Urea Derivative Catalyzed Enantioselective Hydroxyalkylation of Hydroxyindoles with Isatins


Department of Pharmacy, Jilin Medical University, Jilin 132013, China
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(21), 3944; https://doi.org/10.3390/molecules24213944
Submission received: 19 September 2019
/
Revised: 21 October 2019
/
Accepted: 21 October 2019
/
Published: 31 October 2019
Abstract
:The enantioselective transformations of indoles preferentially take place in the more-reactive azole ring. However, the methods for the enantioselective functionalization of the indole benzene ring are scarce. In this paper, a series of bifunctional (thio)urea derivatives were used to organocatalyze the enantioselective Friedel-Crafts hydroxyalkylation of indoles with isatins. The resulting products were obtained in good yields (65–90%) with up to 94% enantiomer excess (ee). The catalyst type and the substrate scope were broadened in this methodology.
1. Introduction
The indole scaffolds are privileged skeletons, as they have been widely found in many bioactive natural products, pharmaceuticals, and material molecules [1,2,3,4,5,6]. The synthesis and modification of indoles have attracted intensive interest for a long time. Accordingly, the enantioselective functionalization of indoles has been one of the most studied reactions in asymmetric catalysis [7,8,9,10,11,12,13,14]. Indoles show a high nucleophilic reactivity in the azole ring, which preferentially reacts with electrophilic aromatic substitution at the C-3 position [15,16,17,18,19,20,21,22]. Additionally, these Friedel-Crafts (F-C) reactions also selectively take place at the positions C-2 [23,24,25,26,27,28,29,30] and N-1 [31,32,33,34,35,36,37] by using different strategies. However, the functionalization in the benzene ring of indole is still difficult, which generally requires the presence of directing or blocking group in the azole ring [38,39,40,41,42,43,44,45,46] and employs transition metal catalysts [47,48,49].
In 2015, Jørgensen reported the first example of catalytic asymmetric F-C alkylation of 4-hydroxyindole occurring at the C-5 position [50]. Subsequently, Pedro and coworkers developed the first asymmetric F-C reaction of hydroxyindoles with isatin-derived ketimines [51] or isatins [52] organocatalyzed by cinchona-derived squaramide. These methodologies allow the functionalization of indoles in every position of the benzene ring in a regioselective and enantioselective fashion, by utilizing the activating/directing ability of hydroxyl group. Moreover, the OH group was removed smoothly upon hydrogenolysis of the corresponding triflate. Very recently, Zhao [53] and Deng [54], respectively, reported the cinchona-derived squaramide organocatalyzed the enantioselective F-C transformation in the benzene ring of indoles, employing other electrophiles with hydroxyindoles. In particular, the resulting hydroxyindole moiety is of high importance in medicinal chemistry and natural products [51,55,56,57], showing great potential in diversity-oriented synthesis and drug discovery.
In spite of the significant developments, there is a high demand for the enantioselective functionalization in the benzene ring of indole by using new types of catalysts. Takemoto’s catalyst is the commercially available chiral organocatalyst, which was first synthesized by Takemoto in 2003 [58]. Then, they were used efficiently for a wide range of diastereoselective and enantioselective reactions [59,60,61,62,63,64,65]. We herein first reported the enantioselective hydroxyalkylation of hydroxyindoles with isatins by employing Takemoto-type catalysts 1a–1h bearing (thio)urea-tertiary amine moiety (Figure 1).
2. Results and Discussion
According to the optimized conditions reported by Pedro [52], we first screened the bifunctional catalysts in the reaction of 4-hydroxyindole and isatin (2a) with Et2O as a solvent in the presence of 10 mol% of (thio)urea catalysts 1a–1h (Table 1). Initially, widely used thiourea 1a was examined at room temperature. However, the desired 5-alkylated indole 3a was afforded in 70% yield with 68%ee (entry 1). Gratifyingly, when (S, S)-urea catalyst 1b was used to induce the reaction, the obviously improved yield and ee value were observed (entry 2). Based on a comparison of the optical rotation of the product with a value from the literature [52], the absolute configuration of the major product was determined to be R. The enantiomeric (R, R)-urea catalyst 1d gave the S major isomer with slightly decreased ee of 76% (entry 4). Surprisingly, piperidine-based thiourea 1e could not catalyze the reaction. Moreover, the further endeavor to improve ee by increasing the steric bulk of the basic moiety of bifunctional catalysts 1f–1h was not successful (entries 6–8). Therefore, the catalyst 1b, containing N, N-dimethyl tertiary amine and urea moiety, showed the best yield and enantioselectivity.
To optimize the enantioselectivity of the transformation, we investigated a variety of different reaction conditions (Table 2). The survey of solvents showed that Et2O was the optimal solvent in terms of the yield and enantioselectivity (entries 1–4). The screening of catalyst loading exhibited that 10 mol% equivalent of 1b was optimal 5 mol% loading of catalyst led to reduction both in the yield and ee (entry 6), and 20 mol% loading offered no improvement in the asymmetric induction, albeit with a slightly improved yield (entry 7 vs. entry 1). When the reaction temperature was lowered from rt to 0 °C, an improved ee of 83% was afforded (entry 8 vs. entry 1). The further temperature decreases to −20 °C and −40 °C were detrimental for both yield and enantiocontrol (entries 10, 11 vs. entry 8). Furthermore, diluting the reaction concentration by half produced a slightly lower ee value and decreased yield (entry 5). Based on these experiments, the optimized conditions were determined to be Et2O as the solvent with a 10 mol% loading of catalyst 1b at 0 °C.
With the optimized conditions in hand, the substrate scope of this protocol was investigated. As shown in Table 3, the corresponding 5-alkylated products were obtained in good yield (65–90%) with >20:1 regioselectivities determined by 1H NMR. A wide range of isatins bearing various substituents on the phenyl ring such as halogens, methyl and methoxyl were tolerated in good to excellent yields with 71–94%ee except 4-bromo substituted isatin which produced 3b with only 50%ee in low yield (entry 2). It might be caused by the adjacent substituent to the carbonyl of isatin. Therefore, the enantioselectivities were obviously affected by the substituted position on the phenyl ring of isatins. The 5- and 7-substituted isatins appeared to favor higher enantioselectivities (entries 3–6 and entries 8–9). In the case of N-methylisatin and N-benzylisatin, moderate ee value was obtained (entry 10–11). Of all the different substrates listed in Table 3, the reaction of 5-Cl-substituted isatin afforded the optimal enantiomeric excess (94%ee, entry 5). Compared with the results of the reported reference [52], the similar enantioselectivities were achieved with N-unsubstituted isatins and N-Methylisatin as the reactants (entries 1, 6, and 10), whereas the reaction of N-benzylisatin with 4-Hydroxyindole got the significantly decreased ee value (entry 11). However, the obviously high yields were observed with N-unsubstituted isatins as the substrate (entries 1, 6). The reported literature [52] mainly focused on the reaction of N-benzylisatins and only tried three isatins containing the free NH group. We broadened the substrate scope by investigating nine N-unsubstituted isatins.
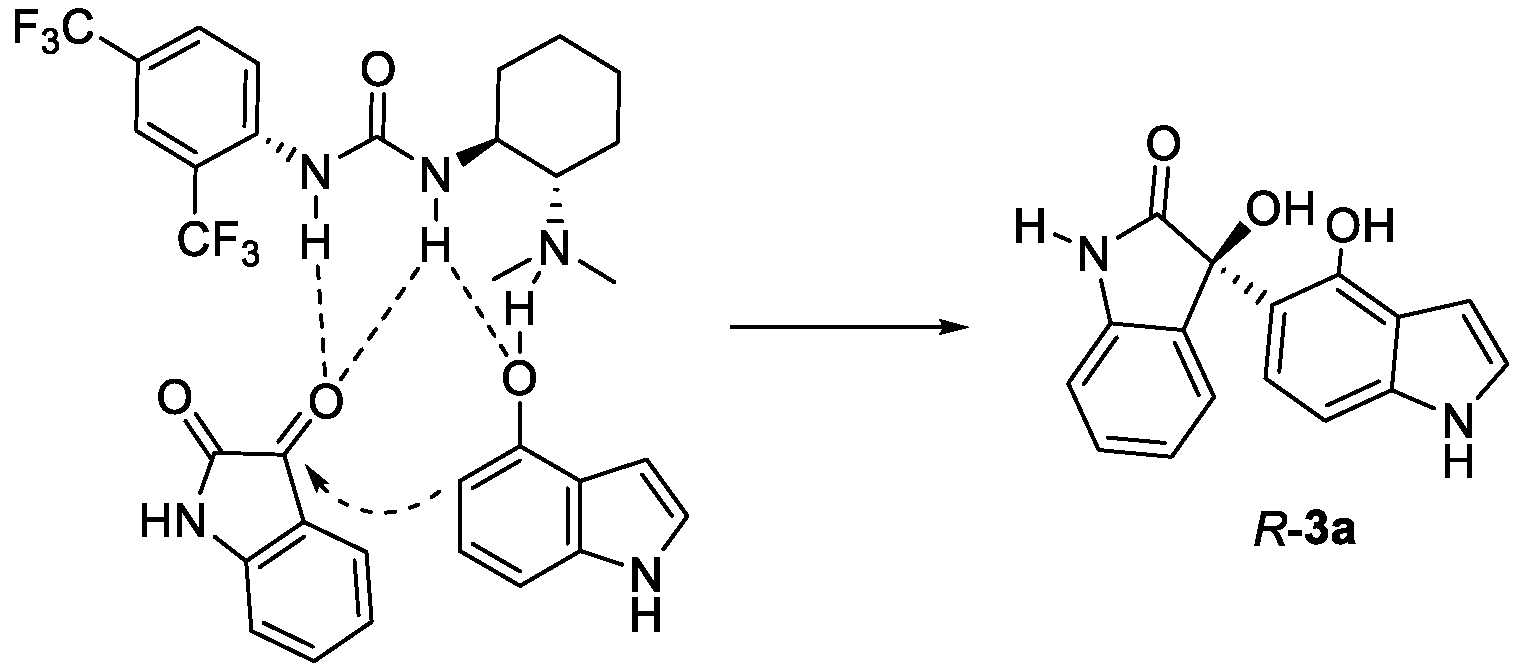
Based on the obtained absolute configuration described above and previously reported enantioselective organocatalytic reactions of isatin-derived ketimines [51], a proposed transition-state model is depicted in Figure 2. Both hydroxyindole and isatin are activated through hydrogen bonding with bifunctional urea catalyst 1b. Then, the reaction proceeds with a Re-face addition of hydroxyindoles to isatin 2a, affording the desired product R-3a.
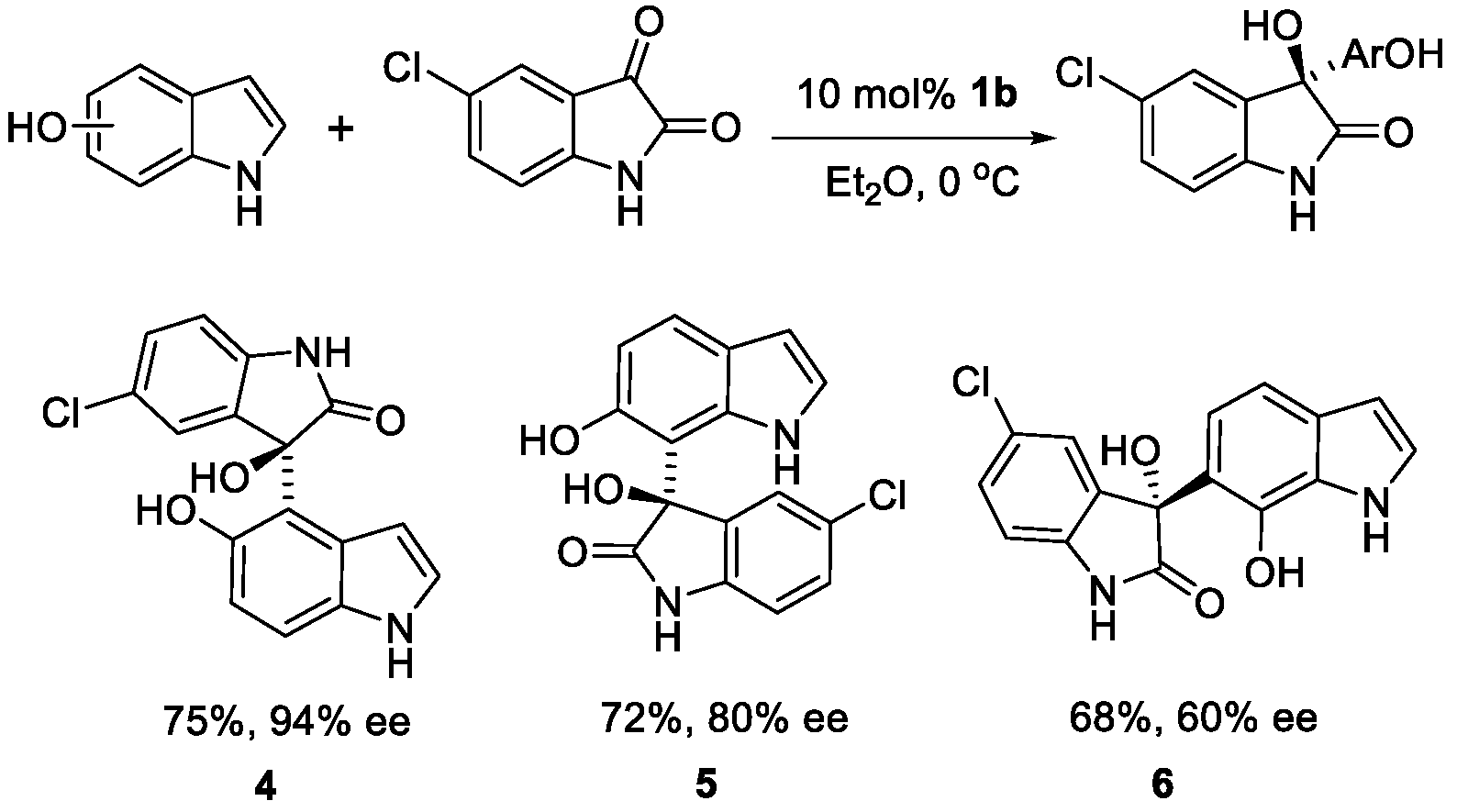
In order to achieve the functionalization of every position in the carbocyclic ring, we respectively examined the reaction of 5-, 6-, and 7-hydroxyindole and 5-Cl substituted isatin (Scheme 1). As we expected, the corresponding alkylated indoles were isolated with excellent regioselectivity in all cases. The 5-hydroxyindole showed optimal reactivity (75% yield) and enantioselectivity (94%ee) under the optimized reaction conditions to give a 4-alkylated product. In a similar manner, 6-hydroxyindole was functionalized selectively in the C-7 position with good enantioselectivity (80%ee). When we tried the reaction of 7-hydroxyindole, the 6-alkylated product was obtained with low enantioselectivity (60%ee).
3. Conclusion
In summary, we described how the first urea derivative catalyst promoted the enantioselective hydroxyalkylation of hydroxyindoles with isatins. The enantioselective modification happened in the benzene ring rather than in the azole ring to give the desired hydroxyalkylated indoles with high enantioselectivity (up to 94%ee). Furthermore, we used our optimized conditions to expand upon the substrate scope of this transformation.
4. Experimental
4.1. Chemistry
The 1H NMR spectra were recorded on a 500 MHz spectrometer, using CD3OD–d4 as a solvent. The chemical shifts were reported in ppm, and the residual CD3OD signal was used as a reference (3.31 and 4.87 ppm). The splitting patterns of the signals were reported as s, singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublets; and m, multiplet. The 13C NMR spectra were recorded on a 125 MHz instrument using CD3OD–d4 as a solvent. The chemical shifts were reported in ppm, and the residual CD3OD signal was used as a reference (49.0 ppm). High-resolution mass spectra (HRMS) were measured on a triple TOF 5600+ mass spectrometer equipped with an electrospray ionization (ESI) source in the negative-ion mode. The enantiomeric excess (ee) values of the products were determined by chiral HPLC, using Daicel Chiralpak AD-H columns (4.6 mm*250 mm). The reactions were monitored by thin layer chromatography (TLC). Purifications by column chromatography were conducted over silica gel (200–300 mesh). The organocatalysts 1a–h were purchased from Daicel chiral technologies (China) company.
4.2. General Procedure for the Enantioselective Friedel-Crafts Reaction of Hydroxyindole and Isatins
To a tube containing hydroxyindole (13.3 mg, 0.1 mmol) and isatin (2, 23.7, 0.1 mmol) and catalyst 1b (4.0 mg, 0.01 mmol), Et2O (1.5 mL) was added. The resulting mixture was stirred at room temperature for 7 h (TLC). After the reaction was finished, the reaction directly poured into a column chromatography on silica gel with hexane/EtOAc (3:1) as eluent to afford the products 3a–k. Experimental data can be found in Supplementary Materials.
(+)-(R)-3-Hydroxy-3-(4-hydroxy-1H-indol-5-yl)indolin-2-one (3a): brown oil (85% yield); 1H NMR (500 MHz, CD3OD) δ 7.28–7.19 (m, 2H), 7.10–7.05 (m, 1H), 7.01 (td, J = 7.5, 1.0 Hz, 1H), 6.93 (d, J = 8.0 Hz, 1H), 6.82 (d, J = 1.0 Hz, 2H), 6.53 (d, J = 3.0 Hz, 1H) ppm; HRMS (ESI) m/z: HRMS (ESI): calcd for C16H11N2O3− [M-1]−: 279.0775, found: 279.0770; [α]D25 = +46.85 (c 0.5, MeOH). Enantiomeric excess (83%) was determined by chiral HPLC (ChiralpakAD-H), hexane:iPrOH = 80:20, 1.5 mL/min, major enantiomer tR = 14.1 min, minor enantiomer tR = 17.3 min.
(+)-(S)-4-bromo-3-hydroxy-3-(4-hydroxy-1H-indol-5-yl)indolin-2-one (3b): brown oil (72% yield); 1H NMR (500 MHz, CD3OD) δ 7.16 (t, J = 8.0 Hz, 1H), 7.11 (d, J = 8.5 Hz, 1H), 7.07 (d, J = 3.5 Hz, 1H), 6.91 (dd, J = 7.5, 1.0 Hz, 1H), 6.82 (d, J = 8.0 Hz, 1H), 6.50 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ 181.8, 150.2, 145.6, 139.3, 132.9, 131.9, 127.8, 124.1, 122.3, 121.3, 120.1, 110.1, 103.8, 99.7, 99.6, 91.5; HRMS (ESI): calcd for C16H10BrN2O3− [M-1]−: 356.9880, found: 356.9874; [α]D25 = +5.10 (c 0.3, MeOH). Enantiomeric excess (50%) was determined by chiral HPLC (Chiralpak AD-H), hexane:iPrOH = 80:20, 1.5 mL/min, major enantiomer tR = 13.2 min, minor enantiomer tR = 17.3 min.
(+)-(R)-5-methyl-3-hydroxy-3-(4-hydroxy-1H-indol-5-yl)indolin-2-one (3c): brown oil (85% yield); 1H NMR (500 MHz, CD3OD) δ 7.08–7.05 (m, 3H), 6.83–6.80 (m, 2H), 6.78 (d, J = 8.5 Hz, 1H), 6.53 (d, J = 3.0 Hz, 1H), 2.26 (s, 3H); 13C NMR (125 MHz, CD3OD) δ 182.7, 150.5, 140.6, 139.4, 134.7, 133.4, 130.7, 126.9, 124.4, 121.7, 120.4, 114.1, 110.9, 103.9, 99.7, 80.7, 21.1; HRMS (ESI): calcd for C17H13N2O3− [M-1]−: 293.0932, found: 293.0927; [α]D25 = +64.42 (c 0.60, MeOH). Enantiomeric excess (85%) was determined by chiral HPLC (Chiralpak AD-H), hexane:iPrOH = 80:20, 1.5 mL/min, major enantiomer tR = 12.8 min, minor enantiomer tR = 14.3 min.
(+)-(R)-5-methoxyl-3-hydroxy-3-(4-hydroxy-1H-indol-5-yl)indolin-2-one (3d): brown oil (78% yield); 1H NMR (500 MHz, CD3OD) δ 7.09 (d, J = 3.5 Hz,1H), 6.87–6.78 (m, 5H), 6.53 (d, J = 3.0 Hz, 1H), 3.71 (s, 3H). 13C NMR (125 MHz, CD3OD) δ 182.6, 157.6, 150.5, 139.5, 136.3, 135.9, 124.4, 121.7, 120.4, 115.4, 114.0, 112.8, 111.7, 104.0, 99.8, 81.0, 56.2; HRMS (ESI): calcd for C17H13N2O4−[M-1]−: 309.0881, found: 309.0870; [α]D25 = +38.52 (c 0.65, MeOH). Enantiomeric excess (82%) was determined by chiral HPLC (Chiralpak AD-H), hexane:iPrOH = 80:20, 1.5 mL/min, minor enantiomer tR = 20.8 min, major enantiomer tR = 22.6 min.
(+)-(R)-5-chloro-3-hydroxy-3-(4-hydroxy-1H-indol-5-yl)indolin-2-one (3e): brown oil (81% yield); 1H NMR (500 MHz, CD3OD) δ 7.22 (dd, J = 8.0, 2.0 Hz, 1H), 7.13 (d, J = 3.0 Hz, 1H), 7.09 (d, J = 3.5 Hz, 1H), 7.03 (d, J = 8.5 Hz, 1H), 6.90 (dd, J = 13.5, 8.5 Hz, 2H), 6.53 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ 182.3, 149.6, 142.0, 139.5, 136.8, 130.1, 128.7, 126.1, 124.5, 121.3, 120.2, 114.3, 112.2, 104.1, 99.6, 79.7; HRMS (ESI): calcd for C16H10ClN2O3− [M-1]−: 313.0385, found: 313.0380; [α]D25 = +90.51 (c 0.46, MeOH). Enantiomeric excess (94%) was determined by chiral HPLC (Chiralpak AD-H), hexane:iPrOH = 80:20, 1.5 mL/min, major enantiomer tR = 13.4 min, minor enantiomer tR = 14.8 min.
(+)-(R)-5-bromo-3-hydroxy-3-(4-hydroxy-1H-indol-5-yl)indolin-2-one (3f): brown oil (80% yield); 1H NMR (500 MHz, CD3OD) δ 7.37 (dd, J = 8.0, 3.0 Hz, 1H), 7.24 (d, J = 2.0 Hz, 1H), 7.09 (d, J = 3.0 Hz, 1H), 7.03 (d, J = 8.5 Hz, 1H), 6.91 (d, J = 8.5 Hz, 1H), 6.85 (d, J = 8.5 Hz, 1H), 6.51 (d, J = 2.5 Hz, 1H); HRMS (ESI): calcd for C16H10BrN2O3− [M-1]−: 356.9880, found: 356.9873; [α]D25 = +80.27 (c 0.67, MeOH). Enantiomeric excess (90%) was determined by chiral HPLC (Chiralpak AD-H), hexane:iPrOH = 80:20, 1.5 mL/min, major enantiomer tR = 21.5 min, minor enantiomer tR = 23.7 min.
(+)-(R)-6-bromo-3-hydroxy-3-(4-hydroxy-1H-indol-5-yl)indolin-2-one (3g): brown oil (83% yield); 1H NMR (500 MHz, CD3OD) δ 7.13 (dd, J = 8.0, 1.5 Hz, 1H), 7.09 (d, J = 1.5 Hz, 1H), 7.03 (d, J = 3.5 Hz, 2H), 6.88 (dd, J = 10.0, 7.5 Hz, 1H), 6.50 (d, J = 3.0 Hz, 1H). 13C NMR (125 MHz, CD3OD) δ 182.4, 149.6, 145.0, 139.6, 134.1, 127.4, 126.3, 124.4, 123.5, 121.3, 120.2, 114.4, 114.2, 104.0, 99.6, 79.3 ppm; HRMS (ESI): calcd for C16H10BrN2O3− [M-1]−: 356.9880, found: 356.9873; [α]D25 = +8.73 (c 0.76, MeOH). Enantiomeric excess (71%) was determined by chiral HPLC (Chiralpak AD-H), hexane:iPrOH = 80:20, 1.5 mL/min, major enantiomer tR = 13.0 min, minor enantiomer tR = 17.2 min.
(+)-(R)-7-fluoro-3-hydroxy-3-(4-hydroxy-1H-indol-5-yl)indolin-2-one (3h): brown oil (88% yield); 1H NMR (500 MHz, CD3OD) δ 7.10–6.94 (m, 4H), 7.01 (d, J = 8.6 Hz, 1H), 6.95 (d, J = 8.0, 4.0 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.51 (d, J = 2.5 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ 182.2, 149.6 (d, J = 59.5 Hz), 147.6, 139.5, 137.8 (d, J = 9.0 Hz), 130.4 (d, J = 49.5 Hz), 124.4, 124.2 (d, J = 22.0 Hz), 121.8 (d, J = 10.5 Hz), 121.4, 120.2, 117.0 (d, J = 69.5 Hz), 114.4, 104.0 (d, J = 25.5 Hz), 99.6 (d, J = 22.5 Hz), 79.8 (d, J = 7.5 Hz) ppm; HRMS (ESI): calcd for C16H10FN2O3−[M-1]−: 297.0681, found: 297.0677; [α]D25 = +43.06 (c 0.82, MeOH). Enantiomeric excess (92%) was determined by chiral HPLC (Chiralpak AD-H), hexane:iPrOH = 80:20, 1.5 mL/min, major enantiomer tR = 10.5 min, minor enantiomer tR = 12.7 min.
(+)-(R)-7-chloro-3-hydroxy-3-(4-hydroxy-1H-indol-5-yl)indolin-2-one (3i): brown oil (90% yield); 1H NMR (500 MHz, CD3OD) δ 7.22 (dd, J = 8.0, 1.0 Hz, 1H), 7.06 (dd, J = 7.5, 1.0 Hz, 2H), 7.02 (d, J = 8.5 Hz, 1H), 6.96–6.91 (m, 1H), 6.87 (d, J = 8.5 Hz, 1H), 6.48 (d, J = 2.5 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ 182.2, 149.5, 141.2, 139.6, 136.7, 130.1, 124.5, 124.4, 124.3, 121.3, 120.1, 116.1, 114.5, 104.0, 99.6, 80.2; HRMS (ESI): calcd for C16H10ClN2O3− [M-1]−: 313.0385, found: 313.0380; [α]D25 = +14.45 (c = 0.49, MeOH). Enantiomeric excess (83%) was determined by chiral HPLC (Chiralpak AD-H), hexane:iPrOH = 80:20, 1.5 mL/min, major enantiomer tR = 11.2 min, minor enantiomer tR = 14.7 min.
(+)-(R)-3-hydroxy-3-(4-hydroxy-1H-indol-5-yl)-1-methylindolin-2-one (3j): brown oil (82 % yield); 1H NMR (500 MHz, CD3OD) δ 7.31 (dt, J = 7.5, 1.0 Hz, 1H), 7.21 (d, J = 7.5 Hz, 1H), 7.07 (t, J = 4.0 Hz, 1H), 7.03 (dt, J = 7.5, 0.5 Hz, 1H), 6.97 (dd, J = 12.5, 8.0 Hz, 2H), 6.86 (dd, J = 8.5, 0.5 Hz, 1H), 6.50 (dd, J = 3.5, 1.0 Hz, 1H), 3.23 (s, 3H); HRMS (ESI): calcd for C17H13N2O3− [M-1]−: 293.0932, found: 293.0922; [α]D25 = +52.23 (c = 0.90, MeOH). Enantiomeric excess (78%) was determined by chiral HPLC (Chiralpak AD-H), hexane:iPrOH = 80:20, 1.5 mL/min, major enantiomer tR = 12.5 min, minor enantiomer tR = 15.3 min.
(+)-(R)-1-benzyl-3-hydroxy-3-(4-hydroxy-1H-indol-5-yl)indolin-2-one (3k): colorless oil (87% yield); 1H NMR (500 MHz, CD3OD) δ 7.43 (d, J = 7.0 Hz, 2H), 7.34 (t, J = 7.5 Hz, 2H), 7.26 (t, J = 7.5 Hz, 1H), 7.19–7.15 (m, 2H), 7.09 (d, J = 3.0 Hz, 1H), 7.03–6.96 (m, 2H), 6.89 (dd, J = 8.5, 1.0 Hz, 1H), 6.80 (d, J = 8.0 Hz, 1H), 6.53 (dd, J = 3.5, 1.0 Hz, 1H), 5.02 (d, J = 16.0 Hz, 1H), 4.93 (d, J = 16.0 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ 180.8, 149.6, 144.3, 139.6, 137.5, 134.3, 130.2, 129.8, 128.6, 128.4, 125.7, 124.4, 124.2, 121.5, 120.23, 114.7, 110.7, 104.0, 99.7, 79.5, 44.7 ppm; HRMS (ESI): calcd for C23H17N2O3− [M-1]−: 369.1245, found: 369.1239; [α]D25 = +25.51 (c = 0.58, MeOH). Enantiomeric excess (74%) was determined by chiral HPLC (Chiralpak AD-H), hexane:iPrOH = 90:10, 1.5 mL/min, minor enantiomer tR = 17.5 min, major enantiomer tR = 19.8 min.
(-)-(R)-5-chloro-3-hydroxy-3-(5-hydroxy-1H-indol-4-yl)indolin-2-one (4): brown oil (75% yield); 1H NMR (500 MHz, CD3OD) δ 7.23 (d, J = 8.5 Hz, 1H), 7.20 (d, J = 8.5 Hz, 1H), 7.12 (s, 1H), 7.08 (s, 1H), 6.92 (d, J = 8.0 Hz, 2H), 6.66 (d, J = 8.5 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ 181.2, 142.0, 136.4, 133.1, 128.8, 126.2, 125.8, 113.7, 113.0, 112.2, 94.6, 80.8; HRMS (ESI): calcd for C16H10ClN2O3−[M-1]−: 313.0385, found: 313.0380; [α]D25= −14.58 (c 0.43, MeOH). Enantiomeric excess (94%) was determined by chiral HPLC (Chiralpak AD-H), hexane:iPrOH = 80:20, 1.5 mL/min, major enantiomer tR = 15.1 min, minor enantiomer tR = 24.1 min.
(-)-(R)-5-chloro-3-hydroxy-3-(6-hydroxy-1H-indol-7-yl)indolin-2-one (5): brown oil (72% yield); 1H NMR (500 MHz, CD3OD) δ 7.30 (d, J = 8.5 Hz, 1H), 7.19 (dd, J = 2.0, 8.0 Hz, 1H), 7.15 (s, 1H), 6.99 (d, J = 2.0 Hz, 1H), 6.87 (d, J = 8.5 Hz, 2H), 6.49 (d, J = 8.5 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ182.0, 150.2, 142.3, 137.2, 136.7, 129.9, 128.3, 125.4, 124.6, 124.0, 121.2, 111.9, 110.5, 109.7, 94.6, 79.6; HRMS (ESI): calcd for C16H10ClN2O3−[M-1]−: 313.0385, found: 313.0379; [α]D25 = −11.50 (c 0.56, MeOH). Enantiomeric excess (80%) was determined by chiral HPLC (Chiralpak AD-H), hexane:iPrOH = 80:20, 1.5 mL/min, major enantiomer tR = 11.6 min, minor enantiomer tR = 33.6 min.
(+)-(R)-5-chloro-3-hydroxy-3-(7-hydroxy-1H-indol-6-yl)indolin-2-one (6): brown oil (68% yield); 1H NMR (500 MHz, CD3OD) δ 7.29 (dd, J = 8.0, 2.0 Hz, 1H), 7.24 (d, J = 1.5 Hz, 1H), 7.20 (d, J = 3.0 Hz, 1H), 6.97 (d, J = 8.5 Hz, 1H), 6.93 (d, J = 8.5 Hz, 1H), 6.54 (d, J = 8.0 Hz, 1H), 6.35 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ 182.0, 144.1, 141.8, 136.3, 131.6, 130.5, 128.5, 126.6, 126.2, 118.8, 115.6, 112.6, 112.5, 102.6, 81.0; HRMS (ESI): calcd for C16H10ClN2O3− [M-1]−: 313.0385, found: 313.0386; [α]D25 = +9.07 (c 0.55, MeOH). Enantiomeric excess (60%) was determined by chiral HPLC (Chiralpak AD-H), hexane:iPrOH = 80:20, 1.5 mL/min, minor enantiomer tR = 10.1 min, major enantiomer tR = 12.8 min.
Supplementary Materials
Copies of 1H and 13C-NMR spectra and HPLC trace of products are available online at https://www.mdpi.com/1420-3049/24/21/3944/s1. Figures S1–S25: NMR spectra of compounds 3a–3k and 4–6; Figures S26–29: HPLC trace of enantiomeric 3a in different solvent. S30–S57: HPLC trace of compounds 3a–3k and 4–6.
Author Contributions
H.W. and L.W. are coauthors; they contributed equally to this work. All the authors have read and approved the final manuscript.
Funding
We thank the National Natural Science Foundation of China (No. 21102055), the Natural Science Foundation of Jilin province (No. 20190201077JC), and the Department of Education of Jilin province (No. JJKH20170409KJ).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Horton, D.A.; Bourne, G.T.; Smythe, M.L. The Combinatorial Synthesis of Bicyclic Privileged Structures or Privileged Substructures. Chem. Rev. 2003, 103, 893–930. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wu, D.; Snyder, B.; Ptak, R.G.; Kaur, H.; Gochin, M. Development of indole compounds as small molecule fusion inhibitors targeting HIV-1 glycoprotein-41. J. Med. Chem. 2011, 54, 7220–7231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Z.; Chen, Q.; Yang, G.-F. A review on recent developments of indole-containing antiviral agents. Eur. J. Med. Chem. 2015, 89, 421–441. [Google Scholar] [CrossRef] [PubMed]
- Sherer, C.; Snape, T. Heterocyclic scaffolds as promising anticancer agents against tumours of the central nervous system: Exploring the scope of indole and carbazole derivatives. Eur. J. Med. Chem. 2015, 97, 552–560. [Google Scholar] [CrossRef]
- Lancianesi, S.; Palmieri, A.; Petrini, M. Synthetic Approaches to 3-(2-Nitroalkyl) Indoles and Their Use to Access Tryptamines and Related Bioactive Compounds. Chem. Rev. 2014, 114, 7108–7149. [Google Scholar] [CrossRef]
- Ishikura, M.; Abe, T.; Choshi, T.; Hibino, S. Simple indole alkaloids and those with a nonrearranged monoterpenoid unit. Nat. Prod. Rep. 2015, 32, 1389–1471. [Google Scholar] [CrossRef]
- Bandini, M.; Umani-Ronchi, A. (Eds.) Catalytic Asymmetric Friedel–Crafts Alkylations||Industrial Friedel–Crafts. Chemistry 2009. [Google Scholar] [CrossRef]
- Jørgensen, K.A. Microwave-Assisted Rapid and Selective Synthesis of cis- and trans-2,4,5-Triarylimidazolines from Aromatic Aldehydes. Synlett 2003, 1117–1120. [Google Scholar] [CrossRef]
- Bandini, M.; Melloni, A.; Umani-Ronchi, A. New Catalytic Approaches in the Stereoselective Friedel–Crafts Alkylation Reaction. Angew. Chem. Int. Ed. 2004, 43, 550–556. [Google Scholar] [CrossRef]
- Poulsen, T.B.; Jørgensen, K.A. Catalytic Asymmetric Friedel-Crafts Alkylation Reactions—Copper Showed the Way. Chem. Rev. 2008, 108, 2903–2915. [Google Scholar] [CrossRef]
- Bandini, M.; Eichholzer, A. Catalytic Functionalization of Indoles in a New Dimension. Angew. Chem. Int. Ed. 2009, 48, 9608–9644. [Google Scholar] [CrossRef] [PubMed]
- You, S.L.; Cai, Q.; Zeng, M. Chiral Bronsted acid catalyzed Friedel-Crafts alkylation reactions. Chem. Soc. Rev. 2009, 38, 2190–2201. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, G.; Bencivenni, G.; Dalpozzo, R. Organocatalytic strategies for the asymmetric functionalization of indoles. Chem. Soc. Rev. 2010, 39, 4449–4465. [Google Scholar] [CrossRef] [PubMed]
- Dalpozzo, R. Strategies for the asymmetric functionalization of indoles: An update. Chem. Soc. Rev. 2015, 44, 742–778. [Google Scholar] [CrossRef]
- Wu, H.; He, Y.P.; Shi, F. Recent Advances in Chiral Phosphoric Acid Catalyzed Asymmetric Reactions for the Synthesis of Enantiopure Indole Derivatives. Synthesis 2015, 47, 1990–2016. [Google Scholar] [CrossRef]
- Cruz, F.A.; Zhu, Y.; Tercenio, Q.D.; Shen, Z.; Dong, Y.M. Alkyne Hydroheteroarylation: Enantioselective Coupling of Indoles and Alkynes via Rh-Hydride Catalysis. J. Am. Chem. Soc. 2017, 139, 10641–10644. [Google Scholar] [CrossRef]
- Zhao, Y.L.; Wang, L.S.; Zhao, J.L. Chiral phosphoric acid catalyzed aza-Friedel-Crafts alkylation of indoles with cyclicaryl αketiminoesters. Tetrahedron Lett. 2017, 58, 213217. [Google Scholar] [CrossRef]
- Zhang, H.H.; Wang, C.S.; Li, C.; Mei, G.J.; Li, Y.; Shi, F. Design and Enantioselective Constructionof Axially Chiral Naphthyl-Indole Skeletons. Angew. Chem. Int. Ed. 2017, 56, 116–121. [Google Scholar] [CrossRef]
- Qi, L.W.; Mao, J.H.; Zhang, J.; Tan, B. Organocatalytic asymmetric arylation of indoles enabled by azo groups. Nat. Chem. 2018, 10, 58–64. [Google Scholar] [CrossRef]
- Nakamura, S.; Furukawa, T.; Hatanaka, T.; Funahashi, Y. Enantioselective aza-Friedel–Crafts reaction of cyclic ketimines with indoles using chiral imidazoline–phosphoric acid catalysts. Chem. Commun. 2018, 54, 3811–3814. [Google Scholar] [CrossRef]
- Vetica, F.; Chauhan, P.; Mahajan, S.; Raabe, G.; Enders, D. Asymmetric Organocatalytic Friedel–Crafts Hydroxyalkylation of Indoles Using Electrophilic Pyrazole-4,5-diones. Synthesis 2018, 50, 1039–1046. [Google Scholar] [CrossRef]
- Lee, J.; Ko, K.M.; Kim, S.G. Ni(ClO4)2− Catalyzed Friedel–Crafts Reaction of Coumarin-Fused Donor–Acceptor Cyclopropanes with Indoles: Stereoselective Synthesis of trans-3,4-Disubstituted-3,4-dihydrocoumarins. Eur. J. Org. Chem. 2018, 4166–4170. [Google Scholar] [CrossRef]
- Evans, D.A.; Fandrick, K.R. Catalytic Enantioselective Pyrrole Alkylations of α,β-Unsaturated 2-Acyl Imidazoles. Org. Lett. 2006, 8, 2249–2252. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Fandrick, K.R.; Song, H.J.; Scheidt, K.A.; Xu, R. Enantioselective Friedel–Crafts Alkylations Catalyzed by Bis(oxazolinyl)pyridine–Scandium(III) Triflate Complexes. J. Am. Chem. Soc. 2007, 129, 10029–10034. [Google Scholar] [CrossRef]
- Blay, G.; Fernández, I.; Pedro, J.R.; Vila, C. Catalytic enantioselective Friedel–Crafts alkylation at the 2-position of indole with simpleenones. Tetrahedron Lett. 2007, 48, 6731–6734. [Google Scholar] [CrossRef]
- Kang, Q.; Zheng, X.J.; You, S.L. Highly Enantioselective Friedel–Crafts Reaction of 4,7-Dihydroindoles with Imines by Chiral Phosphoric Acids: Facile Access to 2-Indolyl Methanamine Derivatives. Chem. Eur. J. 2008, 14, 3539–3542. [Google Scholar] [CrossRef]
- Hong, L.; Liu, C.; Sun, W.; Wang, L.; Wong, K.; Wang, R. Organocatalytic Enantioselective Friedel–Crafts Alkylation of 4,7-Dihydroindoles with α,β-Unsaturated Aldehydes: An Easy Access to 2-Substituted Indoles. Org. Lett. 2009, 11, 2177–2180. [Google Scholar] [CrossRef]
- Sheng, Y.F.; Li, G.Q.; Kang, Q.; Zhang, A.-J.; You, S.L. Asymmetric Friedel–Crafts Reaction of 4,7-Dihydroindoles with Nitroolefins by Chiral Brønsted Acids under Low Catalyst Loading. Chem. Eur. J. 2009, 15, 3351–3354. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, G.W.; Teng, Y.; Nie, J.; Zheng, Y.; Ma, J.A. Chiral Brønsted Acid-Catalyzed Enantioselective Friedel–Crafts Reaction of 4,7-Dihydroindoles with Trifluoromethyl Ketones. Adv. Synth. Catal. 2010, 352, 2773–2777. [Google Scholar] [CrossRef]
- Blay, G.; Fernández, I.; Muñoz, M.C.; Pedro, J.R.; Recuenco, A.; Vila, C. Enantioselective Synthesis of Tertiary Alcohols through a Zirconium-Catalyzed Friedel–Crafts Alkylation of Pyrroles with α-Ketoesters. J. Org. Chem. 2011, 76, 6286–6294. [Google Scholar] [CrossRef]
- Cui, H.L.; Feng, X.; Peng, J.; Lei, J.; Jiang, K.; Chen, Y.C. Chemoselective Asymmetric N-Allylic Alkylation of Indoles with Morita–Baylis–Hillman Carbonates. Angew. Chem. Int. Ed. 2009, 48, 5737–5740. [Google Scholar] [CrossRef] [PubMed]
- Stanley, L.M.; Hartwig, J.F. Iridium-Catalyzed Regio- and Enantioselective N-Allylation of Indoles. Angew. Chem. Int. Ed. 2009, 48, 7841–7844. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Osipov, M.; Dong, G. Palladium-Catalyzed Dyamic Kinetic Asymmetric Transformations of Vinyl Aziridines with Nitrogen Heterocycles: Rapid Access to Biologically Active Pyrroles and Indoles. J. Am. Chem. Soc. 2010, 132, 15800–15807. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhao, Y.; Qian, B.; Yang, L.; Xia, C.; Huang, H. Enantioselective N–H Functionalization of Indoles with α,β-Unsaturated γ-Lactams Catalyzed by Chiral Brønsted Acids. Angew. Chem. Int. Ed. 2011, 50, 5682–5686. [Google Scholar] [CrossRef]
- Cheng, H.G.; Lu, L.Q.; Wang, T.; Yang, Q.Q.; Liu, X.P.; Li, Y.; Deng, Q.H.; Chen, J.R.; Xiao, W.J. Highly Enantioselective Friedel–Crafts Alkylation/N-Hemiacetalization Cascade Reaction with Indoles. Angew. Chem. Int. Ed. 2013, 52, 3250–3254. [Google Scholar] [CrossRef]
- Sevov, C.S.; Zhou, J.; Hartwig, J.F. Iridium-Catalyzed Intermolecular Hydroamination of Unactivated Alkenes with Indoles. J. Am. Chem. Soc. 2014, 136, 3200–3207. [Google Scholar] [CrossRef]
- Bera, K.; Schneider, C. Brønsted Acid Catalyzed [3+2]-Cycloaddition of Cyclic Enamides with in Situ Generated 2-Methide-2H-indoles: Enantioselective Synthesis of Indolo[1,2-a]indoles. Org. Lett. 2016, 18, 5660–5663. [Google Scholar] [CrossRef]
- Shi, Z.; Yang, Y. Regioselective direct arylation of indoles on the benzenoid moiety. Chem. Commun. 2018, 54, 1676–1685. [Google Scholar] [CrossRef]
- Sandtorv, A.H. Catalyzed Transition Metal-Catalyzed C?H Activation of Indoles. Adv. Synth. Catal. 2015, 357, 2403–2435. [Google Scholar] [CrossRef]
- Lanke, V.; Prabhu, K.R. Regioselective Synthesis of 4-Substituted Indoles via C–H Activation: A Ruthenium Catalyzed Novel Directing Group Strategy. Org. Lett. 2013, 15, 6262–6265. [Google Scholar] [CrossRef]
- Liu, Q.; Li, Q.; Ma, Y.; Jia, Y. Direct Olefination at the C-4 Position of Tryptophan via C–H Activation: Application to Biomimetic Synthesis of Clavicipitic Acid. Org. Lett. 2013, 15, 4528–4531. [Google Scholar] [CrossRef] [PubMed]
- Hartung, C.G.; Fecher, A.; Chapell, B.; Snieckus, V. Directed ortho Metalation Approach to C-7-Substituted Indoles. Suzuki–Miyaura Cross Coupling and the Synthesis of Pyrrolophenanthridone Alkaloids. Org. Lett. 2003, 5, 1899–1902. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Holte, D.; Zoller, J.; Umemiya, S.; Simke, L.R.; Baran, P.S. Total Synthesis of Verruculogen and Fumitremorgin A Enabled by Ligand-Controlled C–H Borylation. J. Am. Chem. Soc. 2015, 137, 10160–10163. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Qiu, X.; Zhao, Y.; Mu, Y.; Shi, Z. Palladium-Catalyzed C–H Arylation of Indoles at the C7 Position. J. Am. Chem. Soc. 2016, 138, 495–498. [Google Scholar] [CrossRef]
- Vila, C.; Amr, F.I.; Blay, G.; Muňoz, M.C.; Pedro, J.R. Organocatalytic Enantioselective Synthesis of Pyrazoles Bearing a Quaternary Stereocenter. Chem. Asian J. 2016, 11, 1532–1536. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Wang, S.B.; Zheng, C.; You, S.L. Asymmetric Synthesis of Spiropyrazolones by Rhodium-Catalyzed C (sp2)–H Functionalization/Annulation Reactions. Angew. Chem. Int. Ed. 2017, 56, 4540–4544. [Google Scholar] [CrossRef]
- Xu, Q.L.; Dai, L.X.; You, S.L. Diversity oriented synthesis of indole-based peri-annulated compounds via allylic alkylation reactions. Chem. Sci. 2013, 4, 97–102. [Google Scholar] [CrossRef]
- Liu, H.; Zheng, C.; You, S.L. Catalytic C6 Functionalization of 2,3-Disubstituted Indoles by Scandium Triflate. J. Org. Chem. 2014, 79, 1047–1054. [Google Scholar] [CrossRef]
- Zhou, L.J.; Zhang, Y.C.; Zhao, J.J.; Shi, F.; Tu, S.J. Organocatalytic Arylation of 3-Indolylmethanols via Chemo- and Regiospecific C6-Functionalization of Indoles. J. Org. Chem. 2014, 79, 10390–10398. [Google Scholar] [CrossRef]
- Poulsen, P.H.; Feu, K.S.; Paz, B.M.; Jensen, F.; Jørgensen, K.A. Organocatalytic Asymmetric 1,6-Addition/1,4-Addition Sequence to 2,4-Dienals for the Synthesis of Chiral Chromans. Angew. Chem. Int. Ed. 2015, 54, 8203–8207. [Google Scholar] [CrossRef]
- Montesinos-Magraner, M.; Vila, C.; Rendón-Patiño, A.; Blay, G.; Fernández, I.; Muñoz, M.C.; Pedro, J.R. Organocatalytic Enantioselective Friedel–Crafts Aminoalkylation of Indoles in the Carbocyclic Ring. ACS Catal. 2016, 6, 2689–2693. [Google Scholar] [CrossRef]
- Montesinos-Magraner, M.; Vila, C.; Blay, G.; Fernández, I.; Muñoz, M.C.; Pedro, J.R. Hydroxy-Directed Enantioselective Hydroxyalkylation in the Carbocyclic Ring of Indoles. Org. Lett. 2017, 19, 1546–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, M.; Xu, D.; Liang, W.; Wu., W.; Chan, A.S.C.; Zhao, J. Organocatalytic Enantioselective Friedel-Crafts Alkylation/Lactonization Reaction of Hydroxyindoles with Methylene Oxindoles. Adv. Synth. Catal. 2018, 360, 917–924. [Google Scholar] [CrossRef]
- Yang, Z.T.; Yang, W.L.; Chen, L.; Sun, H.; Deng, W.P. Organocatalytic Enantioselective aza-Friedel-Crafts Reactions of Pyrazolinone Ketimines with Hydroxyindoles and Electron-Rich Phenols. Adv. Synth. Catal. 2018, 360, 2049–2205. [Google Scholar] [CrossRef]
- Clark, M.P.; Laughlin, S.K.; Laufersweiler, M.J.; Bookland, R.G.; Brugel, T.A.; Golebiowski, A.; Sabat, M.P.; Townes, J.A.; VanRens, J.C.; Djung, J.F.; et al. Development of Orally Bioavailable Bicyclic Pyrazolones as Inhibitors of Tumor Necrosis Factor-α Production. J. Med. Chem. 2004, 47, 2724–2727. [Google Scholar] [CrossRef] [PubMed]
- Hadi, V.; Koh, Y.H.; Sanchez, T.W.; Barrios, D.; Neamati, N.; Jung, K.W. Development of the next generation of HIV-1 integrase inhibitors: Pyrazolone as a novel inhibitor scaffold. Bioorg. Med. Chem. Lett. 2010, 20, 6854–6857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, Z.V.; Wu, N.C.; Kadam, R.U.; Wilson, I.A.; Wolan, D.W. Structure-based optimization and synthesis of antiviral drug Arbidol analogues with significantly improved affinity to influenza hemagglutinin. Bioorg. Med. Chem. Lett. 2017, 27, 3744–3748. [Google Scholar] [CrossRef] [Green Version]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael Reaction of Malonates to Nitroolefins Catalyzed by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef]
- Okino, T.; Nakamura, S.; Furukawa, T.; Takemoto, Y. Enantioselective aza-Henry Reaction Catalyzed by a Bifunctional Organocatalyst. Org. Lett. 2004, 6, 625–627. [Google Scholar] [CrossRef]
- Hoashi, Y.; Okino, T.; Takemoto, Y. Enantioselective Michael Addition to α, β-unsaturated Imides Catalyzed by a Bifunctional Organocatalyst. Angew. Chem. Int. Ed. Engl. 2005, 44, 4032–4035. [Google Scholar] [CrossRef]
- Zea, A.; Valero, G.; Alba, A.R.; Moyano, A.; Rios, R. Development of Diphenylamine-linked Bis(imidazoline) Ligands and Their Application in Asymmetric Friedel-Crafts Alkylation of Indole Derivatives with Nitroalkenes. Adv. Synth. Catal. 2010, 352, 1102–1106. [Google Scholar] [CrossRef]
- Raimondi, W.; Baslé, O.; Constantieux, T.; Bonne, D.; Rodriguez, J. Activation of 1, 2-Ketoesters with Takemoto’s Catalyst toward Michael Addition to Nitroalkenes. Adv. Synth. Catal. 2012, 354, 563–568. [Google Scholar] [CrossRef]
- Ansari, S.; Raabe, G.; Enders, D. Asymmetric Michael Addition of 1, 3-Bis(phenylthio)propan-2-one to Nitroalkenes Employing Takemoto’s Thiourea Catalyst. Monatsh. Chem. 2013, 144, 641–646. [Google Scholar] [CrossRef]
- Wang, Y.; Mo, M.; Zhu, K.; Zheng, C.; Zhang, H.; Wang, W.; Shao, Z. Asymmetric Synthesis of Syn-propargylamines and Unsaturated β-amino Acids under Brønsted Base Catalysis. Nat. Commun. 2015, 6, 8544–8582. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, H.-F.; Zhao, J.-Z.; Du, Z.-H.; Da, C.-S. Organocatalytic Enantioselective Cross-aldol Reaction of o-Hydroxyarylketones and Trifluoromethyl Ketones. Org. Lett. 2017, 19, 2634–2637. [Google Scholar] [CrossRef]
Figure 1.
(Thio)urea derivatives screened as organocatalysts (1a–1h).

Figure 2.
Proposed stereochemical model.

Scheme 1.
Scope for the enantioselective hydroxyalkylation between different hydroxyindoles and isatin. Reaction conditions: hydroxyindole (0.1 mmol), isatin (0.1 mmol), 1b (10 mol %) in Et2O (1.5 mL) at 0 °C. Isolated yields after column chromatography. >20:1 regioselectivity determined by 1H NMR.
Scheme 1.
Scope for the enantioselective hydroxyalkylation between different hydroxyindoles and isatin. Reaction conditions: hydroxyindole (0.1 mmol), isatin (0.1 mmol), 1b (10 mol %) in Et2O (1.5 mL) at 0 °C. Isolated yields after column chromatography. >20:1 regioselectivity determined by 1H NMR.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Asymmetric hydroxyalkylation of 4-hydroxyindole with isatin a.

| Entry | Catalyst a | Yield (%) b | %ee c | Configuration d |
|---|---|---|---|---|
| 1 | 1a | 70 | 68 | R |
| 2 | 1b | 88 | 79 | R |
| 3 | 1c | 75 | 72 | S |
| 4 | 1d | 85 | 76 | S |
| 5 | 1e | - | - | - |
| 6 | 1f | 72 | 55 | S |
| 7 | 1g | 65 | 35 | S |
| 8 | 1h | 66 | 46 | S |
a Reaction conditions: isatin (0.1 mmol), 4-hydroxyindole (0.1 mmol), catalyst (0.01 mmol), and Et2O (1.5 mL) at rt. b Isolated yield. cDetermined by HPLC analysis (Chiralpak AD-H). dAbsolute configuration was determined according to the literature [52].
Table 2.
Screening of the reaction conditions for the asymmetric hydroxyalkylation a.
| Entry. | Solvent | Temperature | Catalyst. Amount (% mmol) | Yield (%) b | %ee c |
|---|---|---|---|---|---|
| 1 | Et2O | rt | 10 | 88 | 79 |
| 2 | DCM | rt | 10 | 69 | 32 |
| 3 | toluene | rt | 10 | 70 | 28 |
| 4 | THF | rt | 10 | 76 | 69 |
| 5 d | Et2O | rt | 10 | 79 | 74 |
| 6 | Et2O | rt | 5 | 79 | 77 |
| 7 | Et2O | rt | 20 | 91 | 76 |
| 8 | Et2O | 0 °C | 10 | 85 | 83 |
| 9 | Et2O | 0 °C | 20 | 83 | 80 |
| 10 | Et2O | −20 °C | 10 | 72 | 68 |
| 11 | Et2O | −40 °C | 10 | 68 | 73 |
a Reaction conditions: isatin (0.1 mmol), 4-hydroxyindole (0.1 mmol), catalyst 1b in solvent (1.5 mL). b Isolated yield. c Determined by HPLC analysis (Chiralpak AD-H). d Reaction performed in Et2O (3.0 mL).
Table 3.
Scope of the enantioselective hydroxyalkylation between 4-hydroxyindole and isatins a.

| Entry | R1, R2 | Product | Yield (%) b | %ee c |
|---|---|---|---|---|
| 1 | R1 = H, R2 = H, 2a | 3a | 85(70) d | 83(85) d |
| 2 | R1 = H, R2 = 4-Br, 2b | 3b | 65 | 50 |
| 3 | R1 = H, R2 = 5-Me, 2c | 3c | 85 | 85 |
| 4 | R1 = H, R2 = 5-OMe, 2d | 3d | 78 | 82 |
| 5 | R1 = H, R2 = 5-Cl, 2e | 3e | 92 | 94 |
| 6 | R1 = H, R2 = 5-Br, 2f | 3f | 80(69) d | 90(84) d |
| 7 | R1 = H, R2 = 6-Br, 2g | 3g | 83 | 71 |
| 8 | R1 = H, R2 = 7-F, 2h | 3h | 90 | 92 |
| 9 | R1 = H, R2 = 7-Cl, 2i | 3i | 81 | 84 |
| 10 | R1 = Me, R2 = H, 2j | 3j | 82(77) d | 78(80) d |
| 11 | R1 = Bn, R2 = H, 2k | 3k | 87(91) d | 74(90) d |
a Reaction conditions: isatin (0.1 mmol), 4-hydroxyindole (0.1 mmol), and catalyst (0.01 mmol) in Et2O (1.5 mL). bIsolated yield. c Determined by HPLC analysis (Chiralpak AD-H). d The data in parentheses are the ee values and yield in [52].
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wu, H.; Wang, L.; Zhang, J.; Jin, Y. Urea Derivative Catalyzed Enantioselective Hydroxyalkylation of Hydroxyindoles with Isatins. Molecules 2019, 24, 3944. https://doi.org/10.3390/molecules24213944
AMA Style
Wu H, Wang L, Zhang J, Jin Y. Urea Derivative Catalyzed Enantioselective Hydroxyalkylation of Hydroxyindoles with Isatins. Molecules. 2019; 24(21):3944. https://doi.org/10.3390/molecules24213944
Chicago/Turabian StyleWu, Hao, Liming Wang, Junwei Zhang, and Ying Jin. 2019. "Urea Derivative Catalyzed Enantioselective Hydroxyalkylation of Hydroxyindoles with Isatins" Molecules 24, no. 21: 3944. https://doi.org/10.3390/molecules24213944