Hits-to-Lead Optimization of the Natural Compound 2,4,6-Trihydroxy-3-geranyl-acetophenone (tHGA) as a Potent LOX Inhibitor: Synthesis, Structure-Activity Relationship (SAR) Study, and Computational Assignment

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Results and Discussion
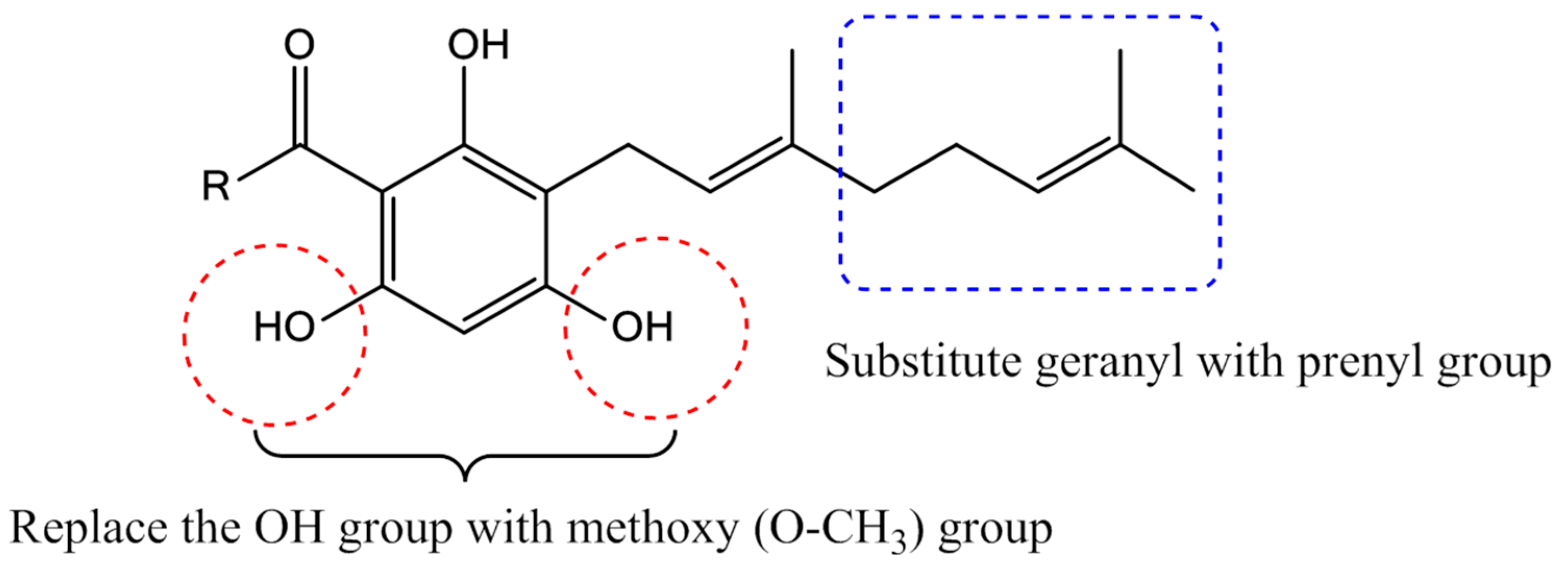
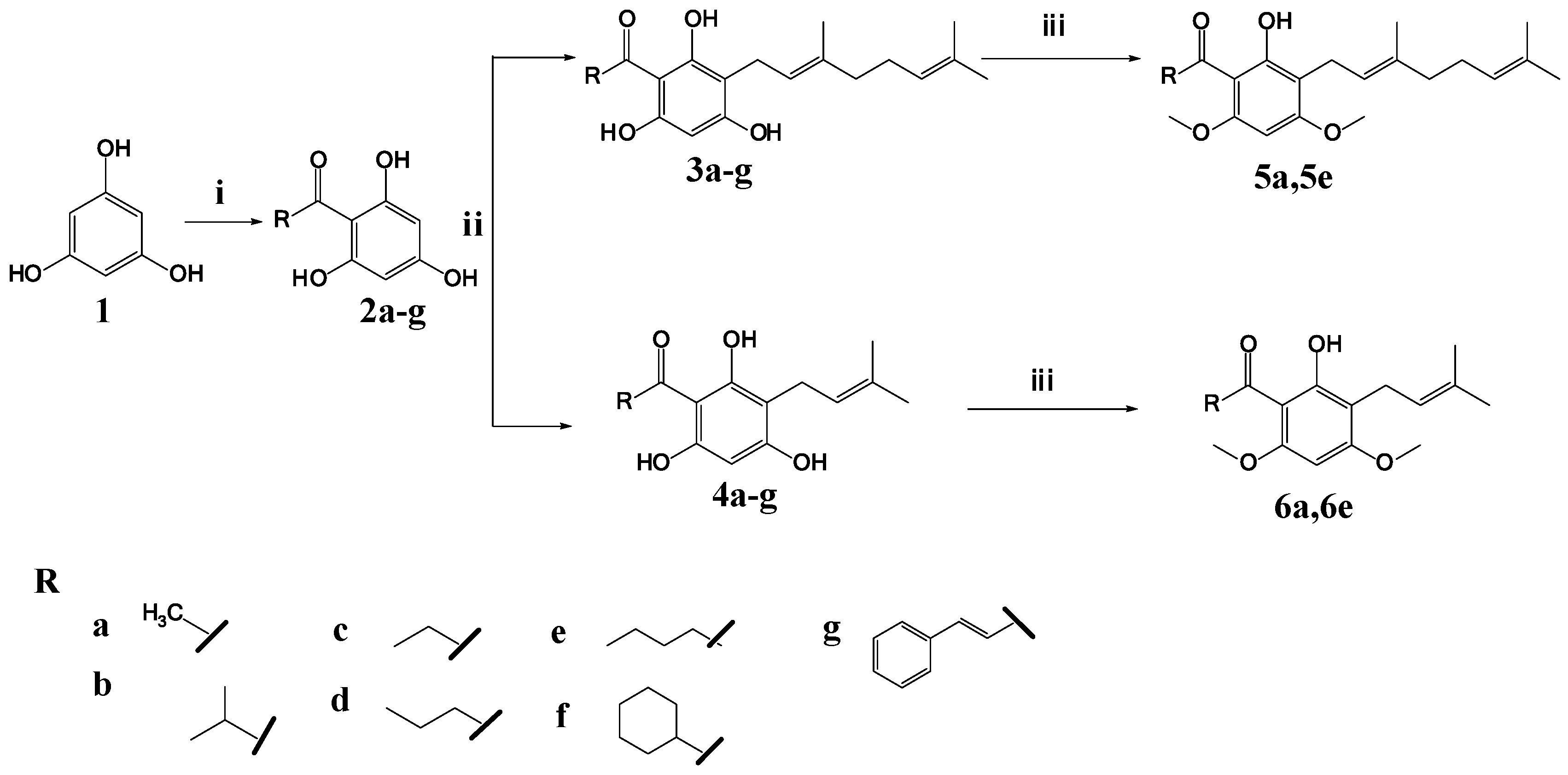
2.1. Synthesis of 2,4,6-Trihydroxy-3-Geranylacetophenone (tHGA) Analogues
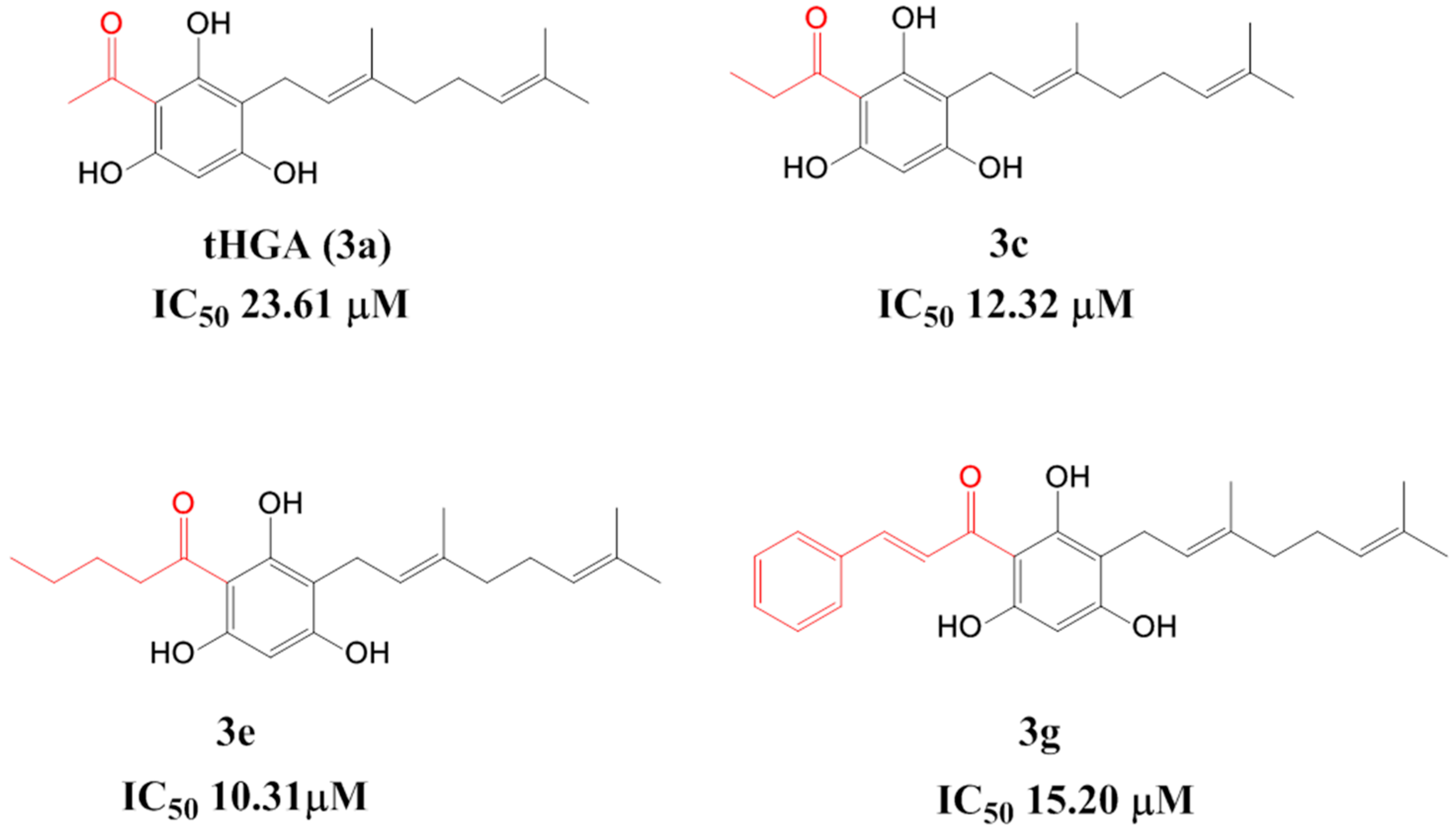
2.2. Soybean LOX-1 Inhibition Assays
2.3. Homology Modeling of Soybean LOX-1
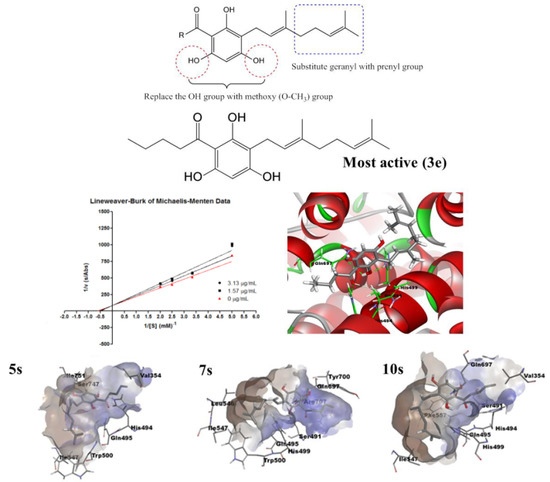
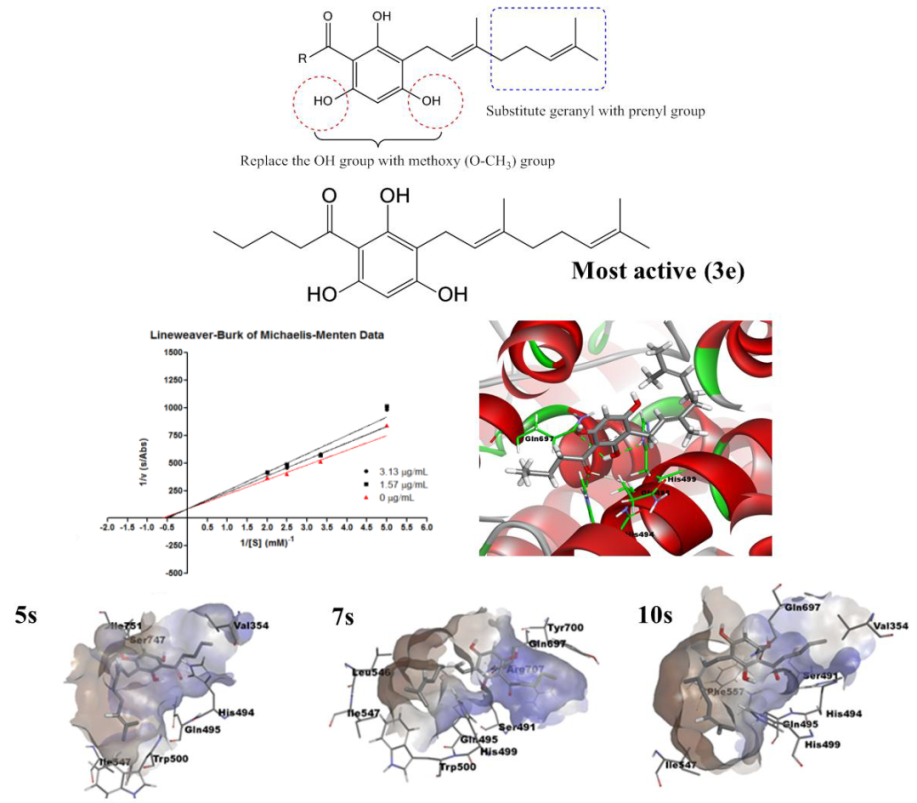
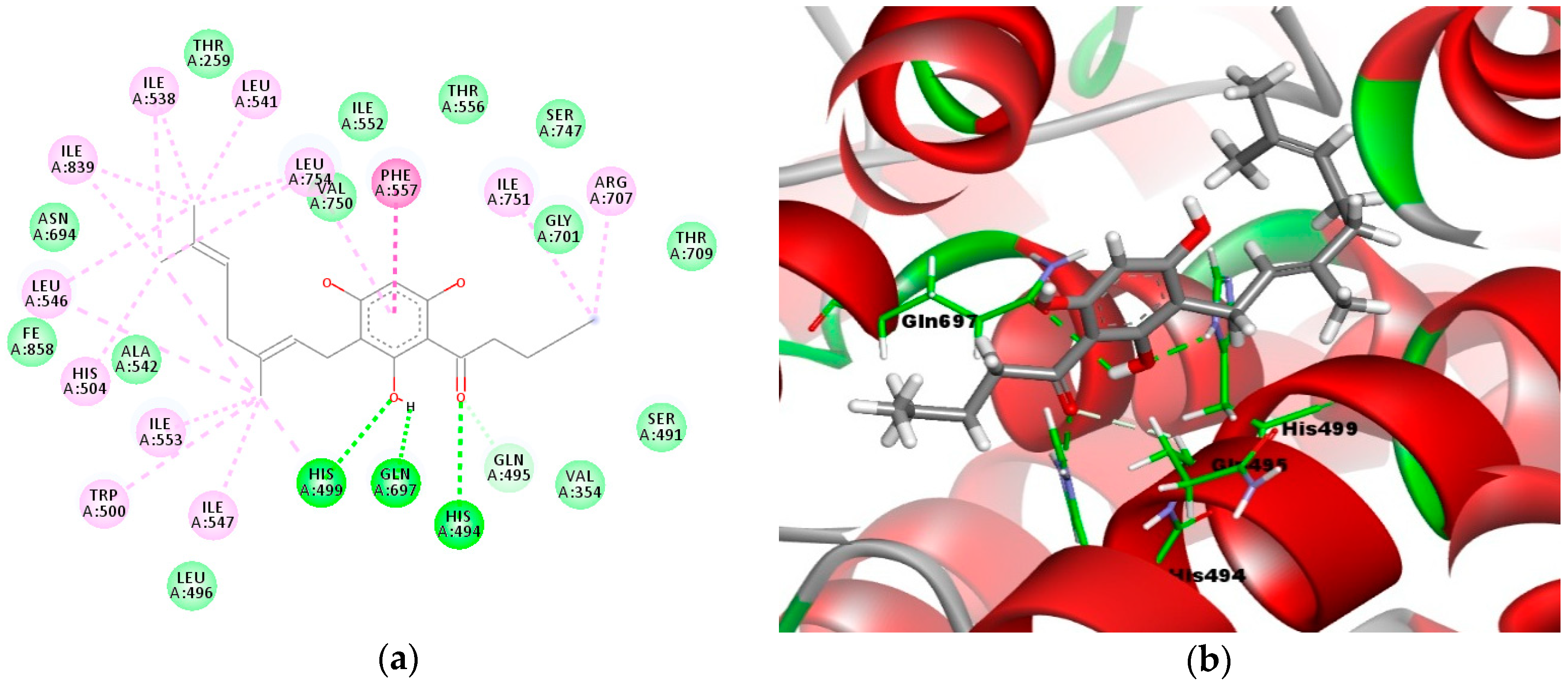
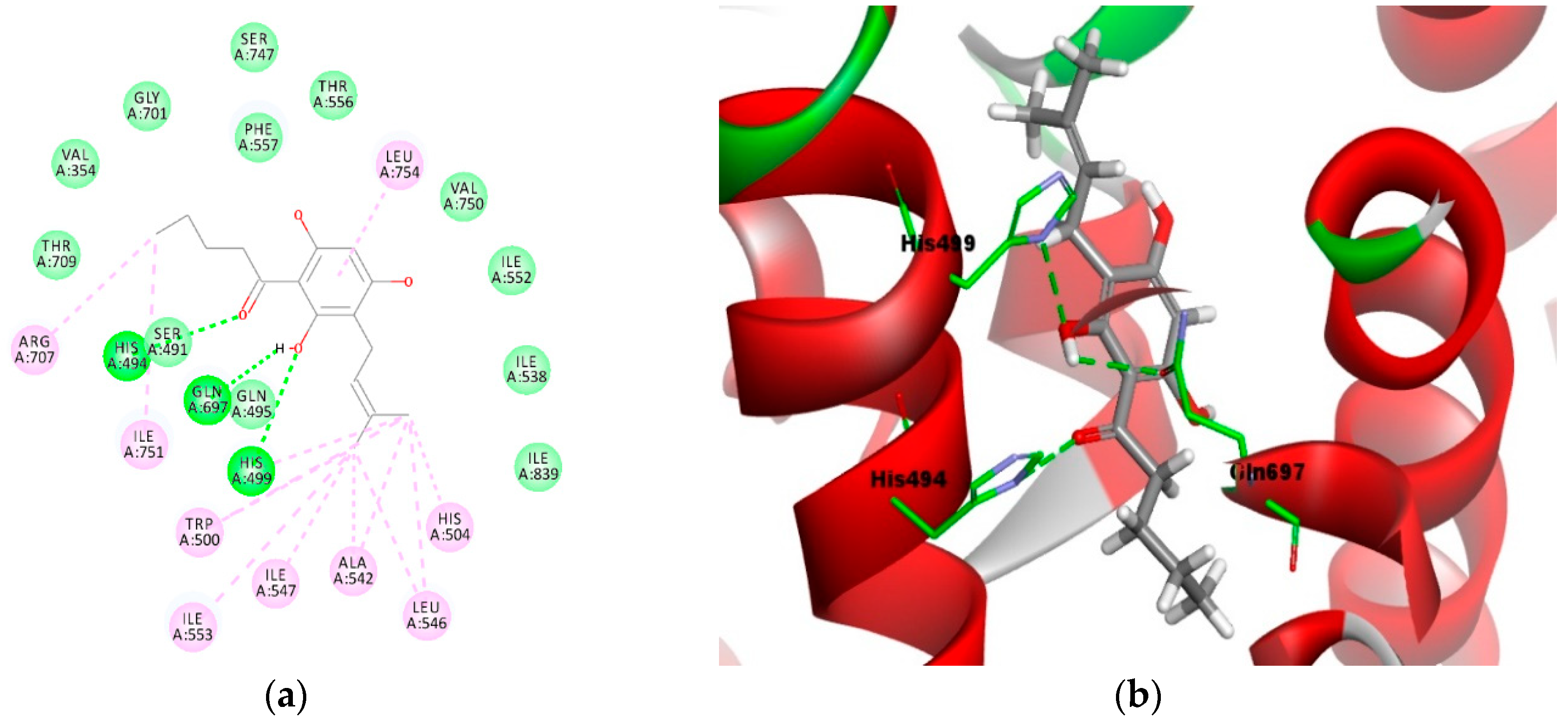
2.4. Molecular Docking Analysis
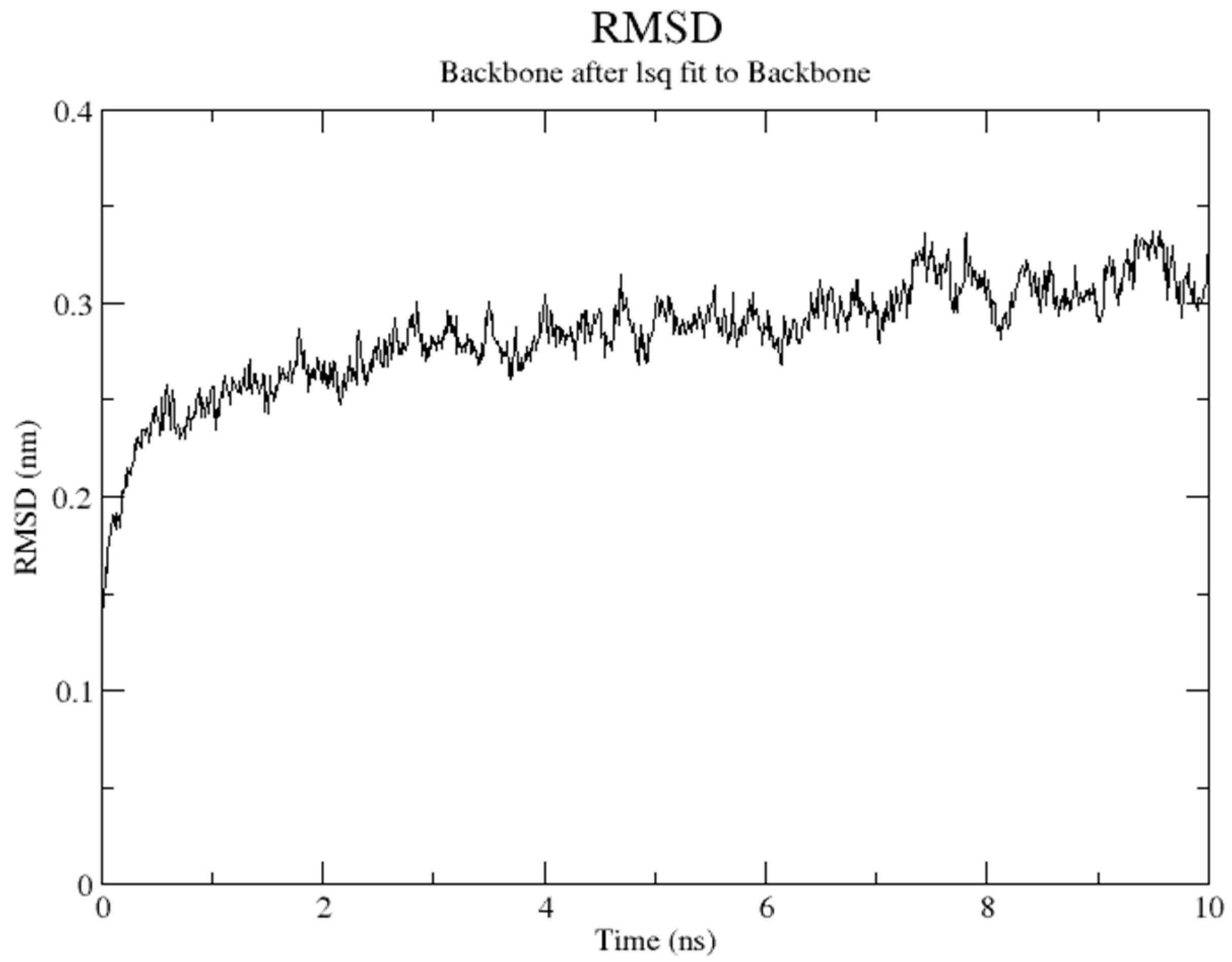
2.5. Molecular Dynamics Simulation
2.6. ADMET Analysis
2.7. TOPKAT Analysis
3. Materials and Methods
3.1. Synthesis of 2,4,6-Trihydroxy-3-Geranylacetophenone (tHGA) Analogues
3.1.1. General Methods
3.1.2. Two-Step Reaction: Friedel–Crafts Acylation and Direct C-Alkylation (3d, 4a–g)
3.1.3. Three-Step Reaction: Friedel–Crafts Acylation, Direct C-Alkylation, and Methylation (5a, 5e, 6a, and 6e)
3.2. In Vitro Soybean 15-Lipoxygenase (LOX) Inhibition Assay
3.3. Homology Modeling
3.4. Molecular Docking
3.5. Molecular Dynamic Simulation
3.6. ADMET and TOPKAT Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gardner, H.W. Recent investigations into the lipoxygenase pathway of plants. Biochim. Biophys. Acta 1991, 1084, 221–239. [Google Scholar] [CrossRef]
- Rapoport, S.M.; Schewe, T.; Wiesner, R.; Halangk, W.; Ludwig, P.; Janicke, H.M.; Tannert, C.; Hiebsch, C.; Klatt, D. The lipoxygenase of reticulocytes: Purification, characterization and biological dynamics of the lipoxygenase; its identity with the respiratory inhibitors of the reticulocyte. Eur. J. Biochem. 1979, 96, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Burnett, B.P.; Jia, Q.; Zhao, Y.; Levy, R.M. A medicinal extract of Scutellaria baicalensis and Acacia catechu acts as a dual inhibitor of cyclooxygenase and 5-lipoxygenase to reduce inflammation. J. Med. Food. 2007, 10, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R. Lipoxygenases: Occurrence, functions, catalysis, and acquisition of substrate. J. Biol. Chem. 1999, 274, 23679–23682. [Google Scholar] [CrossRef] [PubMed]
- Bigby, T.D.; Levy, B.D.; Serhan, C.N. Cell biology of the 5-lipoxygenase pathway: Amplification and generation of leukotrienes and lipoxins by transcellular biosynthesis. Lung Biol. Health Dis. 1998, 120, 125–173. [Google Scholar]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [PubMed]
- Reddanna, P.; Sandeep, P.K.; Whelan, J.; Reddy, C.C. Carboxypeptidase A-catalyzed direct conversion of leukotriene C4 to leukotriene F4. Arch. Biochem. Biophys. 2003, 413, 158–163. [Google Scholar] [CrossRef]
- Skrzypczak-Jankun, E.; Zhou, K.; Jankun, J. Inhibition of lipoxygenase by (-)-epigallocatechin gallate: X-ray analysis at 2.1 A reveals degradation of EGCG and shows soybean LOX-3 complex with EGC instead. Int. J. Mol. Med. 2003, 12, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Dahlen, S.E. Treatment of asthma with antileukotrienes: First line or last resort therapy? Eur. J. Pharmacol. 2006, 533, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Shaari, K.; Suppaiah, V.; Lam, K.W.; Stanlas, J.; Tejo, B.A.; Israf, D.A.; Abas, F.; Safri, S.; Shuaib, H.; Zareen, S.; et al. Bioassay-guided identification of an anti-inflammatory prenylated acylphloroglucinol from Melicope ptelefolia and molecular insights into its interaction with 5-lipoxygenase. Bioorg. Med. Chem. 2011, 19, 6340–6347. [Google Scholar] [CrossRef] [PubMed]
- Ismail, N.; Jambari, N.N.; Zareen, S.; Akhtar, M.N.; Shaari, K.; Zamri-Saad, M.; Ling, T.C.; Sulaiman, M.R.; Lajis, N.H.; Israf, D.A. A geranyl acetophenone targeting cysteinyl leukotriene synthesis prevents allergic airway inflammation in ovalbumin-sensitized mice. Toxicol. Appl. Pharm. 2012, 259, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Shaari, K.; Johnson, S.; Velan, S.; Seema, Z.; Faridah, A.; Daud, I.A.; Nordin, L. Leukotriene Inhibitor and Method for Producing the Same. Patent PI2010000968, 31 December 2010. Available online: http://psasir.upm.edu.my/31549/ (accessed on 30 November 2012).
- Ng, C.H.; Rullah, K.; Mohd Fadhlizil Fasihi, M.A.; Lam, K.W.; Ismail, I.S.; Narayanaswamy, R.; Jamaludin, F.; Shaari, K. Synthesis and docking studies of 2, 4, 6-trihydroxy-3-geranylacetophenone analogs as potential lipoxygenase inhibitor. Molecules 2014, 19, 11645–11659. [Google Scholar] [CrossRef] [PubMed]
- Hartl, A.; Reininger, W. Method of Acylation of Phloroglucinol. U.S. Patent 4,053,517, 11 October 1977. [Google Scholar]
- Smith, M.J.H.; Ford-Hutchinson, A.W.; Bray, M.A. Leukotriene B: A potential mediator of inflammation. J. Phar. Pharmacol. 1980, 32, 517–518. [Google Scholar] [CrossRef]
- Abdellatif, K.R.; Dond, Y.; Chen, Q.H.; Chowdhury, M.A.; Knaus, E.E. Novel (E)-2-(aryl)-3-(4-methanesulfonylphenyl) acrylic ester prodrugs possessing a diazen-1-ium-1, 2-diolate moiety: Design, synthesis, cyclooxygenase inhibition, and nitric oxide release studies. Bioorg. Med. Chem. 2007, 15, 6796–6801. [Google Scholar] [CrossRef] [PubMed]
- Pontiki, E.; Hadjipavlou-Litina, D. Synthesis, antioxidant and anti-inflammatory activity of novel aryl-acetic and aryl-hydroxamic acids. Arzneim. Forsch./Drug Res. 2003, 53, 780–785. [Google Scholar]
- Pontiki, E.; Hadjipavlou-Litina, D. Antioxidant and Anti-inflammatory activity of aryl-acetic and hydroxamic acids as Novel Lipoxygenase Inhibitors. Med. Chem. 2006, 2, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Pontiki, E.; Hadjipavlou-Litina, D. Synthesis of phenyl-substituted amides with antioxidant and antiinflammatory activity as Novel Lipoxygenase Inhibitors. Med. Chem. 2007, 3, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Das, D. Enzymes, 8th ed.; Academic Publishers: Calcutta, India, 1993; pp. 89–111. [Google Scholar]
- Kuhn, H.; Saam, J.; Eibach, S.; Holzhütter, H.G.; Ivanov, I.; Walther, M. Structural biology of mammalian lipoxygenases: Enzymatic consequences of targeted alterations of the protein structure. Biochem. Biophys. Res. Commun. 2005, 338, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Skrzypczak-Jankun, E.; Borbulevych, O.Y.; Zavodszky, M.I.; Baranski, M.R.; Padmanabhan, K.; Petricek, V.; Jankun, J. Effect of crystal freezing and small-molecule binding on internal cavity size in a large protein: X-ray and docking studies of lipoxygenase at ambient and low temperature at 2.0 A resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswamy, R.; Lam, K.W.; Ismail, I.S. Molecular docking analysis of natural compounds as Human neutrophil elastase (HNE) inhibitors. J. Chem. Pharm. Res. 2013, 5, 337–341. [Google Scholar]
- Kubinyi, H. Structure-based design of enzyme inhibitors and receptor ligands. Curr. Opin. Drug Discov. Devel. 1998, 1, 4–15. [Google Scholar] [PubMed]
- Smith, K.A. Structure and Synthesis of Phloroglucinol Derivatives from Hypericum roeperianum. Ph.D. Thesis, University of KwaZulu-Natal, KwaZulu-Natal, South Africa, 2010. [Google Scholar]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertonim, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. SFCHECK: A unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmann, J.A.C.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Rullah, K.; Aluwi, M.F.F.M.; Yamin, B.M.; Bahari, M.N.A.; Leong, S.W.; Ahmad, S.; Abas, F.; Ismail, N.H.; Jantan, I.; Lam, K.W. Inhibition of prostaglandin E2 production by synthetic minor prenylated chalcones and flavonoids: Synthesis, biological activity, crystal structure, and in silico evaluation. Bioorg. Med. Chem. Lett. 2014, 24, 3826–3834. [Google Scholar] [CrossRef] [PubMed]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An automated force field topology builder (ATB) and repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.V.; van Gunsteren, W.F.; DiNola, A.R.H.J.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % Inhibition (100 μg/mL) | IC50 Value (μM) Mean ± SEM |
|---|---|---|
| 3a (tHGA) # | 94.3 ± 3.4 | 23.6 ± 1.7 |
| 3b# | 90.3 ± 4.1 | 27.6 ± 3.6 |
| 3c# | 94.4 ± 3.0 | 12.3 ± 0.6 |
| 3d | 90.7 ± 5.0 | 15.3 ± 0.5 |
| 3e# | 90.3 ± 4.4 | 10.3 ± 1.5 |
| 3f# | 88.8 ± 5.9 | 26.3 ± 1.3 |
| 3g# | 90.8 ± 7.6 | 15.2 ± 1.2 |
| 4a | 94.8 ± 5.2 | 72.1 ± 1.2 |
| 4b | 82.5 ± 7.3 | 53.7 ± 0.7 |
| 4c | 78.1 ± 3.9 | 61.0 ± 1.1 |
| 4d | 86.2 ± 6.9 | 39.5 ± 1.2 |
| 4e | 79.4 ± 6.7 | 35.1 ± 1.3 |
| 4f | 93.7 ± 3.8 | 52.6 ± 1.0 |
| 4g | 95.2 ± 3.2 | 95.4 ± 4.0 |
| NDGA | 100.0 ± 0.0 | 0.1 ± 0.0 |
| Compounds | Concentrations (μg/mL) | Ki (μM) | |||||
|---|---|---|---|---|---|---|---|
| 0 | 1.57 | 3.13 | |||||
| Km | Vmax | Km | Vmax | Km | Vmax | ||
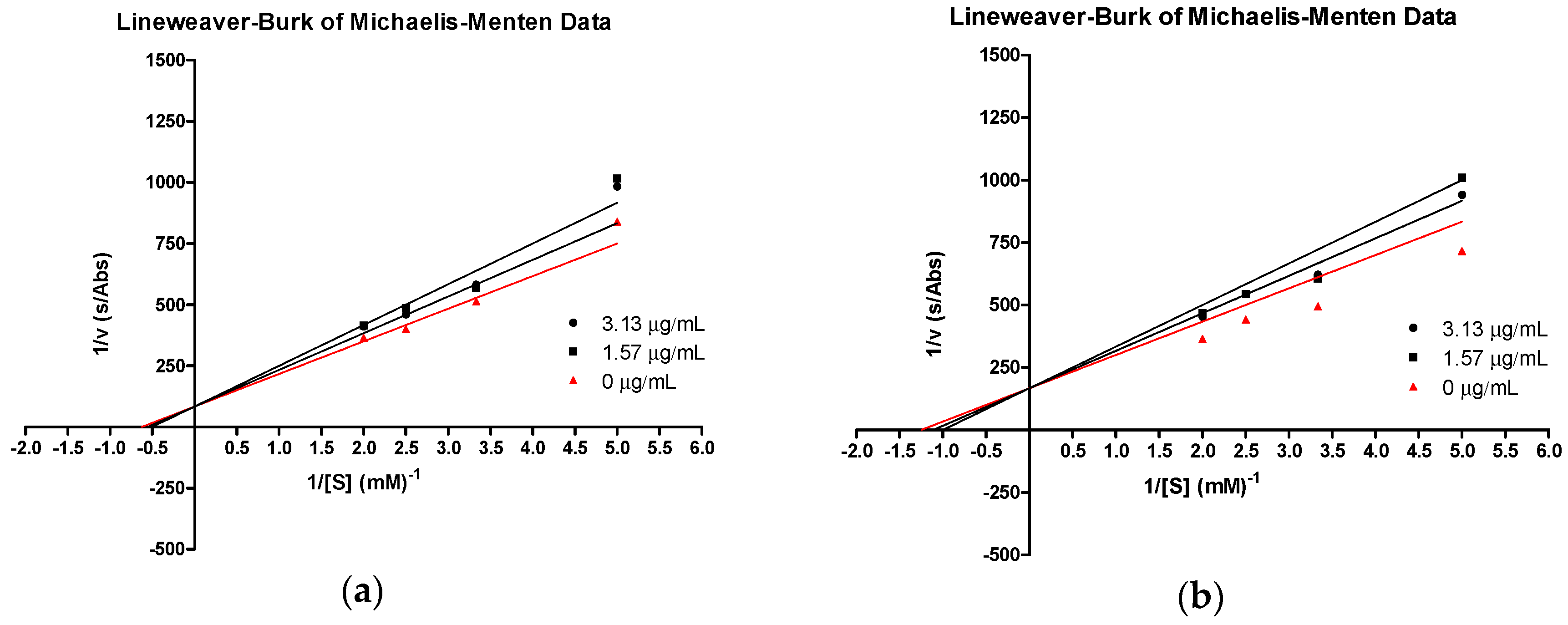
| 3e | 4.62 | 0.035 | 5.20 | 0.035 | 5.78 | 0.035 | 36.1 |
| tHGA (3a) | 2.63 | 0.020 | 2.96 | 0.020 | 3.29 | 0.020 | 41.1 |
| Compound | Van der Waals Energy | Electrostatic Energy | Polar Solvation Energy | Nonpolar Solvation Energy | Total Binding Energy |
|---|---|---|---|---|---|
| 3e | −200.44 (8.75) | −16.93 (8.53) | 87.65 (13.26) | −20.99 (1.02) | −150.71 (11.30) |
| 4e | −175.29 (7.67) | −7.39 (5.74) | 89.10 (11.91) | −18.21 (0.99) | −111.79 (12.59) |
| tHGA (3a) | −171.62 (9.16) | −14.40 (4.19) | 57.28 (9.08) | −19.30 (0.93) | −148.04 (10.89) |
| Compound | HIA | AS | BBB | PBB | CYP2D6 | HT |
|---|---|---|---|---|---|---|
| 3a ** | Good | Good | Medium | Bound | Inhibitor | Nontoxic |
| 3b | Moderate | Low | – | Bound | Inhibitor | Nontoxic |
| 3c | Moderate | Low | – | Bound | Inhibitor | Nontoxic |
| 3d | Moderate | Low | – | Bound | Inhibitor | Nontoxic |
| 3e * | Poor | Low | – | Bound | Inhibitor | Nontoxic |
| 3f | Poor | Low | – | Bound | Noninhibitor | Nontoxic |
| 3g | Poor | Low | – | Bound | Noninhibitor | Nontoxic |
| 4a | Good | Good | Good | Bound | Noninhibitor | Toxic |
| 4b | Good | Good | Medium | Bound | Noninhibitor | Toxic |
| 4c | Good | Good | Medium | Bound | Noninhibitor | Toxic |
| 4d | Good | Good | Medium | Bound | Inhibitor | Toxic |
| 4e | Good | Good | Medium | Bound | Inhibitor | Toxic |
| 4f | Good | Low | High | Bound | Noninhibitor | Toxic |
| 4g | Good | Low | High | Bound | Noninhibitor | Toxic |
| Compound | AB | AM | RC | OI | SI | SS |
|---|---|---|---|---|---|---|
| 3a ** | Degradable | Nonmutagen | Noncarcinogen | Nonirritant | Nonirritant | Sensitizer |
| 3b | Degradable | Nonmutagen | Noncarcinogen | Nonirritant | Nonirritant | Sensitizer |
| 3c | Degradable | Nonmutagen | Noncarcinogen | Nonirritant | Nonirritant | Sensitizer |
| 3d | Degradable | Nonmutagen | Noncarcinogen | Nonirritant | Nonirritant | Sensitizer |
| 3e * | Degradable | Nonmutagen | Noncarcinogen | Nonirritant | Nonirritant | Sensitizer |
| 3f | Degradable | Nonmutagen | Noncarcinogen | Nonirritant | Nonirritant | Sensitizer |
| 3g | Degradable | Nonmutagen | Noncarcinogen | Nonirritant | Nonirritant | Sensitizer |
| 4a | Degradable | Nonmutagen | Noncarcinogen | Nonirritant | Nonirritant | Sensitizer |
| 4b | Degradable | Nonmutagen | Noncarcinogen | Irritant | Nonirritant | Sensitizer |
| 4c | Degradable | Nonmutagen | Noncarcinogen | Irritant | Nonirritant | Sensitizer |
| 4d | Degradable | Nonmutagen | Noncarcinogen | Irritant | Nonirritant | Sensitizer |
| 4e | Degradable | Nonmutagen | Noncarcinogen | Irritant | Nonirritant | Sensitizer |
| 4f | Degradable | Nonmutagen | Noncarcinogen | Irritant | Nonirritant | Sensitizer |
| 4g | Nondegradable | Nonmutagen | Noncarcinogen | Nonirritant | Nonirritant | Sensitizer |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ng, C.H.; Rullah, K.; Abas, F.; Lam, K.W.; Ismail, I.S.; Jamaludin, F.; Shaari, K. Hits-to-Lead Optimization of the Natural Compound 2,4,6-Trihydroxy-3-geranyl-acetophenone (tHGA) as a Potent LOX Inhibitor: Synthesis, Structure-Activity Relationship (SAR) Study, and Computational Assignment. Molecules 2018, 23, 2509. https://doi.org/10.3390/molecules23102509
Ng CH, Rullah K, Abas F, Lam KW, Ismail IS, Jamaludin F, Shaari K. Hits-to-Lead Optimization of the Natural Compound 2,4,6-Trihydroxy-3-geranyl-acetophenone (tHGA) as a Potent LOX Inhibitor: Synthesis, Structure-Activity Relationship (SAR) Study, and Computational Assignment. Molecules. 2018; 23(10):2509. https://doi.org/10.3390/molecules23102509
Chicago/Turabian StyleNg, Chean Hui, Kamal Rullah, Faridah Abas, Kok Wai Lam, Intan Safinar Ismail, Fadzureena Jamaludin, and Khozirah Shaari. 2018. "Hits-to-Lead Optimization of the Natural Compound 2,4,6-Trihydroxy-3-geranyl-acetophenone (tHGA) as a Potent LOX Inhibitor: Synthesis, Structure-Activity Relationship (SAR) Study, and Computational Assignment" Molecules 23, no. 10: 2509. https://doi.org/10.3390/molecules23102509