3.1. Formation Reaction
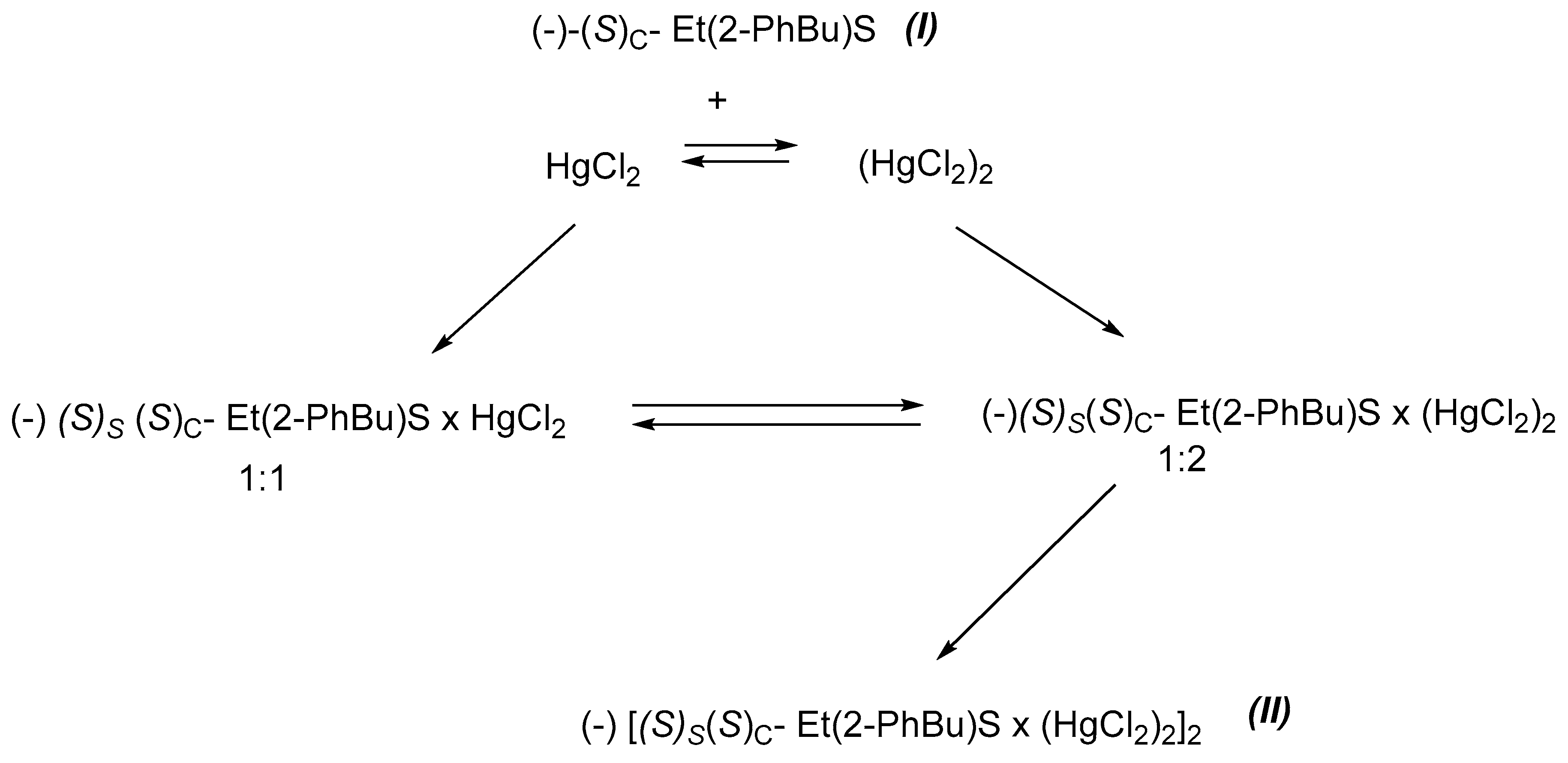
Upon addition of the chiral non-symmetric sulphide (I) to a concentrated solution of mercury(II) chloride in ethyl alcohol at room temperature, the new diastereoisomeric complex with mercury(II) chloride, (−)[(S)S(S)C-Et(PhBu)S.(HgCl2)2]2, (II), was instantaneously formed. The molar optical activity of the isolated complex in ethanol was [α]D = –2.4. This value grows upon addition to the solution of an excess of mercury chloride, thus showing the progressive formation of a new stereogenic centre in the molecule.
It can, therefore, be supposed that in a solution complex (
II) is in an equilibrium with the free ligand (
I) and HgCl
2, as shown in
Scheme 1. The solid state IR spectra, analytical results and X-ray diffractometric characterisation of the complex indicated that its stoichiometry was 1:2. The formation of the complex can reasonably be explained if one assumes that in a concentrated solution mercury (II) chloride exists in the dimeric form and adding it to one of the diastereotopic faces of the prochiral sulphur atom of the optically active sulphide (
I) gives an intermediate complex, which crystallises in polymeric 1:2 form:
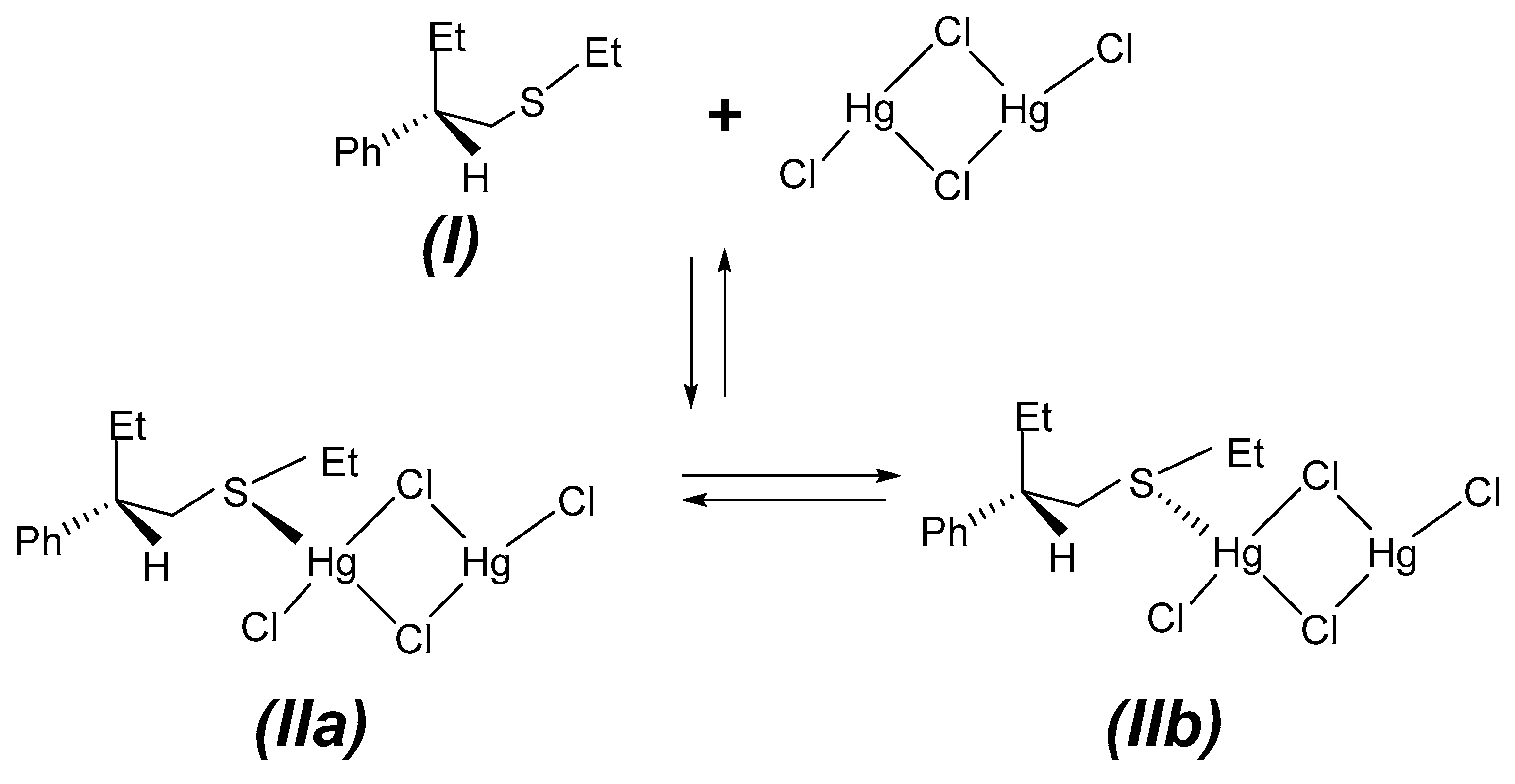
The addition of mercury(II) chloride to the optically active sulphide (
I) may yield two diastereoisomers, (
S)
S(
S)
C -, (
IIa), and (
R)
S(
S)
C-Et(PhBu)S∙(HgCl
2)
2, (
IIb), where
R or
S refers to the absolute configuration of the sulphur and carbon atom, C(3), of the sulphide. The metal could bind to the prochiral sulphur atom from two different directions and create the two diastereoisomers, as shown in
Scheme 2, crystallizing by an asymmetric second order process as (
S)
S(
S)
C-Et(PhBu)S.(HgCl
2)
2, (
IIa), examined by X-ray.
3.3. Molecular and Crystal Structure by X-ray Diffractometry
The adduct (Et(EtCHPhCH
2)S)Hg.(HgCl
2)Cl is polymeric in the crystal, and is constituted by SHgCl
+∙HgCl
2 units, assembled with the assistance of Cl
− anions.
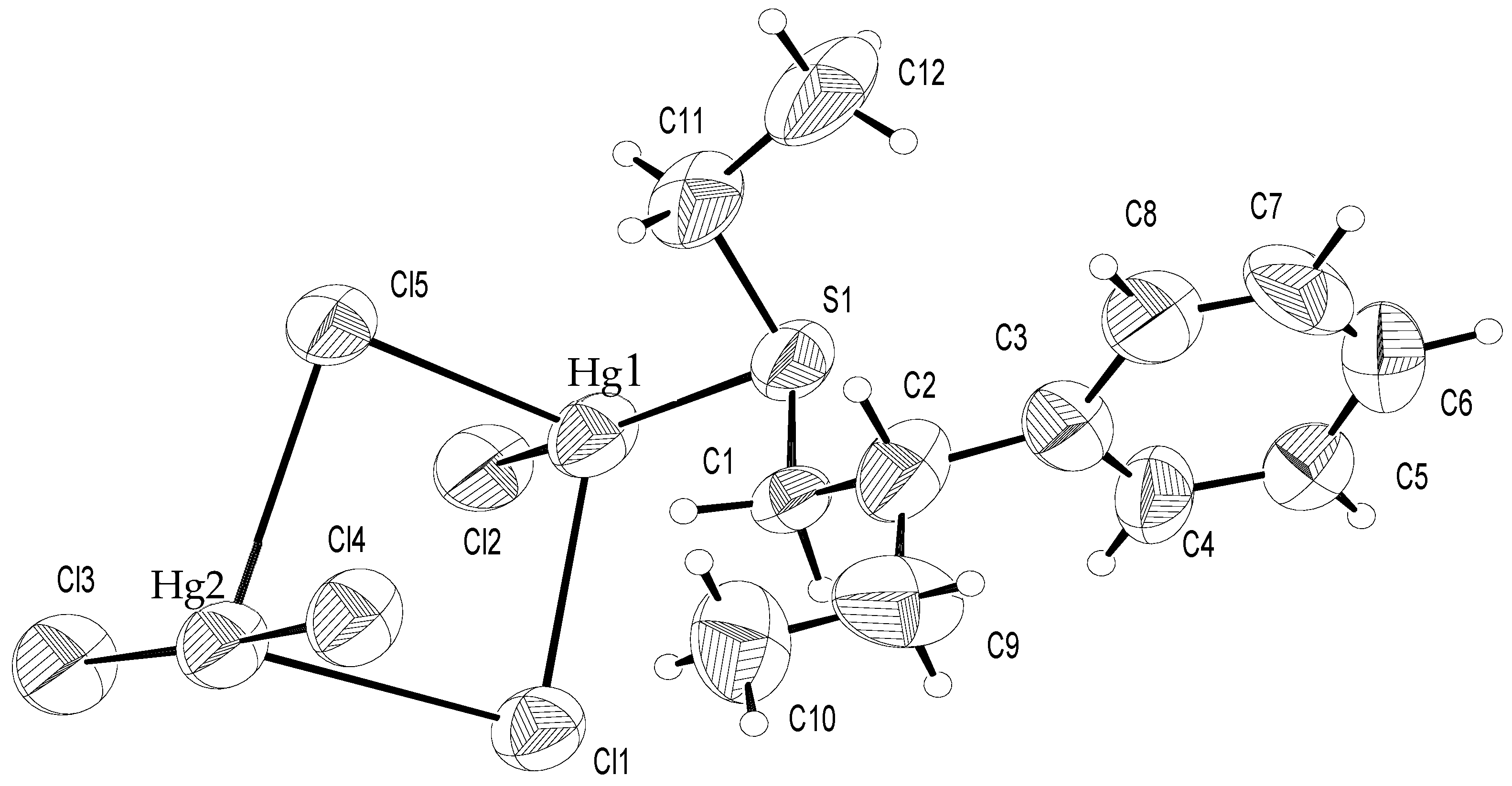
Figure 1 shows the contents of the asymmetric unit along with the atom labelling, and
Table S2 (see SI) lists bond distances and angles. Both metals are surrounded by six donors in a highly distorted octahedral arrangement very similar to the one found in Et2S∙2HgCl
2 [
2].
While only Hg1 is bonded to the sulphur ligand, for both Hg1 and Hg2 the coordination environment is characterised by two short bonds, to Cl2 (2.366(7)Å) and S (2.397(7)Å) for Hg1, and to Cl3 (2.276(8)Å) and Cl4 (2.300(7)Å) for Hg2, which originate digonal units, more distorted for Hg1 (154.9(3)°), rather regular for Hg2 (173.2(4)°). The electroneutrality of the adduct is achieved by a further chlorine, which can be considered prevalently ionic [
2]. In the crystal, this atom is placed in two independent positions on the two-fold axis, Cl1 and Cl5. The anions are bonded to Hg1 and Hg2, at distances falling in the range 2.767–3.146 Å, as observed for similar compounds forming square bridging systems nearly perpendicular to the digonal molecules, with internal angles ranging from 85.1(2)° to 96.9(2)° [
6]. The tetraatomic ring Hg1–Cl1–Hg2–Cl5 is slightly folded (8.5°) along the Cl1–Cl5 vector. Since both anions lie on symmetry elements, two equivalent adjacent rings, reaching a distorted tetrahedral coordination, with angles Hg–Cl–Hg ranging from 80.3(3) to 145.9(2)° for Cl1 and from 77.8(2) to 141.3(2)° for Cl5 share each anion. Both metals complete hexa-coordination by means of long interactions ranging between 3.162 and 3.333 Å with atoms Cl2 and Cl4 belonging to symmetry related molecules. The coordination polyhedra can be interpreted as 2 + 4 tetragonally compressed octahedra with the four longer contacts lying in the equatorial plane. However, the real picture is more complex in both cases, with atoms deviating up to 0.45 Å and 0.39 Å from the least-squares average equatorial plane containing Hg1 and Hg2, respectively. It is convenient to describe the crystal polymeric packing on the basis of the interactions between digonal molecules Et(EtCHPhCH
2)SHgCl
+ and HgCl
2, and of the role played by the different chlorines in these interactions.
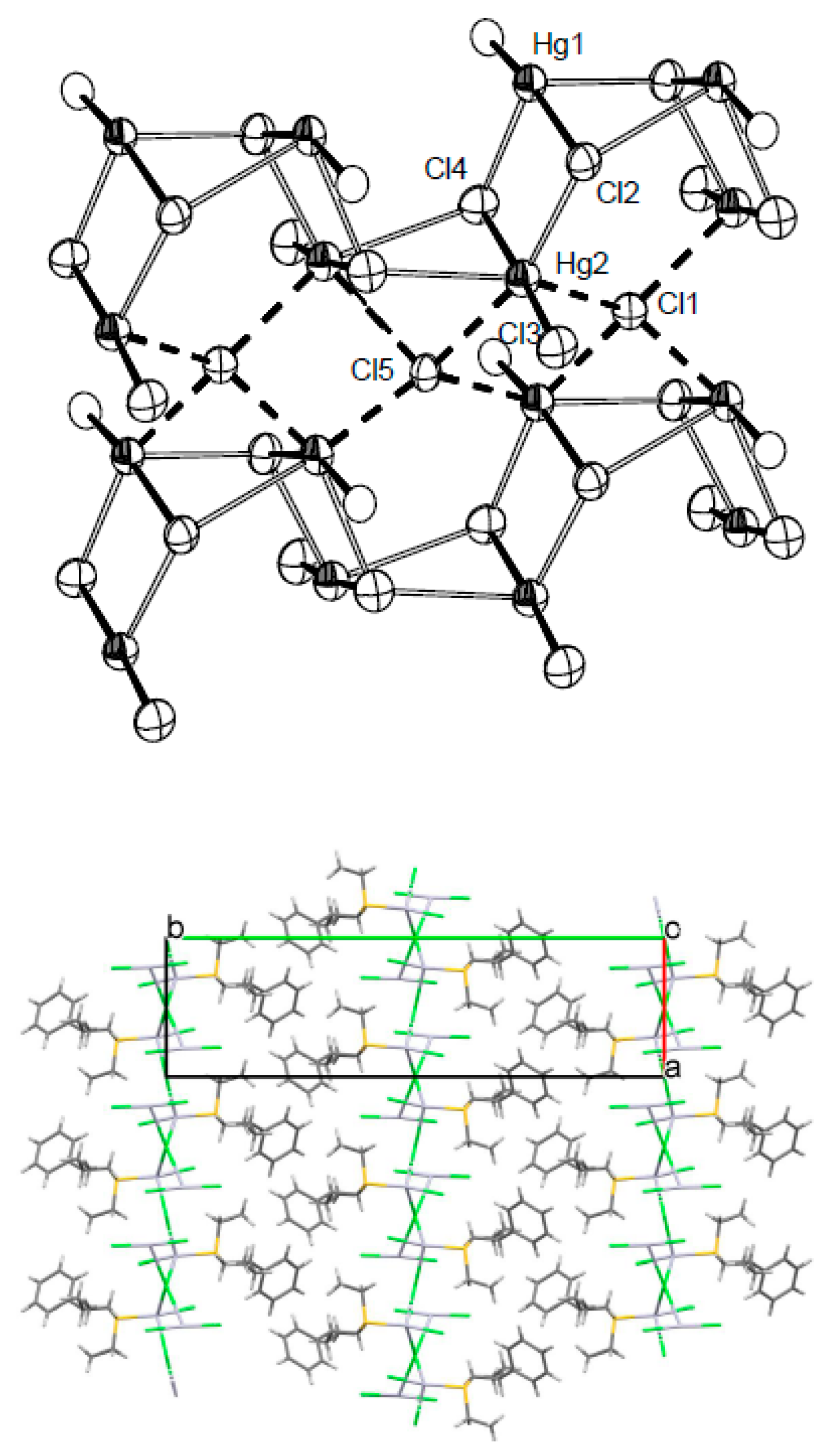
As shown in
Figure 2, Cl2 and Cl4 are strongly bonded to Hg1 and Hg2, respectively (digonal molecules, indicated with thick bonds), and reach a trigonal coordination by bridging two other neighbours metals, namely Hg1 (−x, −y + 1, z) and Hg2 (x, y, z − 1) for Cl2 and Hg1 (x, y, z + 1) and Hg2 (−x + 1, −y + 1, z) for Cl4. This originates tetraatomic rings in which the metals occupy opposite vertices and in which the edges corresponding to the short bond are shared between adjacent rings. The polymeric structure can be viewed as the joining of Et(EtCHPhCH
2)S HgCl
+∙HgCl
2 dimers in a head-to-head, tail-to-tail fashion. The resulting cationic ribbons are highly puckered since the interactions between dimers occur perpendicularly to the dimer plane. The anions Cl1 and Cl5 are intercalated in tetracoordinated sites and have the two-fold function of stabilising the puckering pattern by bridging pairs of metals which delimitate the convex sections of the ribbon, and of favouring the stacking of the ribbons in the
a direction, forming inorganic layers in the
ac plane. On the basis of their role in the crystal packing, it is likely that Cl1 and Cl5 act as templates in determining the organisation of the packing motif. The remaining non-ionic chlorine, Cl3, does not participate to any intermolecular contact, and it is topologically equivalent to the sulphur ligand in trimming the ribbon lateral edges. (
Figure 3) Lipophilic interactions due to the aromatic part of the ligand occur between layers in the
b direction.
The above motif is partially present also in the packing of the achiral Et2S∙2HgCl
2 where short five-ring ribbons can be found organised in layers [
2]. The main differences between the crystal arrangements of the two compounds involve the anionic chlorine, which in Et2S∙2HgCl
2 is tricoordinated and loses part of its functionality of inter-ribbons stabiliser, and the HgCl
2 linear molecule, which in Et2S∙2HgCl
2 employs both its chlorines in the coordination of neighbouring metals.
Another interesting comparison can be made with the 1:1 adduct Me(EtCHPhCH2)S∙HgCl2[[6],5 containing a chiral diastereotopic thioether ligand very similar to the present one. The complex contains two chiral centres, the carbon C2, whose configuration is (S)c, and the coordinated sulphur, which can result either (S)S or (R)S depending on the stereochemistry of the coordination reaction. In both compounds the attack of the prochiral sulphur to the metal occurs in the direction which induces at the sulphur the same absolute configuration as the stereogenic carbon has. In the 1:1 adduct this was explained by the fact that the observed stereochemistry allowed to attain a suitable ligand conformation which stabilises the complex by means of π interaction, retained in the crystal structure, between the metal and the phenyl electronic cloud. The X-ray analysis shows that these interactions stabilize the (S)S(S)C diastereoisomer in respect of the (R)S(S)C one. The differential energy of the two possible diastereoisomers and stereochemistry of the intermolecular association interactions are the driving forces of the stereoselective synthesis and stabilization of the complex.
Assignment of the carbon absolute configuration was made on the basis of chemical arguments based on the synthesis of (
S)(−)Et(PhBu)S, (
I), starting from optically pure (S)(+)-2-phenylbutanoic acid [
5]. Since subsequent replacement on the chiral centre occurred with retention of the initial configuration, this knowledge allows the (
S)
C configuration to be assigned at C(3). At the same time, after crystallographic resolution, the sulphur chirality can be fixed accordingly as (
S)
S. In the present compound,
IIa, the ligand adopts an extended conformation with a C11–S1–C1–C2 torsion angle of −69(2)°, compared with the value of −165(1)° found in the 1:1 adduct. The interaction with the phenyl ring is replaced by the intra-ribbon bond to Cl2 (−x, −y − 1, z), which is reinforced by the action of Cl1, bridging Hg1 and Hg1 (−x, −y − 1, z). This does not rule out the possibility that the aromatic ring is important in favouring the formation in solution of the (
S)
S(
S)
C diastereoisomer, but suggests that the association between Et(EtCHPhCH
2SHgCl
+∙HgCl
2 dimers might occur via an “in line” nucleophilic substitution of Cl2 on Hg1 (−x, −y + 1, z), and Cl2 (−x, −y + 1, z) on Hg1, assisted by the bridging action of Cl1.
3.4. Absorbance and Circular Dichroism Spectra
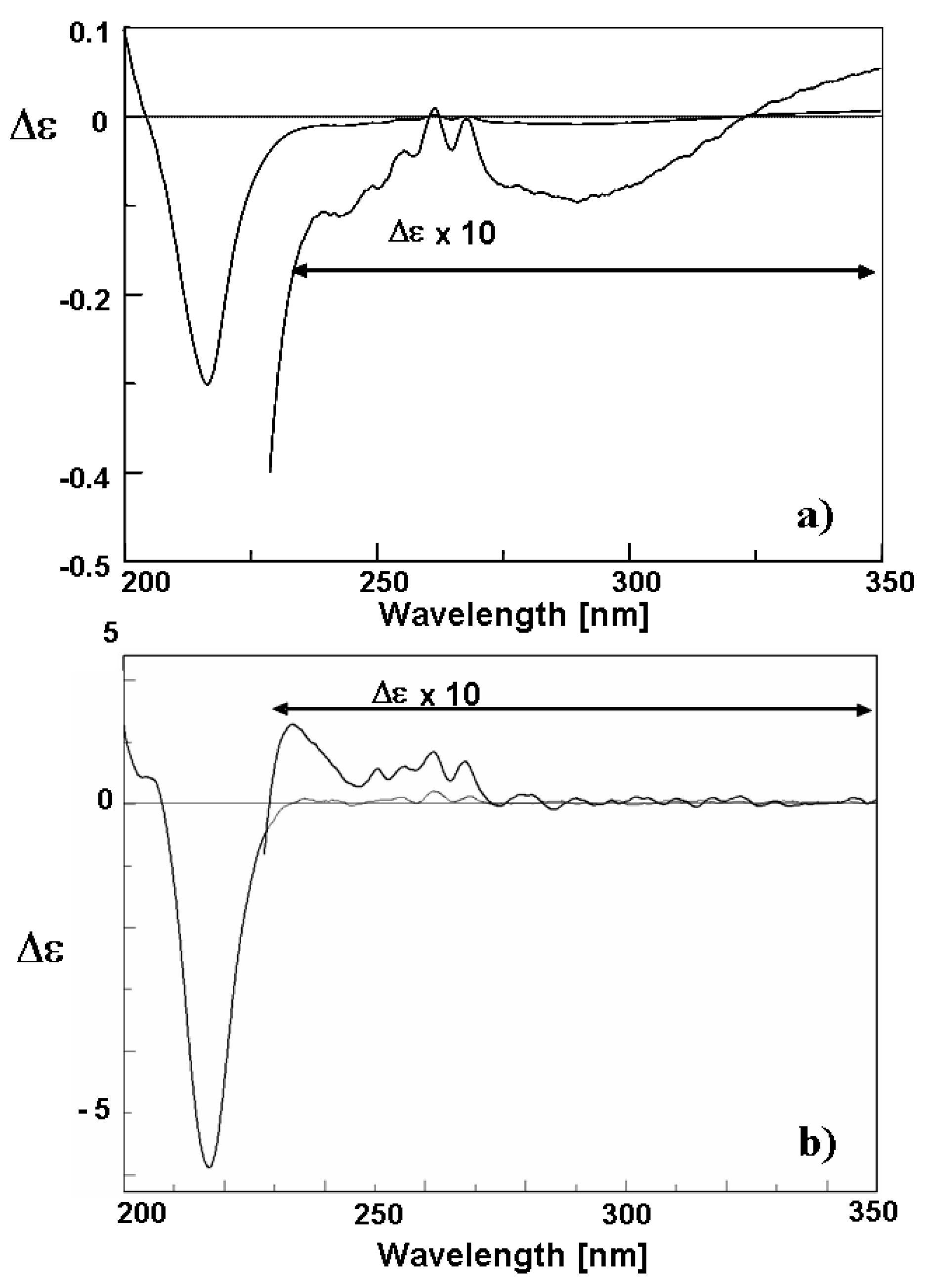
The formation of the chiral diastereopure complex (
II) according to
Scheme 1 is supported by the CD spectrum. The absorbance spectra of monosubstituted benzene derivatives show a weak first vibrationally structured band at ca. 260 nm assigned to the B
2u benzene-like transition. Additionally, less structured bands are observed at higher energies. The CD spectrum closely corresponds to the absorbance spectra, showing more clearly the energies and intensities of the expected transitions [
10]. Three bands and one inflection corresponding to the four electronic transitions can be detected also in the CD spectrum of the sulphide-mercury(II) chloride complex (
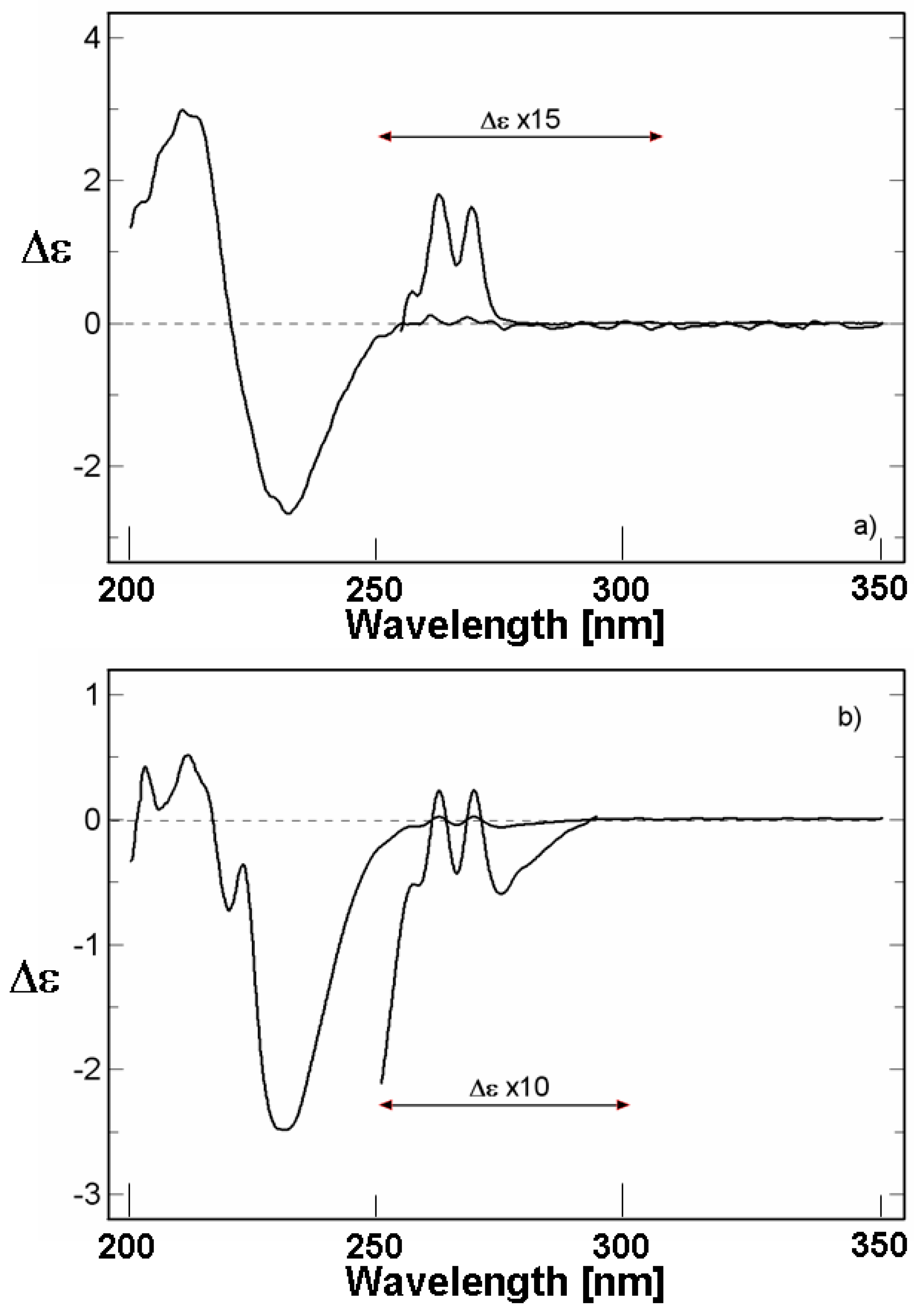
II). In diluted solutions, this spectrum as given in
Figure 4a is very similar to that of the sulphide (
I) [
17]. It shows a positive Cotton effect with a vibronic structure, assigned to the B
2u transition of the phenyl group of the ligand at about 260 nm, a negative band at 232 nm related with the A
1 and A
2 electrically forbidden transition of the sulphide chromophore, a positive one at 210 nm related to the B
1u benzene-like transition, and an inflection at about 205 nm related to the allowed sulphide transition.
As for the sulphide (
I), the (
S)
C absolute configuration can be correlated with the sign of these three bands. In a diluted solution, complex (
II) seems to be strongly dissociated and it is not possible to establish the Cotton effect due to the formation of the S–Hg bond. However, as it is shown in
Figure 4b, by the addition of an excess of mercury(II) chloride the Cotton effect at 260 nm progressively passes into the negative part of the spectrum, being lowered by another band which grows at 230 nm as a strong negative Cotton effect, g = −2 × 10
−3 for the ratio sulphide/HgCl
2, 1/200. This new negative band can be tentatively assigned to a CT transition which grows upon addition of an excess of mercury(II) chloride, while, at the same time, the A
1 and A
2 CD band at 232 nm disappears; this is indicative of mercury(II) chloride addition to the sulphide and formation of a new S–Hg bond. These changes in the CD spectra also demonstrate the formation of a new stereogenic centre on the pseudo-tetrahedral sulphur atom bonding the metal.
A very similar Cotton effect related with a CT transition was observed earlier in the CD spectrum of the 1:1 complex of optically active (−)(
S)
S(
S)
C methyl 2-phenylbutyl sulphide with mercury (II)chloride for which the (
S) absolute configuration at the newly created chiral sulphur atom was established [
6]. This similarity clearly indicates that the isolated diastereoisomer of the complex (
II) also has the (
S)
S absolute configuration at the sulphur atom as shown in the solid state by the molecular structure description.
The molecular structure of ethyl-2-phenylbutyl sulphide-mercury(II) chloride complex, 1/2, (
II), shows that the (
S)s absolute configuration at the sulphur atom has been obtained. In the CD spectra, the negative Cotton effect due to the CT of the 1:2 complex at ~230 nm is related with the (
S)
S absolute configuration on a new stereogenic sulphur centre. This correlation demonstrates that the diastereoisomer (−)[(
S)
S(
S)
C-Et(PhBu)S∙2HgCl
2]
2, (
IIa), (
Scheme 2), has preferentially been formed and that a stereoselective addition of mercury to the prochiral sulphide sulphur atom has been achieved. The complex can be crystallized in the presence of a large excess of mercury chloride. In solutions near to saturation, the equilibria shown on
Scheme 1 are shifted to the formation of the complex, which crystallizes following an asymmetric second order crystallization process. The same configuration is maintained in the solid state, as shown by molecular structure X-ray determination.
The crystal structure determination shows that the (S)S configuration is always thermodynamically more stable and the stabilisation of the S–Hg bond is maintained in spite of the rotation of the aryl-substituted butyl chain, which thus establishes new interactions with the same groups of another parallel chain of the polymeric complex during crystal packing. The crystal packing energy is sufficiently high to overcome the phenyl-Hg interactions established during the formation process, and to determine the rotation of the phenyl-butyl chain around the C1-S bond, but not sufficient to break the S–Hg bond resulting from the first attack during the formation of the complex in solution.
3.5. Oxidation
The oxidation of dialkyl-sulphides R
1R
2S with hydrogen peroxide and H
2SO
4 as a catalyst in presence of mercury(II)chloride are chemoselective giving complexes of mercury(II)chloride with pure sulphoxides as ligands without formation of the corresponding sulfones [
3].
The oxidation of the chiral sulphide (
I) with hydrogen peroxide and H
2SO
4 as a catalyst (Equation (1)) gives diastereoisomeric sulphoxide (−)(
S)
S(
S)
C -Et(PhBu)SO, (
III) having a de = 18.2%. This result shows that the oxidation of sulphide (
I) is asymmetrically driven by the (
S)
C absolute configuration of the carbon atom of the alkyl chain.
The oxidation of the 1:2 complex alone and in the presence of an excess of mercury (II) chloride (Equations (2) and (3)) shows that a huge excess of mercury (II) chloride (1:200) induces a substantial increase in the diastereoselectivity of the reaction with inversion of configuration at a stereogenic sulphur atom. The oxidation of the 1:2 complex, without an excess of mercury (II) chloride gives sulphoxide (
S)
S(
S)
C, (
III) with d.e = 18%, and with 200 molar excess of HgCl
2, sulphoxide (
R)
S(
S)
C, (
IV) with d.e = 31%, respectively (
Scheme 3).
The last oxidation shows that mercury chloride bonding sulphur atom with the (S)S absolute configuration cooperates significantly to the asymmetric synthesis of (−)(R)S(S)C-Et(PhBu)SO, (IV), driving the oxidation on the enantiotopic site of prochiral sulphur atom.
The (
R)
S absolute configuration at the sulfinyl sulphur atom of this sulphoxide can be deduced from the CD spectrum (
Figure 5) where a new positive Cotton effect at ~235 nm followed by another negative one at 220, a positive one at 205 and a strong positive one at ~198 nm are observed. When compared with the spectra of the corresponding sulphide, the CD spectra of sulphoxides show, a new positive or negative Cotton effect at ~235 nm. The presence of this new Cotton effect is commonly considered as an indicator of the absolute configuration at the sulphur atom, (
S)
S if negative, (
R)
S if positive [
17]. In the examined case the configuration (
S)
S in the complex of sulphide (
II) becomes (
R)
S in sulphoxides (
IV), as shown by the positive sign of the Cotton effect at ca. 235 nm.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
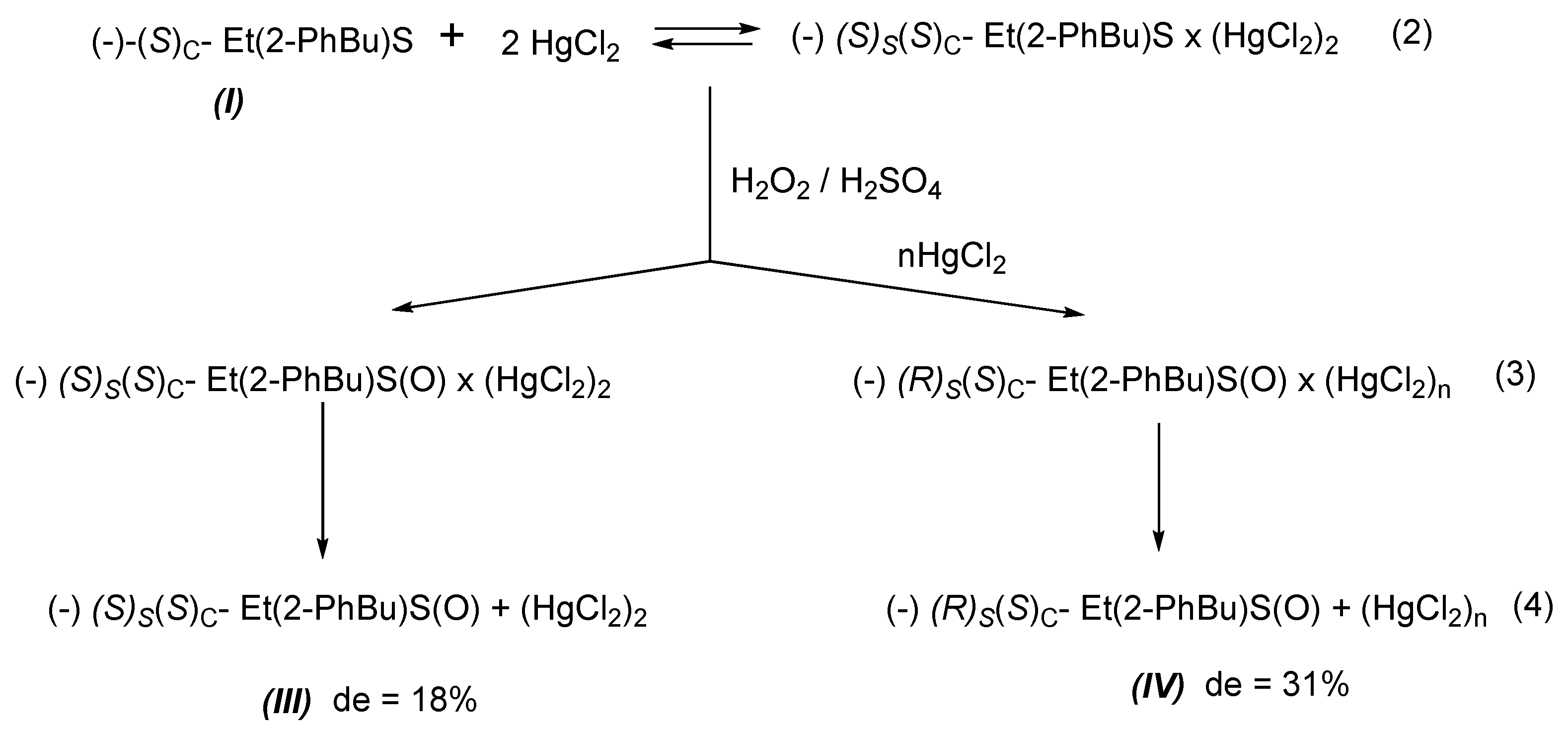{kind=link}