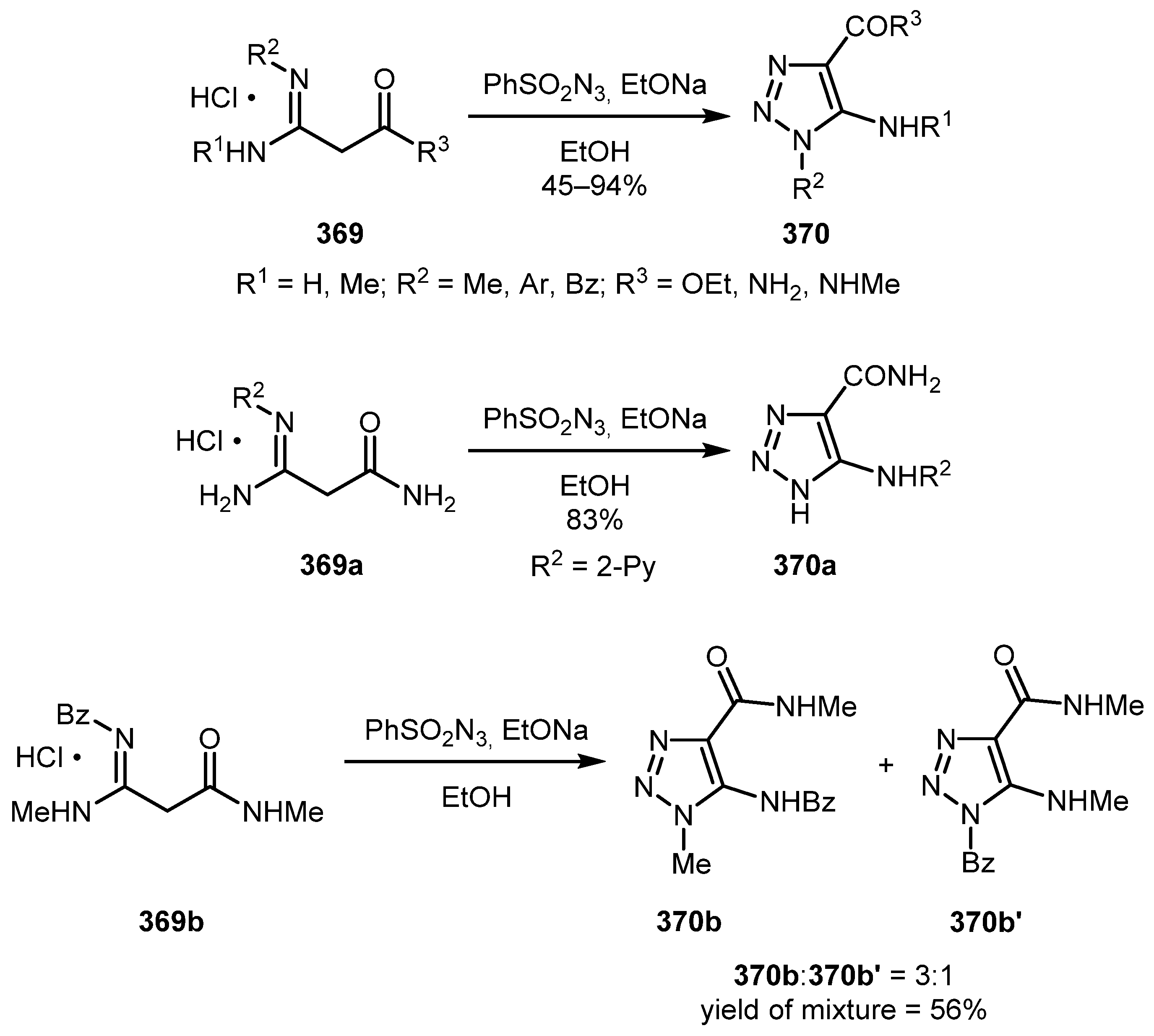
5-Aminotriazoles 370 can be obtained by performing diazo transfer on amidine hydrochlorides 369 as was demonstrated by Dankova and co-authors [182]. The reaction proceeded regioselectively with asymmetric amidines, except for 369b when a 3:1 mixture of isomeric triazoles 370b and 370b’ was obtained (Scheme 91).
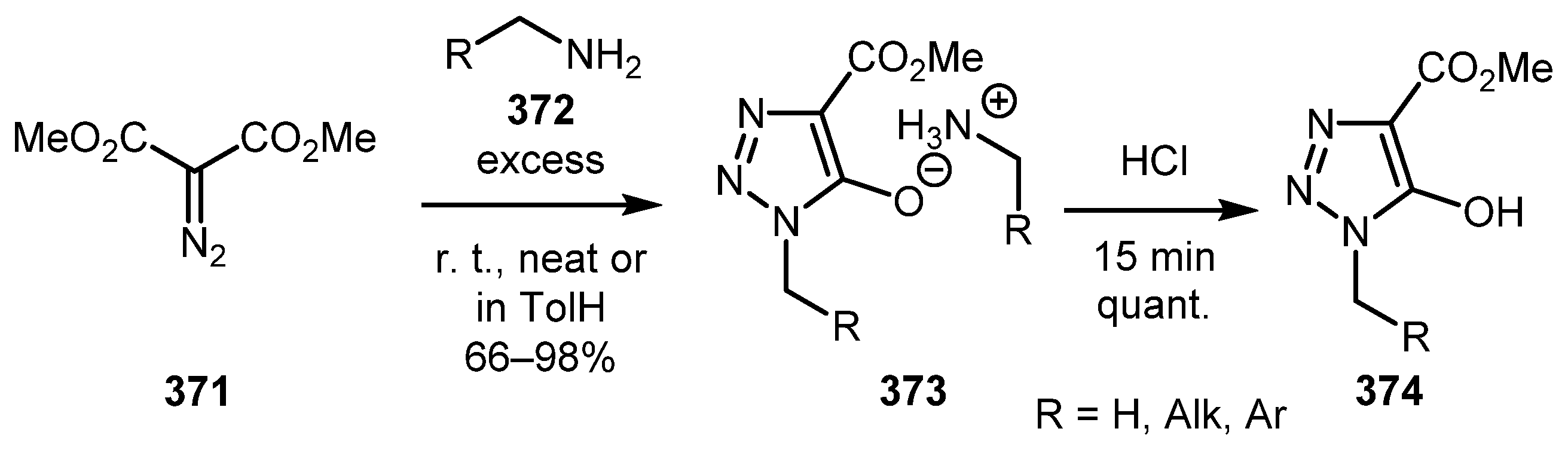
α-Diazoamides are known to undergo cyclization under basic conditions to give salts of 5-hydroxy-1,2,3-triazoles [183]. In 1984, Murrey-Rust and coworkers developed a simple method for the preparation of 5-hydroxytriazole ammonium salts 373 by treatment of dimethyl diazomalonate 371 with an excess nuber of amines 372—neat or in toluene solution. (Scheme 92) [184]. The role of amine in this reaction is both as a nucleophile for the formation of diazo amide and as a base for the subsequent cyclization. The limitation of this approach is that aromatic amines fail to react, which is consistent with reduced nucleophilicity (and basicity) of the amino group in aromatic amines. This methodology has been used recently for the preparation of 5-hydroxytriazole derivatives acting as a potential carboxylic acid bioisosters [185,186]. Hydroxytriazole salts can be acidified to afford free 5-hydroxytriazoles 374, but these compounds are often unstable and undergo ring opening to form parent diazo amide species [187]. Some mechanistic investigations of these equilibrium processes have been reported elsewhere [188,189].
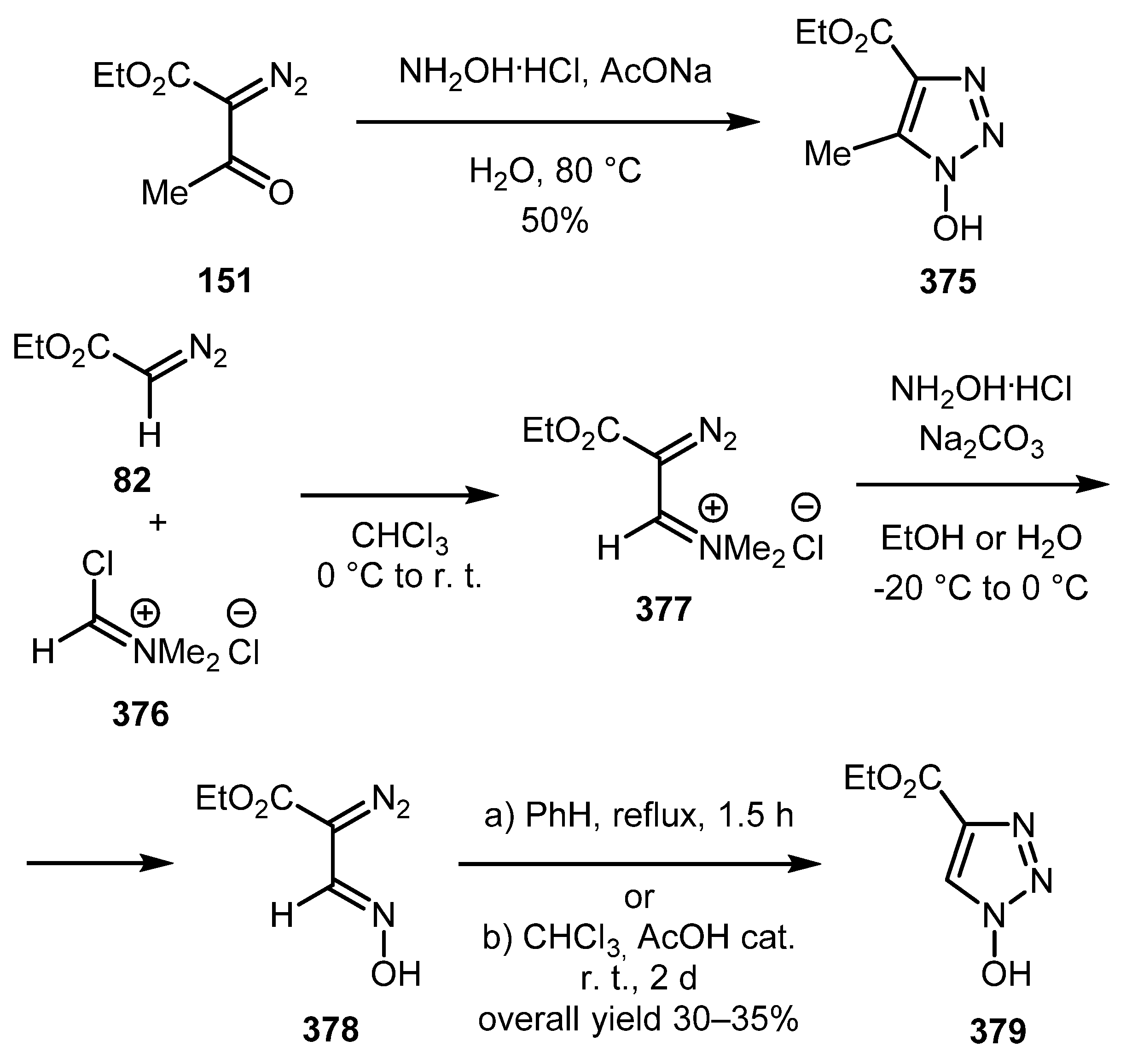
The information on synthesis of N-hydroxytriazoles from diazo compounds is scarce. In a report by Jiang and co-workers devoted to the synthesis of some N-hydroxytriazoles and their evaluation as peptide coupling reagents, 1-hydroxy-5-methyltriazole-4-carboxylate 375 was prepared by the Wolff condensation of α-diazo-β-ketobutanoate 151 with hydroxylamine [190]. This procedure, however, did not work well for the synthesis of 5-unsubstituted 1-hydroxytriazole 379, so an alternative route had to be devised. Ethyl diazoacetate 82 was coupled with Vilsmeier reagent 376 and the product 377 was treated with hydroxylamine. The cyclization of diazo oxime 378 thus obtained was carried out either by refluxing it in benzene or by storing its solution in chloroform at room temperature for 2 days. The overall yield of the target N-hydroxytriazole 379 was fair (Scheme 93).
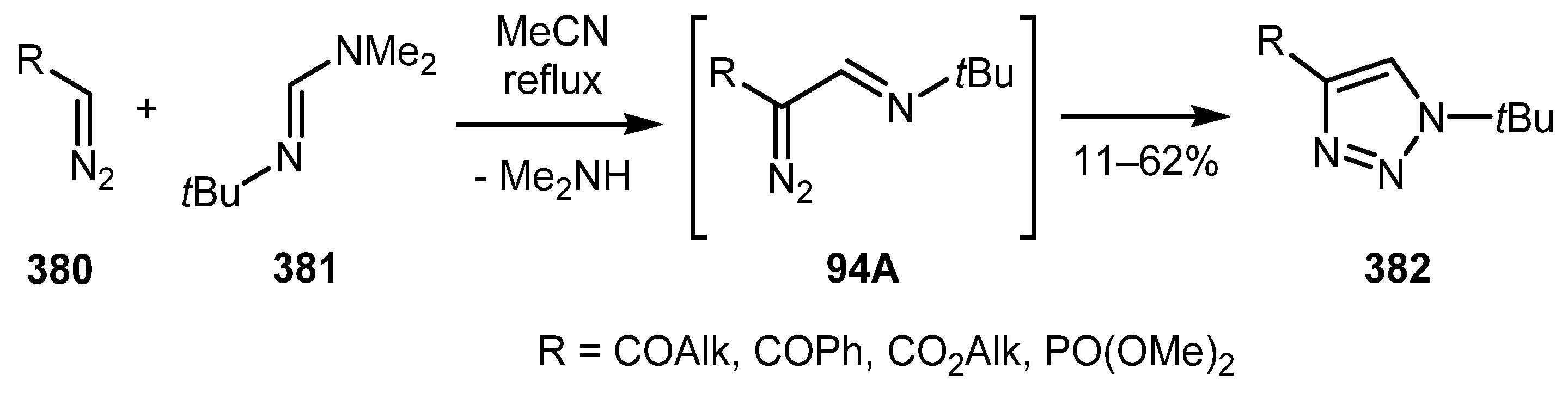
Several approaches to the synthesis of intermediate diazo imines have been reported to date. For example, Regitz and Schoder reported the synthesis of triazoles 382 in which diazo imine intermediate 94A was prepared by iminomethylation of terminal diazo compounds 380 with formamidine 381 under reflux in acetonitrile (Scheme 94) [191].
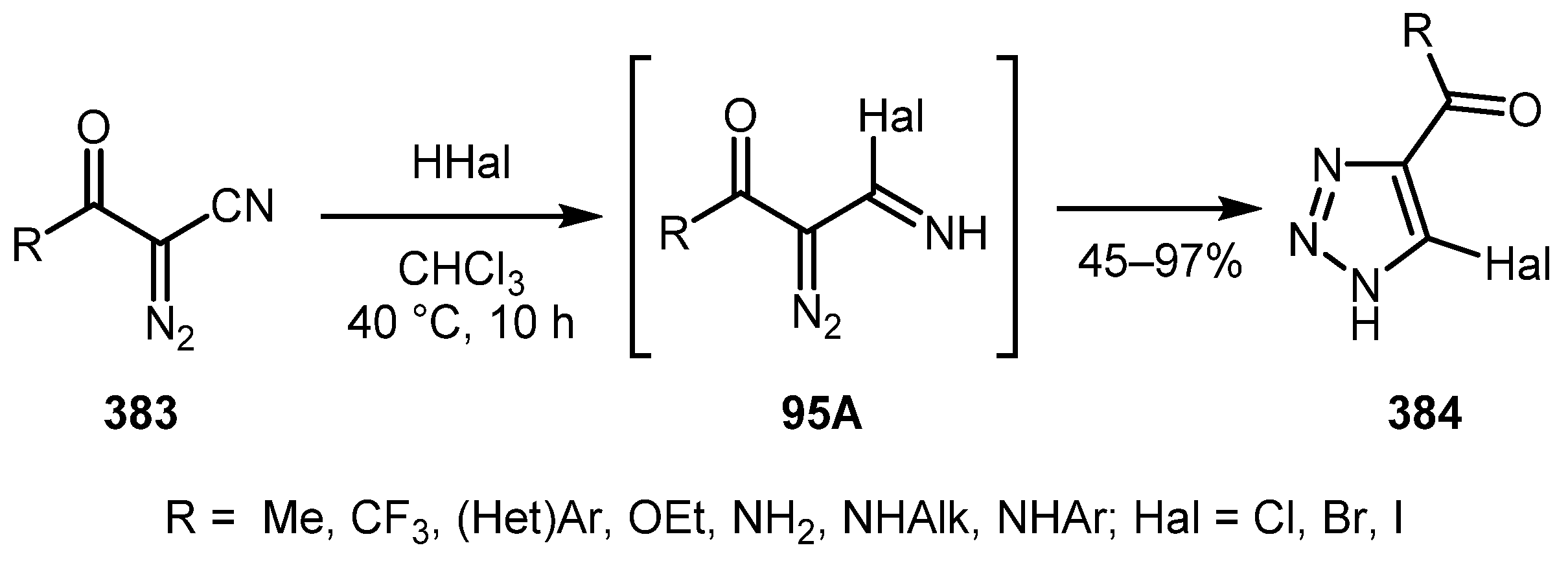
5-Halo-substituted triazoles 384 can be prepared from α-diazo-β-ketonitriles 383 by treatment of the latter with HHal, as was described in a work by the Mokrushin group [192]. The reaction is believed to proceed via diazoimidoyl chloride intermediate 95A. The presence of an acyl group at the α-position of diazo nitrile proved to be crucial for this reaction (Scheme 95). α-Phosphonyl α-diazoacetonitriles was also shown to undergo this transformation.
3.2.2. 1,2,3-Triazoles via Formal Cycloaddition to Multiple CN bonds
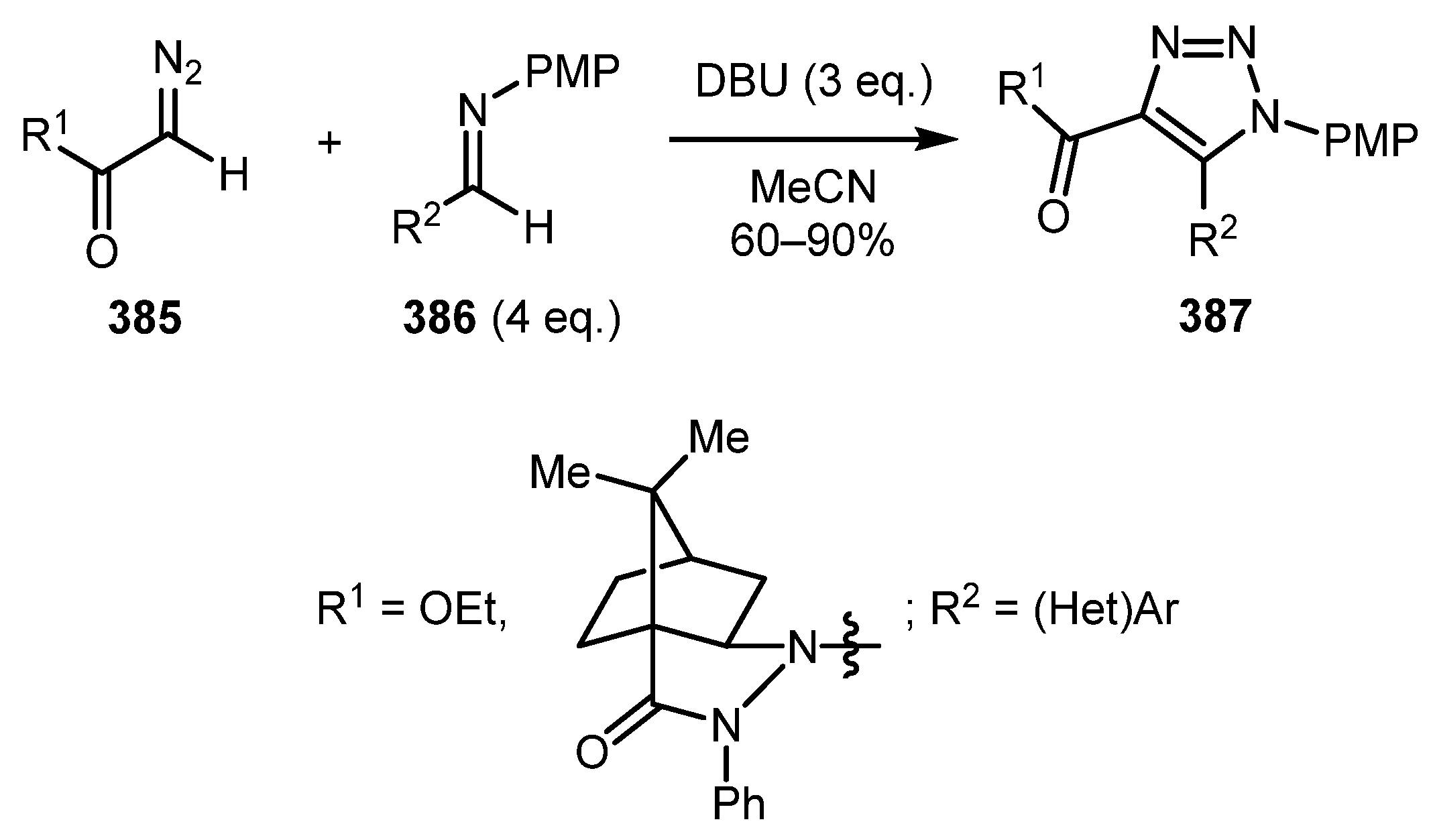
This section describes methods that do not rely on the formation of diazo imine intermediates. In 2010, Chen and co-workers reported a method for the construction of fully substituted triazoles 387 by a DBU-mediated reaction of diazo compounds 385 with N-PMP aryl imines 386 (Scheme 96) [193]. The best yields were obtained with EWG-substituted aryl imines, diminishing in the case of halogen or EDG-substituted ones.
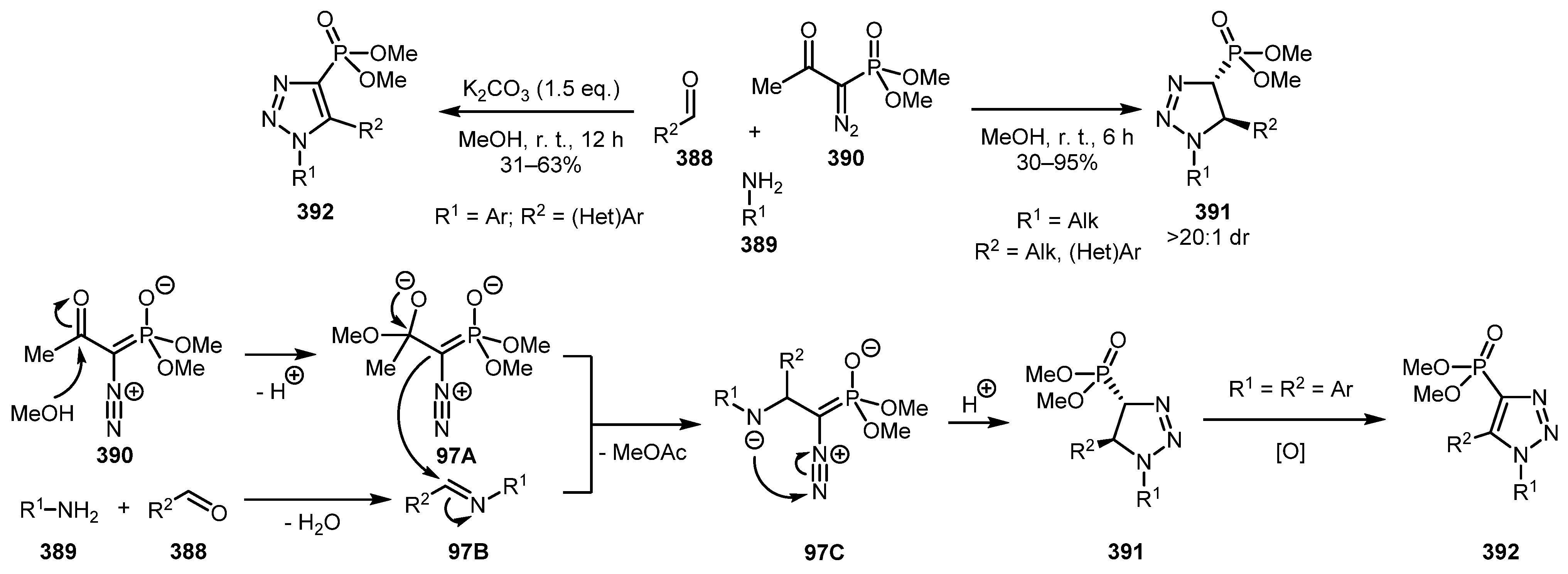
This reaction can be performed in a three-component fashion using aldehyde 388, amine 389, and BOR (390) as the diazo component as was shown by the Mohanan group [194]. The method allows the diastereoselective synthesis of either triazolines 391 or triazoles 392, depending on the nature of the amine and the aldehyde used. The tentative mechanism is as follows. BOR (390), upon deacylation, reacted with imine 97B to give the intermediate 97C. Subsequent 5-endo-dig cyclization and proton transfer furnished the triazoline 391. When the aldehyde and the amine are both aromatic, 391 can undergo a spontaneous air oxidation to form triazole 392 (Scheme 97). Interestingly, in contrast with the previous example, aldehydes bearing EWG-substituents in the aryl moiety failed to deliver the product. An analogous protocol for the preparation of triazole-4-sulfones have been reported by this group [195].
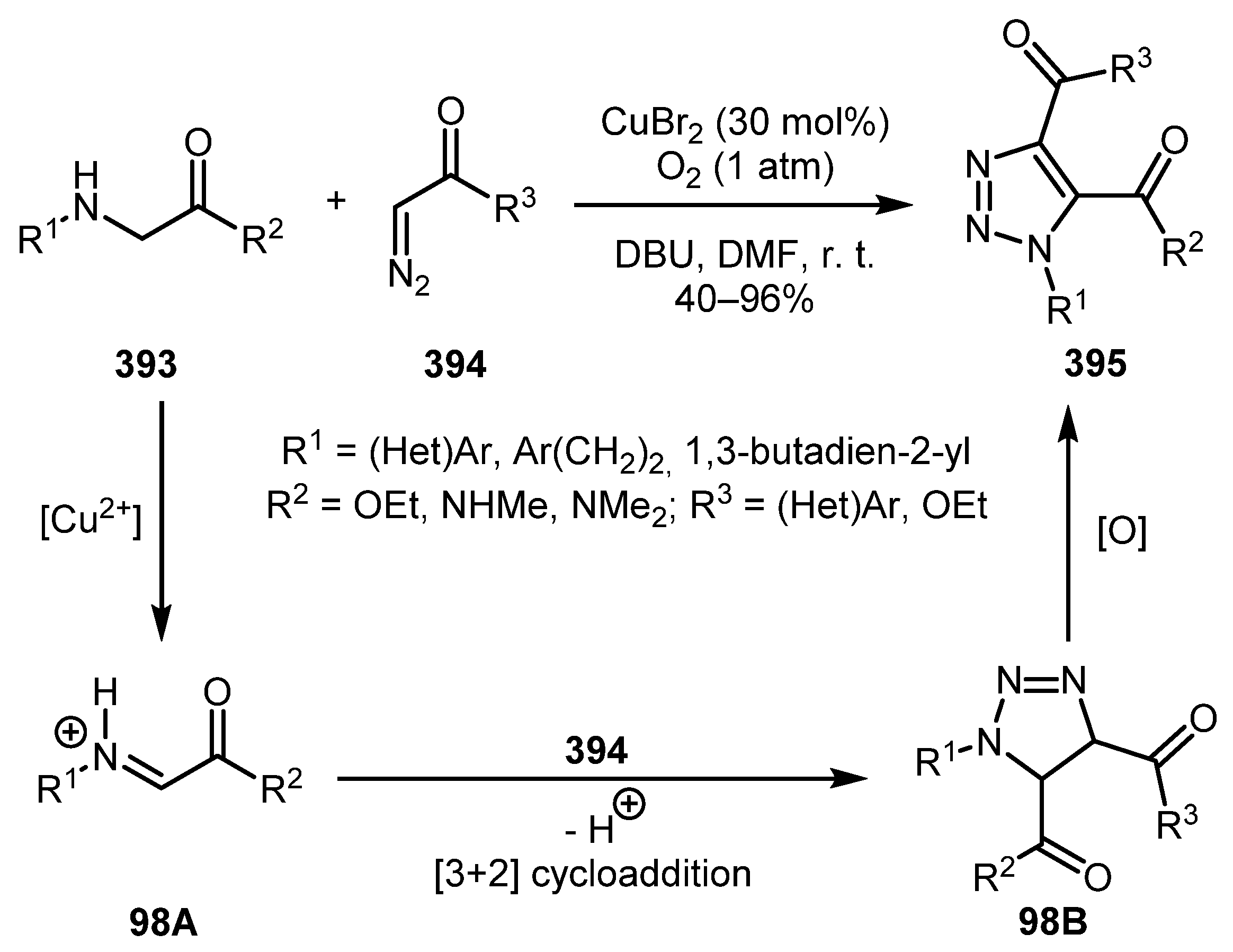
Li and co-workers used CuBr2 oxidation of secondary amines, namely N-arylglycine derivatives 393, for the in situ generation of imines [196]. Their reaction with diazo compounds 394 afforded fully decorated triazoles 395 in moderate to high yields. Mechanistically, the reaction proceeds through iminium salt 98A, which upon cycloaddition with diazo compound 394 forms intermediate 98B. Subsequent oxidation, probably by copper ions, produces the triazole product 395 (Scheme 98).
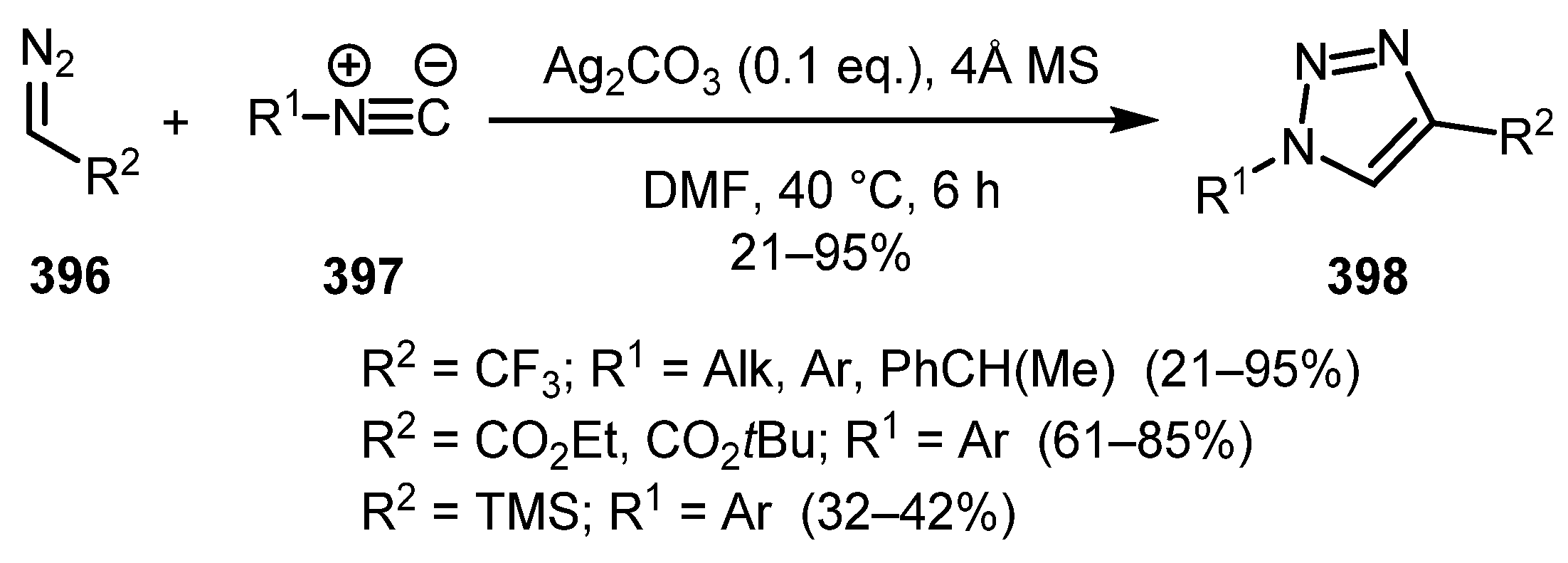
Silver-catalyzed [3+2] cycloaddition of diazo compounds 396 to isocyanides 397 was reported by Wang and co-workers [197]. The reaction proceeded smoothly to give 1,4-disubstituted triazoles 398 with complete regioselectivity (Scheme 99). This protocol is compatible with both EWG- and EDG-substituted aryl isocyanides. However, when aliphatic isocyanides were used, the yields were decreased. As for the diazo compounds, trifluorodiazoethane, diazo esters, and trimethylsilyl diazomethane can be used, although the latter provides the desired triazoles only in moderate yields.
3.3. 1,2,4-Triazoles
1,2,4-Triazoles possess the biological profile that is somewhat similar to that displayed by 1,2,3-triazoles. Moreover, medicinal agents containing 1,2,4-triazole moiety exhibit numerous biological activities such as antifungal, antiparasitic, anxiolytic, antidepressant, anticonvulsant, etc. [198]. Besides, 1,2,4-triazole derivatives are used as ligand in coordination chemistry [199] and as a building blocks for the construction of coordination polymers and metal organic frameworks [200]. Classical methods for the synthesis of 1,2,4-triazoles include condensation of substituted hydrazines with imides, reaction of amides with hydrazides, and coupling of amidrazones with acyl chlorides, although many other methodologies have been developed [201]. However, there are only few reports on the synthesis of 1,2,4-triazoles using diazo compounds.
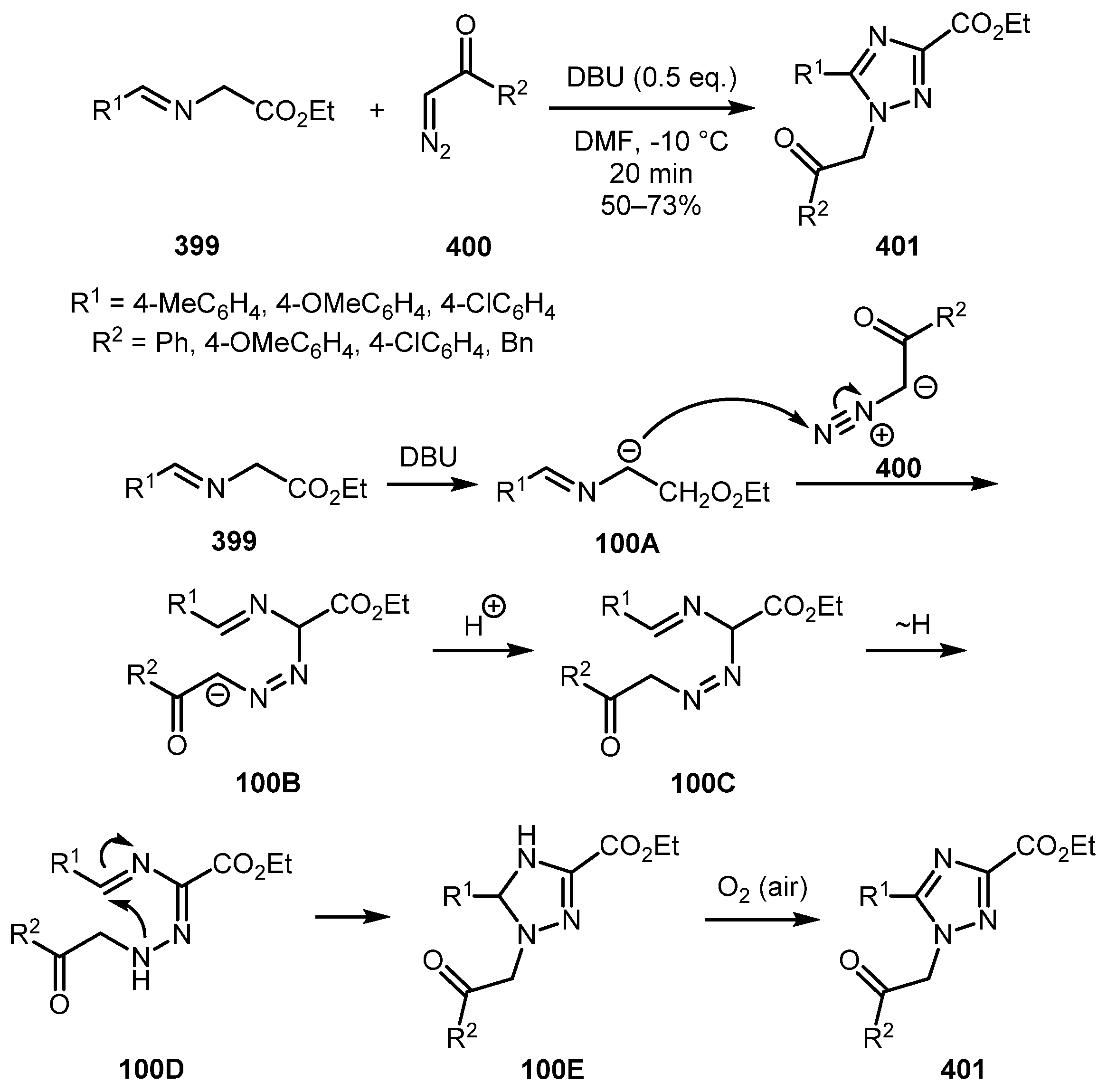
A protocol for the synthesis of 1,2,4-triazole-3-carboxylates 401 from imines 399 and diazo ketones 400 was devised by Zhao and co-wrokers [202]. The reaction involved a nucleophilic attack of anion 100A, generated upon deprotonation of imine 399 with DBU, onto a terminal diazo nitrogen of 400 followed by proton transfer and tautomerization to give the hydrazone intermediate 100D. Subsequent cyclization and air oxidation furnishes the final triazole product 401 (Scheme 100). Interestingly, only terminal α-diazoketones produced 1,2,4-triazoles, whereas diazo esters gave tetrahydro-1,2,4-triazines.
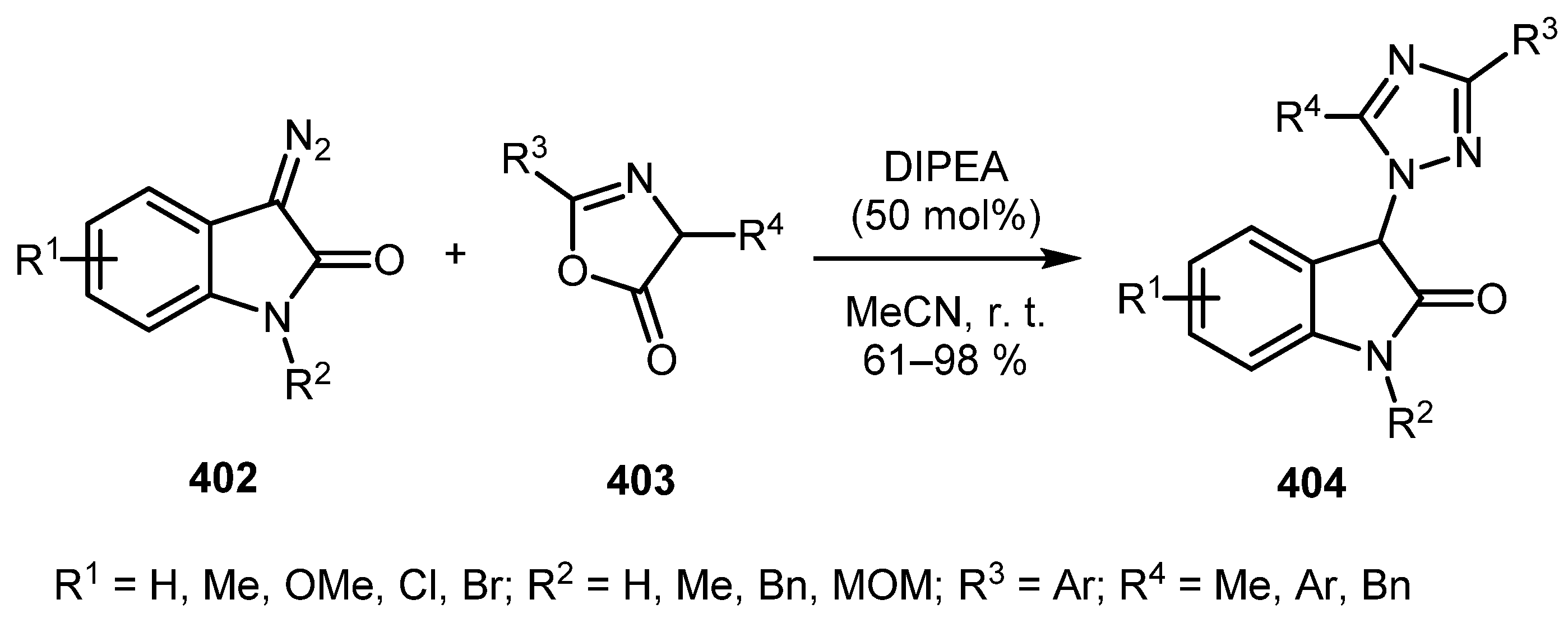
In 2018, the Chen group described a base-mediated reaction of diazo oxindoles 402 with oxazol-5-(4H)-ones 403 affording 3-(1H-1,2,4-triazol-1-yl)indolin-2-ones 404 in reasonable yields (Scheme 101) [203]. The best results were obtained with electron-donating R1 groups on the phenyl ring of 402. As for the oxazolones 403, electron-poor aryl rings as the R3 substituents provided the desired product in higher yields in comparison with electron-rich ones. Noteworthily, when 4-unsubstituted oxazolone 403 (R4 = H) was used, the reaction failed to give any triazole product. Attempts to perform the reaction with diazo dicarbonyl compounds and a vinyl diazo compound did not work as well. The mechanism of the reaction is presumably similar to the previous example.
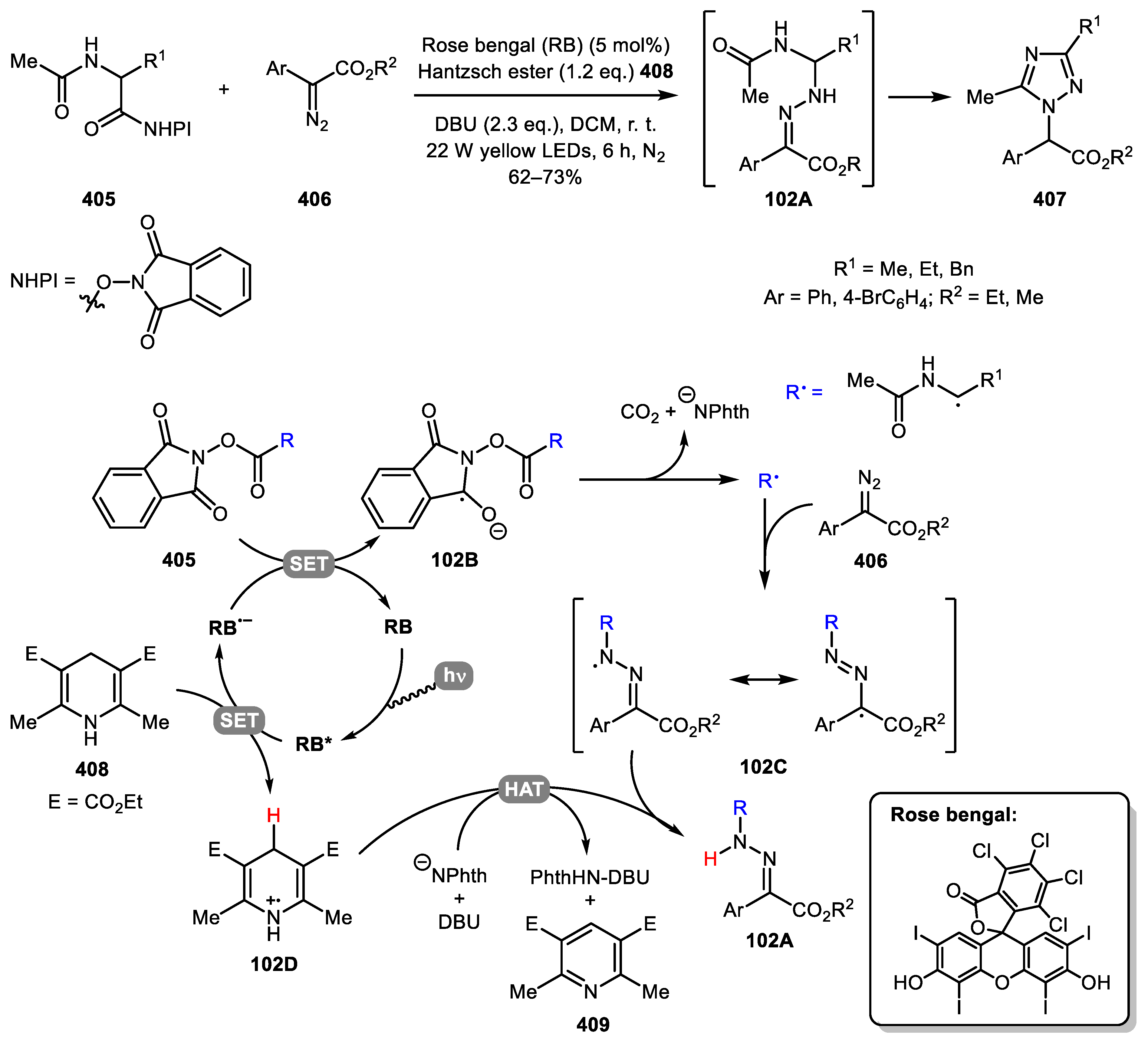
Radical reactions of diazo compounds have also been employed for the assembly of hydrazones with subsequent cyclization to 1,2,4-triazoles. Chan and co-workers developed a photoredox decarboxylative C(sp3)−N coupling of N-hydroxyphthalimide (NHPI) esters with α-diazoacetates 406 for the synthesis of hydrazones [204]. When N-acetyl amino acids derived NHPI esters 405 were used, the initially formed hydrazones 102A underwent cyclization to give 1,2,4-triazoles 407 in good yields. Mechanistically, the reaction begins with photoexcitation of Rose bengal (RB) with 585 nm yellow LED irradiation. An excited state RB* reacts with Hantzsch ester 408 by single-electron transfer (SET) process to generate the Rose bengal radical anion (RB•−), which reduces the NHPI ester 405 to form radical anion 102B. Subsequent C–C bond fragmentation gives alkyl radical R•, which reacts with the terminal diazo nitrogen of 406 to produce intermediate 102C. Following hydrogen atom transfer (HAT) from the cationic radical, Hantzsch ester 102D provides hydrazone 102A. Finally, 102A undergoes cyclization to afford the desired 1,2,4-triazole product 407 (Scheme 102).
A three-component protocol for the preparation of 1,2,4-triazoles 413 from aryl diazonium salts 410, diazo esters 411, and nitriles 412 was developed by the Wan group [205]. EDG-substituted aryl diazonium salts performed better than EWG-substituted ones. Based on preliminary mechanistic studies and DFT-calculations, the authors suggest the following mechanism for the reaction. Nitrile 412 most likely attacks copper carbene 103A to form intermediate 103B, which then undergoes a highly regioselective [3+2] cycloaddition to diazonium salt 410 to afford cyclic intermediate 103C. The latter suffers base-mediated isomerization to deliver the desired 1,2,4-triazole 413 (Scheme 103).
Diazo azoles, namely diazo pyrazoles 414 and diazo imidazoles 417, were employed for the synthesis of N-azolyl-1,2,4-triazoles 416 and 418 in a study reported by Sadchikova and co-workers [206]. The reaction of 414 and 417 with ethyl isocyanoacetate 415 proceeded smoothly to afford the desired products in good to excellent yields (Scheme 104). Interestingly, when a primary or secondary amide moiety was present in the diazo heterocyclic substrate, the reaction gave azolotriazinones instead of triazoles.
4. Azoles with Four Heteroatoms
Tetrazoles
Tetrazole derivatives are known to be high-energy compounds. They have found application in explosives, propellants, and pyrotechnics. In these fields, tetrazoles are used either as individual compounds [207] or as complexes with metals, which are called energetic metal-organic frameworks (EMOFs) [208]. Several tetrazoles have been prepared which outperform classic high performance explosives, such as RDX [207]. As for medicinal chemistry, 5-substituted tetrazoles are commonly considered as carboxylic acids bioisosteres whereas 1,5-disubstituted ones are used as amide surrogates. Moreover, tetrazoles possess remarkable metabolic stability. According to the Drug Bank, there are more than 40 drugs that exhibit antimicrobial, anticancer [209], antiallergic, hypertensive, nootropic, and other biological activities [210]. The general approach for the synthesis of 1-unsubstituted and 1,5-disubstituted tetrazoles is the reaction of nitriles or imidoyl derivatives with inorganic or organic azides, respectively [210,211]. In contrast to these methodologies, the synthesis of tetrazoles using diazo compounds provides access to an alternative regioisomer, namely, 2,5-disubstituted tetrazole, with high regioselectivity.
In 1982, Aoyama and Shioiri reported a reaction of a 2-fold excess of lithium trimethylsilyl diazomethane 419 with methyl esters 420, which afforded 2-substituted-5-TMS-tetrazoles 421 in generally good yields [212]. The authors propose the following mechanism for the reaction. Lithium derivative 419 initially attacks the ester carbonyl carbon of 420 producing intermediate 105A. Either the 1,3-dipolar cycloaddition of 105A to 419 or the nucleophilic attack at the terminal diazo nitrogen of 105A with a second molecule of 419, followed by cyclization, leads to the formation of intermediate 105B. The latter is hydrolyzed during the aqueous work-up to give the desired tetrazole products 421. Similar «dimerization» of α-diazophosphonates has also been reported (Scheme 105) [213].
In recent years, an alternative «diazo» approach for the preparation of tetrazoles, which is based on the reaction of terminal diazo compounds with aryl diazonium salts, has been extensively studied. In 2015, the Ma group for the first time described a silver acetate-catalyzed [3+2]-cycloaddition of diversely substituted aryl diazonium tetrafluoroborates 422 and 2,2,2-trifluorodiazoethane 423 for the synthesis of 2-aryl-5-trifluoromethyltetrazoles 424 [214]. The reaction tolerated a wide range of electron-donating and electron-withdrawing substituents in the aryl ring. Some heterocyclic diazonium salts were also found to be compatible with this protocol. Moreover, the reaction could be performed in a one-pot diazotization/cycloaddition sequence starting from anilines 425. In this case, electron-deficient substrates showed better yields in comparison with electron-rich ones (Scheme 106).
Further developing the above methodology, the same group demonstrated its utility for the synthesis of CF2-functionalized tetrazoles. ((2-Diazo-1,1-difluoroethyl)sulfonyl)benzene 426 was used as a diazo reagent to afford the desired tetrazoles 427 in good to excellent yields [215]. As in the previous example, the reaction can also be performed in a one-pot fashion. More importantly, the obtained tetrazoles 427 can be easily converted to the medicinally relevant CHF2-substituted tetrazoles 428 either by treatment with tBuOK or by reduction with LiAlH4 (Scheme 107).
In a 2016 study by Patouret and Kamenecka, trimethylsilyl diazomethane 42 was involved into this reaction in the presence of CsF to obtain 5-unsubstituted 2-aryltetrazoles 429 [216]. In this case, CF3CO2Ag was found to be the silver source of choice. The use of a stoichiometric amount of silver salt (1.2 eq.) and low temperature (−78 °C) proved to be crucial for a successful reaction. When the reaction was carried out in the absence of CsF, 5-TMS-substituted tetrazole 430 could be isolated. The latter can be used for the preparation of 5-bromotetrazole 431, which has the potential for further modifications via cross-coupling reactions (Scheme 108).
α-Diazoketones along with α-diazoamides (433) were first employed for this reaction by Krasavin and co-workers to obtain 2-aryl-5-acyltetrazoles 434 in moderate to good yields (Scheme 109) [217]. In this case, silver nitrate as a catalyst showed the best results. As a diazonium reagent, aryl diazonium tosylates 432 were used as they are more stable and convenient to handle compared to the respective tetrafluoroborates. The reaction was found to be not particularly sensitive to substituent effects in the diazonium moiety. As for the diazo portion, aromatic and heteroaromatic diazo ketones performed better in comparison with aliphatic diazo ketone and diazo amides. Recently, α-diazoesters [218] and the Seyferth–Gilbert reagent [219] have been involved in this reaction by the Ma group.
In a 2020 work by the Krasavin group, it was demonstrated that (hetero)aryl-, alkylsulfonyl, and aminosulfonyl diazomethanes 435 are also applicable for this reaction [220]. Thus, the rare 2-aryltetrazol-5-yl sulfones and previously not described in the literature 2-aryltetrazol-5-yl sulfonamides 436 were prepared in generally good yields. However, diminished yields were observed with aryl diazonium tosylates 432 bearing electron-donating or ortho-substituents. The following plausible mechanism was suggested (which can be presumably extended also to the examples discussed above). Diazo substrate 435 was deprotonated by the base and bound by the Ag cation to give intermediate 110A. The latter coordinated with diazonium salt in a way that unambiguously determines the regiochemistry of the final product, i.e., providing intermediate 110C. Subsequent silver-assisted cycloaddition with the liberation of the catalyst delivers the desired tetrazole 436 (Scheme 110).
5. Conclusions
As we have demonstrated in this review, diazocarbonyl and related compounds provide rapid and efficient access to a wide range of various azoles. Among the most thoroughly studied are approaches to 1,3-oxazoles, pyrazoles, and 1,2,3-triazoles. Some of these have even become common synthetic methodologies in the organic chemist’s toolbox. On the other hand, the synthesis of 1,3-thiazoles, imidazoles, 1,2,3-thiadiazoles, and 1,2,4-triazoles is less developed. To date, information on the synthesis of isoxazoles from diazocarbonyl compounds has been limited to only a few reports. Most recently, great progress has been achieved in the synthesis of tetrazoles from various diazo compounds. However, it is obvious that the synthetic potential of diazo compounds in this area is far from its full disclosure. In addition, despite the extensive research, some mechanistic details are still not clear and need further investigation. The main advantage of «diazo» approaches to azoles is their high, often absolute regioselectivity. The utility of these methods is emphasized by numerous examples of the use of diazo compounds at key steps of the total synthesis of azole-containing natural products. Given the easy availability of diazo substrates, the described methodologies represent a valuable alternative to more conventional methods.
Author Contributions
Conceptualization, A.B., D.D. and M.K.; literature search, A.B. and D.D.; figure drawing, A.B.; writing—original draft preparation, A.B.; writing—review and editing, D.D., G.K. and M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Russian Science Foundation (project grant 20-13-00024).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ford, A.; Miel, H.; Ring, A.; Slattery, C.N.; Maguire, A.R.; McKervey, M.A. Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev. 2015, 115, 9981–10080. [Google Scholar] [CrossRef]
- Medvedev, J.J.; Nikolaev, V.A. Recent advances in the chemistry of Rh carbenoids: Multicomponent reactions of diazocarbonyl compounds. Russ. Chem. Rev. 2015, 84, 737–757. [Google Scholar] [CrossRef]
- Ciszewski, Ł.W.; Rybicka-Jasińska, K.; Gryko, D. Recent developments in photochemical reactions of diazo compounds. Org. Biomol. Chem. 2019, 17, 432–448. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, J. Gold-catalyzed transformations of α-diazocarbonyl compounds: Selectivity and diversity. Chem. Soc. Rev. 2016, 45, 506–516. [Google Scholar] [CrossRef]
- Carreras, V.; Tanbouza, N.; Ollevier, T. The Power of Iron Catalysis in Diazo Chemistry. Synthesis 2021, 53, 79–94. [Google Scholar]
- Da Silva, F.D.C.; Jordao, A.K.; da Rocha, D.R.; Ferreira, S.B.; Cunha, A.C.; Ferreira, V.F. Recent Advances on the Synthesis of Heterocycles from Diazo Compounds. Curr. Org. Chem. 2012, 16, 224–251. [Google Scholar] [CrossRef]
- Burtoloso, A.C.B.; Dias, R.M.P.; Bernardim, B. α,β-Unsaturated Diazoketones as Useful Platforms in the Synthesis of Nitrogen Heterocycles. Acc. Chem. Res. 2015, 48, 921–934. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Ha, S.; Park, C.-M. α-Diazo oxime ethers for N-heterocycle synthesis. Chem. Commun. 2017, 53, 6054–6064. [Google Scholar] [CrossRef]
- Xiang, Y.; Wang, C.; Ding, Q.; Peng, Y. Diazo Compounds: Versatile Synthons for the Synthesis of Nitrogen Heterocycles via Transition Metal-Catalyzed Cascade C–H Activation/Carbene Insertion/Annulation Reactions. Adv. Synth. Catal. 2019, 361, 919–944. [Google Scholar] [CrossRef]
- Cheng, Q.-Q.; Deng, Y.; Lankelma, M.; Doyle, M.P. Cycloaddition reactions of enoldiazo compounds. Chem. Soc. Rev. 2017, 46, 5425–5443. [Google Scholar] [CrossRef]
- Cheng, Q.-Q.; Yu, Y.; Yedoyan, J.; Doyle, M.P. Vinyldiazo Reagents and Metal Catalysts: A Versatile Toolkit for Heterocycle and Carbocycle Construction. ChemCatChem 2018, 10, 488–496. [Google Scholar] [CrossRef]
- Jamali, M.F.; Vaishanv, N.K.; Mohanan, K. The Bestmann-Ohira Reagent and Related Diazo Compounds for the Synthesis of Azaheterocycles. Chem. Record 2020, 20, 1394–1408. [Google Scholar] [CrossRef] [PubMed]
- Bresciani, S.; Tomkinson, N.C.O. Transition Metal-Mediated Synthesis of Oxazoles. Heterocycles 2014, 89, 2479–2543. [Google Scholar] [CrossRef] [Green Version]
- Baiju, T.V.; Namboothiri, I.N.N. Synthesis of Functionalized Pyrazoles via 1,3-Dipolar Cycloaddition of α-Diazo-β-ketophosphonates, Sufones and Esters with Electron-Deficient Alkenes. Chem. Record 2017, 17, 939–955. [Google Scholar] [CrossRef] [PubMed]
- Levashova, E.Y.; Zhukovsky, D.D.; Dar’in, D.V.; Krasavin, M.Y. Synthesis of pyrazoles from α-diazo-β-ketosulfones and α-diazo-β-ketophosphonates. Chem. Heterocycl. Compd. 2020, 56, 806–808. [Google Scholar] [CrossRef]
- Shafran, Y.; Glukhareva, T.; Dehaen, W.; Bakulev, V. Chapter Three - Recent Developments in the Chemistry of 1,2,3-Thiadiazoles. In Advances in Heterocyclic Chemistry; Scriven, E.F.V., Ramsden, C.A., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 126, pp. 109–172. [Google Scholar]
- Zhang, H.-Z.; Zhao, Z.-L.; Zhou, C.-H. Recent advance in oxazole-based medicinal chemistry. Eur. J. Med. Chem. 2018, 144, 444–492. [Google Scholar] [CrossRef]
- Kaur, R.; Palta, K.; Kumar, M.; Bhargava, M.; Dahiya, L. Therapeutic potential of oxazole scaffold: A patent review (2006–2017). Expert Opin. Ther. Pat. 2018, 28, 783–812. [Google Scholar] [CrossRef]
- Jin, Z. Muscarine, imidazole, oxazole, and thiazole alkaloids. Nat. Prod. Rep. 2011, 28, 1143–1191. [Google Scholar] [CrossRef]
- Ibrar, A.; Khan, I.; Abbas, N.; Farooq, U.; Khan, A. Transition-metal-free synthesis of oxazoles: Valuable structural fragments in drug discovery. RSC Adv. 2016, 6, 93016–93047. [Google Scholar] [CrossRef]
- Moody, C.J.; Doyle, K.J. Chapter 1 The synthesis of oxazoles from diazocarbonyl compounds. In Progress in Heterocyclic Chemistry; Gribble, G.W., Gilchrist, T.L., Eds.; Elsevier: Amsterdam, The Netherlands, 1997; Volume 9, pp. 1–16. [Google Scholar]
- Huisgen, R.; König, H.; Binsch, G.; Sturm, H.J. 1.3-Dipolare Additionen der Ketocarbene. Angew. Chem. 1961, 73, 368–371. [Google Scholar] [CrossRef]
- Loy, N.S.Y.; Choi, S.; Kim, S.; Park, C.-M. The synthesis of pyrroles and oxazoles based on gold α-imino carbene complexes. Chem. Commun. 2016, 52, 7336–7339. [Google Scholar] [CrossRef] [PubMed]
- Sakharov, P.A.; Novikov, M.S.; Khlebnikov, A.F. 2-Diazoacetyl-2H-azirines: Source of a Variety of 2H-Azirine Building Blocks with Orthogonal and Domino Reactivity. J. Org. Chem. 2018, 83, 8304–8314. [Google Scholar] [CrossRef] [PubMed]
- Kumaraswamy, G.; Gangadhar, M. A Metal-Free, Organo Lewis Acid Catalyzed Synthesis of Highly Substituted Oxazoles. ChemistrySelect 2019, 4, 8973–8977. [Google Scholar] [CrossRef]
- Kantin, G.; Dar’in, D.; Krasavin, M. RhII-Catalyzed Cycloaddition of α-Diazo Homophthalimides and Nitriles Delivers Oxazolo[5,4-c]isoquinolin-5(4H)-one Scaffold. Eur. J. Org. Chem. 2018, 2018, 4857–4859. [Google Scholar] [CrossRef]
- Dar’in, D.; Kantin, G.; Krasavin, M. A ‘sulfonyl-azide-free’ (SAFE) aqueous-phase diazo transfer reaction for parallel and diversity-oriented synthesis. Chem. Commun. 2019, 55, 5239–5242. [Google Scholar] [CrossRef]
- Gecht, M.; Kantin, G.; Dar’in, D.; Krasavin, M. A novel approach to biologically relevant oxazolo[5,4-d]pyrimidine-5,7-diones via readily available diazobarbituric acid derivatives. Tetrahedron Lett. 2019, 60, 151120. [Google Scholar] [CrossRef]
- Fan, C.; He, X.; Zuo, Y.; Shang, Y. Synthesis of oxazole and furan derivatives via Rh2(OAc)4-catalyzed C≡X bond insertion of cyclic 2-diazo-1,3-diketones with nitriles and arylacetylenes. Synth. Commun. 2018, 48, 2782–2792. [Google Scholar] [CrossRef]
- Vasin, V.A.; Fadin, M.V.; Tarasova, I.V. Synthesis and some transformations of polybrominated quinone diazides. Russ. J. Org. Chem. 2017, 53, 1815–1821. [Google Scholar] [CrossRef]
- Kim, K.S.; Kimball, S.D.; Misra, R.N.; Rawlins, D.B.; Hunt, J.T.; Xiao, H.-Y.; Lu, S.; Qian, L.; Han, W.-C.; Shan, W.; et al. Discovery of Aminothiazole Inhibitors of Cyclin-Dependent Kinase 2: Synthesis, X-ray Crystallographic Analysis, and Biological Activities. J. Med. Chem. 2002, 45, 3905–3927. [Google Scholar] [CrossRef]
- Epple, R.; Cow, C.; Xie, Y.; Azimioara, M.; Russo, R.; Wang, X.; Wityak, J.; Karanewsky, D.S.; Tuntland, T.; Nguyêñ-Trân, V.T.B.; et al. Novel Bisaryl Substituted Thiazoles and Oxazoles as Highly Potent and Selective Peroxisome Proliferator-Activated Receptor δ Agonists. J. Med. Chem. 2010, 53, 77–105. [Google Scholar] [CrossRef] [PubMed]
- Barceló, M.; Raviña, E.; Varela, M.J.; Brea, J.; Loza, M.I.; Masaguer, C.F. Potential atypical antipsychotics: Synthesis, binding affinity and SAR of new heterocyclic bioisosteric butyrophenone analogues as multitarget ligands. Med. Chem. Comm. 2011, 2, 1194–1200. [Google Scholar] [CrossRef]
- Bagley, M.C.; Buck, R.T.; Hind, S.L.; Moody, C.J.; Slawin, A.M.Z. A New Route to Functionalised Oxazoles. Synlett 1996, 1996, 825–826. [Google Scholar] [CrossRef]
- Bagley, M.C.; Buck, R.T.; Hind, S.L.; Moody, C.J. Synthesis of functionalised oxazoles and bis-oxazoles 1. J. Chem. Soc. Perkin Trans. 1 1998, 591–600. [Google Scholar] [CrossRef]
- Reddy, M.R.; Reddy, G.N.; Mehmood, U.; Hussein, I.A.; Rahman, S.U.; Harrabi, K.; Subba Reddy, B.V. Copper(II) Triflate Catalyzed Synthesis of 2,4-Disubstituted Oxazoles from α-Diazoketones. Synthesis 2015, 47, 3315–3320. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, X.; Huang, J.; Zeng, W. Rhodium(I)-Catalyzed Coupling–Cyclization of C=O Bonds with α-Diazoketones. Org. Lett. 2018, 20, 3980–3983. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Blake, A.J.; Lewis, W.; Campbell, I.B.; Judkins, B.D.; Moody, C.J. Rhodium Carbene Routes to Oxazoles and Thiazoles. Catalyst Effects in the Synthesis of Oxazole and Thiazole Carboxylates, Phosphonates, and Sulfones. J. Org. Chem. 2010, 75, 152–161. [Google Scholar] [CrossRef]
- Nagaraju, A.; Sandeep, K.; Kumara Swamy, K.C. Copper catalyzed access to functionalized oxazoles from oximes via carbenoids. Tetrahedron Lett. 2018, 59, 2238–2242. [Google Scholar] [CrossRef]
- Trujillo, J.I.; Arnold, E.P.; Kortum, S.; Robinson, R.P. Facile Synthesis of 2,4-Disubstituted Thiooxazoles and 2,4-Disubstituted Oxazole Sulfonyl Chlorides via Acyl Isothiocyanates and TMS-Diazomethane. Synlett 2015, 26, 1764–1768. [Google Scholar] [CrossRef] [Green Version]
- Linder, J.; Moody, C.J. The total synthesis of siphonazole, a structurally unusual bis-oxazole natural product. Chem. Commun. 2007, 1508–1509. [Google Scholar] [CrossRef]
- Dexter, H.L.; Williams, H.E.L.; Lewis, W.; Moody, C.J. Total Synthesis of the Post-translationally Modified Polyazole Peptide Antibiotic Goadsporin. Angew. Chem. Int. Ed. 2017, 56, 3069–3073. [Google Scholar] [CrossRef] [Green Version]
- Inman, M.; Dexter, H.L.; Moody, C.J. Total Synthesis of the Cyclic Dodecapeptides Wewakazole and Wewakazole, B. Org. Lett. 2017, 19, 3454–3457. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Barco, A.; Benetti, S.; Pollini, G.P.; Simoni, D. Synthesis of Natural Products via Isoxazoles. Synthesis 1987, 1987, 857–869. [Google Scholar] [CrossRef]
- Galenko, E.E.; Khlebnikov, A.F.; Novikov, M.S. Isoxazole-azirine isomerization as a reactivity switch in the synthesis of heterocycles. Chem. Heterocycl. Compd. 2016, 52, 637–650. [Google Scholar] [CrossRef]
- Hu, F.; Szostak, M. Recent Developments in the Synthesis and Reactivity of Isoxazoles: Metal Catalysis and Beyond. Adv. Synth. Catal. 2015, 357, 2583–2614. [Google Scholar] [CrossRef]
- Zhu, J.; Mo, J.; Lin, H.-z.; Chen, Y.; Sun, H.-p. The recent progress of isoxazole in medicinal chemistry. Bioorg. Med. Chem. 2018, 26, 3065–3075. [Google Scholar] [CrossRef]
- Morita, T.; Yugandar, S.; Fuse, S.; Nakamura, H. Recent progresses in the synthesis of functionalized isoxazoles. Tetrahedron Lett. 2018, 59, 1159–1171. [Google Scholar] [CrossRef]
- Duffy, K.J.; Tennant, G.; Wallis, C.J.; Weaver, G.W. Aspects of heterocyclisation reactions mediated by nucleophilic interaction of aromatic nitro groups with ortho heterocumulene side chains. ARKIVOC 2002, 2002, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Ogunlana, A.A.; Fang, S.; Long, W.; Sun, H.; Bao, X.; Wan, X. In situ generation of nitrile oxides from copper carbene and tert-butyl nitrite: Synthesis of fully substituted isoxazoles. Org. Biomol. Chem. 2018, 16, 4683–4687. [Google Scholar] [CrossRef]
- Wang, X.-D.; Zhu, L.-H.; Liu, P.; Wang, X.-Y.; Yuan, H.-Y.; Zhao, Y.-L. Copper-Catalyzed Cascade Cyclization Reactions of Diazo Compounds with tert-Butyl Nitrite and Alkynes: One-Pot Synthesis of Isoxazoles. J. Org. Chem. 2019, 84, 16214–16221. [Google Scholar] [CrossRef]
- Lin, Y.; Fan, H.; Li, Y.; Zhan, X. Thiazole-Based Organic Semiconductors for Organic Electronics. Adv. Mater. 2012, 24, 3087–3106. [Google Scholar] [CrossRef]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent applications of 1,3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef]
- Narasimhamurthy, K.H.; Sajith, A.M.; Joy, M.N.; Rangappa, K.S. An Overview of Recent Developments in the Synthesis of Substituted Thiazoles. ChemistrySelect 2020, 5, 5629–5656. [Google Scholar] [CrossRef]
- Seed, A.J.; Sampson, P. A review of self-organising 2,5- and 2,4-disubstituted 1,3-thiazole-containing materials: Synthesis, mechanisms and tactics. Liq. Cryst. 2017, 44, 1894–1910. [Google Scholar] [CrossRef]
- Kim, H.-S.; Lee, J.-Y.; Kwon, I.-C.H.Z.O. Synthesis of Thiazole Derivatives via Lewis Acid Promoted Reactions of Diazopyruvate with Thioamides. Bull. Korean Chem. Soc. 1995, 16, 4–5. [Google Scholar] [CrossRef]
- Kim, H.-S.; Kwon, I.-C.; Kim, O.-H. Synthesis of 2-Substituted-4-carbethoxythiazoles. J. Heterocycl. Chem. 1995, 32, 937–939. [Google Scholar] [CrossRef]
- Kim, H.-S.; Kwon, I.-C.; Choi, J.-H. Synthesis of crown ethers containing a thiazole subcyclic unit. J. Heterocycl. Chem. 1999, 36, 1285–1289. [Google Scholar] [CrossRef]
- Capuano, L.; Bolz, G.; Burger, R.; Burkhardt, V.; Huch, V. Neue Synthesen von 1,3-Oxazinen, Thiazolen und 1,2-Dithiolen. Liebigs Ann. Chem. 1990, 1990, 239–243. [Google Scholar] [CrossRef]
- Fontrodona, X.; Díaz, S.; Linden, A.; Villalgordo, J.M. Copper(I) Bromide-Mediated Synthesis of Novel 2-Arylthiazole-5-carboxylates from α-Diazo-β-Keto Esters and Aromatic Thioamides. Synthesis 2001, 2001, 2021–2027. [Google Scholar] [CrossRef] [Green Version]
- Obijalska, E.; Błaszczyk, M.; Kowalski, M.K.; Mlostoń, G.; Heimgartner, H. A novel access to 4-trifluoromethyl-1,3-thiazole derivatives via an intermediate thiocarbonyl ylide. J. Fluorine Chem. 2019, 220, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Yadav, J.S.; Reddy, B.V.S.; Rao, Y.G.; Narsaiah, A.V. First example of the coupling of α-diazoketones with thiourea: A novel route for the synthesis of 2-aminothiazoles. Tetrahedron Lett. 2008, 49, 2381–2383. [Google Scholar] [CrossRef]
- Babu, B.H.; Vijay, K.; Murali Krishna, K.B.; Sharmila, N.; Ramana, M.B. An efficient PEG-400 mediated catalyst free green synthesis of 2-amino-thiazoles from α-diazoketones and thiourea. J. Chem. Sci. 2016, 128, 1475–1478. [Google Scholar] [CrossRef] [Green Version]
- Davies, J.R.; Kane, P.D.; Moody, C.J. N–H Insertion reactions of rhodium carbenoids. Part 5: A convenient route to 1,3-azoles. Tetrahedron 2004, 60, 3967–3977. [Google Scholar] [CrossRef]
- Hughes, R.A.; Thompson, S.P.; Alcaraz, L.; Moody, C.J. Total synthesis of the thiopeptide amythiamicin D. Chem. Commun. 2004, 946–948. [Google Scholar] [CrossRef]
- Hughes, R.A.; Thompson, S.P.; Alcaraz, L.; Moody, C.J. Total Synthesis of the Thiopeptide Antibiotic Amythiamicin D. J. Am. Chem. Soc. 2005, 127, 15644–15651. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Dethe, D.H.; Leung, G.Y.C.; Zou, B.; Chen, D.Y.-K. Total Synthesis of Thiopeptide Antibiotics GE2270A, GE2270T, and GE2270C1. Chem. Asian J. 2008, 3, 413–429. [Google Scholar] [CrossRef]
- Srivastava, A.; Shukla, G.; Yadav, D.; Singh, M.S. Access to Fully Substituted Thiazoles and 2,3-Dihydrothiazoles via Copper-Catalyzed [4+1] Heterocyclization of α-(N-Hydroxy/aryl)imino-β-oxodithioesters with α-Diazocarbonyls. J. Org. Chem. 2017, 82, 10846–10854. [Google Scholar] [CrossRef]
- Zhang, L.; Peng, X.-M.; Damu, G.L.V.; Geng, R.-X.; Zhou, C.-H. Comprehensive Review in Current Developments of Imidazole-Based Medicinal Chemistry. Med. Res. Rev. 2014, 34, 340–437. [Google Scholar] [CrossRef]
- Chen, W.-C.; Zhu, Z.-L.; Lee, C.-S. Organic Light-Emitting Diodes Based on Imidazole Semiconductors. Adv. Opt. Mater. 2018, 6, 1800258. [Google Scholar] [CrossRef]
- Heravi, M.M.; Daraie, M.; Zadsirjan, V. Current advances in the synthesis and biological potencies of tri- and tetra-substituted 1H-imidazoles. Mol. Divers. 2015, 19, 577–623. [Google Scholar] [CrossRef]
- Daraji, D.G.; Prajapati, N.P.; Patel, H.D. Synthesis and Applications of 2-Substituted Imidazole and Its Derivatives: A Review. J. Heterocycl. Chem. 2019, 56, 2299–2317. [Google Scholar] [CrossRef]
- Lee, S.-H.; Clapham, B.; Koch, G.; Zimmermann, J.; Janda, K.D. Rhodium Carbenoid N−H Insertion Reactions of Primary Ureas: Solution and Solid-Phase Synthesis of Imidazolones. Org. Lett. 2003, 5, 511–514. [Google Scholar] [CrossRef]
- Lee, S.-H.; Yoshida, K.; Matsushita, H.; Clapham, B.; Koch, G.; Zimmermann, J.; Janda, K.D. N−H Insertion Reactions of Primary Ureas: The Synthesis of Highly Substituted Imidazolones and Imidazoles from Diazocarbonyls. J. Org. Chem. 2004, 69, 8829–8835. [Google Scholar] [CrossRef]
- Liao, W.; Hu, G.; Guo, Z.; Sun, D.; Zhang, L.; Bu, Y.; Li, Y.; Liu, Y.; Gong, P. Design and biological evaluation of novel 4-(2-fluorophenoxy)quinoline derivatives bearing an imidazolone moiety as c-Met kinase inhibitors. Bioorg. Med. Chem. 2015, 23, 4410–4422. [Google Scholar] [CrossRef]
- Tokuhara, H.; Imaeda, Y.; Fukase, Y.; Iwanaga, K.; Taya, N.; Watanabe, K.; Kanagawa, R.; Matsuda, K.; Kajimoto, Y.; Kusumoto, K.; et al. Discovery of benzimidazole derivatives as orally active renin inhibitors: Optimization of 3,5-disubstituted piperidine to improve pharmacokinetic profile. Bioorg. Med. Chem. 2018, 26, 3261–3286. [Google Scholar] [CrossRef]
- Bu, X.-B.; Yu, Y.; Li, B.; Zhang, L.; Chen, J.-J.; Zhao, Y.-L. Copper-Catalyzed Cascade Cyclization Reactions of Isocyanides with α-Diazocarbonyls as N-Terminal Electrophiles: Efficient Synthesis of 2-Imidazolines and 1,1′-Biimidazoles. Adv. Synth. Catal. 2017, 359, 351–356. [Google Scholar] [CrossRef]
- Kuruba, B.K.; Shariff, N.; Vasanthkumar, S.; Emmanuvel, L. NaOH/Et3N-Promoted Stereoselective One-Pot Synthesis of α-Diazo Oxime Ethers via Diazo Transfer Reaction. Synth. Commun. 2015, 45, 2454–2461. [Google Scholar] [CrossRef]
- Kuruba, B.K.; Vasanthkumar, S.; Emmanuvel, L. Cu(OTf)2-catalyzed synthesis of highly substituted 1-methoxy imidazoles via (3+2) cycloaddition between imino carbenoids and nitriles. Synth. Commun. 2016, 46, 799–804. [Google Scholar] [CrossRef]
- Binnemans, K. Ionic Liquid Crystals. Chem. Rev. 2005, 105, 4148–4204. [Google Scholar] [CrossRef]
- Welton, T. Room-Temperature Ionic Liquids. Solvents for Synthesis and Catalysis. Chem. Rev. 1999, 99, 2071–2084. [Google Scholar] [CrossRef]
- Herrmann, W.A. N-Heterocyclic Carbenes: A New Concept in Organometallic Catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Pawar, S.K.; Yang, M.-C.; Su, M.-D.; Liu, R.-S. Gold-Catalyzed Oxidative [2+2+1] Annulations of Aryldiazo Nitriles with Imines To Yield Polyarylated Imidazolium Salts. Angew. Chem. Int. Ed. 2017, 56, 5035–5039. [Google Scholar] [CrossRef]
- Bagdi, A.K.; Santra, S.; Monir, K.; Hajra, A. Synthesis of imidazo[1,2-a]pyridines: A decade update. Chem. Commun. 2015, 51, 1555–1575. [Google Scholar] [CrossRef]
- Delgado, O.; Delgado, F.; Vega, J.A.; Trabanco, A.A. N-bridged 5,6-bicyclic pyridines: Recent applications in central nervous system disorders. Eur. J. Med. Chem. 2015, 97, 719–731. [Google Scholar] [CrossRef]
- Yadav, J.S.; Subba Reddy, B.V.; Gopal Rao, Y.; Srinivas, M.; Narsaiah, A.V. Cu(OTf)2-catalyzed synthesis of imidazo[1,2-a]pyridines from α-diazoketones and 2-aminopyridines. Tetrahedron Lett. 2007, 48, 7717–7720. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Shen, L.-L.; Cheon, H.-G.; Xu, Y.-N.; Jeong, J.-H. Synthesis and biological evaluation of glucagon-like peptide-1 receptor agonists. Arch. Pharmacal Res. 2014, 37, 588–599. [Google Scholar] [CrossRef] [Green Version]
- Gulevich, A.V.; Helan, V.; Wink, D.J.; Gevorgyan, V. Pyridine Group Assisted Addition of Diazo-Compounds to Imines in the 3-CC Reaction of 2-Aminopyridines, Aldehydes, and Diazo-Compounds. Org. Lett. 2013, 15, 956–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, N.B.; Sharma, S.; Chetry, R.L.; Baishya, G. A Green Protocol for the Synthesis of α-Diazo-β-hydroxyesters and One-Pot Conversion to β-Keto-Esters and Imidazo[1,2-a]pyridine-3-carboxylates. ChemistrySelect 2019, 4, 5817–5822. [Google Scholar] [CrossRef]
- Park, S.; Kim, H.; Son, J.-Y.; Um, K.; Lee, S.; Baek, Y.; Seo, B.; Lee, P.H. Synthesis of Imidazopyridines via Copper-Catalyzed, Formal Aza-[3+2] Cycloaddition Reaction of Pyridine Derivatives with α-Diazo Oxime Ethers. J. Org. Chem. 2017, 82, 10209–10218. [Google Scholar] [CrossRef]
- Ueda, T.; Yatsuzuka, N.; Nagai, S.-I.; Okada, K.; Takeichi, E.; Segi, H.; Sakakibara, J. Oxidation of 7,8-diaminotheophylline with lead tetraacetate and reaction of the oxidation product, 6-cyanoimino-5-diazo-1,3-dimethylpyrimidine-2,4-dione with alcohols or amines. J. Heterocycl. Chem. 2001, 38, 141–145. [Google Scholar] [CrossRef]
- Faria, J.V.; Vegi, P.F.; Miguita, A.G.C.; dos Santos, M.S.; Boechat, N.; Bernardino, A.M.R. Recently reported biological activities of pyrazole compounds. Bioorg. Med. Chem. 2017, 25, 5891–5903. [Google Scholar] [CrossRef]
- Küçükgüzel, Ş.G.; Şenkardeş, S. Recent advances in bioactive pyrazoles. Eur. J. Med. Chem. 2015, 97, 786–815. [Google Scholar] [CrossRef]
- Gaikwad, D.D.; Chapolikar, A.D.; Devkate, C.G.; Warad, K.D.; Tayade, A.P.; Pawar, R.P.; Domb, A.J. Synthesis of indazole motifs and their medicinal importance: An overview. Eur. J. Med. Chem. 2015, 90, 707–731. [Google Scholar] [CrossRef]
- Zhang, S.G.; Liang, C.G.; Zhang, W.H. Recent Advances in Indazole-Containing Derivatives: Synthesis and Biological Perspectives. Molecules 2018, 23, 2783. [Google Scholar] [CrossRef] [Green Version]
- Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. From 2000 to Mid-2010: A Fruitful Decade for the Synthesis of Pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-aizari, F.A.; Ansar, M.h. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.W. Chapter Two—Recent Developments in the Chemistry of Pyrazoles. In Advances in Heterocyclic Chemistry; Scriven, E.F.V., Ramsden, C.A., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 126, pp. 55–107. [Google Scholar]
- Brewbaker, J.L.; Hart, H. Cyclization of 3-diazoalkenes to pyrazoles. J. Am. Chem. Soc. 1969, 91, 711–715. [Google Scholar] [CrossRef]
- Supurgibekov, M.B.; Zakharova, V.M.; Sieler, J.; Nikolaev, V.A. Stereochemistry and reactivity of F- and H-vinyldiazocarbonyl compounds and their phosphazines: Synthesis of pyrazoles and pyridazines. Tetrahedron Lett. 2011, 52, 341–345. [Google Scholar] [CrossRef]
- Supurgibekov, M.B.; Cantillo, D.; Kappe, C.O.; Surya Prakash, G.K.; Nikolaev, V.A. Effect of configuration of 2-vinyldiazocarbonyl compounds on their reactivity: Experimental and computational study. Org. Biomol. Chem. 2014, 12, 682–689. [Google Scholar] [CrossRef]
- Nguyen, T.Q.; Alqurafi, M.; Edwards, C.; Nguyen, P.; Kim, J.; Casco, S.; Bennet, M.; Chiang, C.; Lohry, M.; Cox, M.; et al. Functionalizing the γ-position of α-diazo-β-ketoesters. Tetrahedron Lett. 2016, 57, 3330–3333. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Zhang, D.; Zhu, C.; Li, J.; Xu, G.; Sun, J. When Aryldiazonium Salts Meet Vinyl Diazoacetates: A Cobalt-Catalyzed Regiospecific Synthesis of N-Arylpyrazoles. Org. Lett. 2014, 16, 3110–3113. [Google Scholar] [CrossRef] [PubMed]
- Drikermann, D.; Kerndl, V.; Görls, H.; Vilotijevic, I. Intramolecular Cyclization of Vinyldiazoacetates as a Versatile Route to Substituted Pyrazoles. Synlett 2020, 31, 1158–1162. [Google Scholar] [CrossRef]
- Zhao, M.-N.; Zhang, M.-N.; Ren, Z.-H.; Wang, Y.-Y.; Guan, Z.-H. Base-mediated formal [3+2] cycloaddition of β,γ-alkenyl esters and p-TsN3 for the synthesis of pyrazoles. Sci. Bull. 2017, 62, 493–496. [Google Scholar] [CrossRef]
- Cui, S.; Zhang, Y.; Wang, D.; Wu, Q. Rh(iii)-catalyzed C–H activation/[4 + 3] cycloaddition of benzamides and vinylcarbenoids: Facile synthesis of azepinones. Chem. Sci. 2013, 4, 3912–3916. [Google Scholar] [CrossRef]
- Wang, J.; Rainier, J.D. Reactivity of Vinyl Phosphonate Containing Diazoesters: Formation, Reactivity, and Utility. Org. Lett. 2015, 17, 266–269. [Google Scholar] [CrossRef]
- O’Connor, N.R.; Bolgar, P.; Stoltz, B.M. Development of a simple system for the oxidation of electron-rich diazo compounds to ketones. Tetrahedron Lett. 2016, 57, 849–851. [Google Scholar] [CrossRef] [Green Version]
- Roizen, J.L.; Jones, A.C.; Smith, R.C.; Virgil, S.C.; Stoltz, B.M. Model Studies To Access the [6,7,5,5]-Core of Ineleganolide Using Tandem Translactonization–Cope or Cyclopropanation–Cope Rearrangements as Key Steps. J. Org. Chem. 2017, 82, 13051–13067. [Google Scholar] [CrossRef]
- Mata, S.; González, M.J.; González, J.; López, L.A.; Vicente, R. Zinc-Catalyzed Synthesis of Conjugated Dienoates through Unusual Cross-Couplings of Zinc Carbenes with Diazo Compounds. Chem. Eur. J. 2017, 23, 1013–1017. [Google Scholar] [CrossRef]
- Craig, R.A.; Smith, R.C.; Roizen, J.L.; Jones, A.C.; Virgil, S.C.; Stoltz, B.M. Development of a Unified Enantioselective, Convergent Synthetic Approach Toward the Furanobutenolide-Derived Polycyclic Norcembranoid Diterpenes: Asymmetric Formation of the Polycyclic Norditerpenoid Carbocyclic Core by Tandem Annulation Cascade. J. Org. Chem. 2018, 83, 3467–3485. [Google Scholar] [CrossRef]
- Deng, G.; Wang, F.; Lu, S.; Cheng, B. Synthesis of Pyrano[3,2-c]pyrazol-7(1H)-one Derivatives by Tandem Cyclization of 2-Diazo-3,5-dioxo-6-ynoates (Ynones). Org. Lett. 2015, 17, 4651–4653. [Google Scholar] [CrossRef]
- Babinski, D.J.; Aguilar, H.R.; Still, R.; Frantz, D.E. Synthesis of Substituted Pyrazoles via Tandem Cross-Coupling/Electrocyclization of Enol Triflates and Diazoacetates. J. Org. Chem. 2011, 76, 5915–5923. [Google Scholar] [CrossRef] [Green Version]
- Babinski, D.J.; Bao, X.; El Arba, M.; Chen, B.; Hrovat, D.A.; Borden, W.T.; Frantz, D.E. Synchronized Aromaticity as an Enthalpic Driving Force for the Aromatic Cope Rearrangement. J. Am. Chem. Soc. 2012, 134, 16139–16142. [Google Scholar] [CrossRef]
- Xu, X.; Zavalij, P.Y.; Hu, W.; Doyle, M.P. Vinylogous Reactivity of Enol Diazoacetates with Donor–Acceptor Substituted Hydrazones. Synthesis of Substituted Pyrazole Derivatives. J. Org. Chem. 2013, 78, 1583–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Zhu, C.; Gu, W.; Li, J.; Sun, J. Gold(I)-Catalyzed Diazo Cross-Coupling: A Selective and Ligand-Controlled Denitrogenation/Cyclization Cascade. Angew. Chem. Int. Ed. 2015, 54, 883–887. [Google Scholar] [CrossRef]
- Raj, A.S.K.; Liu, R.-S. Gold-Catalyzed Bicyclic Annulations of 2-Alkynylbenzaldehydes with Vinyldiazo Carbonyls that Serve as Five-atom Building Units. Angew. Chem. Int. Ed. 2019, 58, 10980–10984. [Google Scholar] [CrossRef] [PubMed]
- Van Alphen, J. Pyrazolines and their rearrangement to form pyrazoles. II. (Pyrazole and pyrazoline derivatives, IV). Recl. Trav. Chim. Pays-Bas 1943, 62, 491–496. [Google Scholar] [CrossRef]
- Hüttel, R.; Franke, K.; Martin, H.; Riedl, J. Zur Kenntnis der Umlagerung 3.3-disubstituierter Pyrazolenine. Chem. Ber. 1960, 93, 1433–1446. [Google Scholar] [CrossRef]
- Sammes, M.P.; Katritzky, A.R. The 3H-Pyrazoles. In Advances in Heterocyclic Chemistry; Katritzky, A.R., Ed.; Academic Press: Cambridge, MA, USA, 1983; Volume 34, pp. 1–52. [Google Scholar]
- Sultanova, R.M.; Lobov, A.N.; Spirikhin, L.V. Synthesis of dimethyl esters of 7-oxo-4,5,6,7-tetrahydropyrazolo[1,5-c]pyrimidine-2,3-dicarboxylic acid. Chem. Heterocycl. Compd. 2015, 51, 1048–1051. [Google Scholar] [CrossRef]
- Marichev, K.O.; Qiu, H.; Offield, A.C.; Arman, H.; Doyle, M.P. Catalyst-Free Rearrangement of Allenyl Aryldiazoacetates into 1,5-Dihydro-4H-pyrazol-4-ones. J. Org. Chem. 2016, 81, 9235–9246. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.; Zheng, Y.; He, C.; Chen, J.; Zhen, J.; Qiu, L.; Xu, X. Synthesis of spiro-4H-pyrazole-oxindoles and fused 1H-pyrazoles via divergent, thermally induced tandem cyclization/migration of alkyne-tethered diazo compounds. Org. Biomol. Chem. 2018, 16, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Mertens, L.; Hock, K.J.; Koenigs, R.M. Fluoroalkyl-Substituted Diazomethanes and Their Application in a General Synthesis of Pyrazoles and Pyrazolines. Chem. Eur. J. 2016, 22, 9542–9545. [Google Scholar] [CrossRef]
- Gladow, D.; Doniz-Kettenmann, S.; Reissig, H.-U. 1,3-Dipolar Cycloadditions of Ethyl 2-Diazo-3,3,3-trifluoropropanoate to Alkynes and [1,5] Sigmatropic Rearrangements of the Resulting 3H-Pyrazoles: Synthesis of Mono-, Bis- and Tris(trifluoromethyl)-Substituted Pyrazoles. Helv. Chim. Acta 2014, 97, 808–821. [Google Scholar] [CrossRef]
- Vasin, V.A.; Razin, V.V.; Bezrukova, E.V.; Popkova, Y.A.; Somov, N.V. Rearrangements of 3H-Pyrazoles—Adducts of Dimethyl Acetylenedicarboxylate with Diphenyldiazomethane and 9-Diazofluorene. Russ. J. Org. Chem. 2018, 54, 892–900. [Google Scholar] [CrossRef]
- Strakova, I.; Strakovs, A.; Petrova, M.; Belyakov, S. 5-Diazo-6,6-dimethyl-4-oxo-4,5,6,7-tetrahydroindazoles in [3+2] cycloaddition reactions. Chem. Heterocycl. Compd. 2007, 43, 1512–1518. [Google Scholar] [CrossRef]
- Santos, B.S.; Nunes, S.C.C.; Pais, A.A.C.C.; Pinho e Melo, T.M.V.D. Chiral spiro-β-lactams from 6-diazopenicillanates. Tetrahedron. 2012, 68, 3729–3737. [Google Scholar] [CrossRef]
- Shelke, A.M.; Suryavanshi, G. An efficient one pot regioselective synthesis of a 3,3′-spiro-phosphonylpyrazole-oxindole framework via base mediated [1,3]-dipolar cycloaddition reaction of the Bestmann–Ohira reagent with methyleneindolinones. Org. Biomol. Chem. 2015, 13, 8669–8675. [Google Scholar] [CrossRef] [PubMed]
- Friscourt, F.; Fahrni, C.J.; Boons, G.-J. Fluorogenic Strain-Promoted Alkyne–Diazo Cycloadditions. Chem. Eur. J. 2015, 21, 13996–14001. [Google Scholar] [CrossRef] [Green Version]
- Abdou, W.M.; Khidre, M.D.; Khidre, R.E. Diverse reactivity of α-carbanions derived from alkylidenephosphoranes toward 2-(1λ5-diazynylidene)-1H-indene-1,3-(2H)dione. General approach to conjugated oxadiazines, pyridazines and spiro[3]pyrazoles. J. Heterocycl. Chem. 2008, 45, 1571–1577. [Google Scholar] [CrossRef]
- Jin, T.; Yamamoto, Y. An Efficient, Facile, and General Synthesis of 1H-Indazoles by 1,3-Dipolar Cycloaddition of Arynes with Diazomethane Derivatives. Angew. Chem. Int. Ed. 2007, 46, 3323–3325. [Google Scholar] [CrossRef]
- Liu, Z.; Shi, F.; Martinez, P.D.G.; Raminelli, C.; Larock, R.C. Synthesis of Indazoles by the [3+2] Cycloaddition of Diazo Compounds with Arynes and Subsequent Acyl Migration. J. Org. Chem. 2008, 73, 219–226. [Google Scholar] [CrossRef]
- Phatake, R.S.; Mullapudi, V.; Wakchaure, V.C.; Ramana, C.V. Fluoride-Mediated Dephosphonylation of α-Diazo-β-carbonyl Phosphonates. Org. Lett. 2017, 19, 372–375. [Google Scholar] [CrossRef]
- Ikawa, T.; Masuda, S.; Takagi, A.; Akai, S. 1,3- and 1,4-Benzdiyne equivalents for regioselective synthesis of polycyclic heterocycles. Chem. Sci. 2016, 7, 5206–5211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Hu, M.; Peng, Y. Switchable Synthesis of 3-Substituted 1H-Indazoles and 3,3-Disubstituted 3H-Indazole-3-phosphonates Tuned by Phosphoryl Groups. J. Org. Chem. 2018, 83, 1591–1597. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Xiao, X.; Hoye, T.R. Photochemical Hexadehydro-Diels–Alder Reaction. J. Am. Chem. Soc. 2017, 139, 8400–8403. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Hoye, T.R. The domino hexadehydro-Diels–Alder reaction transforms polyynes to benzynes to naphthynes to anthracynes to tetracynes (and beyond?). Nat. Chem. 2018, 10, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Ikawa, T.; Takagi, A.; Goto, M.; Aoyama, Y.; Ishikawa, Y.; Itoh, Y.; Fujii, S.; Tokiwa, H.; Akai, S. Regiocomplementary Cycloaddition Reactions of Boryl- and Silylbenzynes with 1,3-Dipoles: Selective Synthesis of Benzo-Fused Azole Derivatives. J. Org. Chem. 2013, 78, 2965–2983. [Google Scholar] [CrossRef]
- Bronner, S.M.; Mackey, J.L.; Houk, K.N.; Garg, N.K. Steric Effects Compete with Aryne Distortion To Control Regioselectivities of Nucleophilic Additions to 3-Silylarynes. J. Am. Chem. Soc. 2012, 134, 13966–13969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikawa, T.; Masuda, S.; Nakajima, H.; Akai, S. 2-(Trimethylsilyl)phenyl Trimethylsilyl Ethers as Stable and Readily Accessible Benzyne Precursors. J. Org. Chem. 2017, 82, 4242–4253. [Google Scholar] [CrossRef]
- Kitamura, T.; Gondo, K.; Oyamada, J. Generation and Reactions of Heteroaromatic Arynes Using Hypervalent Iodine Compounds. Heterocycles 2015, 90, 681. [Google Scholar] [CrossRef]
- Kwon, Y.-J.; Jeon, Y.-K.; Sim, H.-B.; Oh, I.-Y.; Shin, I.; Kim, W.-S. 3-Hydroxy-2-(trialkylsilyl)phenyl Triflate: A Benzyne Precursor Triggered via 1,3-C-sp2-to-O Silyl Migration. Org. Lett. 2017, 19, 6224–6227. [Google Scholar] [CrossRef]
- Bronner, S.M.; Goetz, A.E.; Garg, N.K. Overturning Indolyne Regioselectivities and Synthesis of Indolactam V. J. Am. Chem. Soc. 2011, 133, 3832–3835. [Google Scholar] [CrossRef] [Green Version]
- Morita, T.; Yoshida, S.; Kondo, M.; Matsushita, T.; Hosoya, T. Facile Diversification of Simple Benzo[b]thiophenes via Thienobenzyne Intermediates. Chem. Lett. 2017, 46, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Simonin, J.; Vernekar, S.K.V.; Thompson, A.J.; Hothersall, J.D.; Connolly, C.N.; Lummis, S.C.R.; Lochner, M. High-affinity fluorescent ligands for the 5-HT3 receptor. Bioorg. Med. Chem. Lett. 2012, 22, 1151–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muruganantham, R.; Namboothiri, I. Phosphonylpyrazoles from Bestmann−Ohira Reagent and Nitroalkenes: Synthesis and Dynamic NMR Studies. J. Org. Chem. 2010, 75, 2197–2205. [Google Scholar] [CrossRef]
- Tandon, V.K.; Yadav, D.B.; Chaturvedi, A.K.; Shukla, P.K. Synthesis of (1,4)-naphthoquinono-[3,2-c]-1H-pyrazoles and their (1,4)-naphthohydroquinone derivatives as antifungal, antibacterial, and anticancer agents. Bioorg. Med. Chem. Lett. 2005, 15, 3288–3291. [Google Scholar] [CrossRef]
- Yamazaki, T.; Shechter, H. Dipolar addition and acyl migration in reactions of benzyne and acetylenes with 5-membered ring α-diazo ketones. Tetrahedron Lett. 1972, 13, 4533–4536. [Google Scholar] [CrossRef]
- Cheng, B.; Zu, B.; Bao, B.; Li, Y.; Wang, R.; Zhai, H. Synthesis of Spiro[indazole-3,3′-indolin]-2′-ones via [3+2] Dipolar Cycloaddition of Arynes with 3-Diazoindolin-2-ones and Indazolo[2,3-c]quinazolin-6(5H)-ones by Subsequent Thermal Isomerization. J. Org. Chem. 2017, 82, 8228–8233. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Li, Y.; Zu, B.; Wang, T.; Wang, R.; Li, Y.; Zhai, H. Syntheses of spiro[indazole-3,3′-indolin]-2′-ones and spiro[indazole-3,3′-indolin]-2′-imines via 1,3-dipolar cycloadditions of arynes and studies on their isomerization reactions. Tetrahedron 2019, 75, 130775. [Google Scholar] [CrossRef]
- Subba Reddy, B.V.; Gopi Reddy, R.R.; Reddy Thummaluru, V.; Sridhar, B. [3+2] Cycloaddition of 3-Diazooxindole with Arynes for the Synthesis of Spiro[indazole-3,3′-indolin]-2′-ones. ChemistrySelect 2017, 2, 4290–4293. [Google Scholar] [CrossRef]
- Cheng, B.; Bao, B.; Zu, B.; Duan, X.; Duan, S.; Li, Y.; Zhai, H. Synthesis of spiro-3H-indazoles via 1,3-dipolar cycloaddition of arynes with 6-diazocyclohex-2-en-1-one derivatives and fused-2H-indazoles by subsequent rearrangement. RSC Adv. 2017, 7, 54087–54090. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-D.; Liu, R.-S. Silver-catalyzed [3+2]-cycloaddition of benzynes with diazocarbonyl species via a postulated (1H-indazol-1-yl)silver intermediate. Org. Biomol. Chem. 2012, 10, 8948–8952. [Google Scholar] [CrossRef]
- Yong, W.-S.; Park, S.; Yun, H.; Lee, P.H. Synthesis of 2H-Indazoles via Tandem Palladium-Catalyzed Deacylative Cross-Coupling and Denitrogenative Cyclization of 2-Iodoazoarenes and 2-Iodoaryltriazenes with Acyldiazoacetates in One-Pot. Adv. Synth. Catal. 2016, 358, 1958–1967. [Google Scholar] [CrossRef]
- Long, Z.; Wang, Z.; Zhou, D.; Wan, D.; You, J. Rh(III)-Catalyzed Regio- and Chemoselective [4+1]-Annulation of Azoxy Compounds with Diazoesters for the Synthesis of 2H-Indazoles: Roles of the Azoxy Oxygen Atom. Org. Lett. 2017, 19, 2777–2780. [Google Scholar] [CrossRef] [PubMed]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Meldal, M.; Diness, F. Recent Fascinating Aspects of the CuAAC Click Reaction. Trends Chem. 2020, 2, 569–584. [Google Scholar] [CrossRef]
- Kantheti, S.; Narayan, R.; Raju, K.V.S.N. The impact of 1,2,3-triazoles in the design of functional coatings. RSC Adv. 2015, 5, 3687–3708. [Google Scholar] [CrossRef]
- Brunel, D.; Dumur, F. Recent advances in organic dyes and fluorophores comprising a 1,2,3-triazole moiety. New J. Chem. 2020, 44, 3546–3561. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef]
- Johansson, J.R.; Beke-Somfai, T.; Said Stålsmeden, A.; Kann, N. Ruthenium-Catalyzed Azide Alkyne Cycloaddition Reaction: Scope, Mechanism, and Applications. Chem. Rev. 2016, 116, 14726–14768. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.; Razaq, H.; Faisal, M.; Siyal, A.N.; Haider, A. Metal-free and azide-free synthesis of 1,2,3-triazoles derivatives. Synth. Commun. 2017, 47, 1193–1200. [Google Scholar] [CrossRef]
- Chen, Z.; Cao, G.; Song, J.; Ren, H. Recent Developments in Azide-Free Synthesis of 1,2,3-Triazoles. Chin. J. Chem. 2017, 35, 1797–1807. [Google Scholar] [CrossRef]
- Wolff, L. Ueber Diazoanhydride. Justus Liebigs Ann. Chem. 1902, 325, 129–195. [Google Scholar] [CrossRef] [Green Version]
- Costa, M.S.; Boechat, N.; Rangel, É.A.; da Silva, F.d.C.; de Souza, A.M.T.; Rodrigues, C.R.; Castro, H.C.; Junior, I.N.; Lourenço, M.C.S.; Wardell, S.M.S.V.; et al. Synthesis, tuberculosis inhibitory activity, and SAR study of N-substituted-phenyl-1,2,3-triazole derivatives. Bioorg. Med. Chem. 2006, 14, 8644–8653. [Google Scholar] [CrossRef]
- Zhang, H.; Ryono, D.E.; Devasthale, P.; Wang, W.; O’Malley, K.; Farrelly, D.; Gu, L.; Harrity, T.; Cap, M.; Chu, C.; et al. Design, synthesis and structure–activity relationships of azole acids as novel, potent dual PPAR α/γ agonists. Bioorg. Med. Chem. Lett. 2009, 19, 1451–1456. [Google Scholar] [CrossRef] [PubMed]
- Safrygin, A.; Dar’in, D.; Kantin, G.; Krasavin, M. α-Diazo-β-oxosulfones as Partners in the Wolff 1,2,3-Triazole Synthesis and the Wolff Rearrangement in the Presence of Aromatic Amines. Eur. J. Org. Chem. 2019, 2019, 4721–4724. [Google Scholar] [CrossRef]
- Wang, Z.; Bi, X.; Liao, P.; Zhang, R.; Liang, Y.; Dong, D. Intramolecular hydrogen bonding-assisted cyclocondensation of α-diazoketones with various amines: A strategy for highly efficient Wolff 1,2,3-triazole synthesis. Chem. Commun. 2012, 48, 7076–7078. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tao, Y.; Wang, Z.; Yan, J. Synthesis and characterization of poly(N-vinyl-1,2,3-triazole)s derived from monomers obtained by highly efficient Wolff’s cyclocondensation. Polym. Chem. 2016, 7, 3172–3178. [Google Scholar] [CrossRef]
- Fershtat, L.; Radzhabov, M.; Romanova, A.; Ananyev, I.; Makhova, N. Lewis acid-catalyzed Wolff cyclocondensation in the synthesis of (1H-1,2,3-triazolyl)furoxans. ARKIVOC 2017, 2017, 140–150. [Google Scholar] [CrossRef] [Green Version]
- Santiago, J.V.; Burtoloso, A.C.B. Synthesis of Fused Bicyclic [1,2,3]-Triazoles from γ-Amino Diazoketones. ACS Omega 2019, 4, 159–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dar’in, D.; Khoroshilova, O.; Kantin, G.; Krasavin, M. Realizing the Trifunctional Potential of Alkyl 4-Chloro-2-diazo-3-oxobutanoates: Convenient Assembly of 6,7-Dihydro-4H-[1,2,3]triazolo[5,1-c][1,4]thiazine Core. Synthesis 2020, 52, 1266–1272. [Google Scholar] [CrossRef]
- Wolff, L.; Krüche, R. Über Diazoanhydride (1,2,3-Oxydiazole oder Diazoxyde) und Diazoketone. Justus Liebigs Ann. Chem. 1912, 394, 23–59. [Google Scholar] [CrossRef] [Green Version]
- Jordão, A.K.; Afonso, P.P.; Ferreira, V.F.; de Souza, M.C.B.V.; Almeida, M.C.B.; Beltrame, C.O.; Paiva, D.P.; Wardell, S.M.S.V.; Wardell, J.L.; Tiekink, E.R.T.; et al. Antiviral evaluation of N-amino-1,2,3-triazoles against Cantagalo virus replication in cell culture. Eur. J. Med. Chem. 2009, 44, 3777–3783. [Google Scholar] [CrossRef] [PubMed]
- Jordão, A.K.; Ferreira, V.F.; Souza, T.M.L.; de Souza Faria, G.G.; Machado, V.; Abrantes, J.L.; de Souza, M.C.B.V.; Cunha, A.C. Synthesis and anti-HSV-1 activity of new 1,2,3-triazole derivatives. Bioorg. Med. Chem. 2011, 19, 1860–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, T.F.; de Jesus, J.B.; Neufeld, P.M.; Jordão, A.K.; Campos, V.R.; Cunha, A.C.; Castro, H.C.; de Souza, M.C.B.V.; Ferreira, V.F.; Rodrigues, C.R.; et al. Exploring 1,2,3-triazole derivatives by using in vitro and in silico assays to target new antifungal agents and treat Candidiasis. Med. Chem. Res. 2017, 26, 680–689. [Google Scholar] [CrossRef]
- Nagarajan, R.; Jayashankaran, J.; Emmanuvel, L. Transition metal-free steric controlled one-pot synthesis of highly substituted N-amino 1,2,3-triazole derivatives via diazo transfer reaction from β-keto esters. Tetrahedron Lett. 2016, 57, 2612–2615. [Google Scholar] [CrossRef]
- Kuruba, B.K.; Emmanuvel, L.; Sridhar, B.; Vasanthkumar, S. Unprecedented cyclization of α-diazo hydrazones upon N-H functionalization: A Et3N base promoted one-step synthetic approach for the synthesis of N-amino-1,2,3-triazole derivatives from α-diazo hydrazone. Tetrahedron 2017, 73, 2674–2681. [Google Scholar] [CrossRef]
- Cheng, G.; Zeng, X.; Shen, J.; Wang, X.; Cui, X. A Metal-Free Multicomponent Cascade Reaction for the Regiospecific Synthesis of 1,5-Disubstituted 1,2,3-Triazoles. Angew. Chem. Int. Ed. 2013, 52, 13265–13268. [Google Scholar] [CrossRef] [PubMed]
- Dankova, E.F.; Bakulev, V.A.; Krut’ko, D.P. New method for studying the reactivities of α-diazo imines. Investigation of the cyclization of N-substituted 2-diazoacetamidines to 1,2,3-triazoles. Chem. Heterocycl. Compd. 1991, 27, 607–613. [Google Scholar] [CrossRef]
- Dimorth, O. Über intramolekulare Umlagerungen. Fünfte Abhandlung. Der Einfluß des Lösungsmittels auf Reaktionsgeschwindigkeit und Gleichgewicht. Der Einfluß des Lösungsmittels auf Reaktionsgeschwindigkeit und Gleichgewicht. Justus Liebigs Ann. Chem. 1910, 377, 127–163. [Google Scholar] [CrossRef] [Green Version]
- Murray-Rust, P.; McManus, J.; Lennon, S.P.; Porter, A.E.A.; Rechka, J.A. An expeditious synthesis of 4-alkoxycarbonyl-5-hydroxy-1,2,3-triazoles: The crystal and molecular structure of the 2-thienylammonium salt of 5-hydroxy-4-methoxycarbonyl-1-(2-thienyl)-1,2,3-triazole. J. Chem. Soc. Perkin Trans. 1 1984, 713–716. [Google Scholar] [CrossRef]
- Pippione, A.C.; Dosio, F.; Ducime, A.; Federico, A.; Martina, K.; Sainas, S.; Frølund, B.; Gooyit, M.; Janda, K.D.; Boschi, D.; et al. Substituted 4-hydroxy-1,2,3-triazoles: Synthesis, characterization and first drug design applications through bioisosteric modulation and scaffold hopping approaches. Med. Chem. Comm. 2015, 6, 1285–1292. [Google Scholar] [CrossRef]
- Sainas, S.; Pippione, A.C.; Giraudo, A.; Martina, K.; Bosca, F.; Rolando, B.; Barge, A.; Ducime, A.; Federico, A.; Grossert, S.J.; et al. Regioselective N-Alkylation of Ethyl 4-Benzyloxy-1,2,3-triazolecarboxylate: A Useful Tool for the Synthesis of Carboxylic Acid Bioisosteres. J. Heterocycl. Chem. 2019, 56, 501–519. [Google Scholar] [CrossRef]
- Morzherin, Y.Y.; Rozin, Y.A.; Vorob’eva, E.A.; Bakulev, V.A. Synthesis and Properties of 1-Arylsulfonyl-1,2,3-triazol-5-olates. Chem. Heterocycl. Compd. 2001, 37, 560–566. [Google Scholar] [CrossRef]
- Brown, B.R.; Hammick, D.L. 257. The mechanism of the tautomeric change of 5-hydroxy-1:2:3-triazole-4-carboxylic esters into aliphatic diazo-compounds. J. Chem. Soc. 1947, 1384–1386. [Google Scholar] [CrossRef]
- Morzherin, Y.Y.; Kolobov, M.Y.; Mokrushin, V.S.; Brauer, M.; Anders, E.; Bakulev, V.A. Heterocyclization of compounds containing diazo and cyano groups. 6. Theoretical and experimental investigations of cyclization of 2-cyano-2-diazoacetamides to 5-hydroxy-1,2,3-triazole-4-carbonitriles. Chem. Heterocycl. Compd. 2000, 36, 22–36. [Google Scholar] [CrossRef]
- Jiang, L.; Davison, A.; Tennant, G.; Ramage, R. Synthesis and application of a novel coupling reagent, ethyl 1-hydroxy-1H -1,2,3-triazole-4-carboxylate. Tetrahedron 1998, 54, 14233–14254. [Google Scholar] [CrossRef]
- Regitz, M.; Schoder, W. Untersuchungen an Diazo-Verbindungen und Aziden; LVI1. Amino- und Iminomethylierung von Diazomethyl-Verbindungen. Synthesis 1985, 1985, 178–180. [Google Scholar] [CrossRef]
- Bakulev, V.; Morzerin, Y.; Shafran, Y.; Mokrushin, V. Tandem pseudopericyclic processes in the cyclization of α-diazonitriles to 5-halo-1,2,3-triazoles. Scope and limitations. ARKIVOC 2002, 2002, 166–179. [Google Scholar] [CrossRef]
- Chen, J.-H.; Liu, S.-R.; Chen, K. An Efficient and Convenient Synthesis of Ethyl 1-(4-Methoxyphenyl)-5-phenyl-1H-1,2,3-triazole-4-carboxylate. Chem. Asian J. 2010, 5, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Ahamad, S.; Kant, R.; Mohanan, K. Metal-Free Three-Component Domino Approach to Phosphonylated Triazolines and Triazoles. Org. Lett. 2016, 18, 280–283. [Google Scholar] [CrossRef]
- Ahamad, S.; Kumar, A.; Kant, R.; Mohanan, K. Metal-Free Three-Component Assembly of Fully Substituted 1,2,3-Triazoles. Asian J. Org. Chem. 2018, 7, 1698–1703. [Google Scholar] [CrossRef]
- Li, Y.-J.; Li, X.; Zhang, S.-X.; Zhao, Y.-L.; Liu, Q. Copper(ii)-catalyzed oxidative [3+2] cycloaddition reactions of secondary amines with α-diazo compounds: A facile and efficient synthesis of 1,2,3-triazoles. Chem. Commun. 2015, 51, 11564–11567. [Google Scholar] [CrossRef]
- Wang, S.; Yang, L.-J.; Zeng, J.-L.; Zheng, Y.; Ma, J.-A. Silver-catalyzed [3+2] cycloaddition of isocyanides with diazo compounds: New regioselective access to 1,4-disubstituted-1,2,3-triazoles. Org. Chem. Front. 2015, 2, 1468–1474. [Google Scholar] [CrossRef]
- Aggarwal, R.; Sumran, G. An insight on medicinal attributes of 1,2,4-triazoles. Eur. J. Med. Chem. 2020, 205, 112652. [Google Scholar] [CrossRef]
- Haasnoot, J.G. Mononuclear, oligonuclear and polynuclear metal coordination compounds with 1,2,4-triazole derivatives as ligands. Coord. Chem. Rev. 2000, 200-202, 131–185. [Google Scholar] [CrossRef]
- Naik, A.D.; Dîrtu, M.M.; Railliet, A.P.; Marchand-Brynaert, J.; Garcia, Y. Coordination Polymers and Metal Organic Frameworks Derived from 1,2,4-Triazole Amino Acid Linkers. Polymers 2011, 3, 1750–1775. [Google Scholar] [CrossRef] [Green Version]
- Al-Masoudi, I.A.; Al-Soud, Y.A.; Al-Salihi, N.J.; Al-Masoudi, N.A. 1,2,4-Triazoles: Synthetic approaches and pharmacological importance. (Review). Chem. Heterocycl. Compd. 2006, 42, 1377–1403. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, J.-J.; Liu, S.-S.; Liang, Y.-X.; Zhao, Y.-L. DBU-Catalyzed [3+3] and [3+2] Annulation Reactions of Azomethine Ylides with α-Diazocarbonyls as N-Terminal Electrophiles: Modular, Atom-Economical Access to 1,2,4-Triazine and 1,2,4-Triazole Derivatives. Adv. Synth. Catal. 2018, 360, 2172–2177. [Google Scholar] [CrossRef]
- Zhao, H.-W.; Liu, Y.-Y.; Zhao, Y.-D.; Feng, N.-N.; Du, J.; Song, X.-Q.; Pang, H.-L.; Chen, X.-Q. Base-Catalyzed Formal [3+2] Cycloaddition of Diazooxindoles with Oxazol-5-(4H)-ones. Eur. J. Org. Chem. 2018, 2018, 341–346. [Google Scholar] [CrossRef]
- Chan, C.-M.; Xing, Q.; Chow, Y.-C.; Hung, S.-F.; Yu, W.-Y. Photoredox Decarboxylative C(sp3)–N Coupling of α-Diazoacetates with Alkyl N-Hydroxyphthalimide Esters for Diversified Synthesis of Functionalized N-Alkyl Hydrazones. Org. Lett. 2019, 21, 8037–8043. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, X.; Hao, W.; Li, H.; Zhao, Y.; Wang, Y.; Lian, P.; Zheng, Y.; Bao, X.; Wan, X. [3+2] Cycloaddition of Nitrile Ylides with Diazonium Salts: Copper-Catalyzed One-Pot Synthesis of Fully Substituted 1,2,4-Triazoles. Org. Lett. 2018, 20, 5224–5227. [Google Scholar] [CrossRef]
- Sadchikova, E.V.; Alexeeva, D.L.; Nenajdenko, V.G. PASE synthesis of 1-azolyl-1H-1,2,4-triazoles by the reaction of diazoazoles with ethyl isocyanoacetate. Mendeleev Commun. 2019, 29, 653–654. [Google Scholar] [CrossRef]
- Sabatini, J.; Oyler, K. Recent Advances in the Synthesis of High Explosive Materials. Crystals 2016, 6, 5. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Qiao, J.; Xu, S.; Zhao, X.; Gong, W.; Huang, T. Constructing Strategies and Applications of Nitrogen-Rich Energetic Metal–Organic Framework Materials. Catalysts 2020, 10, 690. [Google Scholar] [CrossRef]
- Dhiman, N.; Kaur, K.; Jaitak, V. Tetrazoles as anticancer agents: A review on synthetic strategies, mechanism of action and SAR studies. Bioorg. Med. Chem. 2020, 28, 115599. [Google Scholar] [CrossRef] [PubMed]
- Neochoritis, C.G.; Zhao, T.; Dömling, A. Tetrazoles via Multicomponent Reactions. Chem. Rev. 2019, 119, 1970–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarvary, A.; Maleki, A. A review of syntheses of 1,5-disubstituted tetrazole derivatives. Mol. Divers. 2015, 19, 189–212. [Google Scholar] [CrossRef]
- Aoyama, T.; Shioiri, T. New Methods and Reagents in Organic Synthesis. 31. Lithium Trimethylsilyldiazomethane: A New Synthon for the Preparation of Tetrazoles. Chem. Pharm. Bull. (Tokyo) 1982, 30, 3450–3452. [Google Scholar] [CrossRef] [Green Version]
- Maas, G.; Gümbel, H.; Weise, G.; Regitz, M. Untersuchungen an Diazoverbindungen und Aziden, 58. Dikation-ether, 5. Formamidinium-substituierte Diazophosphoryl-Verbindungen: Synthese, Eigenschaften und Reaktionen zu Tetrazolen. Chem. Ber. 1985, 118, 2105–2117. [Google Scholar] [CrossRef]
- Chen, Z.; Fan, S.-Q.; Zheng, Y.; Ma, J.-A. Silver-catalyzed regioselective [3+2] cycloaddition of arene-diazonium salts with 2,2,2-trifluorodiazoethane (CF3CHN2): A facile access to 2-aryl-5-trifluoromethyltetrazoles. Chem. Commun. 2015, 51, 16545–16548. [Google Scholar] [CrossRef]
- Peng, X.; Xiao, M.-Y.; Zeng, J.-L.; Zhang, F.-G.; Ma, J.-A. Construction of Difluoromethylated Tetrazoles via Silver-Catalyzed Regioselective [3+2] Cycloadditions of Aryl Diazonium Salts. Org. Lett. 2019, 21, 4808–4811. [Google Scholar] [CrossRef]
- Patouret, R.; Kamenecka, T.M. Synthesis of 2-aryl-2H-tetrazoles via a regioselective [3+2] cycloaddition reaction. Tetrahedron Lett. 2016, 57, 1597–1599. [Google Scholar] [CrossRef] [Green Version]
- Chuprun, S.; Dar’in, D.; Kantin, G.; Krasavin, M. [3+2]-Cycloaddition of α-Diazocarbonyl Compounds with Arenediazonium Salts Catalyzed by Silver Nitrate Delivers 2,5-Disubstituted Tetrazoles. Synthesis 2019, 51, 3998–4005. [Google Scholar] [CrossRef] [Green Version]
- Xiao, M.-Y.; Chen, Z.; Zhang, F.-G.; Ma, J.-A. Regioselective synthesis of carboxylic and fluoromethyl tetrazoles enabled by silver-catalyzed cycloaddition of diazoacetates and aryl diazonium salts. Tetrahedron 2020, 76, 131063. [Google Scholar] [CrossRef]
- Zhai, S.-J.; Peng, X.; Zhang, F.-G.; Ma, J.-A. Catalytic Direct Regioselective Synthesis of Phosphonylated Tetrazoles from Aryl Diazonium Salts and Seyferth-Gilbert Reagent. Org. Lett. 2019, 21, 9884–9888. [Google Scholar] [CrossRef]
- Levashova, E.; Bakulina, O.; Dar’in, D.; Bubyrev, A.; Chuprun, S.; Krasavin, M. From Rare Reagents to Rare Products: Regiospecific Silver-Catalyzed [3+2] Cycloaddition of Aryl-, Alkyl- and Aminosulfonyl Diazomethanes with Arenediazonium Tosylates. Eur. J. Org. Chem. 2020, 2020, 4239–4242. [Google Scholar] [CrossRef]
Scheme 1.
Gold-catalyzed oxazole synthesis from α-diazo oxime ethers and nitriles.

Scheme 2.
Utilization of 2-diazoacetyl-2H-azirines for the synthesis of heterocycle-substituted oxazoles.
Scheme 2.
Utilization of 2-diazoacetyl-2H-azirines for the synthesis of heterocycle-substituted oxazoles.

Scheme 3.
(C6F5)3B-catalyzed oxazole synthesis from diazo carbonyls and nitriles.

Scheme 4.
Synthesis of oxazolo[5,4-c]isoquinolin-5(4H)-ones from diazo homophthalimides and nitriles.
Scheme 4.
Synthesis of oxazolo[5,4-c]isoquinolin-5(4H)-ones from diazo homophthalimides and nitriles.

Scheme 5.
Synthesis of oxazolo[5,4-d]pyrimidine-5,7(4H,6H)-diones 18 from diazobarbituric acids 16 and nitriles 17.
Scheme 5.
Synthesis of oxazolo[5,4-d]pyrimidine-5,7(4H,6H)-diones 18 from diazobarbituric acids 16 and nitriles 17.

Scheme 6.
Synthesis of 6,7-dihydrobenzo[d]oxazol-4(5H)-ones 23 from alicyclic diazo diketones and nitriles.
Scheme 6.
Synthesis of 6,7-dihydrobenzo[d]oxazol-4(5H)-ones 23 from alicyclic diazo diketones and nitriles.

Scheme 7.
Synthesis of polybrominated benzoxazoles 27 from o-quinone diazides 25 and nitriles 26.

Scheme 8.
Moody’s approach for the synthesis of oxazoles from amides and diazo compounds.

Scheme 9.
Cu-catalyzed synthesis of oxazoles from terminal diazo carbonyls and amides.

Scheme 10.
Rh(I)-catalyzed synthesis of oxazoles from diazo dicarbonyl compounds and amides.

Scheme 11.
Cu(OAc)2-catalyzed synthesis of oxazoles from α-ketooximes and diazo carbonyl compounds.

Scheme 12.
Synthesis of sulfur derivatives of oxazoles from acyl isothiocyanates and TMSCHN2.

Scheme 13.
Two complementary strategies for the synthesis of oxazoles from diazocarbonyl compounds.

Scheme 14.
Preparation of oxazole building block for the total synthesis of siphonazole.

Scheme 15.
Preparation of oxazole building blocks for the total synthesis of goadsporin.

Scheme 16.
Preparation of oxazole building blocks for the total synthesis of wewakazole and wewakazole B.
Scheme 16.
Preparation of oxazole building blocks for the total synthesis of wewakazole and wewakazole B.

Scheme 17.
Synthesis of 4H-imidazo[4,5-c]isoxazole from nitroimidazolyl diazo ketoester 83.

Scheme 18.
Synthesis of isoxazoles from β-ketoesters 86 and diazo acetates 85.

Scheme 19.
Synthesis of isoxazoles from alkynes and diazocarbonyl compounds.

Scheme 20.
BF3·OEt2-promoted synthesis of thiazoles from ethyl diazopyruvate and thioamides.

Scheme 21.
Synthesis of bisthiazolylbenzene 95 and thiazole-containing crown ethers 98.

Scheme 22.
Thermally or photochemically promoted reactions of 2-diazo-1,3-diketones with thioamides.
Scheme 22.
Thermally or photochemically promoted reactions of 2-diazo-1,3-diketones with thioamides.

Scheme 23.
Copper(I)-mediated synthesis of thiazoles from α-diazo-β-ketoesters and aromatic thioamides.
Scheme 23.
Copper(I)-mediated synthesis of thiazoles from α-diazo-β-ketoesters and aromatic thioamides.

Scheme 24.
Rh2(pfm)4-catalyzed synthesis of thiazoles from 1-diazopropan-2-ones 108 and aromatic thioamides 109.
Scheme 24.
Rh2(pfm)4-catalyzed synthesis of thiazoles from 1-diazopropan-2-ones 108 and aromatic thioamides 109.

Scheme 25.
Synthesis of trifluoromethylated thiazoles and their further derivatization.

Scheme 26.
Cu(OTf)2-catalyzed synthesis of 2-aminothiazoles from diazo ketones and thiourea.

Scheme 27.
Synthesis of fully substituted thiazoles via rhodium carbene NH-insertion into amide with subsequent thionation.
Scheme 27.
Synthesis of fully substituted thiazoles via rhodium carbene NH-insertion into amide with subsequent thionation.

Scheme 28.
Preparation of thiazole 127 from amino acid amide and diazo compound in the total synthesis of amythiamicin D.
Scheme 28.
Preparation of thiazole 127 from amino acid amide and diazo compound in the total synthesis of amythiamicin D.

Scheme 29.
[Cu(CH3CN)4]PF6-catalyzed synthesis of 5-mercaptothiazoles and 5-mercapto-2,3-dihydrothiazoles from diazo carbonyl compounds and α-(N-hydroxy/aryl)imino-β-oxodithioesters.
Scheme 29.
[Cu(CH3CN)4]PF6-catalyzed synthesis of 5-mercaptothiazoles and 5-mercapto-2,3-dihydrothiazoles from diazo carbonyl compounds and α-(N-hydroxy/aryl)imino-β-oxodithioesters.

Scheme 30.
Two-step 2-imidazolone synthesis from α-diazo-β-ketoesters and primary ureas.

Scheme 31.
2-Imidazolone synthesis from α-diazo-β-ketoesters and primary ureas on solid phase.

Scheme 32.
Synthesis of 2-imidazolones from various diazo carbonyls and primary ureas.

Scheme 33.
Derivatization of 2-imidazolones in solution- and solid-phase.

Scheme 34.
Diazo approach for the preparation of 2-imidazolones 152 in the synthesis of c-Met kinase inhibitors 153.
Scheme 34.
Diazo approach for the preparation of 2-imidazolones 152 in the synthesis of c-Met kinase inhibitors 153.

Scheme 35.
Two-step imidazole synthesis from α-diazo-β-ketoesters and primary amides.

Scheme 36.
CuI-catalyzed synthesis of imidazolines 147 and biimidazoles 148 from diazo carbonyls and isocyanides.
Scheme 36.
CuI-catalyzed synthesis of imidazolines 147 and biimidazoles 148 from diazo carbonyls and isocyanides.

Scheme 37.
Mechanism of CuI-catalyzed synthesis of imidazolines 159 and biimidazoles 160 from diazo carbonyls and isocyanides.
Scheme 37.
Mechanism of CuI-catalyzed synthesis of imidazolines 159 and biimidazoles 160 from diazo carbonyls and isocyanides.

Scheme 38.
Copper-mediated synthesis of N-methoxyimidazoles from α-diazo oxime ethers and nitriles.

Scheme 39.
Gold-catalyzed [2+2+1] annulation reaction for synthesis of polyarylated imidazolium salts.
Scheme 39.
Gold-catalyzed [2+2+1] annulation reaction for synthesis of polyarylated imidazolium salts.

Scheme 40.
Mechanism of gold-catalyzed [2+2+1] annulation.

Scheme 41.
Synthesis of imidazo[1,2-a]pyridines from α-diazoketones and 2-aminopyridines.

Scheme 42.
Synthesis of imidazo[1,2-a]pyridines 176 from 2-diazo-3-(pyridin-2-ylamino)propanoate.

Scheme 43.
WEB-DMSO promoted synthesis of imidazo[1,2-a]pyridines.

Scheme 44.
Synthesis of imidazo[1,2-a]pyridines from α-diazooxime ethers and pyridines.

Scheme 45.
Mechanism of formal aza-[3+2] cycloaddition of α-diazooxime ethers and pyridines.

Scheme 46.
Synthesis of theophyllines from diazobarbituric acid derivative and amines.

Scheme 47.
The mechanism of intramolecular cyclization of vinyldiazo compounds.

Scheme 48.
Effect of double bond configuration and fluoroalkyl substituent on 1,5-electrocyclization reaction.
Scheme 48.
Effect of double bond configuration and fluoroalkyl substituent on 1,5-electrocyclization reaction.

Scheme 49.
Formation of N-substituted hydroxypyrazoles from enolizable diazo carbonyls and β-lactam derivatives.
Scheme 49.
Formation of N-substituted hydroxypyrazoles from enolizable diazo carbonyls and β-lactam derivatives.

Scheme 50.
Mechanism of formation of N-substituted hydroxypyrazoles from enolizable diazo carbonyls and β-lactam derivatives.
Scheme 50.
Mechanism of formation of N-substituted hydroxypyrazoles from enolizable diazo carbonyls and β-lactam derivatives.

Scheme 51.
Synthesis of diversely substituted pyrazoles from vinyl diazo compounds.

Scheme 52.
Cascade electrocyclization-Michael addition process for synthesis of pyrano[3,2-c]pyrazol-7(1H)-ones 211 from 2-diazo-3,5-dioxo-6-ynoates (ynones) 210.
Scheme 52.
Cascade electrocyclization-Michael addition process for synthesis of pyrano[3,2-c]pyrazol-7(1H)-ones 211 from 2-diazo-3,5-dioxo-6-ynoates (ynones) 210.

Scheme 53.
Vinyl diazo compound electrocyclic ring closure triggers two alternative sigmatropic rearrangements.
Scheme 53.
Vinyl diazo compound electrocyclic ring closure triggers two alternative sigmatropic rearrangements.

Scheme 54.
Synthesis of pyrazoles from enol diazo acetates and donor-acceptor hydrazones.

Scheme 55.
Gold-catalyzed position-switchable synthesis of pyrazoles by cross-coupling of vinyl diazo compounds with aryl diazo acetates.
Scheme 55.
Gold-catalyzed position-switchable synthesis of pyrazoles by cross-coupling of vinyl diazo compounds with aryl diazo acetates.

Scheme 56.
Synthesis of 4,5-dihydro-benzo[g]indazoles by gold-catalyzed [5+4]-annulation between 2-alkynyl-1-carbonylbenzenes and vinyldiazo ketones.
Scheme 56.
Synthesis of 4,5-dihydro-benzo[g]indazoles by gold-catalyzed [5+4]-annulation between 2-alkynyl-1-carbonylbenzenes and vinyldiazo ketones.

Scheme 57.
Synthesis of 1H-pyrazoles from diazo compounds and alkynes.

Scheme 58.
Synthesis of 7-oxo-4,5,6,7-tetrahydropyrazolo[1,5-c]pyrimidines 234 from 3-diazopyrrolidones and DMAD.
Scheme 58.
Synthesis of 7-oxo-4,5,6,7-tetrahydropyrazolo[1,5-c]pyrimidines 234 from 3-diazopyrrolidones and DMAD.

Scheme 59.
Rearrangement of diazo esters 235 to 1,5-dihydro-4H-pyrazol-4-ones 236-237.

Scheme 60.
Synthesis of spiro-4H-pyrazoles 240 and fused 1H-pyrazoles 242 from alkyne-tethered diazo amides.
Scheme 60.
Synthesis of spiro-4H-pyrazoles 240 and fused 1H-pyrazoles 242 from alkyne-tethered diazo amides.

Scheme 61.
Continuous-flow synthesis of fluoroalkyl-substituted diazomethanes for preparation of 1H-pyrazoles, pyrazolines, and 3H-pyrazoles.
Scheme 61.
Continuous-flow synthesis of fluoroalkyl-substituted diazomethanes for preparation of 1H-pyrazoles, pyrazolines, and 3H-pyrazoles.

Scheme 62.
Synthesis of spiro-3H-pyrazole adducts 252 and 254 from diazo compounds and DMAD.

Scheme 63.
Regio- and stereoselective synthesis of spiro-3H-pyrazole-penicillanates 257 from 6-diazopenicillanates 255 and alkynes 256.
Scheme 63.
Regio- and stereoselective synthesis of spiro-3H-pyrazole-penicillanates 257 from 6-diazopenicillanates 255 and alkynes 256.

Scheme 64.
Synthesis of 3,3′-spiro-phosphonylpyrazole-oxindoles 260 by deacylative 1,3-dipolar cycloaddition of BOR to methylidene indolinones.
Scheme 64.
Synthesis of 3,3′-spiro-phosphonylpyrazole-oxindoles 260 by deacylative 1,3-dipolar cycloaddition of BOR to methylidene indolinones.

Scheme 65.
Strain-promoted cycloaddition reaction of dibenzocyclooctyne derivative 263 and diazo compounds for the synthesis of 3H- and 1H-pyrazoles.
Scheme 65.
Strain-promoted cycloaddition reaction of dibenzocyclooctyne derivative 263 and diazo compounds for the synthesis of 3H- and 1H-pyrazoles.

Scheme 66.
Synthesis of spiro-3H-pyrazoles 272 and 274 from diazo indan-1,3-dione and triphenyl(alkenyl)phosphonium bromides.
Scheme 66.
Synthesis of spiro-3H-pyrazoles 272 and 274 from diazo indan-1,3-dione and triphenyl(alkenyl)phosphonium bromides.

Scheme 67.
Synthesis of N-unsubstituted and N-arylated indazoles from o-silylaryl triflates and diazo compounds.
Scheme 67.
Synthesis of N-unsubstituted and N-arylated indazoles from o-silylaryl triflates and diazo compounds.

Scheme 68.
Synthesis of 1H- and 3H-indazoles from internal diazo compounds and benzynes.

Scheme 69.
Synthesis of 1,3-substituted 1H-indazoles 289 from BOR, Michael acceptors and silylaryl triflates.
Scheme 69.
Synthesis of 1,3-substituted 1H-indazoles 289 from BOR, Michael acceptors and silylaryl triflates.

Scheme 70.
Synthesis of tricyclic indazole from 1,4-benzdiyne equivalent, ethyl diazoacetate, and 1,1-dimethoxyethene.
Scheme 70.
Synthesis of tricyclic indazole from 1,4-benzdiyne equivalent, ethyl diazoacetate, and 1,1-dimethoxyethene.

Scheme 71.
Phosphoryl group-controlled synthesis of 1H- and 3H-indazoles from diazo phosphonates and arynes.
Scheme 71.
Phosphoryl group-controlled synthesis of 1H- and 3H-indazoles from diazo phosphonates and arynes.

Scheme 72.
(a) Photochemical and (b) thermal domino HDDA for the synthesis of indazoles.

Scheme 73.
Synthesis of fused phosphonylindazoles from nitroarenes and BOR.

Scheme 74.
A 1,3-dipolar cycloaddition of naphthoquinones and diazo compounds for the synthesis of naphthohydroquinone-derived indazoles and (1,4)-naphthoquinono-[3,2-c]-1H-pyrazoles.
Scheme 74.
A 1,3-dipolar cycloaddition of naphthoquinones and diazo compounds for the synthesis of naphthohydroquinone-derived indazoles and (1,4)-naphthoquinono-[3,2-c]-1H-pyrazoles.

Scheme 75.
The general pathway for the formation of 2H-indazoles by the reaction of cyclic diazocarbonyl compounds with arynes.
Scheme 75.
The general pathway for the formation of 2H-indazoles by the reaction of cyclic diazocarbonyl compounds with arynes.

Scheme 76.
Synthesis of spiro-3H-indazoles 315 and their thermal rearrangement into fused 2H-indazoles 316.
Scheme 76.
Synthesis of spiro-3H-indazoles 315 and their thermal rearrangement into fused 2H-indazoles 316.

Scheme 77.
Synthesis of spiro-3H-indazoles and fused 2H-indazoles from cyclic diazo ketones.

Scheme 78.
Ag(I)-promoted synthesis of 2-aryl-2H-indazoles from terminal diazo carbonyls and benzynes.
Scheme 78.
Ag(I)-promoted synthesis of 2-aryl-2H-indazoles from terminal diazo carbonyls and benzynes.

Scheme 79.
Pd-catalyzed tandem deacylative cross-coupling/denitrogenative cyclization reaction for the synthesis of 2H-indazoles.
Scheme 79.
Pd-catalyzed tandem deacylative cross-coupling/denitrogenative cyclization reaction for the synthesis of 2H-indazoles.

Scheme 80.
Rh(III)-catalyzed C-H-alkylation/intramolecular decarboxylative cyclization reaction for synthesis of 2H-indazoles.
Scheme 80.
Rh(III)-catalyzed C-H-alkylation/intramolecular decarboxylative cyclization reaction for synthesis of 2H-indazoles.

Scheme 81.
General «diazo» approach to 1,2,3-thiadiazoles.

Scheme 82.
Mechanism of Wolff cyclocondensation.

Scheme 83.
Synthesis of 1,2,3-triazoles from diazo malonaldehyde and aniline hydrochlorides.

Scheme 84.
The Wolff cyclocondensation between α-diazo-β-oxosulfones and anilines.

Scheme 85.
FeCl2-catalyzed Wolff cyclocondensation of amines and α-diazo-β-oxoamides.

Scheme 86.
Synthesis of (1,2,3-triazol-1-yl)furoxans from aminofuroxans and diazo dicarbonyls.

Scheme 87.
Intramolecular Wolff cyclocondensation with γ-N-protected amino diazo ketones.

Scheme 88.
Synthesis of fused triazole systems from α-diazo-β-keto-γ-haloesters and aminothiols.

Scheme 89.
Synthesis of N-(arylamino)triazoles from diazo ketones and arylhydrazines.

Scheme 90.
Synthesis of N-aminotriazoles by imine formation with subsequent diazo transfer.

Scheme 91.
Synthesis of 5-aminotriazoles by diazo transfer on α-acyl acetamidine hydrochlorides.

Scheme 92.
Synthesis of 5-hydroxytriazoles from dimethyl diazomalonate and amines.

Scheme 93.
Synthesis of N-hydroxytriazoles.

Scheme 94.
Iminomethylation of terminal diazo compounds for the synthesis of 1,2,3-triazoles.

Scheme 95.
Synthesis of 5-halo-1,2,3-triazoles from α-acyl α-diazoacetonitriles.

Scheme 96.
DBU-mediated reaction of diazo compounds with aryl imines for the synthesis of fully substituted 1,2,3-triazoles.
Scheme 96.
DBU-mediated reaction of diazo compounds with aryl imines for the synthesis of fully substituted 1,2,3-triazoles.

Scheme 97.
Three-component synthesis of triazole-4-phosphonates.

Scheme 98.
CuBr2-promoted synthesis of triazoles from N-arylglycine derivatives and diazocarbonyl compounds.
Scheme 98.
CuBr2-promoted synthesis of triazoles from N-arylglycine derivatives and diazocarbonyl compounds.

Scheme 99.
Ag-catalyzed [3+2]-cycloaddition of diazo compounds to isocyanides affording 1,4-disubstituted triazoles.
Scheme 99.
Ag-catalyzed [3+2]-cycloaddition of diazo compounds to isocyanides affording 1,4-disubstituted triazoles.

Scheme 100.
DBU-promoted reaction of glycine imines with diazo ketones for the synthesis of 1,2,4-triazole-3-carboxylates.
Scheme 100.
DBU-promoted reaction of glycine imines with diazo ketones for the synthesis of 1,2,4-triazole-3-carboxylates.

Scheme 101.
Synthesis of 3-(1H-1,2,4-triazol-1-yl)indolin-2-ones from diazo oxindoles and oxazol-5-(4H)-ones.
Scheme 101.
Synthesis of 3-(1H-1,2,4-triazol-1-yl)indolin-2-ones from diazo oxindoles and oxazol-5-(4H)-ones.

Scheme 102.
Photoredox decarboxylative C(sp3)−N coupling of N-hydroxyphthalimide (NHPI) esters with α-diazoacetates for the synthesis of 1,2,4-triazoles.
Scheme 102.
Photoredox decarboxylative C(sp3)−N coupling of N-hydroxyphthalimide (NHPI) esters with α-diazoacetates for the synthesis of 1,2,4-triazoles.

Scheme 103.
Three-component synthesis of 1,2,4-triazoles from diazo esters, aryl diazonium salts and nitriles.
Scheme 103.
Three-component synthesis of 1,2,4-triazoles from diazo esters, aryl diazonium salts and nitriles.

Scheme 104.
Synthesis of 1,2,4-triazoles from diazo azoles and ethyl isocyanoacetate.

Scheme 105.
Synthesis of 2,5-disubstituted tetrazoles from lithium trimethylsilyl diazomethane and methyl esters.
Scheme 105.
Synthesis of 2,5-disubstituted tetrazoles from lithium trimethylsilyl diazomethane and methyl esters.

Scheme 106.
AgOAc-catalyzed [3+2]-cycloaddition of 2,2,2-trifluorodiazoethane to aryl diazonium tetrafluoroborates for the synthesis of 2,5-disubstituted tetrazoles.
Scheme 106.
AgOAc-catalyzed [3+2]-cycloaddition of 2,2,2-trifluorodiazoethane to aryl diazonium tetrafluoroborates for the synthesis of 2,5-disubstituted tetrazoles.

Scheme 107.
Synthesis of CF2-substituted tetrazoles.

Scheme 108.
Synthesis of C-unsubstituted and 5-bromo-substituted 2-aryltetrazoles.

Scheme 109.
Silver nitrate-catalyzed [3+2]-cycloaddition of diazo ketones and diazo amides to aryl diazonium tosylates for the synthesis of 2-aryl-5-acyltetrazoles.
Scheme 109.
Silver nitrate-catalyzed [3+2]-cycloaddition of diazo ketones and diazo amides to aryl diazonium tosylates for the synthesis of 2-aryl-5-acyltetrazoles.

Scheme 110.
Synthesis of 2-aryltetrazol-5-yl sulfones and 2-aryltetrazol-5-yl sulfonamides.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Budeev, A.; Kantin, G.; Dar’in, D.; Krasavin, M. Diazocarbonyl and Related Compounds in the Synthesis of Azoles. Molecules 2021, 26, 2530. https://doi.org/10.3390/molecules26092530
AMA Style
Budeev A, Kantin G, Dar’in D, Krasavin M. Diazocarbonyl and Related Compounds in the Synthesis of Azoles. Molecules. 2021; 26(9):2530. https://doi.org/10.3390/molecules26092530
Chicago/Turabian StyleBudeev, Anton, Grigory Kantin, Dmitry Dar’in, and Mikhail Krasavin. 2021. "Diazocarbonyl and Related Compounds in the Synthesis of Azoles" Molecules 26, no. 9: 2530. https://doi.org/10.3390/molecules26092530