Design, Synthesis, and Molecular Docking Study of Novel Heterocycles Incorporating 1,3,4-Thiadiazole Moiety as Potential Antimicrobial and Anticancer Agents


 ,
,  and
and 
Abstract
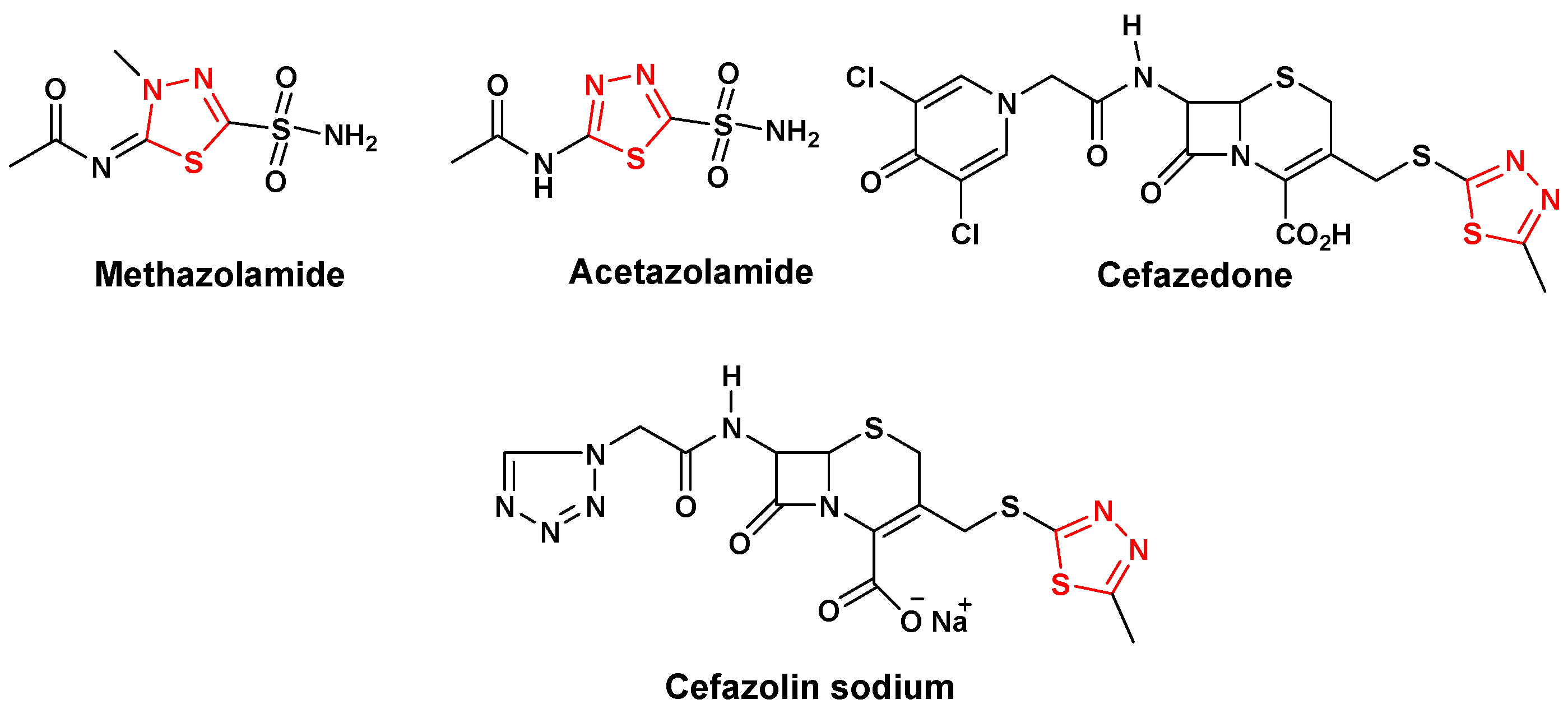
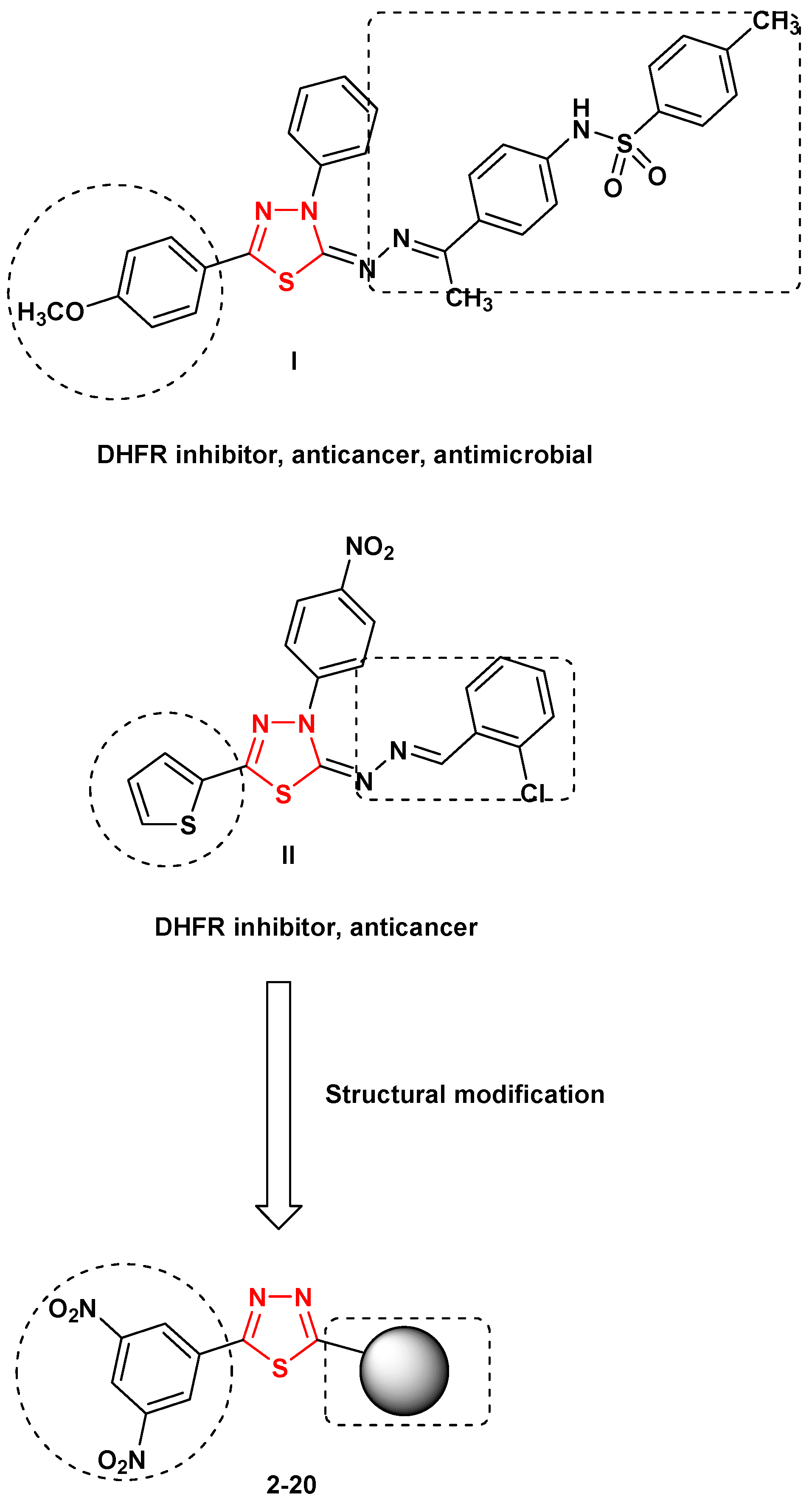
:1. Introduction
2. Results and Discussion
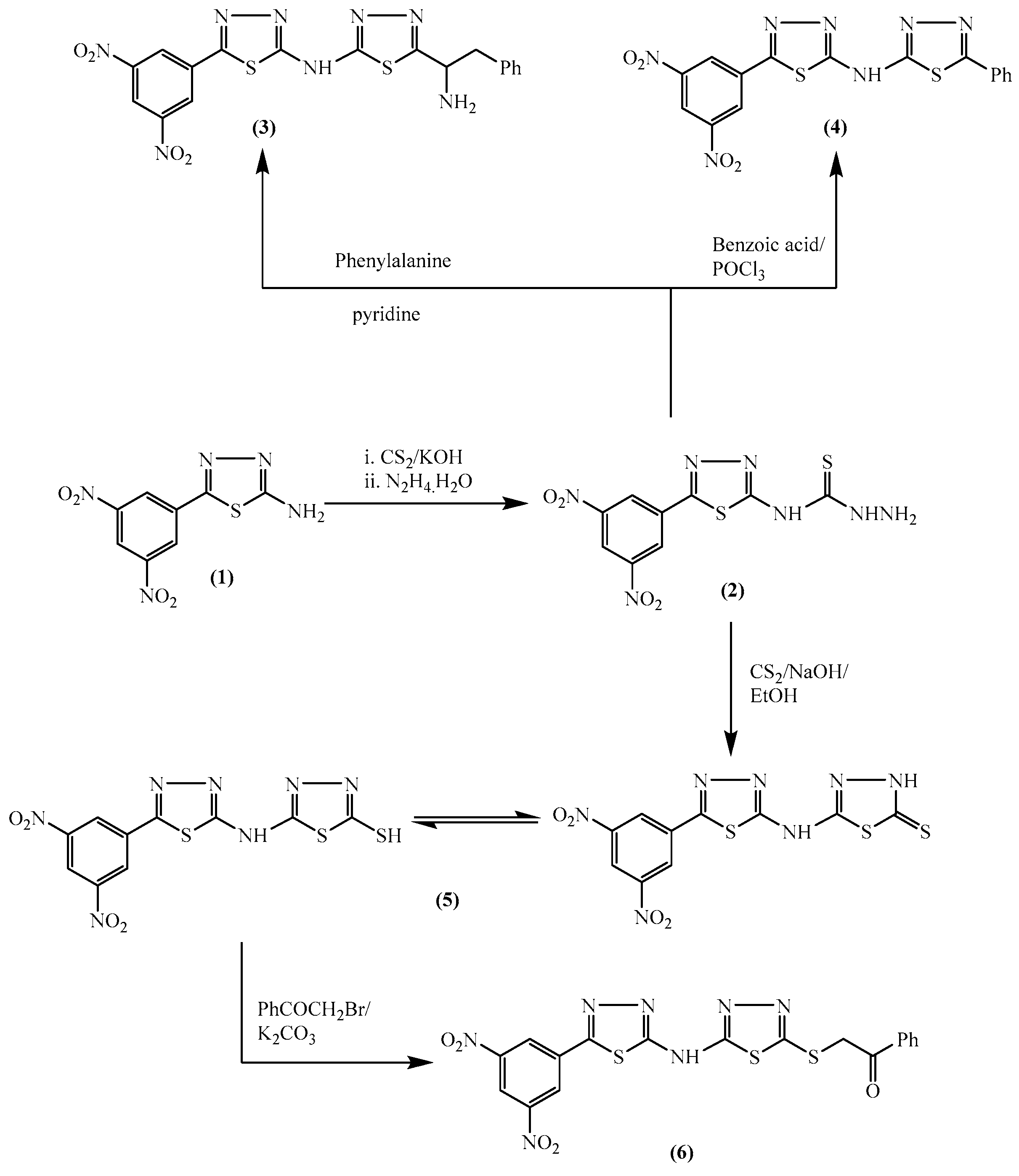
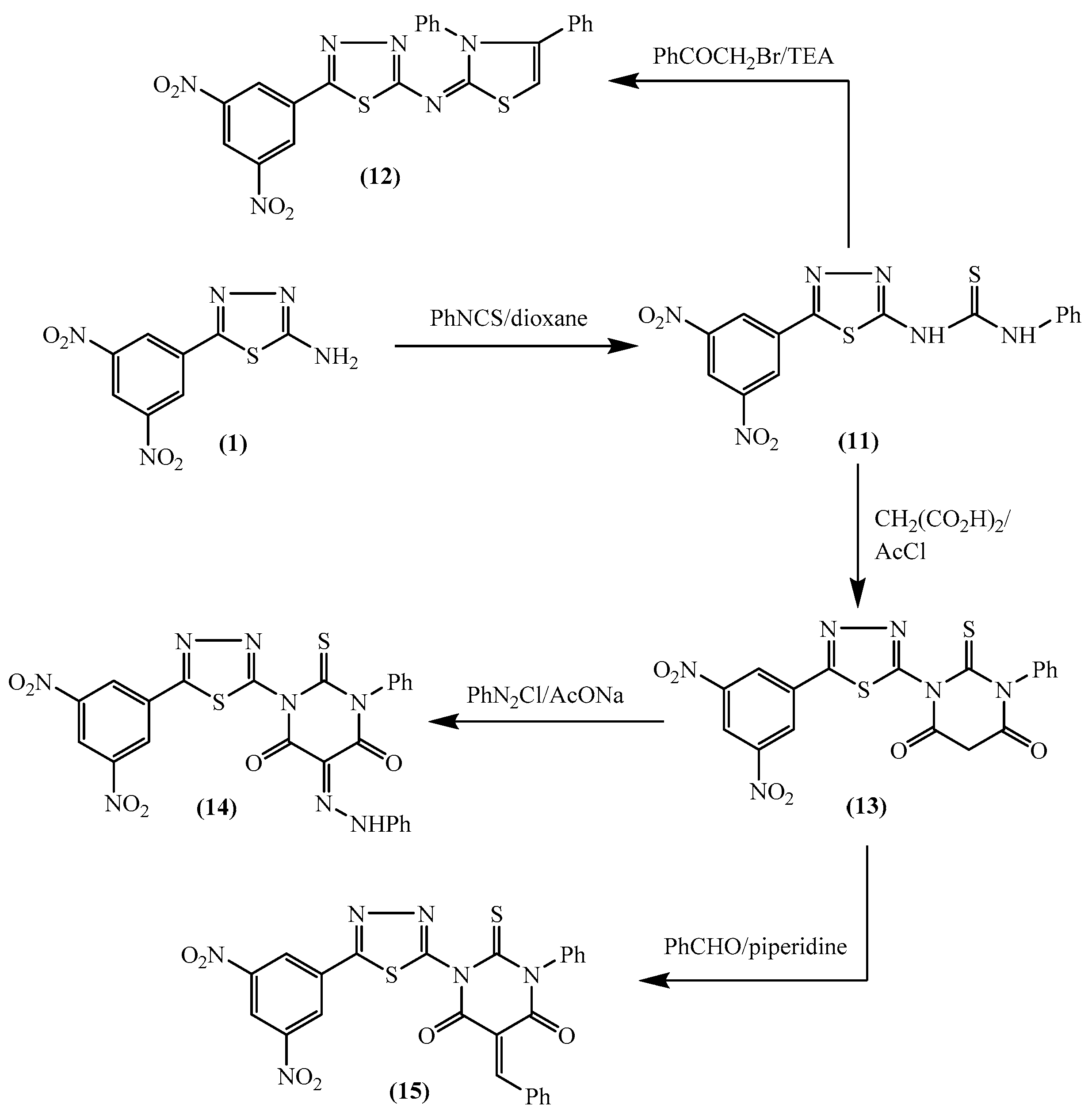
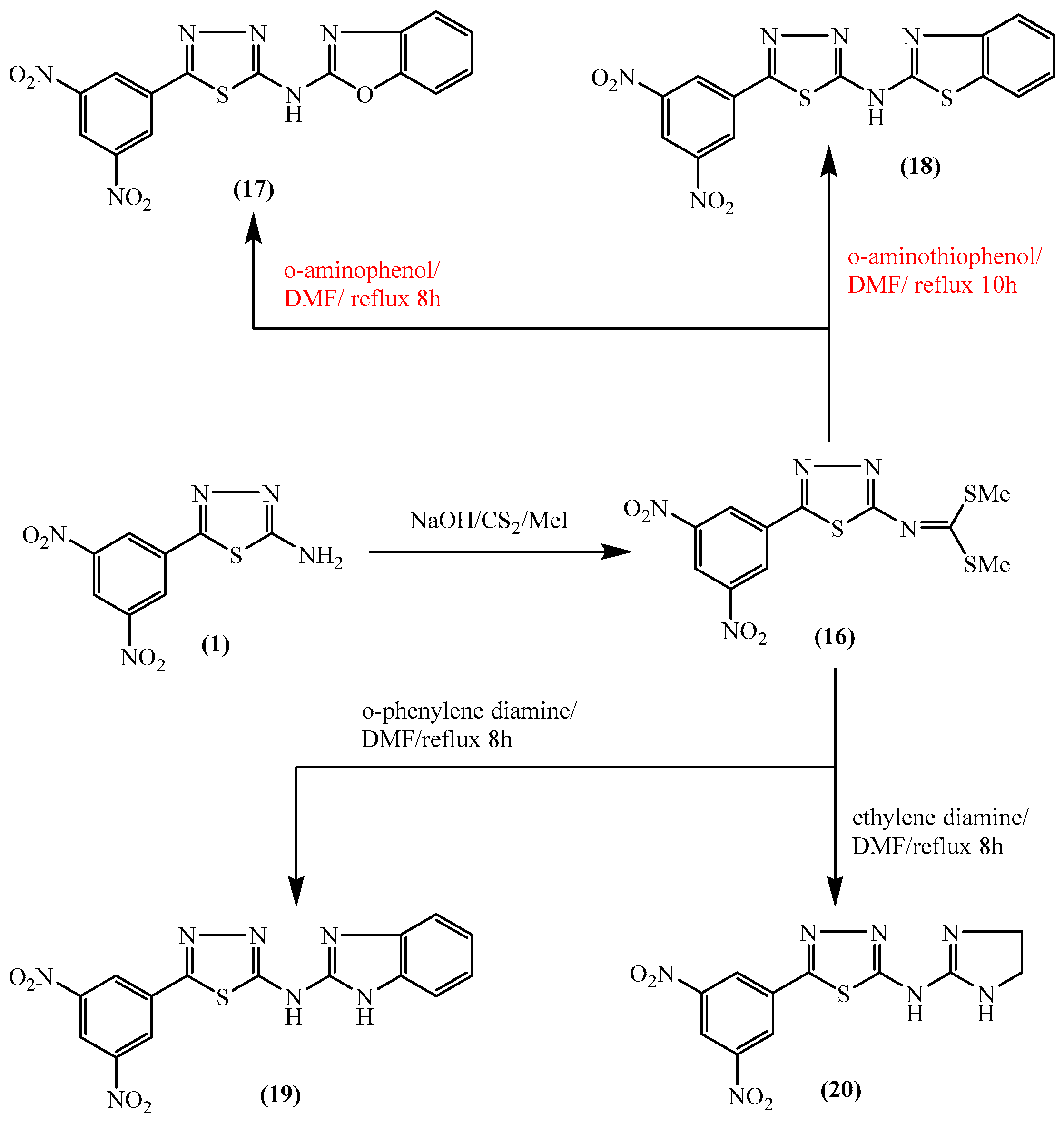
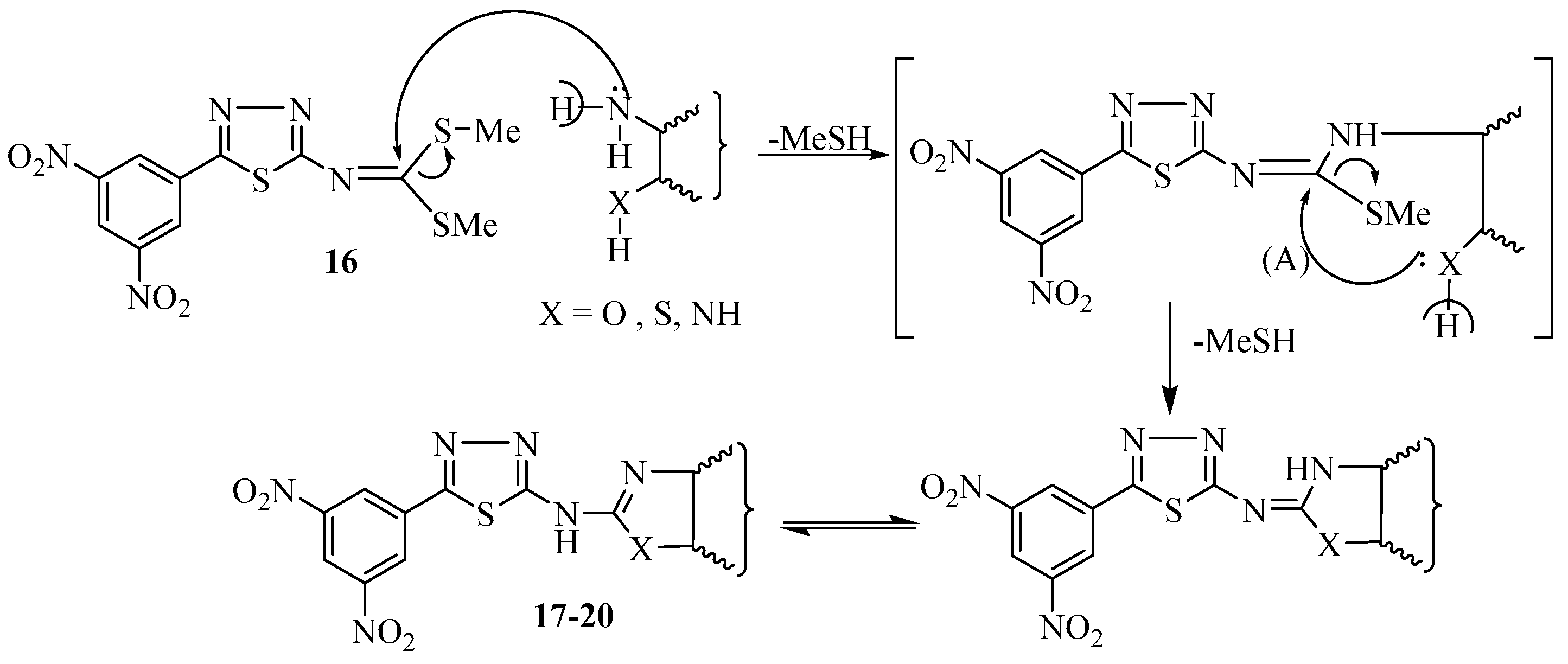
2.1. Chemistry
2.2. Biological Evaluation
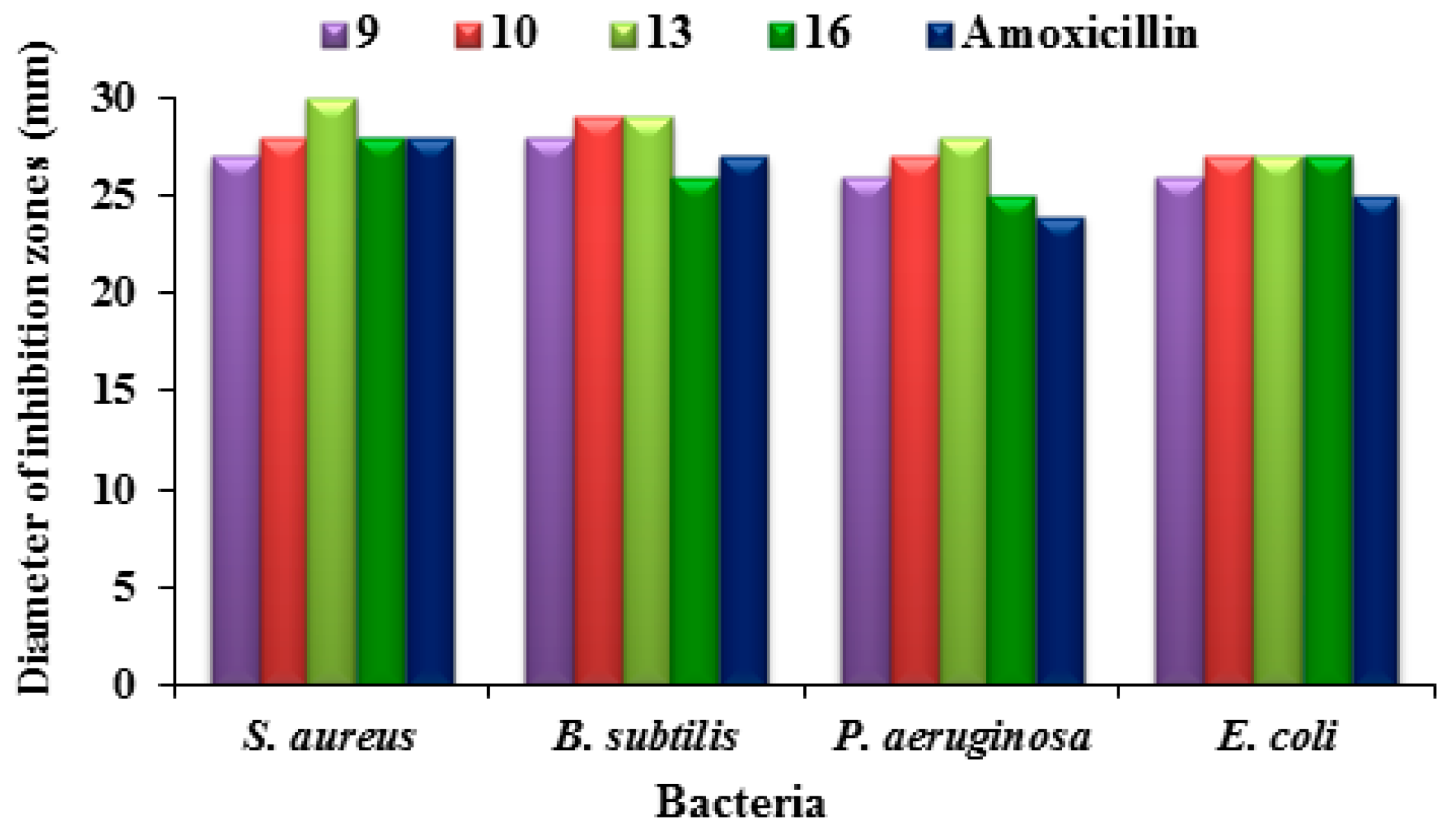
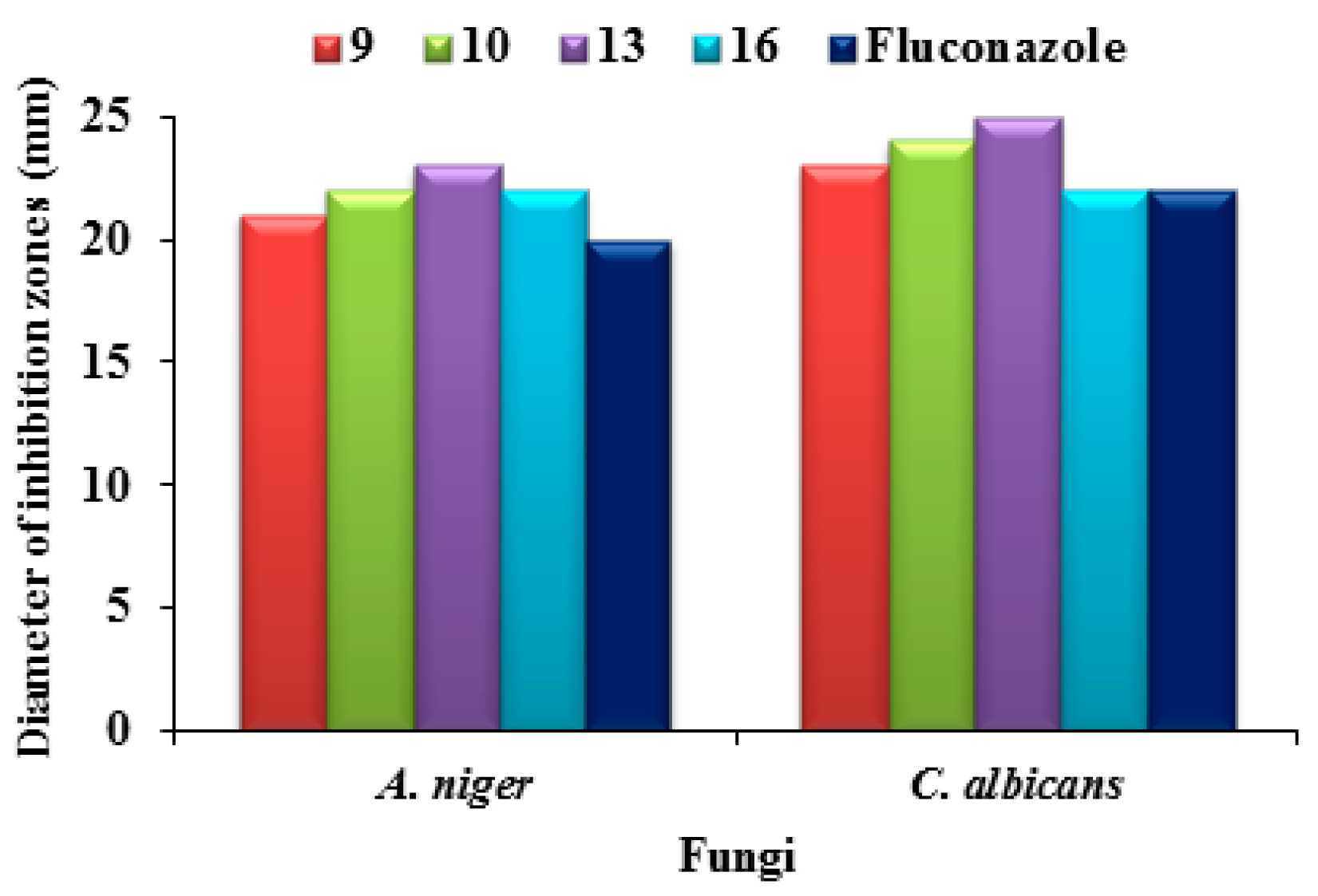
2.2.1. Antimicrobial Sensitivity Assay
2.2.2. Structure–Activity Relationship for Antimicrobial Activity
- a drop in the potency, illustrated in substitution at p-2 of the new 1,3,4-thiadiazole moiety with 1-amino-2-phenylethyl in compound 3 or with phenyl in compound 4;
- elevated potency if the same position in the new 1,3,4-thiadiazole moiety is substituted with a free thioxo group in compound 5; and
- moderate activity but only about half that of compound 5 if the thioxo group is substituted with phenylethane-1-one in compound 6, or p-2 is linked to pyrimidine-2,4,6-trione scaffold in compound 8.
2.2.3. In Vitro Anticancer Activity
2.2.4. Structure–Activity Relationship for Anticancer Activity
2.2.5. Dihydrofolate Reductase (DHFR) Inhibition
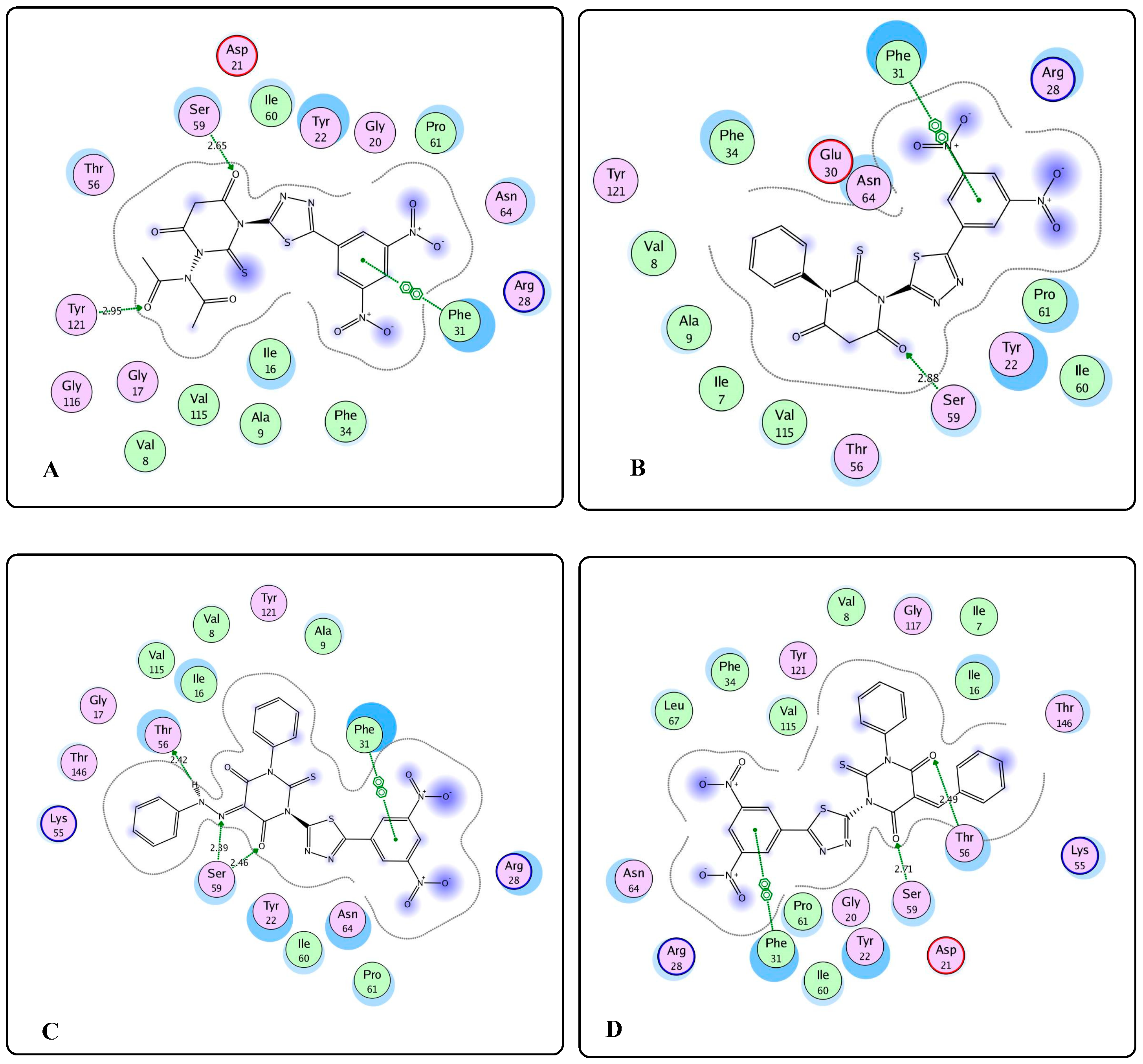
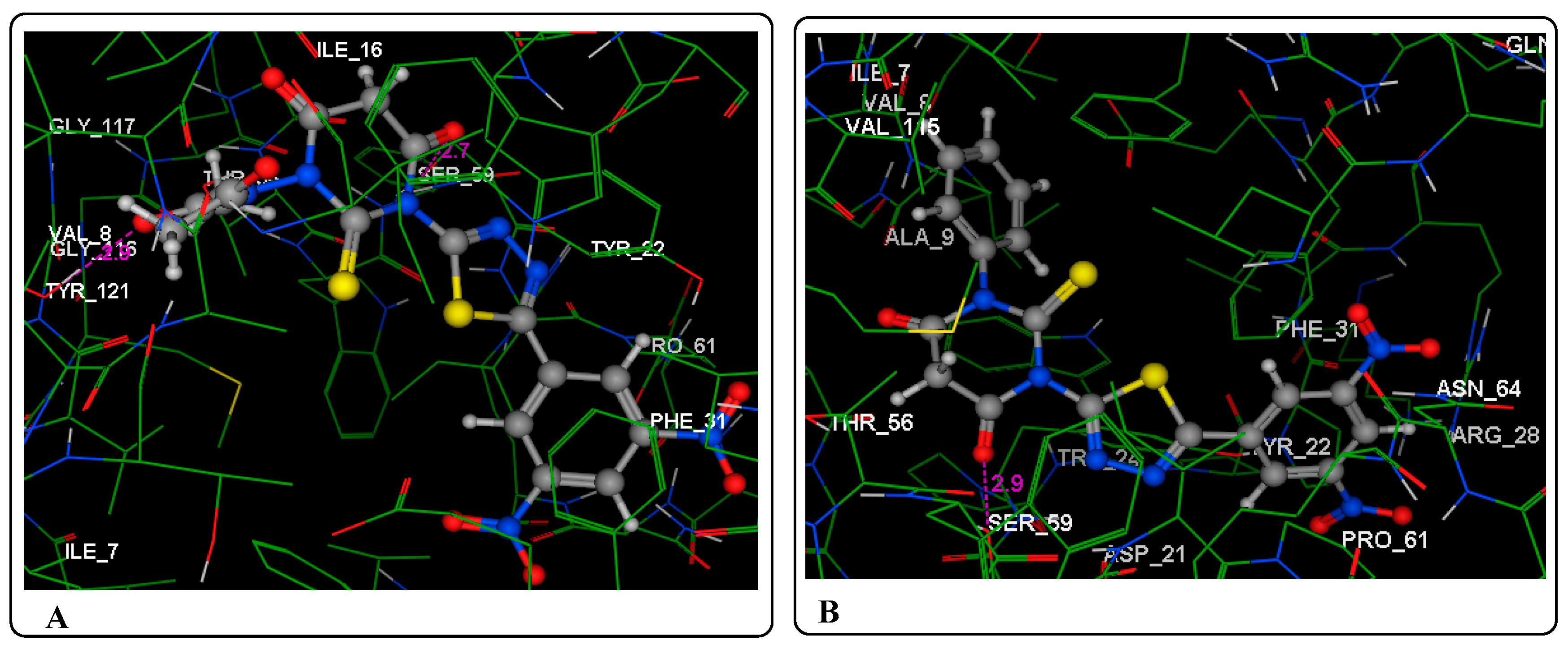
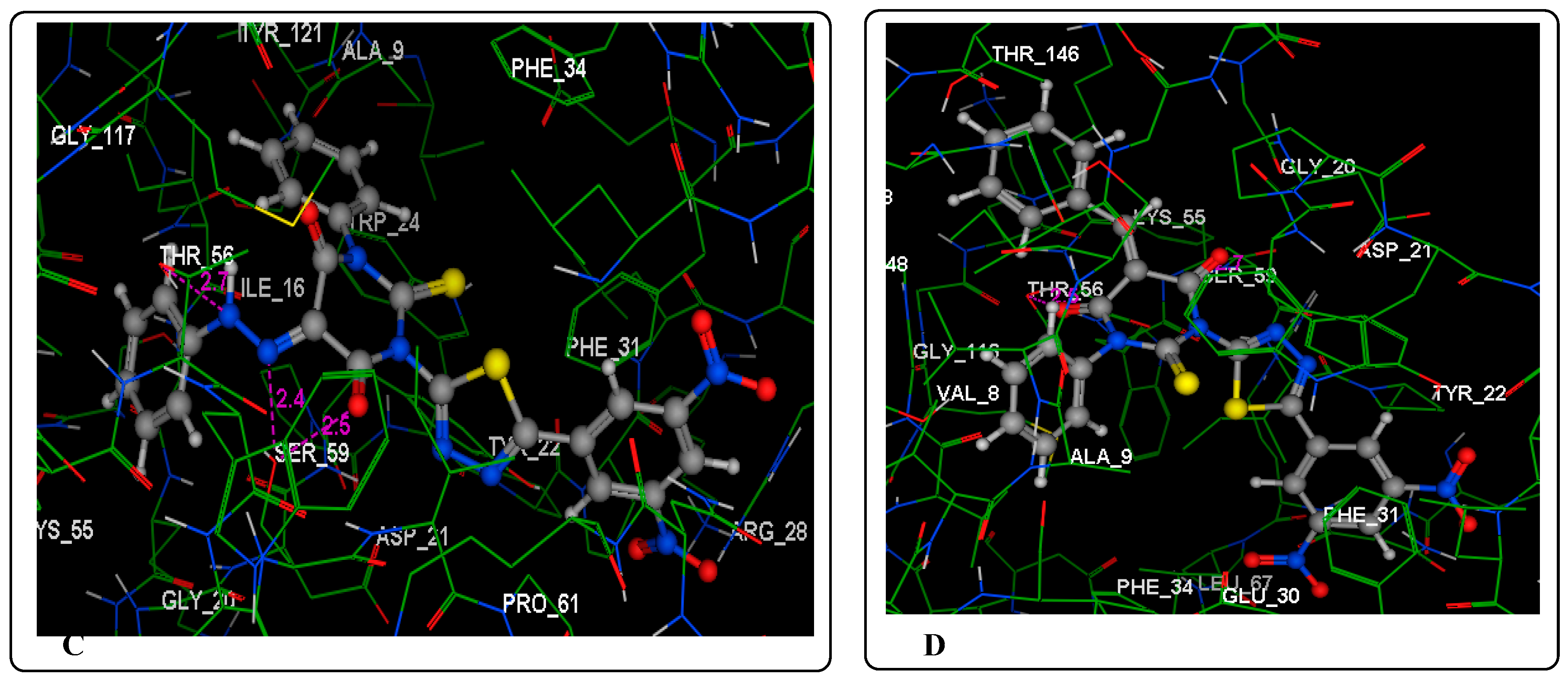
2.3. Molecular Modeling Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. N-(5-(3,5-Dinitrophenyl)-1,3,4-thiadiazol-2-yl)hydrazinecarbothioamide (2)
3.1.2. 5-(1-Amino-2-phenylethyl)-N-(5-(3,5-dinitrophenyl)-1,3,4-thiadiazol-2-yl)-1,3,4-thiadiazol-2-amine (3)
3.1.3. 5-(3,5-Dinitrophenyl)-N-(5-phenyl-1,3,4-thiadiazol-2-yl)-1,3,4-thiadiazol-2-amine (4)
3.1.4. 5-{[5-(3,5-Dinitrophenyl)-1,3,4-thiadiazol-2-yl]amino}-1,3,4-thiadiazole-2-thiol (5)
3.1.5. 2-((5-((5-(3,5-Dinitrophenyl)-1,3,4-thiadiazol-2-yl)amino)-1,3,4-thiadiazol-2-yl)thio)-1-phenylethan-1-one (6)
3.1.6. 5-(5-((5-(3,5-Dinitrophenyl)-1,3,4-thiadiazol-2-yl)amino)-1,3,4-thiadiazol-2(3H)-ylidene)pyrimidine-2,4,6(1H,3H,5H)-trione (8)
3.1.7. 3-[5-(3,5-Dinitrophenyl)-1,3,4-thiadiazol-2-yl]-2-hydrazonothiazolidin-4-one (9)
3.1.8. N-Acetyl-N-[3-(5-(3,5-dinitrophenyl)-1,3,4-thiadiazol-2-yl]-4,6-dioxo-2-thioxotetrahydro-pyrimidin-1(2H)-yl)acetamide (10)
3.1.9. 1-[5-(3,5-Dinitrophenyl)-1,3,4-thiadiazol-2-yl]-3-phenylthiourea (11)
3.1.10. N-[5-(3,5-Dinitrophenyl)-1,3,4-thiadiazol-2-yl]-3,4-diphenylthiazol-2(3H)-imine (12)
3.1.11. 1-[5-(3,5-Dinitrophenyl)-1,3,4-thiadiazol-2-yl]-3-phenyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (13)
3.1.12. 1-[5-(3,5-Dinitrophenyl)-1,3,4-thiadiazol-2-yl]-3-phenyl-5-(2-phenylhydrazono)-2-thioxo-dihydro-pyrimidine-4,6(1H,5H)-dione (14)
3.1.13. 5-Benzylidene-1-[5-(3,5-dinitrophenyl)-1,3,4-thiadiazol-2-yl]-3-phenyl-2-thioxodihydro-pyrimidine-4,6(1H,5H)-dione (15)
3.1.14. Dimethyl (5-(3,5-dinitrophenyl)-1,3,4-thiadiazol-2-yl)carbonimidodithioate (16)
3.1.15. Synthesis of N-[5-(3,5-dinitrophenyl)-1,3,4-thiadiazol-2-yl]benzo[d]azole derivatives 17–19
3.1.16. N-[5-(3,5-Dinitrophenyl)-1,3,4-thiadiazol-2-yl]benzo[d]oxazol-2-amine (17)
3.1.17. N-[5-(3,5-Dinitrophenyl)-1,3,4-thiadiazol-2-yl]benzo[d]thiazol-2-amine (18)
3.1.18. N-(1H-Benzo[d]imidazol-2-yl)-5-(3,5-dinitrophenyl)-1,3,4-thiadiazol-2-amine (19)
3.1.19. N-(4,5-Dihydro-1H-imidazol-2-yl)-5-(3,5-dinitrophenyl)-1,3,4-thiadiazol-2-amine (20)
3.2. Biological Evaluation
3.2.1. Antimicrobial Sensitivity Assay
3.2.2. In Vitro Anticancer Activity
3.2.3. Dihydrofolate Reductase (DHFR) Inhibition
3.3. Molecular Modeling Studies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schnell, J.R.; Dyson, H.J.; Wright, P.E. Structure, dynamics, and catalytic function of dihydrofolate reductase. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 119–140. [Google Scholar] [CrossRef]
- Huang, H.; Lu, W.; Li, X.; Cong, X.; Ma, H.; Liu, X.; Zhang, Y.; Che, P.; Ma, R.; Li, H.; et al. Design and synthesis of small molecular dual inhibitor of falcipain-2 and dihydrofolate reductase as antimalarial agent. Bioorg. Med. Chem. Lett. 2012, 22, 958–962. [Google Scholar] [CrossRef]
- Borst, P.; Quellette, M. New mechanisms of drug resistance in parasitic protozoa. Annu. Rev. Microbiol. 1995, 49, 427–460. [Google Scholar] [CrossRef] [PubMed]
- Foye, W.O.; Lemke, T.L.; Williams, D.A. Principles of Medicinal Chemistry, 4th ed.; Williams and Wilkins: Media, PA, USA, 2005; pp. 442–456. [Google Scholar]
- Champe, P.C.; Harvey, R.A. Lippincott’s Illustrated Reviews: Biochemistry, 2nd ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 1994; pp. 350–356. [Google Scholar]
- Berman, E.M.; Werbel, L.M. The renewed potential for folate antagonists in contemporary cancer chemotherapy. J. Med. Chem. 1991, 34, 479–485. [Google Scholar] [CrossRef] [PubMed]
- El-Gazzar, Y.I.; Georgey, H.H.; El-Messery, S.M.; Ewida, H.A.; Hassan, G.S.; Raafat, M.M.; Ewida, M.A.; El-Subbagh, H.I. Synthesis, biological evaluation and molecular modeling study of new (1,2,4-triazole or 1,3,4-thiadiazole)-methylthio-derivatives of quinazolin-4(3H)-one as DHFR inhibitors. Bioorg. Chem. 2017, 72, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Al-Rashood, S.T.; Aboldahab, I.A.; Nagi, M.N.; Abouzeid, L.A.; Abdel-Aziz, A.A.M.; Abdel-hamide, S.G.; Youssef, K.M.; Al-Obaida, A.M.; El-Subbagha, H.I. Synthesis, dihydrofolate reductase inhibition, antitumor testing, and molecular modeling study of some new 4(3H)-quinazolinone analogs. Bioorg. Med. Chem. 2006, 14, 8608–8621. [Google Scholar] [CrossRef]
- Jain, K.; Sharma, S.; Vaidya, A.; Ravichandran, V.; Agrawal, R.K. 1,3,4-Thiadiazole and its derivatives: A review on recent progress in biological activities. Chem. Biol. Drug Des. 2013, 81, 557–576. [Google Scholar] [CrossRef]
- Kushwaha, N.; Kushwaha, S.K.S.; Rai, A.K. Biological activities of thiadi¬azole derivatives: A review. Int. J. Chem. Res. 2012, 4, 517–531. [Google Scholar]
- Gomha, S.M.; Salah, T.A.; Abdelhamid, A.O. Synthesis, characterization, and pharmacological evaluation of some novel thiadiazoles and thiazoles incorporating pyrazole moiety as anticancer agents. Monatsh. Chem. 2015, 146, 149–158. [Google Scholar]
- Gomha, S.M.; Abdel-aziz, H.M. Synthesis and antitumor activity of 1,3,4-thiadiazole derivatives bearing coumarine ring. Heterocycles 2015, 91, 583–592. [Google Scholar] [CrossRef]
- Siddiqui, N.; Ahuja, P.; Ahsan, W.; Pandeya, S.N.; Alam, M.S. Thiadiazoles: Progress report on biological activities. J. Chem. Pharm. Res. 2009, 1, 19–30. [Google Scholar]
- Bhattacharya, P.; Leonard, J.T.; Roy, K. Exploring QSAR of thiazole and thiadiazole derivatives as potent and selective human adenosine A3 receptor antagonists using FA and GFA techniques. Bioorg. Med. Chem. 2005, 15, 1159–1165. [Google Scholar] [CrossRef]
- Foroumadi, A.; Kargar, Z.; Sakhteman, A.; Sharifzadeh, Z.; Feyzmohammadi, R.; Kazemi, M.; Shafiee, A. Synthesis and antimycobacterial activity of some alkyl[5-(nitroaryl)-1,3,4-thiadiazol-2-ylthio]propionates. Bioorg. Med. Chem. Lett. 2006, 16, 1164–1167. [Google Scholar] [CrossRef]
- Kumar, D.; Kumar, N.M.; Chang, K.H.; Shah, K. Synthesis and anticancer activity of 5-(3-indolyl)-1,3,4-thiadiazoles. Eur. J. Med. Chem. 2010, 45, 4664–4668. [Google Scholar] [PubMed]
- Sharma, B.; Verma, A.; Prajapati, S.; Sharma, U.K. Synthetic methods, chemistry, and the anticonvulsant activity of thiadiazoles. Int. J. Med. Chem. 2013, 2013, 348948. [Google Scholar] [PubMed]
- Mathew, V.; Keshavayya, J.; Vaidya, V.P.; Giles, D. Studies on synthesis and pharmacological activities of 3,6-disubstituted-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles and their dihydro analogues. Eur. J. Med. Chem. 2007, 42, 823–840. [Google Scholar] [CrossRef] [PubMed]
- Masereel, B.; Rolin, S.; Abbate, F.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Anticonvulsant sulfonamides incorporating valproyl and other lipophilic moieties. J. Med. Chem. 2002, 45, 312–320. [Google Scholar] [CrossRef]
- Said, H.M.; Hagemann, C.; Carta, F.; Katzer, A.; Polat, B.; Staab, A.; Scozzafava, A.; Anacker, J.; Vince, G.H.; Flentje, M.; et al. Hypoxia induced CA9 inhibitory targeting by two different sulfonamide derivatives including Acetazolamide in human Glioblastoma. Bioorg. Med. Chem. 2013, 21, 3949–3957. [Google Scholar] [PubMed]
- Li, Y.; Geng, J.; Liu, Y.; Yu, S.; Zhao, G. Thiadiazole—A Promising Structure in Medicinal Chemistry. Chem. Med. Chem. 2013, 8, 27–41. [Google Scholar] [CrossRef]
- Riyadh, S.M.; El-Motairi, S.A.; Ahmed, H.E.A.; Khalil, K.D.; Habibe, E.E. Synthesis, biological evaluation, and molecular docking of novel thiazoles and [1,3,4]thiadiazoles incorporating sulfonamide group as DHFR Inhibitors. Chem. Biodivers. 2018, 15. [Google Scholar] [CrossRef]
- Gomha, S.M.; Edrees, M.M.; Muhammad, Z.A.; El-Reedy, A.A.M. 5-(Thiophen-2-yl)-1,3,4-thiadiazole derivatives: Synthesis, molecular docking and in vitro cytotoxicity evaluation as potential anticancer agents. Drug Des. Dev. Ther. 2018, 12, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- El-Hashash, M.A.; Azab, M.E.; Faty, R.A.; Amr, A.-G. Synthesis, antimicrobial and anti-inflammatory activity of some new benzoxazinone and quinazolinone candidates. Chem. Pharm. Bull. 2016, 64, 263–271. [Google Scholar] [PubMed]
- Azab, M.E.; Rizk, S.A.; Mahmoud, N.F. Facile Synthesis, characterization and antimicrobial evaluation of novel heterocycles, schiff bases and N-nucleosides bearing phthalazine moiety. Chem. Pharm. Bull. 2016, 64, 349–350. [Google Scholar] [CrossRef] [PubMed]
- Sayed, G.H.; Azab, M.E.; Anwer, K.E.; Raouf, M.A.; Negm, N.A. Pyrazole, pyrazolone and enaminonitrile pyrazole derivatives: Synthesis, characterization and potential in corrosion inhibition and antimicrobial applications. J. Mol. Liq. 2018, 252, 329–338. [Google Scholar] [CrossRef]
- Sayed, G.H.; Azab, M.E.; Anwer, K.E.; Negm, N.A. Antimicrobial and cytotoxic activities of some novel heterocycles bearing pyrazole moiety. J. Heterocycl. Chem. 2018, 55, 1615–1625. [Google Scholar]
- Amr, A.E.; Abo-Ghalia, M.H.; Abdalah, M.M. Synthesis of new (Nα-dipicolinoyl)-bis-L-valyl-L- phenylalanyl linear and macrocyclic bridged peptides as anti-inflammatory agents. Arch. Pharm. 2007, 340, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Al-Omar, M.A.; Amr, A.E. Synthesis of some new pyridine-2,6-carboxamide-derived schiff bases as potential antimicrobial agents. Molecules 2010, 15, 4711–4721. [Google Scholar] [CrossRef]
- Amr, A.E.; Abdel-Latif, N.A.; Abdalla, M.M. Synthesis of some new testosterone derivatives fused with substituted pyrazoline ring as promising 5α-reductase inhibitors. Acta Pharm. 2006, 56, 203–218. [Google Scholar]
- Abdel-Wahab, B.F.; Mohamed, S.F.; Amr, A.E.; Abdalla, M.M. Synthesis and reactions of thiosemicarbazides, triazoles, and Schiff bases as antihypertensive α-blocking agents. Monatshefte fur Chemie 2008, 139, 1083–1090. [Google Scholar] [CrossRef]
- Amr, A.E.; Abdulla, M.M. Synthesis and anti-inflammatory activities of new cyanopyrane derivatives fused with steroidal nuclei. Arch. Pharm. 2006, 339, 88–95. [Google Scholar] [CrossRef]
- Khalifa, N.M.; Al-Omar, M.A.; Amr, A.E.; Haiba, M.E. HIV-1 and HSV-1 virus activities of some new polycyclic nucleoside pyrene candidates. Int. J. Biol. Macromol. 2013, 54, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, M.M.; Al-Omar, M.A.; Bhat, M.A.; Amr, A.E.; Al-Mohizea, A.M. Steroidal pyrazolines evaluated as aromatase and quinone reductase-2 inhibitors for chemoprevention of cancer. Int. J. Biol. Macromol. 2012, 50, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Cheesbrough, M. District Laboratory Practice in Tropical Countries; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Pignatello, R.; Sapmpinato, G.; Sorrenti, V.; Vicari, L.; Di-Giacomo, C.; Vanella, A.; Puglisi, G. Aliphatic α-γ-bis(amides) of Methotrexate. Influence of chain length on in-vitro activity against sensitive and resistant tumour cells. Pharm. Pharmacol. Commun. 1999, 5, 299–305. [Google Scholar] [CrossRef]
- Lewis, W.S.; Cody, V.; Galitsky, N.; Luft, J.R.; Pangborn, W.; Chunduru, S.K.; Spencer, H.T.; Appleman, J.R.; Blakley, R.L. Methotrexate-resistant variants of human dihydrofolate reductase with substitutions of leucine 22. Kinetics, crystallography, and potential as selectable markers. J. Biol. Chem. 1995, 270, 5057–5064. [Google Scholar] [CrossRef]
- Amr, A.E.; Abo-Ghalia, M.H.; Moustafa, G.; Al-Omar, M.A.; Nossier, E.S.; Elsayed, E.A. Design, synthesis and docking studies of novel macrocyclic pentapeptides as anticancer multi-targeted kinase inhibitors. Molecules 2018, 23, 2416. [Google Scholar] [CrossRef]
- Elzahabi, H.S.A.; Nossier, E.S.; Khalifa, N.M.; Alasfoury, R.A.; El-Manawat, M.A. Anticancer evaluation and molecular modeling of multi-targeted kinase inhibitors based pyrido[2,3-d]- pyrimidine scaffold. J. Enzym. Inhib. Med. Chem. 2018, 33, 546–557. [Google Scholar] [CrossRef] [PubMed]
- Vachala, S.D.; Bhargavi, B. Design, synthesis and screening of novel 5-substituted-1,3,4-thiadiazol-2- amines and their Schiff bases. J. Chem. Pharm. Res. 2014, 6, 377–389. [Google Scholar]
- Gür, M.; Şener, N.; Muğlu, H.; Çavuş, M.S.; Özkan, O.E.; Kandemirli, F.; Şener, İ. New 1,3,4-thiadiazole compounds including pyrazine moiety: Synthesis, structural properties and antimicrobial features. J. Mol. Struct. 2017, 1139, 111–118. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diameter of Inhibition Zone (mm) | ||||||
|---|---|---|---|---|---|---|
| Compound Number | Gram Positive Bacteria | Gram Negative Bacteria | Fungi | |||
| Staphylococcus aureus | Bacillus subtilis | Pseudomonas aeruginosa | Escherichia coli | Aspergillus niger | Candida albicans | |
| 1 | 12 | 10 | 14 | 8 | 6 | 9 |
| 2 | 25 | 24 | 22 | 23 | 17 | 18 |
| 3 | 0 | 0 | 0 | 2 | 0 | 0 |
| 4 | 0 | 0 | 2 | 3 | 0 | 0 |
| 5 | 20 | 19 | 20 | 21 | 16 | 18 |
| 6 | 10 | 11 | 13 | 12 | 9 | 8 |
| 8 | 9 | 7 | 8 | 10 | 8 | 9 |
| 9 | 27 | 28 | 26 | 26 | 21 | 23 |
| 10 | 28 | 29 | 27 | 27 | 22 | 24 |
| 11 | 13 | 14 | 16 | 15 | 11 | 13 |
| 12 | 0 | 2 | 0 | 2 | 0 | 0 |
| 13 | 30 | 29 | 28 | 27 | 23 | 25 |
| 14 | 18 | 13 | 11 | 9 | 8 | 12 |
| 15 | 16 | 19 | 13 | 10 | 6 | 15 |
| 16 | 28 | 26 | 25 | 27 | 22 | 22 |
| 17 | 8 | 8 | 6 | 9 | 7 | 6 |
| 18 | 4 | 3 | 0 | 3 | 2 | 3 |
| 19 | 5 | 0 | 6 | 4 | 3 | 4 |
| 20 | 21 | 18 | 20 | 22 | 16 | 18 |
| Amoxicillin | 28 | 27 | 24 | 25 | - | - |
| Fluconazole | - | - | - | - | 20 | 22 |
| Compound Number | IC50 (Mean ± SEM) (µM) a | ||||
|---|---|---|---|---|---|
| CCRF-CEM | HCT-15 | PC-3 | UACC-257 | MCF-10A | |
| DOX b | 6.78 ± 0.7 | 5.17 ± 0.2 | 4.56 ± 0.4 | 7.34 ± 0.5 | 377.33 ± 2.55 |
| 1 | 90.7 ± 1.2 | 79.8 ± 2.6 | 88.2 ± 1.6 | 81.6 ± 1.1 | 497.54 ± 5.44 |
| 2 | 74.56 ± 3.8 | 67.20 ± 2.9 | 70.18 ± 3.2 | 71.20 ± 3.9 | 586.43 ± 4.53 |
| 3 | 51.88 ± 1.6 | 30.12 ± 1.8 | 27.24 ± 1.2 | 46.44 ± 1.1 | 464.34 ± 3.47 |
| 4 | 39.28 ± 2.6 | 32.75 ± 2.2 | 29.52 ± 1.5 | 36.33 ± 2.4 | 383.63 ± 2.56 |
| 5 | 77.74 ± 2.6 | 65.66 ± 3.3 | 82.55 ± 1.9 | 80.66 ± 2.2 | 252.74 ± 2.45 |
| 6 | 10.99 ± 0.3 | 9.87 ± 0.5 | 9.92 ± 1.2 | 12.67 ± 0.6 | 461.85 ± 5.58 |
| 8 | 19.19 ± 0.5 | 17.24 ± 0.9 | 15.44 ± 1.4 | 22.66 ± 1.3 | 373.96 ± 6.49 |
| 9 | 76.22 ± 2.6 | 69.65 ± 2.7 | 81.59 ± 1.3 | 83.33 ± 2.9 | 262.07 ± 4.50 |
| 10 | 40.44 ± 1.9 | 36.76 ± 1.8 | 33.88 ± 1.1 | 38.55 ± 2.1 | 351.89 ± 6.68 |
| 11 | 71.26 ± 3.2 | 63.27 ± 2.3 | 66.14 ± 3.6 | 73.34 ± 3.5 | 380.68 ± 5.57 |
| 12 | 15.78 ± 0.8 | 15.01 ± 0.5 | 12.14 ± 1.1 | 18.38 ± 1.6 | 373.47 ± 6.45 |
| 13 | 49.36 ± 1.2 | 52.72 ± 1.2 | 48.88 ± 2.1 | 55.55 ± 0.9 | 460.56 ± 8.64 |
| 14 | 9.66 ± 0.6 | 8.59 ± 0.7 | 6.99 ± 1.2 | 9.20 ± 0.8 | 474.44 ± 8.72 |
| 15 | 6.99 ± 0.4 | 5.28 ± 0.5 | 4.67 ± 0.3 | 7.41 ± 0.5 | 352.35 ± 7.53 |
| 16 | 79.32 ± 3.1 | 68.33 ± 4.3 | 80.59 ± 1.7 | 76.61 ± 1.7 | 473.26 ± 8.44 |
| 17 | 67.45 ± 3.4 | 61.27 ± 2.3 | 62.56 ± 2.6 | 70.77 ± 1.5 | 382.35 ± 7.35 |
| 18 | 58.58 ± 4.4 | 55.67 ± 1.6 | 61.21 ± 1.1 | 65.34 ± 2.2 | 491.24 ± 9.53 |
| 19 | 84.44 ± 2.2 | 69.87 ± 3.3 | 86.95 ± 1.5 | 83.54 ± 2.9 | 382.32 ± 8.72 |
| 20 | 83.34 ± 4.2 | 71.64 ± 4.1 | 92.34 ± 4.2 | 88.64 ± 4.1 | 473.23 ± 9.83 |
| Compound Number | IC50 (Mean ± SEM) (µM) |
|---|---|
| DHFR | |
| 1 | 17.26 ± 0.43 |
| 2 | 14.33 ± 0.81 |
| 3 | 28.54 ± 0.22 |
| 4 | 31.57 ± 0.30 |
| 5 | 15.38 ± 0.12 |
| 6 | 13.45 ± 0.23 |
| 8 | 20.54 ± 0.14 |
| 9 | 8.46 ± 0.13 |
| 10 | 1.00 ± 0.85 |
| 11 | 17.13 ± 0.90 |
| 12 | 36.48 ± 0.72 |
| 13 | 0.09 ± 0.91 |
| 14 | 0.08 ± 0.37 |
| 15 | 0.04 ± 0.82 |
| 16 | 10.24 ± 0.97 |
| 17 | 27.42 ± 0.35 |
| 18 | 24.71 ± 1.26 |
| 19 | 29.00 ± 1.25 |
| 20 | 11.80 ± 0.79 |
| Methotrexate | 0.14 ± 1.38 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Naggar, M.; Sallam, H.A.; Shaban, S.S.; Abdel-Wahab, S.S.; E. Amr, A.E.-G.; Azab, M.E.; Nossier, E.S.; Al-Omar, M.A. Design, Synthesis, and Molecular Docking Study of Novel Heterocycles Incorporating 1,3,4-Thiadiazole Moiety as Potential Antimicrobial and Anticancer Agents. Molecules 2019, 24, 1066. https://doi.org/10.3390/molecules24061066
El-Naggar M, Sallam HA, Shaban SS, Abdel-Wahab SS, E. Amr AE-G, Azab ME, Nossier ES, Al-Omar MA. Design, Synthesis, and Molecular Docking Study of Novel Heterocycles Incorporating 1,3,4-Thiadiazole Moiety as Potential Antimicrobial and Anticancer Agents. Molecules. 2019; 24(6):1066. https://doi.org/10.3390/molecules24061066
Chicago/Turabian StyleEl-Naggar, Mohamed, Hanan A. Sallam, Safaa S. Shaban, Salwa S. Abdel-Wahab, Abd El-Galil E. Amr, Mohammad E. Azab, Eman S. Nossier, and Mohamed A. Al-Omar. 2019. "Design, Synthesis, and Molecular Docking Study of Novel Heterocycles Incorporating 1,3,4-Thiadiazole Moiety as Potential Antimicrobial and Anticancer Agents" Molecules 24, no. 6: 1066. https://doi.org/10.3390/molecules24061066