Reactive Cobalt–Oxo Complexes of Tetrapyrrolic Macrocycles and N-based Ligand in Oxidative Transformation Reactions


Department of Chemistry, The Key Laboratory of Fuel Cell Technology of Guangdong Province, South China University of Technology, Guangzhou 510641, China
*
Author to whom correspondence should be addressed.
Molecules 2019, 24(1), 78; https://doi.org/10.3390/molecules24010078
Submission received: 28 November 2018
/
Revised: 21 December 2018
/
Accepted: 25 December 2018
/
Published: 26 December 2018
(This article belongs to the Special Issue Tetrapyrrolic Macrocycles: Synthesis, Functionalization and Applications 2018)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
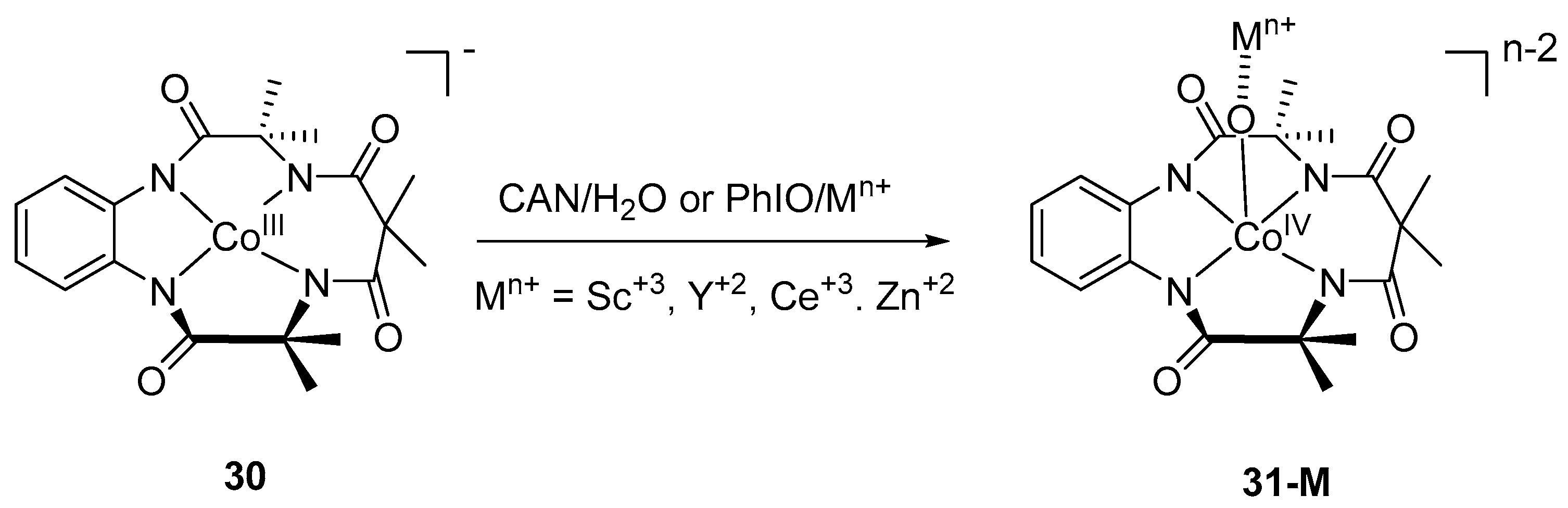{kind=link}
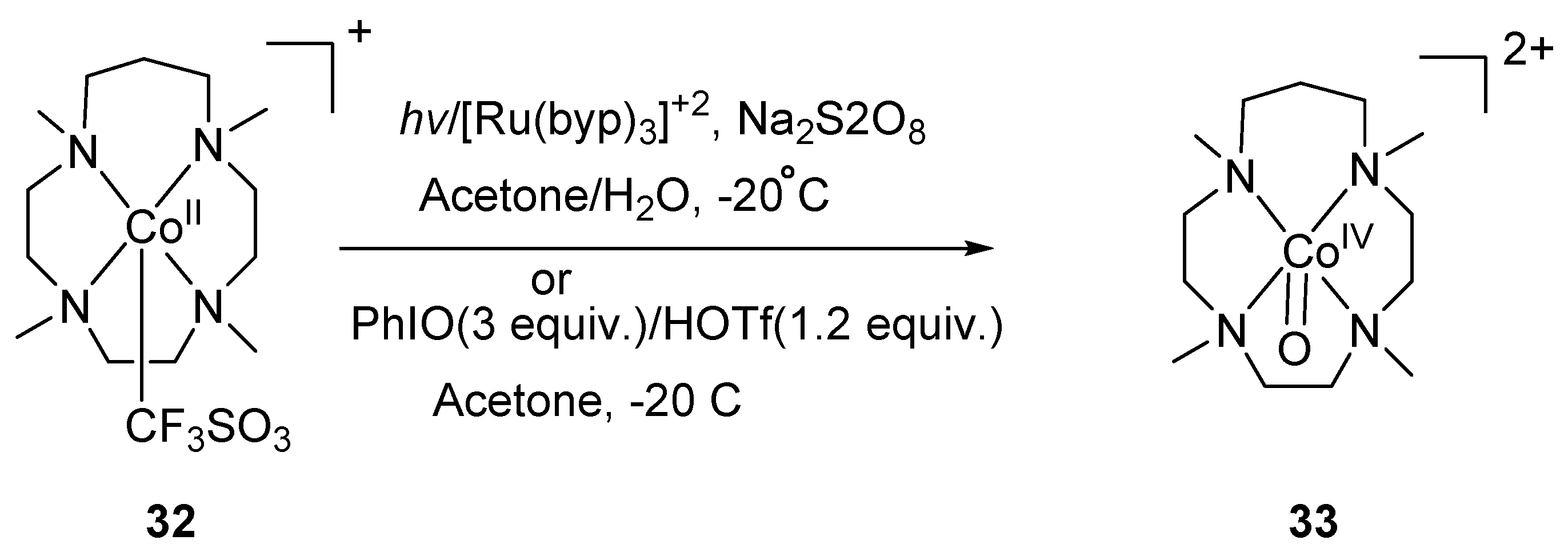{kind=link}
Abstract
:High-valent cobalt–oxo complexes are reactive transient intermediates in a number of oxidative transformation processes e.g., water oxidation and oxygen atom transfer reactions. Studies of cobalt–oxo complexes are very important for understanding the mechanism of the oxygen evolution center in natural photosynthesis, and helpful to replicate enzyme catalysis in artificial systems. This review summarizes the development of identification of high-valent cobalt–oxo species of tetrapyrrolic macrocycles and N-based ligands in oxidation of organic substrates, water oxidation reaction and in the preparation of cobalt–oxo complexes.
1. Introduction
In biological systems, metalloenzymes, typically containing Mn, Fe and Cu centers, are known to catalyze a wide range of reactions including aliphatic and aromatic C–H hydroxylation, epoxidation, desaturation, and heteroatom (S, N or O) dealkylation or oxidation [1,2]. It is well known that iron‒oxo species are the reactive oxidants in the catalytic cycle of heme [3] and non-heme iron enzymes [4]. Similarly, manganese–oxo complex has been suggested the key intermediate in oxygen-evolving center of photo-system II (PSII) [5,6,7]. The transition metal–oxo complexes of iron and manganese involved in artificial oxygen transfer and C–H bond activations reactions have been extensively reviewed [8,9,10,11,12,13]. Except for the early transition metal–oxo complexes, high-valent metal–oxo complexes of late transition metals, particularly cobalt–oxo complexes, are also highly reactive transient intermediates in cobalt-catalyzed C‒H bond activation and O‒O bond formation reactions [14,15,16], and they are considered to be more reactive then related iron‒oxo species due to a weak metal–oxygen bond [17,18]. Currently, clean energy production by maneuvering natural photosynthesis in water oxidation reactions to develop artificial photosynthesis [19,20,21] for efficient water splitting is a hot topic of research [22,23,24]. In particular, the cobalt oxides are often used materials for water oxidation to generate molecular oxygen [25,26,27,28]. The high-valent cobalt–oxo complexes of N-based ligand can be implicated as reactive species in the O–O bond-forming event during water oxidation [29,30]. Furthermore, cobalt complexes based on tetrapyrrolic macrocycles are often used in mimicking the peroxidase-like activity for the selective oxidation of organic substrates via high-valent cobalt(IV)–oxo intermediates [31,32]. Obviously, in the study of the reactive oxidants in these catalytic reactions it is essential to provide insight into their mechanism of reaction, allowing us to probe the critical step in these challenging reactions. However, the isolation and identification of these transient intermediates is considerable challenge. The cobalt–oxo complexes are not stable because cobalt has large number of d-electrons which produces strong electronic repulsion between the rich electron oxygen and cobalt center. Also, the strong oxidative environment will cause oxidative degradation of the ligand, making the high-valent cobalt–oxo complexes unstable. To reduce the electronic repulsion between cobalt and oxygen, and to avoid oxidative degradation, tetrapyrrolic macrocylces and N-based ligands with a different electronic environment were implemented to increase the stability of cobalt–oxo complexes (Figure 1). Mostly, these can only be characterized in situ by electron paramagnetic resonance (EPR) [33], X-ray absorption [34] and time-resolved Fourier-transform infrared (FT-IR) [16] spectroscopic methods. Computational studies were also carried out to understand the nature of species involved in water oxidation [35]. Recently, the isolation and/or identification of high-valent cobalt–oxo complexes has become a key topic in order to develop and understand the mechanism of artificial photosynthesis and to replicate enzymatic process in artificial reactions.
This review comprehends the high-valent cobalt–oxo complexes of tetrapyrrolic macrocycles and N-based ligands reported to date, along with outlooks in this intriguing research area. It has been divided into three sections: identification of cobalt–oxo species involved in oxidation of organic substrates; identification of cobalt–oxo species involved in heterogeneous and homogeneous water oxidation reactions; and preparation of high-valent cobalt–oxo complexes.
2. Cobalt–Oxo Species Involved in Oxidation of Organic Substrates
Cobalt–oxo species are involved in many of oxidative and C‒H bond activation reactions. The ligands used to generate cobalt–oxo species play a key role in stabilizing cobalt–oxo species. Also, to mimic the enzymes-like environment, different types of support are used as protein backbone for example cellulosic fiber and multiwall carbon nanotubes. These supports cannot alter the reaction mechanism however, precisely control the generation of reactive intermediate, which also determines the activity, durability and stability of the complexes [36,37,38].
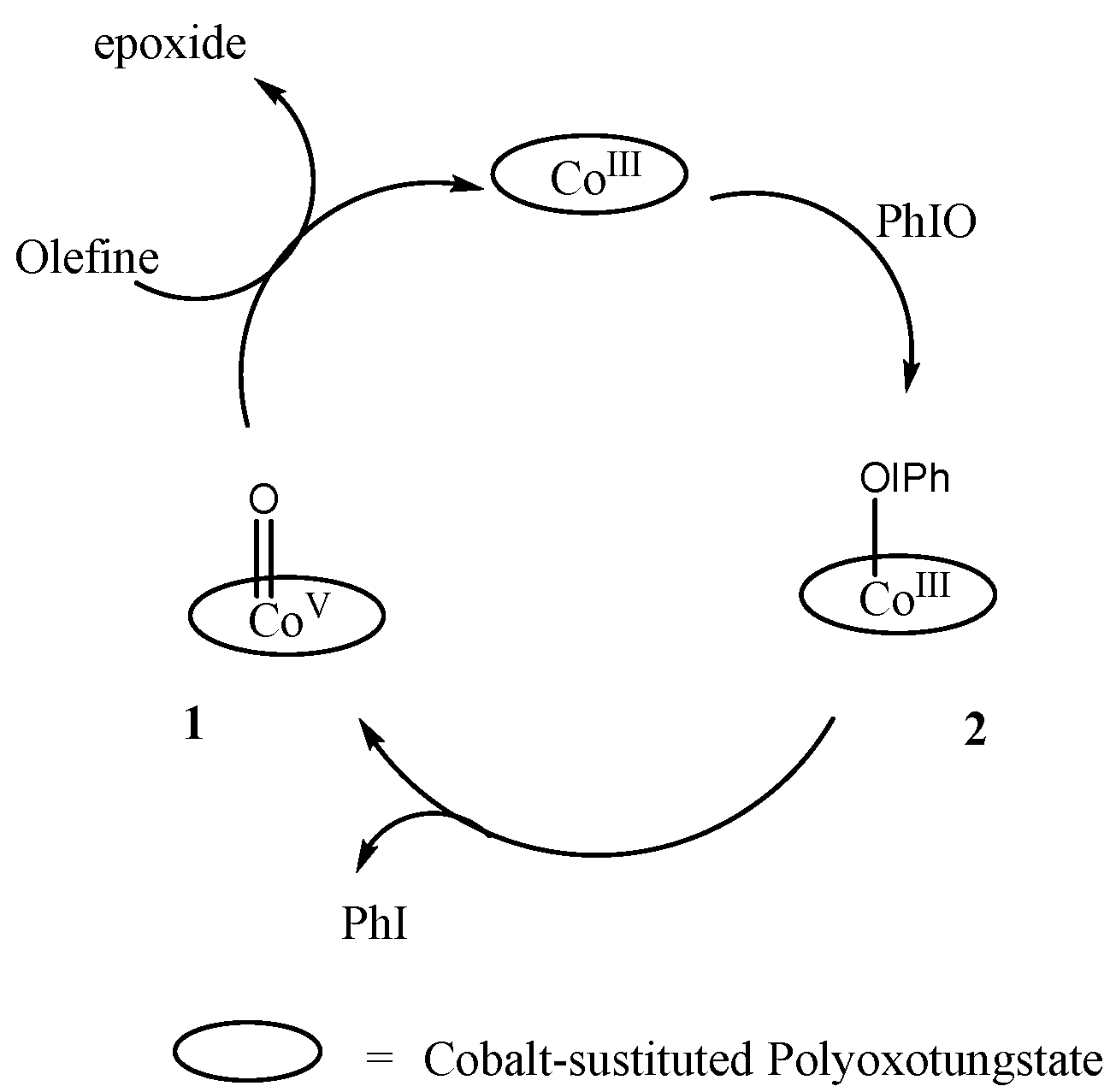
Nam et al. reported [39] the catalytic oxidation of alkene and alkane using cobalt-substituted polyoxotungstate and employed different oxidants such as iodosylbenzene, potassium monopersulfate and m-CPBA. Cobalt-substituted polyoxotungstate was proved to be a good catalyst. They proposed the involvement of different cobalt–oxo species with the different oxidants. Two possible species may form with iodosylbenzene, high-valent cobalt(V)–oxo 1 and cobalt–iodosylbenzene adduct 2 (Scheme 1). They suggested that complex 2 is responsible for oxygen transfer because cobalt cannot be obtained in +5 oxidation state. KHSO5 and m-CPBA predict the involvement of cobalt(III)–oxygen adducts as oxygen transfer complex. Isotopically labeled water (H218O) is a useful experimental tool to investigate the involvement of high-valent cobalt–oxo species in cobalt-mediated oxygen atom transfer reactions, but all the attempts to obtain 18O-labeled products have failed. Furthermore, porphyrins are extensively used to get stable metal–oxo complexes [13]. Therefore, porphyrins with a different electronic environment were used to stabilize cobalt–oxo species [40,41]. Cobalt(IV)‒oxo [40] and cobalt(IV)–oxo porphyrin radical [41] were proposed to be involved in C‒H bond activation reaction. These species are quite reactive towards the oxidation of alkane and alcohol, respectively. However, there is no experimental evidence to support presence of cobalt–oxo species due to instability. Likewise, a cobalt(IV)–oxo species was reported [42], based on the tetraanionic cobalt(II) complex of (BrHBA-Et)H4, N,N′-(ethane-1,2-diyl)bis(5-bromo-2-hydroxybenzamide), that provides a strong ligand field. Consequently, this specie was stable enough to be characterized by EPR and ESI-MS spectroscopy analysis. Also, the presence of high-valent cobalt(IV)–oxo porphyrin was reported during the oxidation of alcohol to benzaldehyde by molecular oxygen in the presence of isobutyraldehyde, using bifunctional hybride catalyst originated from cobalt tetra(4-sulfonatophenyl)porphyrinate anion [43] and a cationic meso-tetrakis (1-methyl-4-pyridyl) cobalt porphyrin immobilized in montmorillonite interlayers [44]. The presence of a cobalt(IV)–oxo specie was predicted by an 18O-labeled experiment of product [43]. The turnover frequency and catalytic yield was higher in the prior case. Later, the cobalt(IV)–oxo porphyrin was generated [45] by the oxidation of cobalt porphyrin Co(TPFPP)(CF3SO3) utilizing m-CPBA as oxidant in solvent mixture of CH3CN and CH2Cl2. Incorporation of H218O in the catalytic oxidation demonstrated the presence of 18O-labeled alcohol in the product, which is evidence for the presence of cobalt–oxo species. Furthermore, cobalt(V)=O and cobalt(IV)=O were generated [46] by the oxidation of a mononuclear non haem cobalt(III) [Co((bpc)Cl2][Et4N] (H2bpc=4,5-dichloro-1,2-bis(2-pyridine-2-carboxamido)benzene) complex of a tetradentate ligand containing two deprotonated amide moieties with PhIO. Oxidation of the cobalt(III) complex generated cobalt acylperoxo intermediate, which on the heterolytic and homolytic cleavage of O‒O bond generated respective cobalt(V)–oxo and cobalt(IV)–oxo species. These species are also not enough stable to be characterized spectroscopically. Similarly, a cobalt(IV)=O specie based on isoindole-core ligand was proposed [47] as a reactive intermediate, during the stereoselective oxidation of alkane using m-CPBA as oxidant. The kinetic isotopic effect and 18O-labeled experiment predict the involvement of cobalt–oxo species. Recently, the involvement of a high-valent cobalt(IV)=O radical cation was proposed [48] during the reduction of O2. The dianionic pentadentate ligand system based on bis-pyrazolyl diaryl borate arms attached to a 2,6-substituted pyridyl frame was used to stabilize the cobalt(IV)–oxo intermediate. Cobalt(IV)=O radical cation was generated by the cleavage of Co‒O bond, and examined theoretically and experimentally. A density functional theory (DFT) calculation suggests the presence of maximum electron density at oxygen 70% with Co‒O bond length of 1.67 Å.
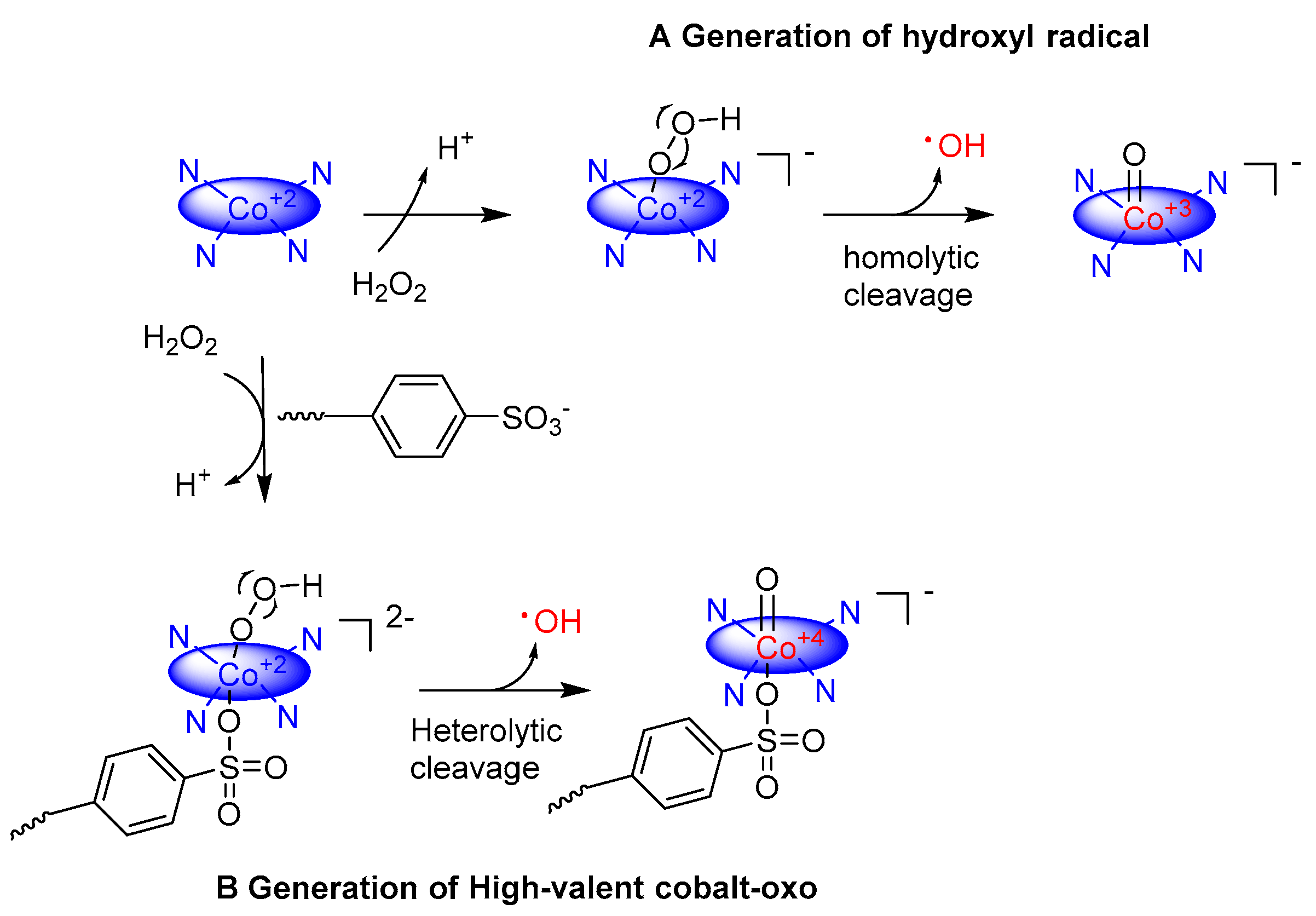
Moreover, to mimic the enzyme activity for controllable catalytic oxidation, researchers made extensive efforts to develop and discover functional materials having properties intrinsic to enzymes. Many transition-metal complexes were prepared [49,50,51] to mimic the expected features of enzymes, such as selectivity and steric accessibility, but these do not present the said features due to the non-natural environment. A catalyst which is a replication of enzyme should possess a suitable cavity or cleft for accessibility of substrates and introduction of functional groups that act as active sites within the cavity [52,53]. Enzymatically inspired catalytic system was prepared by using cobalt tetraaminophthalocyanine (CoTAPc) as a catalyst supported by ordered-mesoporous-carbon (OMC) for controllable activation of hydrogen oxide (H2O2) to generate stable cobalt–oxo intermediate [32]. Ordered-mesoporous-carbon provides the steric environment for a substrate to attach with active sites and protects the active sites against the external interface. However, a disadvantage of hydrogen peroxide is the formation of hydroxyl radical that is highly reactive, so it decreases the selectivity. A fifth ligand dodecylbenzenesulfonate (LAS) is employed to inhibit the production of hydroxyl radical. This fifth ligand also helps to generate high-valent cobalt(IV)–oxo specie by heterolytic cleavage of peroxide O‒O bond. The involvement of cobalt–oxo specie was corroborated by the results of semiempirical quantum-chemical PM6 calculations. Similarly, a modification in the tetrapyrrolic macrocycle of cobalt tetraaminophthalocyanine (CoTAPc) was made by the attachment of epoxy compound 2,3-epoxypropyl triethylammonium chloride (EPTAC), to obtain a new catalyst with positively charged quaternary ammonium salt chain (OMC-CoTAPc-EPTAC) [31]. The modified catalyst displays high catalytic activity especially for negatively charged substrates. The free radical trapping EPR analysis using 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) as a free radical scavenger did not detect DMPO-˙OH and DMPO-˙OOH signal, ruling out the free radical type mechanism. That is why, the cobalt(IV)–oxo complex was proposed as a reactive intermediate due to the heterolytic cleavage of O‒O bond of peroxide. Moreover, cellulosic fiber could play the role of the protein backbone in enzymes, providing an enzyme-like environment with enhanced regioselectivity to remove organic dyes and improve the stability of intermediate generated. A catalyst was developed based on cellulosic fiber-bonded cobalt phthalocyanine catalytic entity to activate hydrogen peroxide in order to generate cobalt–oxo specie [54]. The reaction channel was controlled by linear alkylbenzene sulfonate (LAS). High-valent cobalt(IV)–oxo specie 4 was generated by the heterolytic cleavage of peroxide O‒O bond and homolytic cleavage generate cobalt(III)–oxo specie 3 (Figure 2).
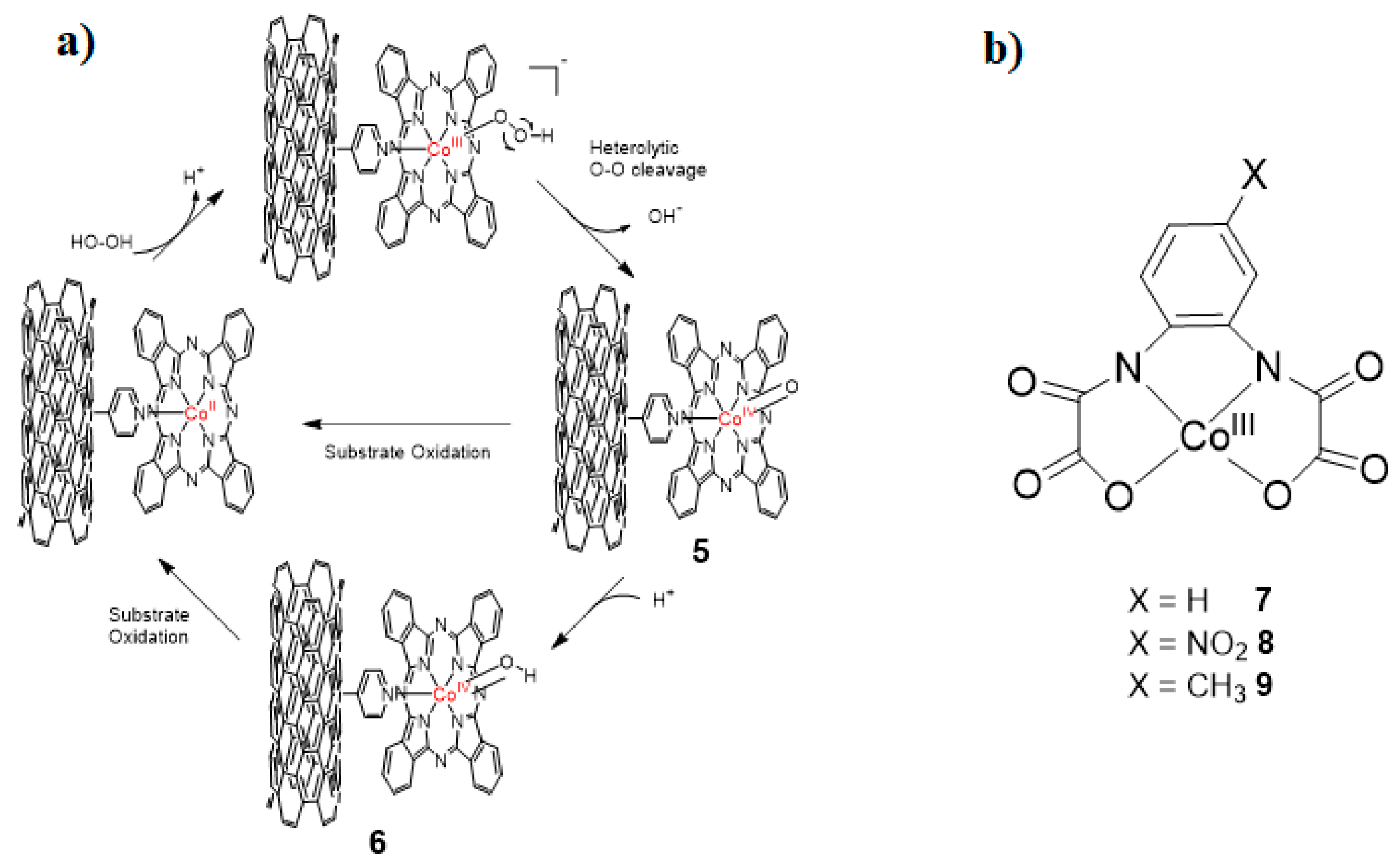
The in situ X-band EPR analysis was conducted at room temperature in the presence of LAS demonstrating a signal at geff = 2.099 identifying the presence of CoIV with spin state (S = ½). Usually, metal–oxo species have been detected at low temperature [55,56]; a high oxidation state of PcCoIV=O was observed at room temperature, presenting high stability of complex, auto-oxidation protected by cellulose matrix. Cellulosic fiber bonded cobalt phthalocyanine (CoPc) can also activate peroxymonosulfate [57]. The oxidation activity of catalyst is remarkably enhanced in the presence of bicarbonate ion (HCO3−) due to the generation of (PcCoIV=O) by the heterolytic cleavage of O–O bond of peroxymonosulfate. Later, the same group [58] employed the multiwall carbon nanotubes (MWCNTs) as protein-like backbone anchored on cobalt phthalocyanine (CoPc) for peroxidase like activation of hydrogen peroxide. The anchoring of catalytic entity on MWCNTs decreases the diffusional mass transfer process (DMTP) and enhances the resistance of CoPc-MWCNTs oxidative decay. The introduction of linear alkylbenzene sulfonates (LAS) facilitates the heterolytic cleavage of O‒O bond of peroxide to generate cobalt(IV)–oxo species. Furthermore, pyridine functionalized MWCNTs were produced and axially coordinated on the cobalt phthalocyanine (CoPc), generating a catalyst with increased catalytic activity and stability [59]. The heterolytic cleavage of O‒O bond of hydrogen peroxide to produce cobalt(IV)–oxo without presence of any fifth ligand. The high-valent cobalt(IV)–oxo was analyzed by in situ ESI-MS and density functional theory. The DFT calculated bond length of Co‒O bond is 1.806 Å and unpaired electron spin populations are mainly on the oxygen. Cobalt(IV)–oxo 5 was generated at pH = 10, the catalytic cycle starts with the coordination of OOH− with CoII, and the heterolytic cleavage of O‒O occurs with the release of OH− (Figure 3a). Moreover, non-haem cobalt(III) oxamate anion could also be used to stabilize high-valent cobalt(IV)–oxo species [60]. The oxidation of industrial contaminants was reported [61] by immobilizing non haem cobalt(III) complex [CoIII(opbaX)]−(opbaX = 4-X-o-phenylenebis(oxamate) on pyridine-modified MWCNTs, where pyridine acts as a fifth ligand. Similarly, cobalt(III) complexes of [CoIII(opbaX)]‒(opbaX = 4-X-o-phenylenebis(oxamate), X = H, NO2, CH3) with different substituents was reported [62] for accelerating heterolytic cleavage of hydrogen peroxide to imitate the essential and general principle of natural enzymes without using any fifth ligand (Figure 3b). An ESI-MS and EPR trapping technique revealed the presence of cobalt(IV)–oxo reactive specie. The generation of cobalt–oxo species depends on the electronic environment of substituent. In Scheme 2 pathway (b) the electron rich cobalt complex favors the homolytical cleavage of the hydroperoxide O‒O bond while the electron deficiency favors the heterolytic cleavage with generation of (CoIV=O)10. The tendency of generation of (CoIV=O) was in order 8 > 7 > 9. Density functional theory also demonstrated that electron withdrawing group helps in pulling electron and lowering the corresponding energy levels. Keeping in mind the concept of the “oxo wall” [63], another pathway (a) also proposed, heterolytic cleavage of O‒O generate the ligand based radical intermediate OH–CoIII(opbaX)11, in which ligand transfers one electron to cobalt and cobalt transfers it to oxygen.
Our group recently [64] reported the catalytic oxidation of alkene using four cobalt(III) corroles of different electronic environment F0C-Co, F5C-Co, F10C-Co and F15C-Co employing various oxidants. The in situ ESI HR-MS analysis of styrene oxidation with KHSO5 predicts the presence of high-valent cobalt(V)–oxo complex as active intermediate. The in situ X-band CW EPR analysis revealed a signal at g = 2.0135 for the presence of cobalt–oxo specie.
3. Cobalt–Oxo Species Involved in Water Oxidation Reaction
Water oxidation is a process that involved the four-electron-four-proton oxidation of water to evolve O2. In natural photosynthesis, sunlight is converted to chemical energy by the oxidation of water [20]. As a consequence, understanding nature’s water oxidation mechanism in photosystem II has been the focus of research for the development of artificial water oxidation catalyst. The development of efficient water oxidation catalysts with minimal cost is a challenge [65,66,67,68]. Various water oxidation catalysts were developed to understand the O‒O bond formation event in natural photosynthesis to evolve oxygen. Cobalt is the most abundant and cheap earth metal. Cobalt oxide materials are among the most promising catalyst for water oxidation [69,70,71] and cobalt–oxo species are involved in the O‒O forming event of water oxidation.
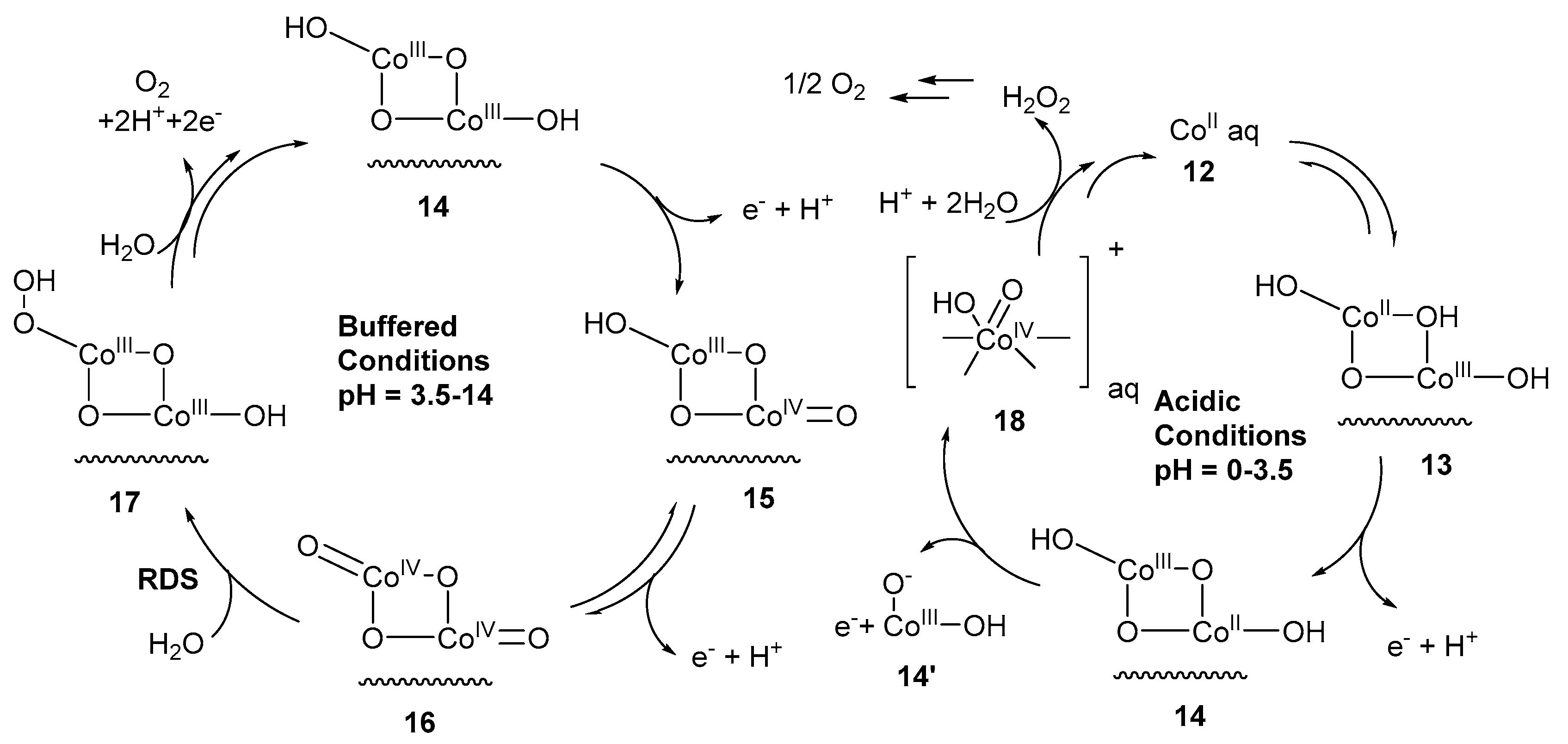
Frei et al. reported [16] the photocatalytic water oxidation using cobalt oxide. The water oxidation was carried out in the presence of photosensitizer [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) that creates a hole and S2SO8−2 as an electron acceptor. The FT–IR characterization revealed the involvement of two intermediates with absorption band at 1013 cm−1 and 840 cm−1. The band at 1013 cm−1 was assigned to Co(III)OO (fast site) group with a neighboring hydroxyl group. Incorporation of H218O shifts the peaks at 995 cm−1 and 966 cm−1. The shifting of frequency agrees well with the presence of superoxide moiety on a metal-oxide surface [72,73]. The superoxide surface intermediate causes the three-electron water oxidation. The band at 840 cm−1 was assigned to CoIV=O (slow site) surface species. No change in the spectrum was observed by the incorporation of H218O, ruling out the presence of any peroxide intermediate. From the mechanistic point of view, CoIV=O was generated by the oxidation of surface group Co(III)–OH. At the fast site catalytic species, catalytic turn over frequency is at least 10 times more than slow site catalytic species, because it has no neighbor hydroxyl group. Furthermore, Stahl et al. reported [74] the water oxidation employing cobalt oxide as an electrocatalyst, and proposed the involvement of (CoIV‒O) as reactive specie. The EPR analysis with signals at g-values 2.59, 2.17 and 1.99 revealed the presence of multiple paramagnetic species during water oxidation, possibly arising from (CoIV‒O) sites in the catalyst with a different coordination environment. The mechanism of water oxidation is pH dependent, at acidic pH homogeneous catalysis leading to H2O2 production, while at pH above 3.5 heterogeneous catalysis takes place, generating O2 from four-electron water oxidation (Scheme 3). The oxidation of 12 produced 14 (12→13→14) corresponds to a 3H+/e− process. Subsequently, 1e− oxidation generated specie 15 corresponding to 7H+/3e− process. Further, oxidation of 15 afforded 16. A key step to evolve oxygen is the nucleophilic attack of water at 16 to produce 17 [75,76,77]. Under the acidic pH, the PCET-mediated formation of 11 was prevented (Scheme 3). The oxidation of 10 produced 18 that dissolve from surface. The intermediate specie 18 invoked the homogeneous oxidation of water to H2O2 [78]. Similarly, bridging cobalt(IV)–oxo [79] and terminal cobalt(IV)–oxo radical [80] species were proposed as reactive catalytic sites for water oxidation, employing amorphous cobalt oxide. X-ray absorption near the edge provides the insight that the generation of high-valent (CoIV‒O) depended on the potential applied and pH. The edge position of the spectra was taken at pH = 7 and pH = 9 differs by about 1.0 eV by keeping potential constant at 0.95 V, and edge position of the spectra were taken at pH = 7 by increasing electrode potential from 0.95 to 1.34 V differs by about 1.2 eV [79]. However, the study of cobalt–oxo species involved in water oxidation was difficult because in oxygen evolving catalysis (OEC), large number of spectroscopically active backgrounds species are present which limits their detection and characterization.
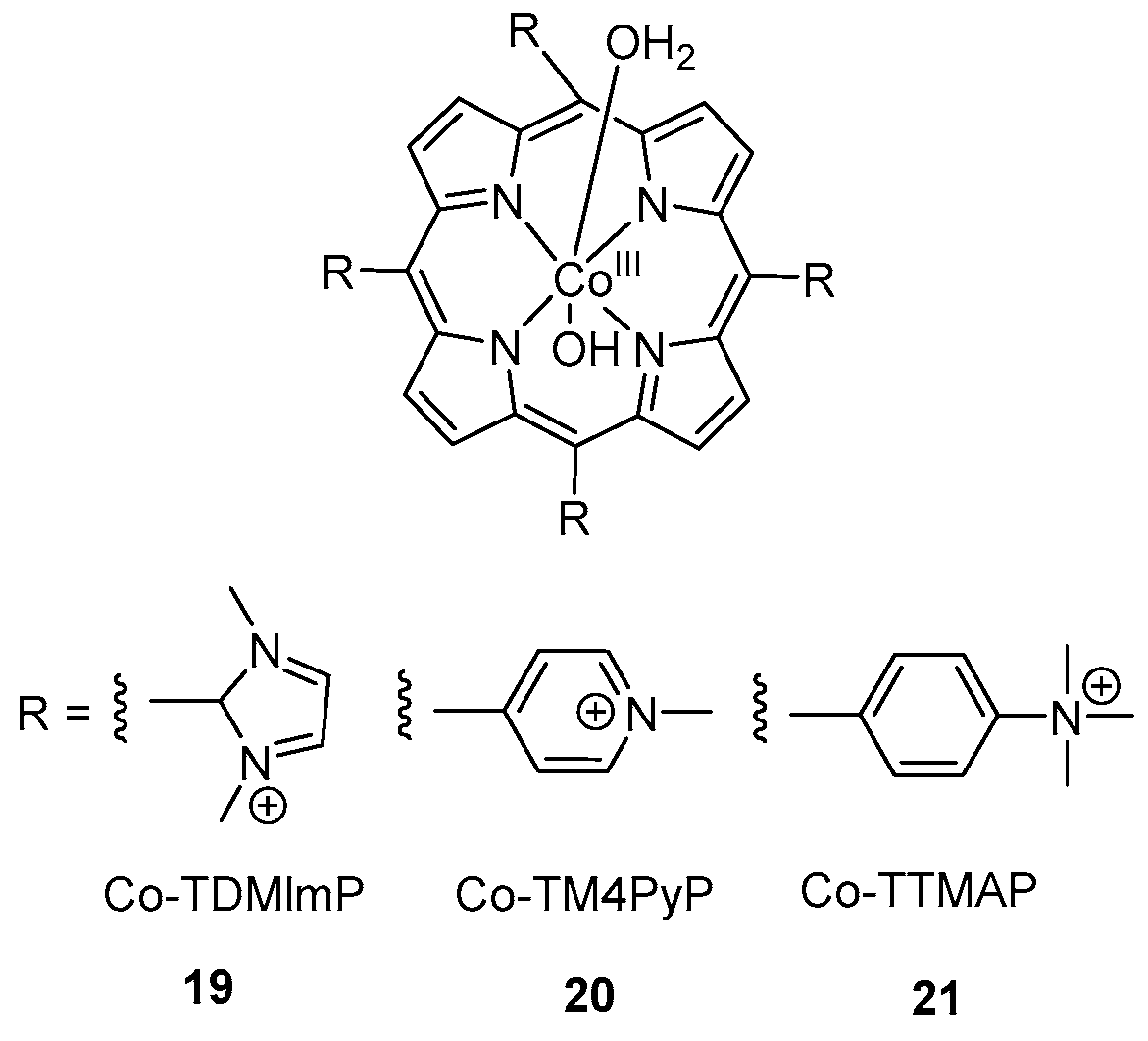
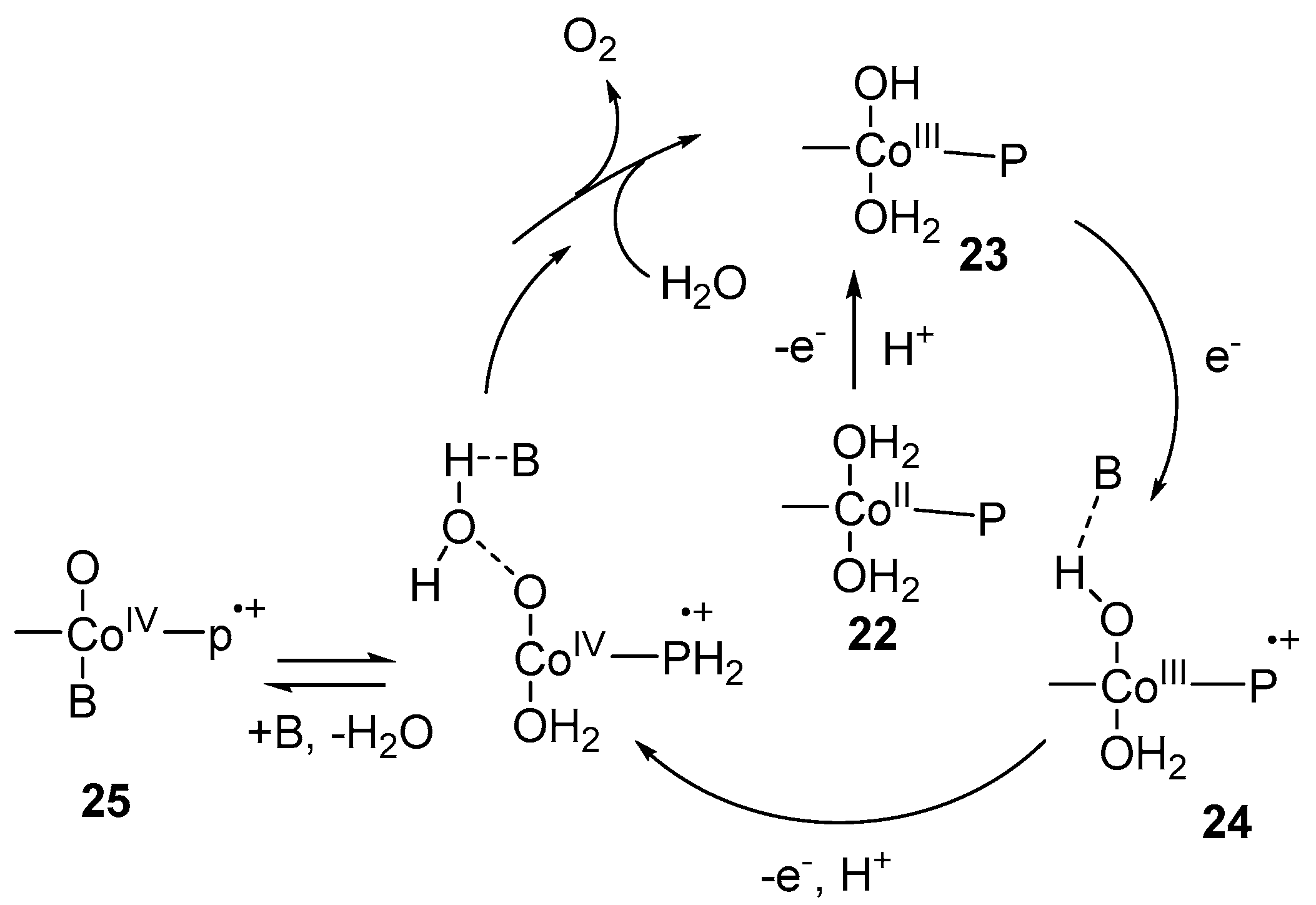
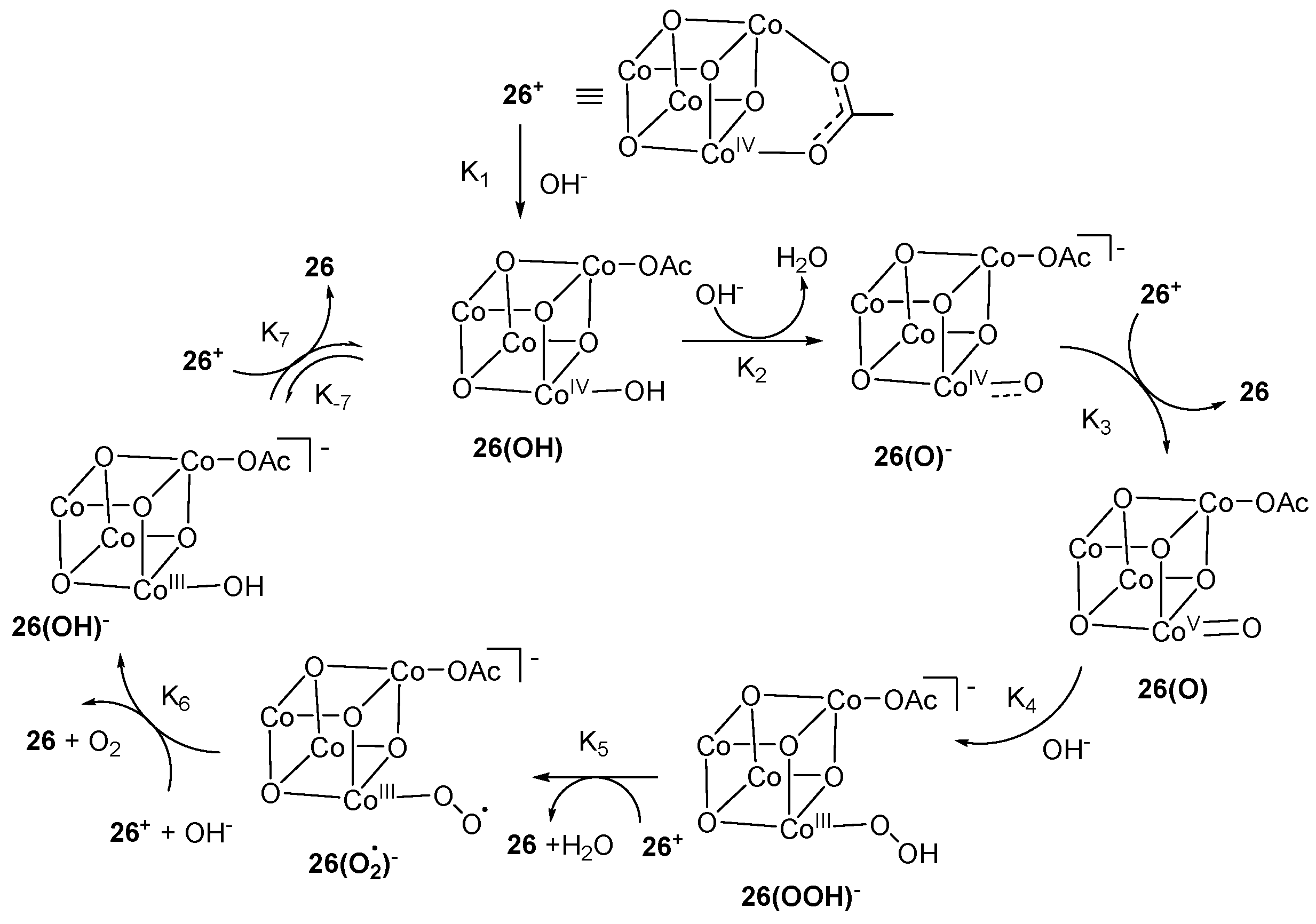
Consequently, suitable catalysts to reduce background active species are needed to design. N-based ligands have attractive properties to be used as homogenous molecular water oxidation catalysts [81]. Recently, a significant number of catalysts are developed based on single site and multinuclear transition metal including Mn, Fe, Co, Cu, Ru and Ir [82,83,84,85,86,87]. The biggest challenge is to find a suitable coordination environment because the metal–ligand bond opposite a metal–oxygen bond can be compromised at higher redox level leading the catalyst to be susceptible to degradation [88]. So, single site N-based ligand homogeneous catalysts of cobalt were developed utilizing stable pentadentate ligand environment of 2,6-(bis(bis-2-pyridyl)methoxy-methane)-pyridine [89] and 6-(bis(bis-2-pyridyl)-methoxymethane)pyridine [30] for water oxidation. The electrochemical studies revealed that over pH range 7.6–10.3 an oxidation event was observed at +1.43 V vs. NHE corresponding to [CoIV–OH]3+/[CoIII–OH]2+ with significant rise in current. This signal is not classified as PCET because E1/2 is static over this pH range. A pH dependent step was observed at pH > 10.3 corresponding to [CoIV=O]2+/[CoIII–OH]2+ which is consistent with PCET. High-valent [CoIV=O]2+ species evolves O2 by the nucleophilic attack of H2O [89,90]. An alternative pathway proposed that the attack of OH- at [CoIV–OH]3+ in the rate determining step will evolve O2 [30]. Likewise, [CoIV‒O] specie was proposed as reactive intermediate during water oxidation at basic pH using cobalt-porphyrins as catalyst [91]. Similarly, Groves and Wang reported [92] the single site homogeneous water oxidation catalyst, employing a series of cobalt porphyrins 19, 20 and 21 (Scheme 4). A high-valent CoIV-porphyrin cation radical acts as reactive intermediate. The electrochemical experiment provides the evidence for the formation of high-valent CoIV–O specie. The redox event at 250 mV vs. Ag/Cl reference represents the resting state of catalyst H2O–CoIII–OH 23. The observed anodic features at ~1 V demonstrates the oxidation of CoIII porphyrin to CoIII porphyrin radical cation (+P-CoIII–OH) 24. As first oxidation occurred before the onset potential of WOC catalytic current, so +P–CoIII–OH is not the reactive oxidant in this system. The second oxidation at 1320 mV generates a reactive high-valent CoIV–O porphyrin radical cation 25. The key step for O‒O bond formation is the nucleophilic addition of H2O to +P–CoIV–O 25 to form Co-hydroperoxo or peroxo which further oxidized to evolve O2 as shown in Scheme 5. Likewise, photo-induced generation of CoIV=O as active oxidant for the water oxidation was reported [93] based on a cobalt(II) complex of salophen ligand. Moreover, a high-valent CoIVO complex isoelectronic to CoVO was reported [29] to act as active specie to generate O2 based on a cobalt(III) complex of N-based ligand bTAML (bTAML = biuret-modified tetraamidomacrocyclic) ligands. The complex [Co(O)(bTAML)]1− cannot be characterized by spectroscopic techniques due to the non-innocent nature of the ligand except UV-vis spectra. The same specie was generated by the one electron oxidation using cerium ammonium nitrate in the presence of ZnCl2. The HR-MS analysis revealed the m/z = 497.026 corresponding to [CoIV(O)(Zn)(bTAML)(H+)]. Further, Nocera et al. reported [94] the dicobalt oxidized site Co(III)2Co(IV)2 during water oxidation using cobalt cubane modified by pyridine ligands that can stabilize tetracobalt core. This pyridine-modified cobalt cubane has molecular nature and termed as molecular cubane. Electrochemical investigation demonstrated two reversible oxidation events at E0(1) = 0.3 V and E0(2) = 1.25 V corresponding to Co(III)3(IV)/Co(III)4 and Co(III)2(IV)2/Co(III)3(IV). X-ray absorption spectroscopy also confirms the presence of Co(III)2(IV)2 specie. The adjacent terminal CoIV=O species in cubane provide a site for direct O–O bond formation by radical coupling to evolve O2. Likewise, the proton-coupled electron transfer generation of (CoIV‒O) was also reported [95] using molecular model cubane, [Co4O4(CO2Me2)2(bpy)4]. Furthermore, molecular cobalt cubane Co4O4(OAc)4py4 26 [96] and a series of modified molecular cobalt cubane with electron rich and electron poor groups [97] were reported to understand the nature of high-valent cobalt–oxo species involved in the water oxidation reaction. The electrochemical studies of 26 revealed the presence of only one fully redox couple from pH 4 to pH 10 at E1/2 = 1.25 V corresponding to Co(III)4/Co(III)3(IV) redox. The increase of pH to 12 produced a significant anode wave current and bubble formation, consistent with the oxidation of hydroxide to oxygen. No change in the current intensity was observed in the presence of EDTA, ruling out the possibility of heterogeneous water oxidation due to the presence of CoII oxide. The ESI-MS analysis by incorporating 97% enriched Na18OH observed the presence of 90% 36O2. No evidence for the exchange of 18O-oxygen between 26 revealing that only terminal oxo/hydroxide specie was involved in O‒O bond formation. The reaction of protonated 26+ and hydroxide showed the importance of the cobalt(IV) oxidation state in O2 formation. The generation of cobalt(V)=O 26(O) was proposed by PCET before the evaluation of O2 as shown in Scheme 6 [96]. The protonated 26+ reacts with hydroxide ion to produce 26(O)− which further oxidized to cobalt(V)=O 26(O). The specie 26(O) had acted as reactive intermediate to evolve O2. Involvement of high-valent cobalt(V)=O complex during water oxidation was also theoretically proposed [98,99,100,101]. Corroles are analogous of porphyrin which have one carbon less than porphyrin and can stabilize metals in a higher oxidation state. A high-valent CoV=O specie suggested [102] to act as reactive specie during water oxidation by using series of cobalt corroles with different axial ligand. Electrochemical study of cobalt corroles represent two reversible oxidation events at E1/2 = 0.75 and E1/2 = 1.32 V vs. NHE corresponding to CoIV/CoIII and CoV/CoIV redox couples, respectively. Nucleophilic attack of the water at Co‒O bond to generate Co-hydroperoxo specie is the key step to evolve O2. Cobalt corroles with electron-donating ligands are more reactive because it causes the Co‒O bond to be weaker and nucleophilic attack become easier.
4. Preparation of Cobalt–Oxo Complexes
The isolated preparation of cobalt–oxo complexes have two major problems (1). Ligands used to stabilize cobalt–oxo complexes are prone to oxidation (2). Electronic repulsion forces between the d-electron of cobalt and electron of the oxygen. Chemists are focusing on how to overcome these problems to prepare cobalt–oxo complexes.
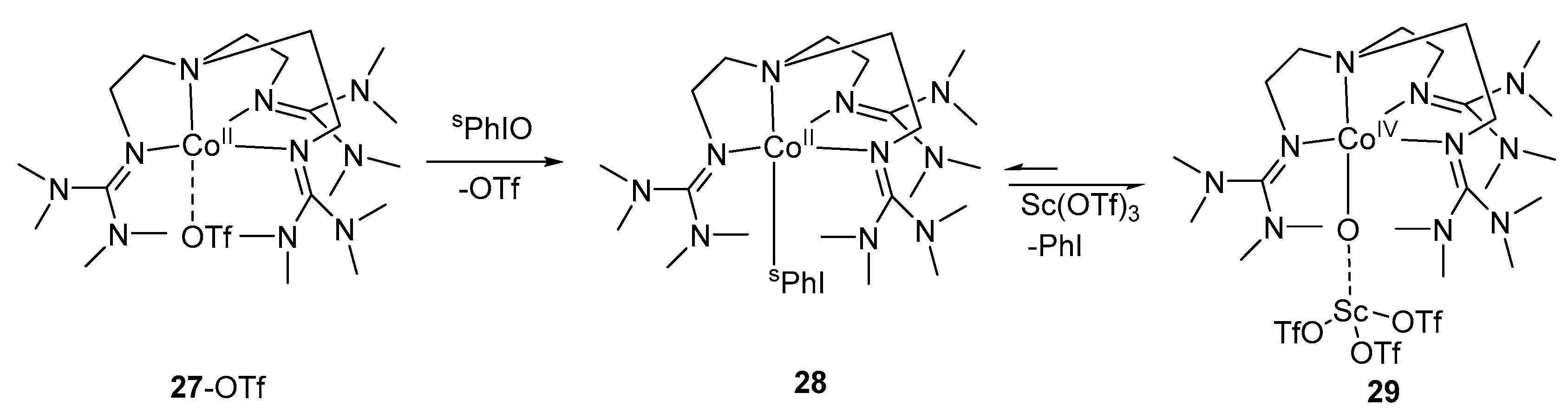
Ray et al. reported [103] the first preparation and isolation of terminal cobalt(IV)–oxo complex using the N-based tetradentate tripodal ligand TMG3tren (tris[2-(N-tetramethylguanidyl)ethyl]amine). The {Co-O} unit was stabilized by the Lewis acid interaction with Sc+3 ion, generating {Co-O-Sc}+3 unit. The complex 29 was obtained by two electron oxidation of 27-OTf in the presence of Sc(OTf)3 (Scheme 7). The complex 29 was characterized by electrospray mass spectrum, EPR and X-ray absorption spectroscopy, and was reactive towards oxidation of triphenylphosphine and dihydroanthracene. The same group two years later reported [104] the square pyramidal cobalt(IV)–oxo with enhanced stability based on the tetraamido macrocyclic ligand (TMAL). The electrochemical study of 30 gave a reversible oxidation peak at 1.00 V vs. a saturated colomel electrode. This reversible oxidation peak suggests that CoIV state is thermally and kinetically accessible. The one electron oxidation of 30 in the presence of cerium ammonium nitrate (CAN) afforded a blue-colored complex 31-Ce with a half-life of 20 min. This blue complex can also be obtained by the oxidation of 30 with PhIO in the presence other redox-inactive metals like Sc+3, Y+3 and Zn+2 (Scheme 8). The complex 31-M was characterized by cold-spray ionization time-of-flight mass spectrometry (CSI-TOF MS), X-band EPR spectrum, and X-ray absorption spectroscopy. All attempts to obtain resonance Raman spectrum have failed. The 31-Sc complex demonstrated high reactivity in the hydrogen abstraction reaction and oxygen atom transfer reactions. The first fully spectroscopically characterized high-valent Cobalt(IV)–oxo complex 33 was generated [105] by the two electron oxidation of a cobalt complex of 13-TMC (2 mM) 32 by PhIO (3 equiv.) following conventional method in the presence of triflic acid (CF3SO3H, HOTf; 1.2 equiv.) in acetone (Scheme 9). The transient complex had a half-life of 3 h and was characterized by CSI-TOF MS, EPR and X-ray absorption spectroscopy. Resonance Raman spectroscopy considered as authentic technique to confirm the presence of metal–oxo complex [106,107]. The resonance Raman spectrum of 33 showed a band at 770 cm−1 which shifts to 736 cm−1 upon 18O-labelling of 33. Recently, preparation of CoIII≡O complex was reported [108] by using tris-(imidazol-2-ylidene)borate ligand PhB(tBuIm)3−. This complex was characterized by infrared (IR) and X-ray diffraction (XRD) spectroscopy. The length of Co‒O bond determined by XRD was 1.68 Å. DFT calculations revealed two Co‒O π* interactions with highest lying dxz and dyz orbitals. These orbitals support the presence of two π-bonds. This complex was thermodynamically unstable with half-life of 8 h.
5. Summary and Outlook
High-valent cobalt–oxo complexes are implicated as key intermediates in many of the oxidative transformation reactions and the water oxidation process. Identification of cobalt–oxo species in water-splitting reactions have been extensively studied. However, the transient nature of cobalt–oxo complexes limits their characterizations to in situ EPR, XAS and mass spectroscopy. Although different strategies, such as using ligands with different electronic environments or MWCNT supports, have been adopted to stabilize cobalt–oxo complexes, until now only one example of Raman characterization for cobalt (IV)=O complex using 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclotridecane ligand has been available. The isolation and identification of high valent cobalt–oxo species remains a great challenge. The design of a suitable N-based ligand which can stabilize coordinated cobalt atom in high oxidation might be the key step for the preparation of higher valent cobalt–oxo complexes, which will allow the full characterization and “slow motion picture” study of the factors controlling its reactivity.
Author Contributions
H.-Y.L. coordinated the whole work and provided technical guidance. A.A. and W.A. collected the references and wrote the paper.
Funding
This work was funded by the National Natural Science Foundation of China (NNSFC) under Grant (21671068).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| TPFPP | Meso-tetrakis(pentafluorophenyl)porphinato dianion |
| CoTAPc | cobalt tetraaminophthalocyanine |
| EPTAC | 2,3-epoxypropyl triethylammonium chloride |
| Pc | Phthalocyanine |
| TAPc | Tetraaminophthalocyanine |
| MWNCTs | Multiwall carbon nanotubes |
| DMPO | 5,5-Dimethyl-1-pyrroline N-oxide |
| F0C-Co | Co(III) complex of 5,10,15-triphenylcorrole |
| F5C-Co | Co(III) complex of 5,15-bis(phenyl)-10-(pentafluorophenyl)corrole |
| F10C-Co | Co(III) complex of 5,15-bis(pentafluorophenyl)-10-phenylcorrole |
| F15C-Co | Co(III) complex of 5,10,15-tris(pentafluorophenyl)corrole |
| TMG3tren | (tris[2-(N-tetramethylguanidyl)ethyl]amine) |
| sPhIO | 2-(tert-butylsulfonyl)iodosylbenzene |
| TMAL | Tetraamido macrocyclic ligand |
| 13-TMC | 1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclotridecane |
| Co-TDMImP | Co(III) complex of 5,10,15,20-tetrakis(1,3-dimethylimidazolium-2-yl)porphyrin |
| Co-TM4PyP | Co(III) complex of 5,10,15,20-tetrakis(N-methylpyridinium-4-yl)porphyrin |
| Co-TTMAP | Co(III) complex of 5,10,15,20-tetrakis(N,N,N-trimethylanilinium-4-yl)porphyrin |
References
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Van Eldik, R. Fascinating inorganic/bioinorganic reaction mechanisms. Coord. Chem. Rev. 2007, 251, 1649–1662. [Google Scholar] [CrossRef]
- Costas, M.; Mehn, M.P.; Jensen, M.P.; Que, L. Dioxygen activation at mononuclear nonheme iron active sites: Enzymes, models, and intermediates. Chem. Rev. 2004, 104, 939–986. [Google Scholar] [CrossRef] [PubMed]
- Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Mullins, C.S.; Pecoraro, V.L. Reflections on small molecule manganese models that seek to mimic photosynthetic water oxidation chemistry. Coord. Chem. Rev. 2008, 252, 416–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEvoy, J.P.; Brudvig, G.W. Water-splitting chemistry of photosystem II. Chem. Rev. 2006, 106, 4455–4483. [Google Scholar] [CrossRef]
- Rebelo, L.S.; Silva, M.A.; Medforth, J.C.; Freire, C. Iron(III) fluorinated porphyrins: Greener chemistry from synthesis to oxidative catalysis reactions. Molecules 2016, 21. [Google Scholar] [CrossRef]
- Pereira, F.C.; Simões, M.M.; Tomé, P.J.; Almeida Paz, A.F. Porphyrin-based metal-organic frameworks as heterogeneous catalysts in oxidation reactions. Molecules 2016, 21. [Google Scholar] [CrossRef]
- de Visser, S.P.; Rohde, J.-U.; Lee, Y.-M.; Cho, J.; Nam, W. Intrinsic properties and reactivities of mononuclear nonheme iron–oxygen complexes bearing the tetramethylcyclam ligand. Coord. Chem. Rev. 2013, 25, 381–393. [Google Scholar] [CrossRef]
- Krebs, C.; Galonić Fujimori, D.; Walsh, C.T.; Bollinger, J.M. Non-heme Fe(IV)-oxo intermediates. Acc. Chem. Res. 2007, 40, 484–492. [Google Scholar] [CrossRef]
- Abu-Omar, M.M.; Loaiza, A.; Hontzeas, N. Reaction mechanisms of mononuclear non-heme iron oxygenases. Chem. Rev. 2005, 105, 2227–2252. [Google Scholar] [CrossRef] [PubMed]
- Baglia, R.A.; Zaragoza, J.P.T.; Goldberg, D.P. Biomimetic reactivity of oxygen-derived manganese and iron porphyrinoid complexes. Chem. Rev. 2017, 117, 13320–13352. [Google Scholar] [CrossRef] [PubMed]
- Fukuzumi, S.; Mandal, S.; Mase, K.; Ohkubo, K.; Park, H.; Benet-Buchholz, J.; Nam, W.; Llobet, A. Catalytic four-electron reduction of O2 via rate-determining proton-coupled electron transfer to a dinuclear cobalt-μ-1,2-peroxo complex. J. Am. Chem. Soc. 2012, 134, 9906–9909. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, F.F.; Heims, F.; Kundu, S.; Mebs, S.; Ray, K. Spectroscopic capture and reactivity of S = 1/2 nickel(III)-oxygen intermediates in the reaction of a Ni(II)-salt with mCPBA. Chem. Commun. 2012, 48, 3730–3732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; de Respinis, M.; Frei, H. Time-resolved observations of water oxidation intermediates on a cobalt oxide nanoparticle catalyst. Nat. Chem. 2014, 6, 362–367. [Google Scholar] [CrossRef]
- Pierpont, A.W.; Cundari, T.R. Computational study of methane C-H activation by first-row late transition metal L(n)M=E (M: Fe, Co, Ni) complexes. Inorg. Chem. 2010, 49, 2038–2046. [Google Scholar] [CrossRef]
- Limberg, C. Was ist wirklich nötig, um Komplexe später Übergangsmetalle mit terminalen oxo-liganden zu stabilisieren? Angew. Chem. 2009, 121, 2305–2308. [Google Scholar] [CrossRef]
- Cox, N.; Pantazis Dimitrios, A.; Neese, F.; Lubitz, W. Artificial photosynthesis: Understanding water splitting in nature. Interface Focus 2015, 5, 20150009. [Google Scholar] [CrossRef]
- Yamada, T.; Domen, K. Development of sunlight driven water splitting devices towards future artificial photosynthetic industry. ChemEngineering 2018, 2, 36. [Google Scholar] [CrossRef]
- Young, K.J.; Brennan, B.J.; Tagore, R.; Brudvig, G.W. Photosynthetic water oxidation: Insights from manganese model chemistry. Acc. Chem. Res. 2015, 48, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Tan, J.M.; Besson, C.; Geletii, Y.V.; Musaev, D.G.; Kuznetsov, A.E.; Luo, Z.; Hardcastle, K.I.; Hill, C.L. A fast soluble carbon-free molecular water oxidation catalyst based on abundant metals. Science 2010, 328, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Bozoglian, F.; Mandal, S.; Stewart, B.; Privalov, T.; Llobet, A.; Sun, L. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 2012, 4, 418. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.D.L.; Prévot, M.S.; Fagan, R.D.; Zhang, Z.; Sedach, P.A.; Siu, M.K.J.; Trudel, S.; Berlinguette, C.P. Photochemical route for accessing amorphous metal oxide materials for water oxidation catalysis. Science 2013, 340, 60–63. [Google Scholar] [CrossRef]
- Kanan, M.W.; Nocera, D.G. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Frei, H. Nanostructured cobalt oxide clusters in mesoporous silica as efficient oxygen-evolving catalysts. Angew. Chem. Int. Ed. 2009, 48, 1841–1844. [Google Scholar] [CrossRef]
- Esswein, A.J.; McMurdo, M.J.; Ross, P.N.; Bell, A.T.; Tilley, T.D. Size-dependent activity of Co3O4 nanoparticle anodes for alkaline water electrolysis. J. Phys. Chem. C 2009, 113, 15068–15072. [Google Scholar] [CrossRef]
- Hans Wedepohl, K. The composition of the continental crust. Geochim. Cosmochim. Acta 1995, 59, 1217–1232. [Google Scholar] [CrossRef]
- Das, D.; Pattanayak, S.; Singh, K.K.; Garai, B.; Sen Gupta, S. Electrocatalytic water oxidation by a molecular cobalt complex through a high valent cobalt oxo intermediate. Chem. Commun. 2016, 52, 11787–11790. [Google Scholar] [CrossRef] [PubMed]
- Wasylenko, D.J.; Palmer, R.D.; Schott, E.; Berlinguette, C.P. Interrogation of electrocatalytic water oxidation mediated by a cobalt complex. Chem. Commun. 2012, 48, 2107–2109. [Google Scholar] [CrossRef]
- Chen, Y.; Gu, Y.; Li, N.; Lu, W.; Chen, W. Efficient oxidative removal of organic pollutants by ordered mesoporous carbon-supported cobalt phthalocyanine. J. Nanomater. 2016, 2016, 27. [Google Scholar] [CrossRef]
- Li, N.; Lu, W.; Pei, K.; Yao, Y.; Chen, W. Ordered-mesoporous-carbon-bonded cobalt phthalocyanine: A bioinspired catalytic system for controllable hydrogen peroxide activation. ACS Appl. Mater. Interfaces 2014, 6, 5869–5876. [Google Scholar] [CrossRef] [PubMed]
- McAlpin, J.G.; Surendranath, Y.; Dincǎ, M.; Stich, T.A.; Stoian, S.A.; Casey, W.H.; Nocera, D.G.; Britt, R.D. EPR evidence for Co(IV) species produced during water oxidation at neutral pH. J. Am. Chem. Soc. 2010, 132, 6882–6883. [Google Scholar] [CrossRef] [PubMed]
- Hadt, R.G.; Hayes, D.; Brodsky, C.N.; Ullman, A.M.; Casa, D.M.; Upton, M.H.; Nocera, D.G.; Chen, L.X. X-ray spectroscopic characterization of Co(IV) and metal–metal interactions in Co4O4: Electronic structure contributions to the formation of high-valent states relevant to the oxygen evolution reaction. J. Am. Chem. Soc. 2016, 138, 11017–11030. [Google Scholar] [CrossRef] [PubMed]
- Retegan, M.; Krewald, V.; Mamedov, F.; Neese, F.; Lubitz, W.; Cox, N.; Pantazis, D.A. A five-coordinate Mn(IV) intermediate in biological water oxidation: Spectroscopic signature and a pivot mechanism for water binding. Chem. Sci. 2016, 7, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Singh, K.K.; Panda, C.; Weitz, A.; Hendrich, M.P.; Collins, T.J.; Dhar, B.B.; Sen Gupta, S. Formation of a room temperature stable FeV(O) complex: Reactivity toward unactivated C–H bonds. J. Am. Chem. Soc. 2014, 136, 9524–9527. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.J. TAML oxidant activators: A new approach to the activation of hydrogen peroxide for environmentally significant problems. Acc. Chem. Res. 2002, 35, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Que Jr, L.; Tolman, W.B. Biologically inspired oxidation catalysis. Nature 2008, 455, 333–340. [Google Scholar]
- Nam, W.; Yang, S.J.; Kim, H. Catalytic oxygenation of alkenes and alkanes by oxygen donors catalyzed by cobalt-substituted polyoxotungstate. Bull. Korean Chem. Soc. 1996, 17, 625–630. [Google Scholar] [CrossRef]
- Tang, H.; Shen, C.; Lin, M.; Sen, A. Cobalt porphyrin-catalyzed alkane oxidations using dioxygen as oxidant. Inorg. Chim. Acta 2000, 300–302, 1109–1111. [Google Scholar] [CrossRef]
- Sun, C.; Hu, B.; Liu, Z. Rapid aerobic oxidation of alcohols to carbonyl compounds with dioxygen using metallodeuteroporphyrin dimethyl esters as catalysts in the presence of isobutylaldehyde. Heteroat. Chem 2012, 23, 295–303. [Google Scholar] [CrossRef]
- Nguyen, A.I.; Hadt, R.G.; Solomon, E.I.; Tilley, T.D. Efficient C–H bond activations via O2 cleavage by a dianionic cobalt(II) complex. Chem. Sci. 2014, 5, 2874–2878. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.-Y.; Tian, P.; Sun, F.A.; He, M.-Y.; Chen, Q. Highly efficient transformation of alcohol to carbonyl compounds under a hybrid bifunctional catalyst originated from metalloporphyrins and hydrotalcite. J. Catal. 2016, 335, 105–116. [Google Scholar] [CrossRef]
- Zhou, X.; Ji, H. Cobalt porphyrin immobilized on montmorillonite: A highly efficient and reusable catalyst for aerobic oxidation of alcohols to carbonyl compounds. Chin. J. Catal. 2012, 33, 1906–1912. [Google Scholar] [CrossRef]
- Nam, W.; Kim, I.; Kim, Y.; Kim, C. Biomimetic alkane hydroxylation by cobalt(III) porphyrin complex and m-chloroperbenzoic acid. Chem. Commun. 2001, 0, 1262–1263. [Google Scholar] [CrossRef]
- Song, Y.J.; Hyun, M.Y.; Lee, J.H.; Lee, H.G.; Kim, J.H.; Jang, S.P.; Noh, J.Y.; Kim, Y.; Kim, S.-J.; Lee, S.J.; et al. Amide-based nonheme cobalt(III) olefin epoxidation catalyst: Partition of multiple active oxidants CoV=O, CoIV=O, and CoIII‒OO(O)CR. Chem. A Eur. J. 2012, 18, 6094–6101. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, O.V.; Kopylovich, M.N.; Nesterov, D.S. Stereoselective oxidation of alkanes with m-CPBA as an oxidant and cobalt complex with isoindole-based ligands as catalysts. RSC Adv. 2016, 6, 93756–93767. [Google Scholar] [CrossRef]
- Nurdin, L.; Spasyuk, D.M.; Fairburn, L.; Piers, W.E.; Maron, L. Oxygen–oxygen bond cleavage and formation in Co(II)-mediated stoichiometric O2 reduction via the potential intermediacy of a Co(IV) oxyl radical. J. Am. Chem. Soc. 2018, 140, 16094–16105. [Google Scholar] [CrossRef]
- Ghosh, A.; Mitchell, D.A.; Chanda, A.; Ryabov, A.D.; Popescu, D.L.; Upham, E.C.; Collins, G.J.; Collins, T.J. Catalase–peroxidase activity of iron(III)−TAML activators of hydrogen peroxide. J. Am. Chem. Soc. 2008, 130, 15116–15126. [Google Scholar] [CrossRef]
- Zhang, R.; Horner, J.H.; Newcomb, M. Laser flash photolysis generation and kinetic studies of porphyrin-manganese-oxo intermediates. Rate constants for oxidations effected by porphyrin-MnV-Oxo species and apparent disproportionation equilibrium constants for porphyrin-MnIV-oxo species. J. Am. Chem. Soc. 2005, 127, 6573–6582. [Google Scholar] [CrossRef]
- Zhang, Z.; Hao, J.; Yang, W.; Lu, B.; Ke, X.; Zhang, B.; Tang, J. Porous Co3O4 nanorods-reduced graphene oxide with intrinsic peroxidase-like activity and catalysis in the degradation of methylene blue. ACS Appl. Mater. Interfaces 2013, 5, 3809–3815. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, J.; Stern, C.L.; Mirkin, C.A. A coordination chemistry approach to a multieffector enzyme mimic. J. Am. Chem. Soc. 2007, 129, 10074–10075. [Google Scholar] [CrossRef] [PubMed]
- Wulff, G. Enzyme-like catalysis by molecularly imprinted polymers. Chem. Rev. 2002, 102, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lu, W.; Pei, K.; Yao, Y.; Chen, W. Formation of high-valent cobalt-oxo phthalocyanine species in a cellulose matrix for eliminating organic pollutants. Appl. Catal. B 2015, 163, 105–112. [Google Scholar] [CrossRef]
- Afanasiev, P.; Kudrik, E.V.; Albrieux, F.; Briois, V.; Koifman, O.I.; Sorokin, A.B. Generation and characterization of high-valent iron oxo phthalocyanines. Chem. Commun. 2012, 48, 6088–6090. [Google Scholar] [CrossRef] [PubMed]
- Ramdhanie, B.; Telser, J.; Caneschi, A.; Zakharov, L.N.; Rheingold, A.L.; Goldberg, D.P. An example of O2 binding in a Cobalt(II) corrole system and high-valent cobalt–cyano and cobalt–alkynyl complexes. J. Am. Chem. Soc. 2004, 126, 2515–2525. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Yao, Y.; Lu, J.; Chen, C.; Lu, W.; Huang, S.; Chen, W. The consortium of heterogeneous cobalt phthalocyanine catalyst and bicarbonate ion as a novel platform for contaminants elimination based on peroxymonosulfate activation. J. Hazard. Mater. 2016, 301, 214–221. [Google Scholar] [CrossRef]
- Li, N.; Lu, W.; Pei, K.; Chen, W. Interfacial peroxidase-like catalytic activity of surface-immobilized cobalt phthalocyanine on multiwall carbon nanotubes. RSC Adv. 2015, 5, 9374–9380. [Google Scholar] [CrossRef]
- Li, N.; Wang, Y.; Wu, C.; Lu, W.; Pei, K.; Chen, W. Bioinspired catalytic generation of high-valent cobalt–oxo species by the axially coordinated CoPc on pyridine-functionalized MWCNTs for the elimination of organic contaminants. Appl. Surf. Sci. 2018, 434, 1112–1121. [Google Scholar] [CrossRef]
- Fernández, I.; Pedro, J.R.; Roselló, A.L.; Ruiz, R.; Castro, I.; Ottenwaelder, X.; Journaux, Y. Alcohol oxidation by dioxygen and aldehydes catalysed by square-planar cobalt(III) complexes of disubstituted oxamides and related ligands. Eur. J. Org. Chem. 2001, 2001, 1235–1247. [Google Scholar]
- Li, N.; Zheng, Y.; Jiang, X.; Zhang, R.; Pei, K.; Chen, W. Carbon-based oxamate cobalt(III) complexes as bioenzyme mimics for contaminant elimination in high backgrounds of complicated constituents. Materials 2017, 10, 1169. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zheng, Y.; Jiang, X.; Zhang, R.; Chen, W. Generation of reactive cobalt oxo oxamate radical species for biomimetic oxidation of contaminants. RSC Adv. 2017, 7, 42875–42883. [Google Scholar] [CrossRef] [Green Version]
- Winkler, J.R.; Gray, H.B. Electronic Structures of Oxo-Metal Ions. In Molecular Electronic Structures of Transition Metal Complexes I; Mingos, D.M.P., Day, P., Dahl, J.P., Eds.; Springer Berlin Heidelberg: Berlin/Heidelberg,Germany, 2012; pp. 17–28. [Google Scholar]
- Huang, L.-T.; Ali, A.; Wang, H.-H.; Cheng, F.; Liu, H.-Y. Catalytic oxidation of alkene by cobalt corroles. J. Mol. Catal. A: Chem. 2017, 426, 213–222. [Google Scholar] [CrossRef]
- Betley, T.A.; Wu, Q.; Van Voorhis, T.; Nocera, D.G. Electronic design criteria for O–O bond formation via metal–oxo complexes. Inorg. Chem. 2008, 47, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Sala, X.; Romero, I.; Rodríguez, M.; Escriche, L.; Llobet, A. Molecular catalysts that oxidize water to dioxygen. Angew. Chem. Int. Ed. 2009, 48, 2842–2852. [Google Scholar] [CrossRef]
- Romain, S.; Vigara, L.; Llobet, A. Oxygen–oxygen bond formation pathways promoted by ruthenium complexes. Acc. Chem. Res. 2009, 42, 1944–1953. [Google Scholar] [CrossRef] [PubMed]
- Cape, J.L.; Hurst, J.K. Detection and mechanistic relevance of transient ligand radicals formed during [Ru(bpy)2(OH2)]2O4+-catalyzed water oxidation. J. Am. Chem. Soc. 2008, 130, 827–829. [Google Scholar] [CrossRef]
- Niishiro, R.; Takano, Y.; Jia, Q.; Yamaguchi, M.; Iwase, A.; Kuang, Y.; Minegishi, T.; Yamada, T.; Domen, K.; Kudo, A. A CoOx-modified SnNb2O6 photoelectrode for highly efficient oxygen evolution from water. Chem. Commun. 2017, 53, 629–632. [Google Scholar] [CrossRef]
- Zhong, M.; Hisatomi, T.; Kuang, Y.; Zhao, J.; Liu, M.; Iwase, A.; Jia, Q.; Nishiyama, H.; Minegishi, T.; Nakabayashi, M.; et al. Surface modification of CoOx loaded BiVO4 photoanodes with ultrathin p-type NiO layers for improved solar water oxidation. J. Am. Chem. Soc. 2015, 137, 5053–5060. [Google Scholar] [CrossRef]
- Hisatomi, T.; Katayama, C.; Moriya, Y.; Minegishi, T.; Katayama, M.; Nishiyama, H.; Yamada, T.; Domen, K. Photocatalytic oxygen evolution using BaNbO2N modified with cobalt oxide under photoexcitation up to 740 nm. Energy Environ. Sci. 2013, 6, 3595–3599. [Google Scholar] [CrossRef]
- Zecchina, A.; Spoto, G.; Coluccia, S. Surface dioxygen adducts on MgO-CoO solid solutions: Analogy with cobalt-based homogeneous oxygen carriers. J. Mol. Catal. 1982, 14, 351–355. [Google Scholar] [CrossRef]
- Barraclough, C.G.; Lawrance, G.A.; Lay, P.A. Characterization of binuclear. mu.-peroxo and. mu.-superoxo cobalt(III) amine complexes from Raman spectroscopy. Inorg. Chem. 1978, 17, 3317–3322. [Google Scholar] [CrossRef]
- Gerken, J.B.; McAlpin, J.G.; Chen, J.Y.C.; Rigsby, M.L.; Casey, W.H.; Britt, R.D.; Stahl, S.S. Electrochemical water oxidation with cobalt-based electrocatalysts from pH 0–14: The thermodynamic basis for catalyst structure, stability, and activity. J. Am. Chem. Soc. 2011, 133, 14431–14442. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Fukuda, K.; Osada, M.; Nakai, I.; Izumi, F.; Dilanian, R.A.; Kato, K.; Takata, M.; Sakurai, H.; Takayama-Muromachi, E.; et al. Chemical composition and crystal structure of superconducting sodium cobalt oxide bilayer-hydrate. J. Mater. Chem. 2004, 14, 1448–1453. [Google Scholar] [CrossRef]
- Amatucci, G.G.; Tarascon, J.M.; Klein, L.C. CoO2, the end member of the Li x CoO2 solid solution. J. Electrochem. Soc. 1996, 143, 1114–1123. [Google Scholar] [CrossRef]
- Motohashi, T.; Katsumata, Y.; Ono, T.; Kanno, R.; Karppinen, M.; Yamauchi, H. Synthesis and properties of CoO2, the x = 0 end member of the LixCoO2 and NaxCoO2 systems. Chem. Mater. 2007, 19, 5063–5066. [Google Scholar] [CrossRef]
- Brunschwig, B.S.; Chou, M.H.; Creutz, C.; Ghosh, P.; Sutin, N. Mechanisms of water oxidation to oxygen: Cobalt(IV) as an intermediate in the aquocobalt(II)-catalyzed reaction. J. Am. Chem. Soc. 1983, 105, 4832–4833. [Google Scholar] [CrossRef]
- Risch, M.; Ringleb, F.; Kohlhoff, M.; Bogdanoff, P.; Chernev, P.; Zaharieva, I.; Dau, H. Water oxidation by amorphous cobalt-based oxides: In situ tracking of redox transitions and mode of catalysis. Energy Environ. Sci. 2015, 8, 661–674. [Google Scholar] [CrossRef]
- Koroidov, S.; Anderlund, M.F.; Styring, S.; Thapper, A.; Messinger, J. First turnover analysis of water-oxidation catalyzed by Co-oxide nanoparticles. Energy Environ. Sci. 2015, 8, 2492–2503. [Google Scholar] [CrossRef] [Green Version]
- Wasylenko, D.J.; Palmer, R.D.; Berlinguette, C.P. Homogeneous water oxidation catalysts containing a single metal site. Chem. Commun. 2013, 49, 218–227. [Google Scholar] [CrossRef]
- Fillol, J.L.; Codolà, Z.; Garcia-Bosch, I.; Gómez, L.; Pla, J.J.; Costas, M. Efficient water oxidation catalysts based on readily available iron coordination complexes. Nat. Chem. 2011, 3, 807. [Google Scholar] [CrossRef] [PubMed]
- Ellis, W.C.; McDaniel, N.D.; Bernhard, S.; Collins, T.J. Fast water oxidation using iron. J. Am. Chem. Soc. 2010, 132, 10990–10991. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, J.J.; Jurss, J.W.; Brennaman, M.K.; Hoertz, P.G.; Patrocinio, A.O.T.; Murakami Iha, N.Y.; Templeton, J.L.; Meyer, T.J. Making oxygen with ruthenium complexes. Acc. Chem. Res. 2009, 42, 1954–1965. [Google Scholar] [CrossRef] [PubMed]
- Hetterscheid, D.G.H.; Reek, J.N.H. Mononuclear water oxidation catalysts. Angew. Chem. Int. Ed. 2012, 51, 9740–9747. [Google Scholar] [CrossRef] [PubMed]
- Sartorel, A.; Bonchio, M.; Campagna, S.; Scandola, F. Tetrametallic molecular catalysts for photochemical water oxidation. Chem. Soc. Rev. 2013, 42, 2262–2280. [Google Scholar] [CrossRef] [PubMed]
- Codolà, Z.; Garcia-Bosch, I.; Acuña-Parés, F.; Prat, I.; Luis, J.M.; Costas, M.; Lloret-Fillol, J. Electronic effects on single-site iron catalysts for water oxidation. Chem. A Eur. J. 2013, 19, 8042–8047. [Google Scholar] [CrossRef] [PubMed]
- Wasylenko, D.J.; Ganesamoorthy, C.; Koivisto, B.D.; Henderson, M.A.; Berlinguette, C.P. Insight into water oxidation by mononuclear polypyridyl Ru catalysts. Inorg. Chem. 2010, 49, 2202–2209. [Google Scholar] [CrossRef] [PubMed]
- Wasylenko, D.J.; Ganesamoorthy, C.; Borau-Garcia, J.; Berlinguette, C.P. Electrochemical evidence for catalyticwater oxidation mediated by a high-valent cobalt complex. Chem. Commun. 2011, 47, 4249–4251. [Google Scholar] [CrossRef]
- Crandell, D.W.; Ghosh, S.; Berlinguette, C.P.; Baik, M.H. How a [Co(IV) a bond and a half O](2+) fragment oxidizes water: Involvement of a biradicaloid [Co(II)-(⋅O⋅)](2+) species in forming the O–O bond. ChemSusChem 2015, 8, 844–852. [Google Scholar] [CrossRef]
- Nakazono, T.; Parent, A.R.; Sakai, K. Cobalt porphyrins as homogeneous catalysts for water oxidation. Chem. Commun. 2013, 49, 6325–6327. [Google Scholar] [CrossRef]
- Wang, D.; Groves, J.T. Efficient water oxidation catalyzed by homogeneous cationic cobalt porphyrins with critical roles for the buffer base. Proc. Natl. Acad. Sci. USA 2013, 110, 15579–15584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzolato, E.; Natali, M.; Posocco, B.; Montellano López, A.; Bazzan, I.; Di Valentin, M.; Galloni, P.; Conte, V.; Bonchio, M.; Scandola, F.; Sartorel, A. Light driven water oxidation by a single site cobalt salophen catalyst. Chem. Commun. 2013, 49, 9941–9943. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, C.N.; Hadt, R.G.; Hayes, D.; Reinhart, B.J.; Li, N.; Chen, L.X.; Nocera, D.G. In situ characterization of cofacial Co(IV) centers in Co4O4 cubane: Modeling the high-valent active site in oxygen-evolving catalysts. Proc. Natl. Acad. Sci. USA 2017, 114, 3855–3860. [Google Scholar] [CrossRef] [PubMed]
- Symes, M.D.; Surendranath, Y.; Lutterman, D.A.; Nocera, D.G. Bidirectional and unidirectional PCET in a molecular model of a cobalt-based oxygen-evolving catalyst. J. Am. Chem. Soc. 2011, 133, 5174–5177. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.I.; Ziegler, M.S.; Oña-Burgos, P.; Sturzbecher-Hohne, M.; Kim, W.; Bellone, D.E.; Tilley, T.D. Mechanistic investigations of water oxidation by a molecular cobalt oxide analogue: Evidence for a highly oxidized intermediate and exclusive terminal oxo participation. J. Am. Chem. Soc. 2015, 137, 12865–12872. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.I.; Wang, J.; Levine, D.S.; Ziegler, M.S.; Tilley, T.D. Synthetic control and empirical prediction of redox potentials for Co4O4 cubanes over a 1.4 V range: Implications for catalyst design and evaluation of high-valent intermediates in water oxidation. Chem. Sci. 2017, 8, 4274–4284. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-P.; Van Voorhis, T. Direct-coupling O2 bond forming a pathway in cobalt oxide water oxidation catalysts. J. Phys. Chem. Lett. 2011, 2, 2200–2204. [Google Scholar] [CrossRef]
- Fernando, A.; Aikens, C.M. Reaction pathways for water oxidation to molecular oxygen mediated by model cobalt oxide dimer and cubane catalysts. J. Phys. Chem. C 2015, 119, 11072–11085. [Google Scholar] [CrossRef]
- Li, X.; Siegbahn, P.E.M. Water oxidation mechanism for synthetic Co–Oxides with small nuclearity. J. Am. Chem. Soc. 2013, 135, 13804–13813. [Google Scholar] [CrossRef]
- Mattioli, G.; Giannozzi, P.; Amore Bonapasta, A.; Guidoni, L. Reaction pathways for oxygen evolution promoted by cobalt catalyst. J. Am. Chem. Soc. 2013, 135, 15353–15363. [Google Scholar] [CrossRef]
- Xu, L.; Lei, H.; Zhang, Z.; Yao, Z.; Li, J.; Yu, Z.; Cao, R. The effect of the trans axial ligand of cobalt corroles on water oxidation activity in neutral aqueous solutions. Phys. Chem. Chem. Phys. 2017, 19, 9755–9761. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, F.F.; Kundu, S.; Risch, M.; Pandian, S.; Heims, F.; Pryjomska-Ray, I.; Haack, P.; Metzinger, R.; Bill, E.; Dau, H.; et al. An oxocobalt(IV) complex stabilized by lewis acid interactions with scandium(III) ions. Angew. Chem. Int. Ed. 2010, 50, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Pfaff, F.F.; Kwon, E.; Wang, Y.; Seo, M.-S.; Bill, E.; Ray, K.; Nam, W. Spectroscopic capture and reactivity of a low-spin cobalt(IV)–oxo complex stabilized by binding redox-inactive metal ions. Angew. Chem. 2014, 126, 10571–10575. [Google Scholar] [CrossRef]
- Wang, B.; Lee, Y.-M.; Tcho, W.-Y.; Tussupbayev, S.; Kim, S.-T.; Kim, Y.; Seo, M.S.; Cho, K.-B.; Dede, Y.; Keegan, B.C.; et al. Synthesis and reactivity of a mononuclear non-haem cobalt(IV)–oxo complex. Nat. Commun. 2017, 8, 14839. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-Y.; Yam, F.; Xie, Y.-T.; Li, X.-Y.; Chang, C.K. A bulky bis-pocket manganese(V)−oxo corrole complex: Observation of oxygen atom transfer between triply bonded MnV≡O and alkene. J. Am. Chem. Soc. 2009, 131, 12890–12891. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; So, H.; Yoon, H.; Cho, K.-B.; Lee, Y.-M.; Fukuzumi, S.; Nam, W. Reactivity comparison of high-valent iron(IV)–oxo complexes bearing N-tetramethylated cyclam ligands with different ring size. Dalton Trans. 2013, 42, 7842–7845. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.K.; Hill, E.A.; Filatov, A.S.; Anderson, J.S. Isolation of a terminal Co(III)–oxo complex. J. Am. Chem. Soc. 2018, 140, 13176–13180. [Google Scholar] [CrossRef] [PubMed]
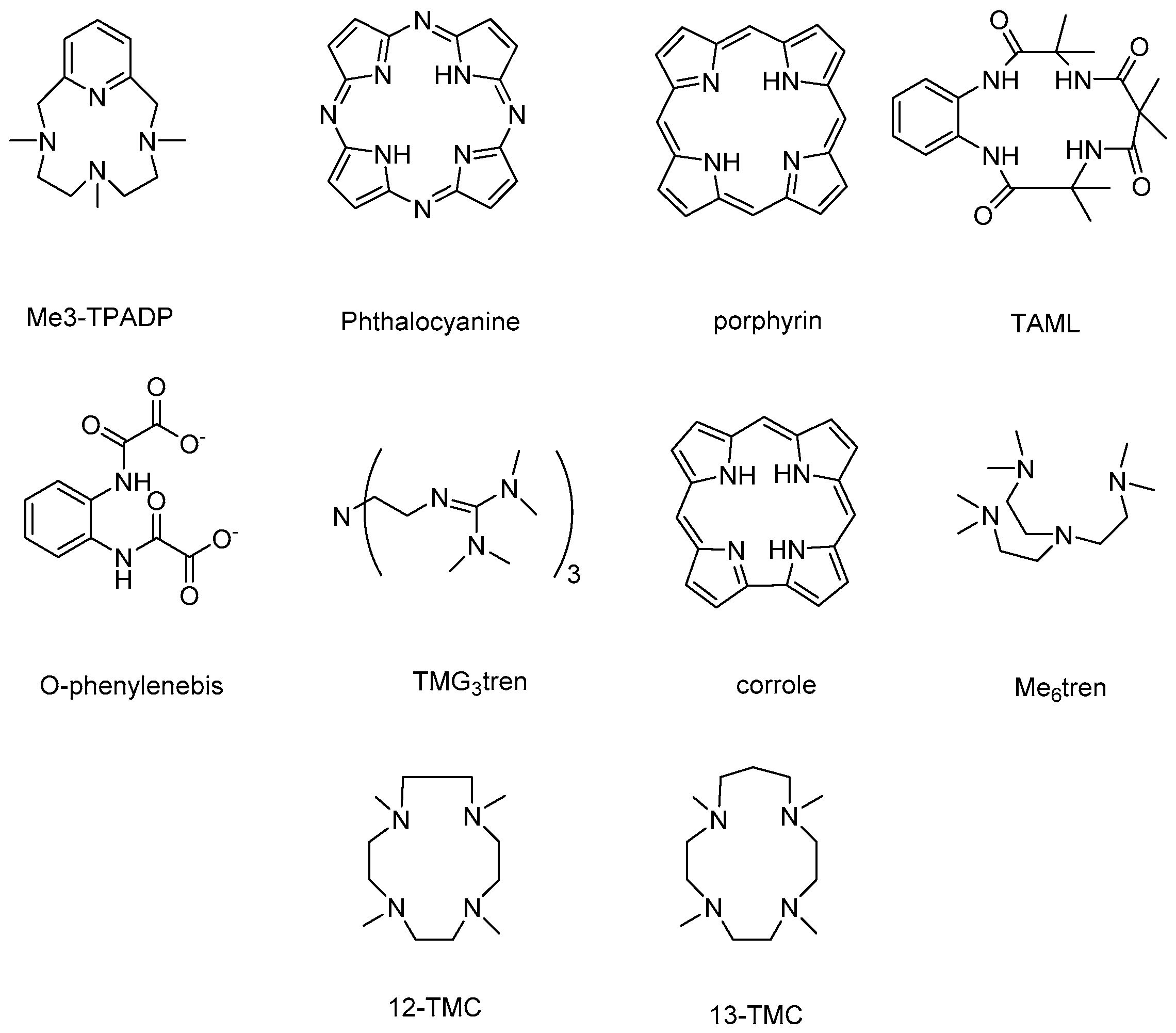
Figure 1.
Some of the most used tetrapyrrolic macrocycles and N-based ligands used to stabilize high-valent cobalt–oxo complexes.
Figure 1.
Some of the most used tetrapyrrolic macrocycles and N-based ligands used to stabilize high-valent cobalt–oxo complexes.

Scheme 1.
Proposed mechanism for oxidation of alkene by cobalt polyoxotungstate using PhIO as oxidant [39].
Scheme 1.
Proposed mechanism for oxidation of alkene by cobalt polyoxotungstate using PhIO as oxidant [39].

Figure 2.
Possible pathway for the formation of active species in cellulosic fiber-bonded cobalt phthalocyanine (CoPc) H2O2 system. (A) Generation of hydroxyl radicals without ligand dodecylbenzenesulfonate (LAS) by the homolytic cleavage of the peroxide O‒O bond; (B) Generation of cobalt–oxo with LAS by the heterolytic cleavage of the peroxide O‒O bond [54].
Figure 2.
Possible pathway for the formation of active species in cellulosic fiber-bonded cobalt phthalocyanine (CoPc) H2O2 system. (A) Generation of hydroxyl radicals without ligand dodecylbenzenesulfonate (LAS) by the homolytic cleavage of the peroxide O‒O bond; (B) Generation of cobalt–oxo with LAS by the heterolytic cleavage of the peroxide O‒O bond [54].

Figure 3.
(a) Proposed mechanism of activation of H2O2 catalyzed by CoPc-PyMWCNTs for oxidation of substrates at pH = 10 [59]. (b) structures of cobalt(III) complexes involved in the activation hydrogen peroxide [62].

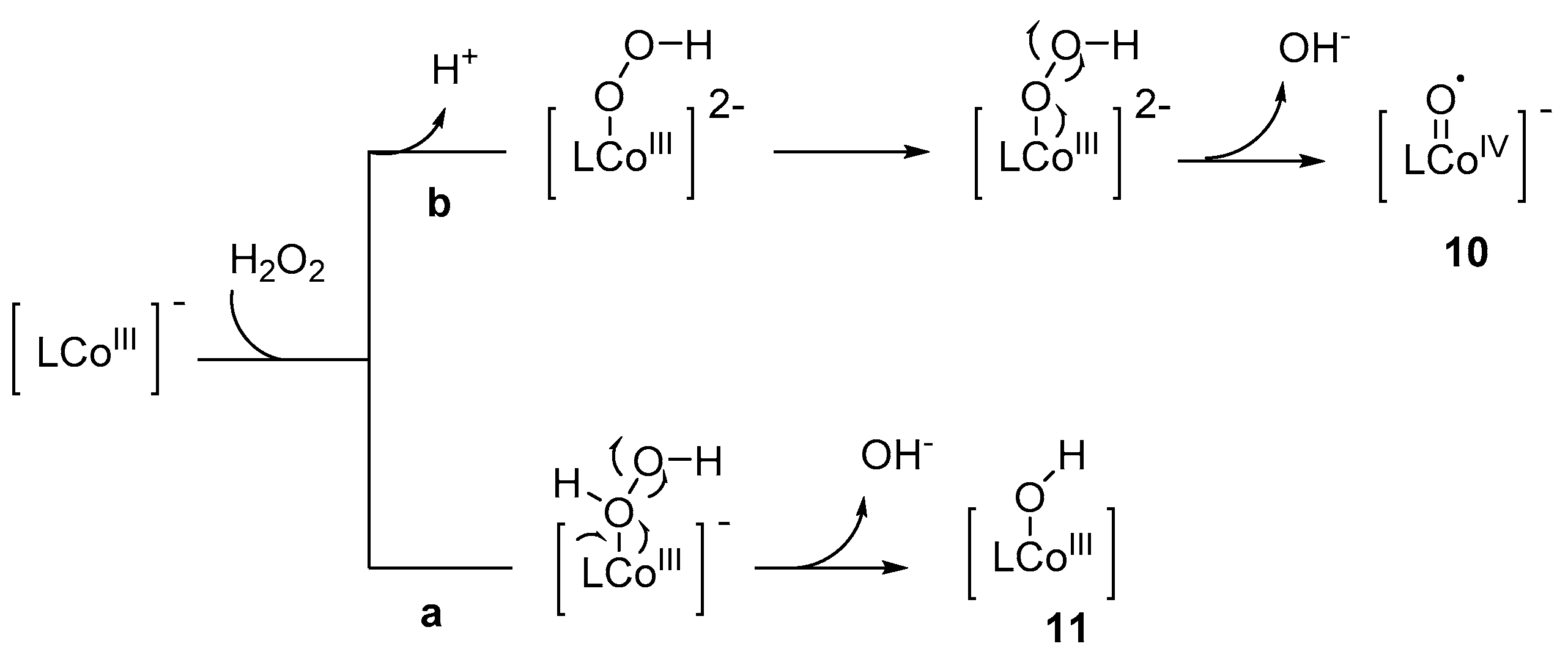
Scheme 2.
Proposed mechanism for the formation of cobalt–oxo species (L = bis-benzoamido) [60].
Scheme 2.
Proposed mechanism for the formation of cobalt–oxo species (L = bis-benzoamido) [60].

Scheme 3.
Mechanism for oxidation of water by cobalt oxide under acidic and basic conditions [74].
Scheme 3.
Mechanism for oxidation of water by cobalt oxide under acidic and basic conditions [74].

Scheme 4.
Molecular structures of cobalt porphyrins employed as water oxidation catalyst.

Scheme 5.
Proposed mechanism of water oxidation catalyzed by cobalt porphyrins [92].
Scheme 5.
Proposed mechanism of water oxidation catalyzed by cobalt porphyrins [92].

Scheme 6.
Generation of high-valent CoV=O 26(O) during water oxidation by molecular cobalt cubane Co4O4(OAc)4py4 [96].
Scheme 6.
Generation of high-valent CoV=O 26(O) during water oxidation by molecular cobalt cubane Co4O4(OAc)4py4 [96].

Scheme 7.
Preparation of high-valent {CoIV-O-Sc3+} complex 29.

Scheme 8.
Oxidation of complex 30 to 31-M by CAN/H2O or PhIO/Mn+.

Scheme 9.
Preparation of a mononuclear non-haem cobalt(IV)–oxo complex [(13-TMC)CoIV(O)]2+.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ali, A.; Akram, W.; Liu, H.-Y. Reactive Cobalt–Oxo Complexes of Tetrapyrrolic Macrocycles and N-based Ligand in Oxidative Transformation Reactions. Molecules 2019, 24, 78. https://doi.org/10.3390/molecules24010078
AMA Style
Ali A, Akram W, Liu H-Y. Reactive Cobalt–Oxo Complexes of Tetrapyrrolic Macrocycles and N-based Ligand in Oxidative Transformation Reactions. Molecules. 2019; 24(1):78. https://doi.org/10.3390/molecules24010078
Chicago/Turabian StyleAli, Atif, Waseem Akram, and Hai-Yang Liu. 2019. "Reactive Cobalt–Oxo Complexes of Tetrapyrrolic Macrocycles and N-based Ligand in Oxidative Transformation Reactions" Molecules 24, no. 1: 78. https://doi.org/10.3390/molecules24010078