General Pathway for a Convenient One-Pot Synthesis of Trifluoromethyl-Containing 2-amino-7-alkyl(aryl/heteroaryl)-1,8-naphthyridines and Fused Cycloalkane Analogues

Abstract
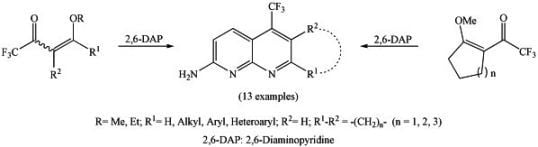
:
{kind=link}
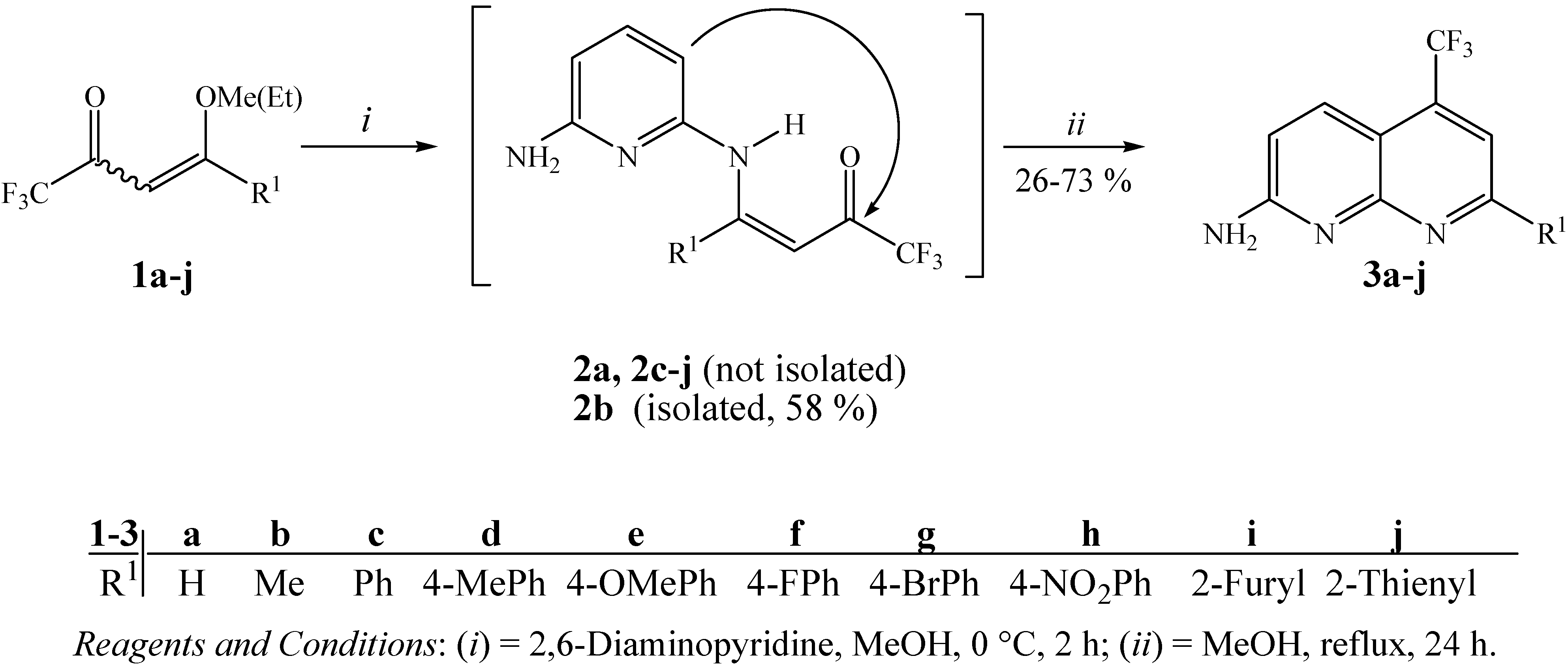{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
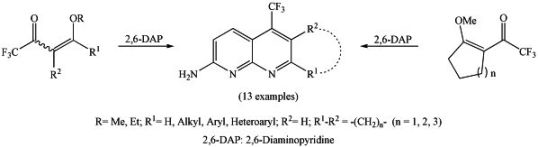
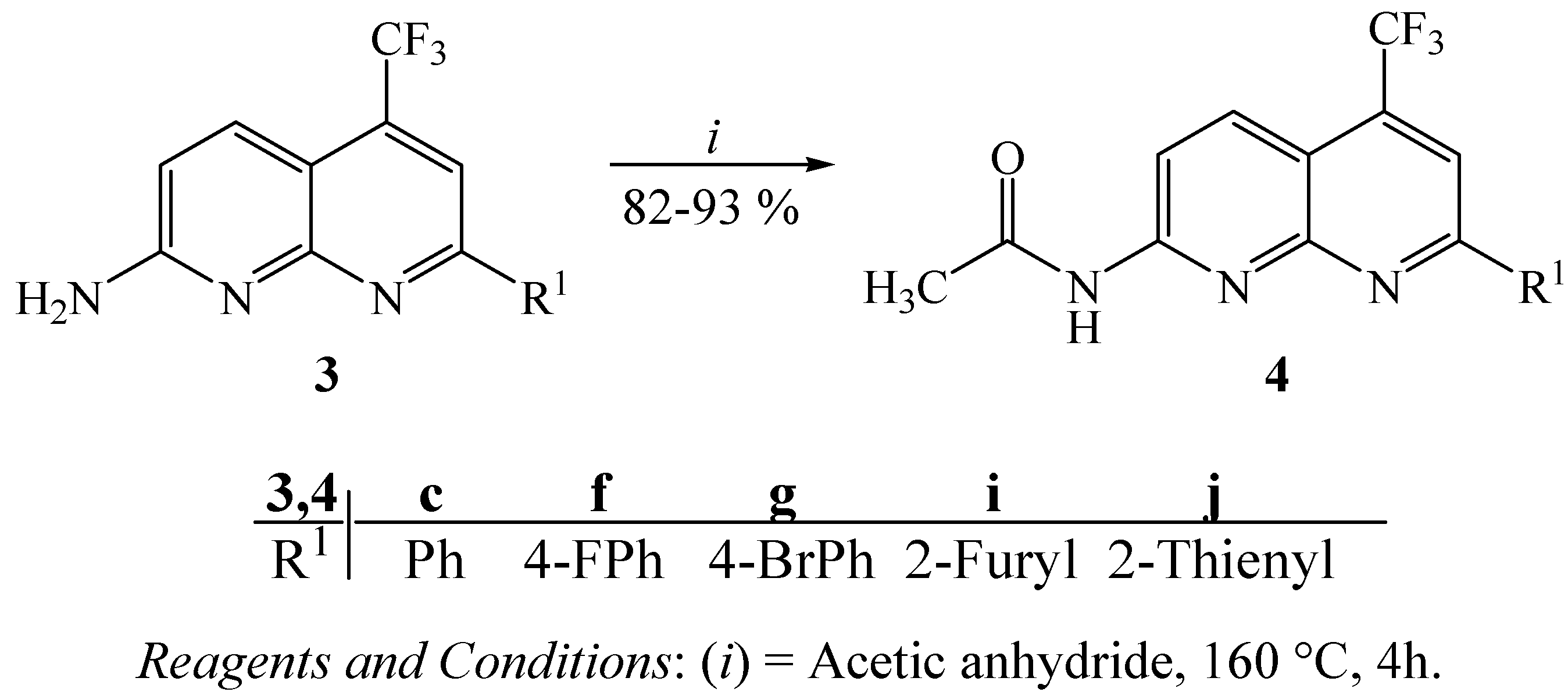
2. Results and Discussion




3. Experimental
3.1. General
3.2. General procedure for the synthesis of (Z)-N(5,5,5-Trifluoro-4-oxo-2-penten-2-yl)-2,6-diamino-pyridine (2b)
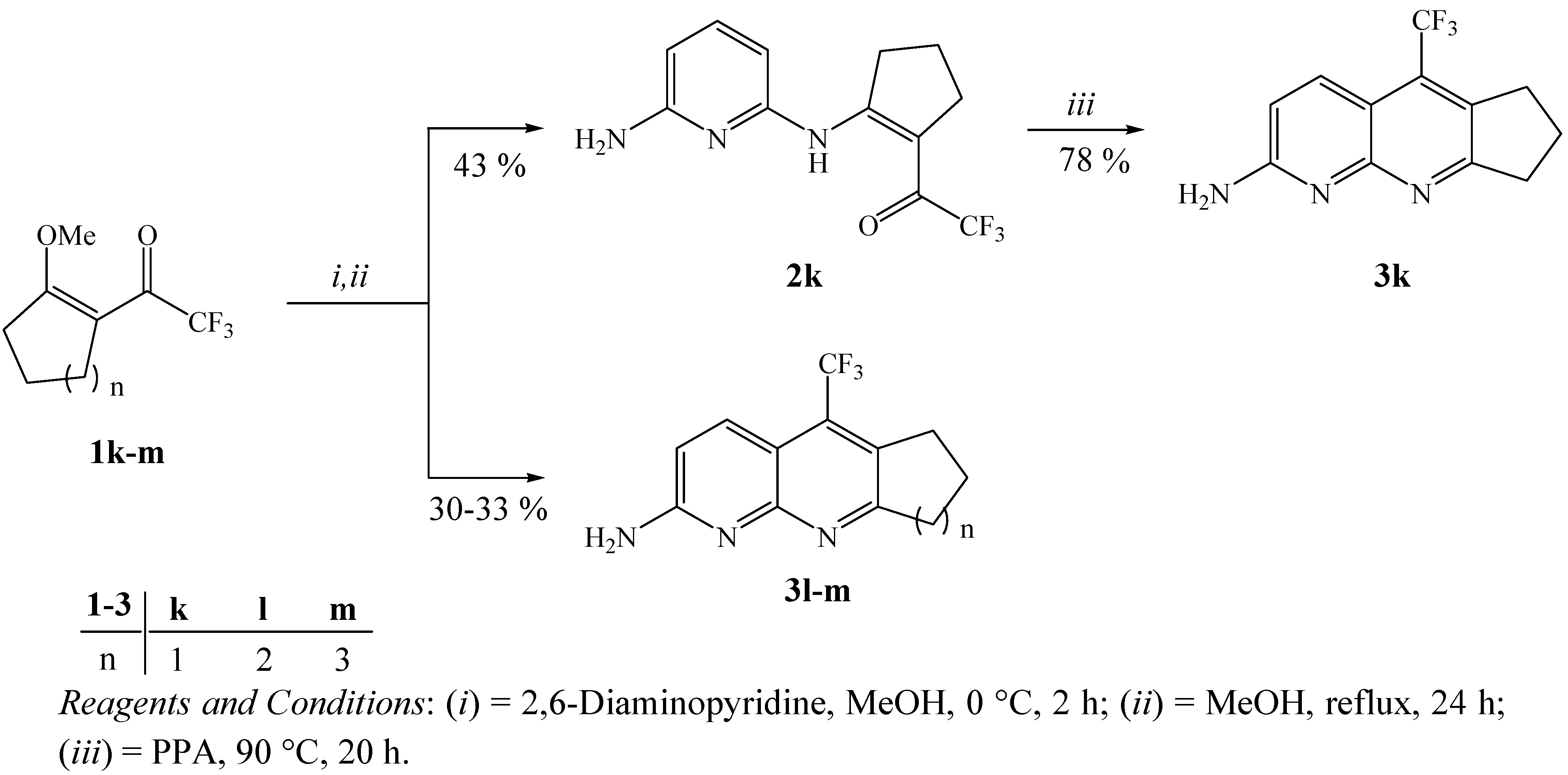
3.3. General procedure for the synthesis of 2-(6-Aminopyridin-2-ylamino)1-trifluoroacetyl-cyclopent-1-ene (2k)
3.4. General procedure for the synthesis of 7-alkyl(aryl/heteroaryl)-2-amino-5-trifluoromethyl-1,8-naphthyridines 3a-j
3.5. General procedure for the synthesis of 2-amino-5-trifluoromethyl-7,8-dihydro-6H-cyclopenta[b][1,8]-naphthyridine (3k)
3.6. General procedure for the synthesis of 2-amino-5-trifluoromethyl-cycloalka[b][1,8]-naphthyridines 3l, 3m
3.7. General procedure for the synthesis of 2-acetylamino-7-(aryl/heteroaryl)-5-trifluoromethyl-1,8-naphthyridines 4
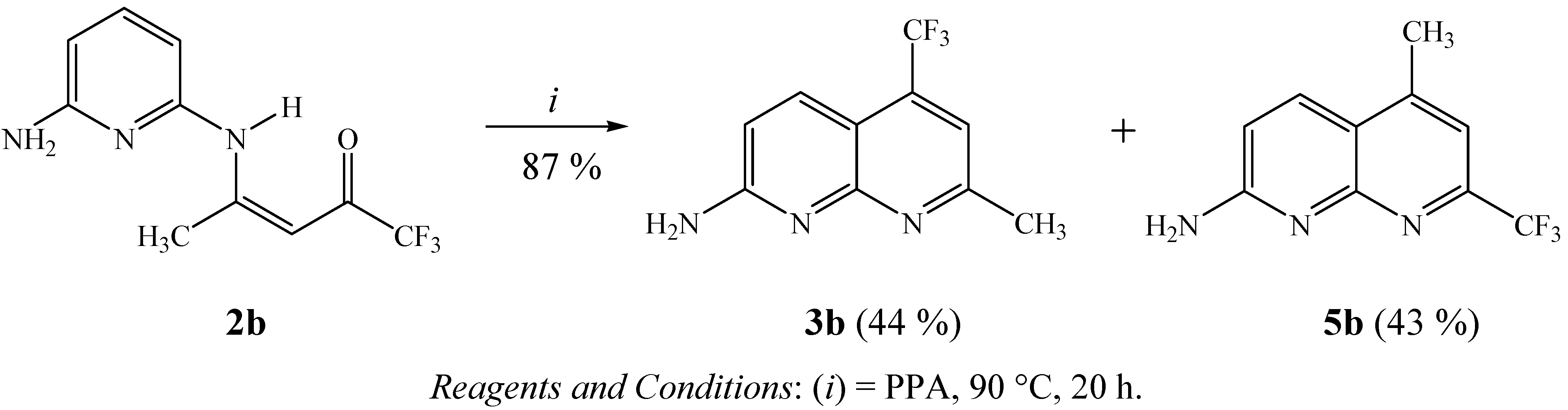
3.8. General procedure for the synthesis of 2-amino-5-trifluoromethyl-7-methyl-1,8-naphthyridine (3b) and 2-amino-7-trifluoromethyl-5-methyl-1,8-naphthyridine (5b)
4. Conclusions
Acknowledgements
References and Notes
- Goswami, S.; Mukherjee, R.; Mukherjee, R.; Jana, S.; Maity, A.C.; Adak, A.K. Simple and efficient synthesis of 2,7-difunctionalized-1,8-naphthyridines. Molecules 2005, 10, 929–936. [Google Scholar] [CrossRef]
- Mekheimer, R.A.; Hameed, A.M.A.; Sadek, K.U. 1,8-Naphthyridines II: Synthesis of novel polyfunctionally substituted 1,8-naphthyridinones and their degradation to 6-aminopyridones. ARKIVOC 2007, xiii, 269–281. [Google Scholar]
- He, C.; Lippard, S.J. Design and synthesis of multidentate dinucleating ligands based on 1,8-naphthyridine. Tetrahedron 2000, 56, 8245–8252. [Google Scholar] [CrossRef]
- Bouzard, D.; DiCesare, P.; Essiz, M.; Jacquet, J.P.; Ledoussal, B.; Remuzon, P.; Kessler, R.E.; Fung-Tomc, J. Fluoronaphthyridines as antibacterial agents. 4. Synthesis and structure-activity relationships of 5-substituted-6-fluoro-7-(cycloalky1amino)-l,4-dihydro-4-oxo-1,8-naphthyridine-3-carboxylic acids. J. Med. Chem. 1992, 35, 518–525. [Google Scholar]
- Ferrarini, P.L.; Manera, C.; Mori, C.; Badawneh, M.; Saccomanni, G. Synthesis and evaluation of antimycobacterial activity of 4-phenyl-1,8-naphthyridine derivatives. Farmaco 1998, 53, 741–746. [Google Scholar] [CrossRef]
- Tsuzuki, Y.; Tomita, K.; Sato, Y.; Kashimoto, S.; Chiba, K. Synthesis and structure–activity relationships of 3-substituted 1,4-dihydro-4-oxo-1-(2-thiazolyl)-1,8-naphthyridines as novel antitumor agents. Bioorg. Med. Chem. Lett. 2004, 14, 3189–3193. [Google Scholar]
- Dianzani, C.; Collino, M.; Gallicchio, M.; Di Braccio, M.; Roma, G.; Fantozzi, R. Effects of anti-inflammatory [1,2,4]triazolo[4,3-a][1,8]naphthyridine derivatives on human stimulated PMN and endothelial cells: An in vitro study. J. Inflamm. 2006, 3, 4. [Google Scholar] [CrossRef]
- Roma, G.; Di Braccio, M.; Grossi, G.; Piras, D.; Ballabeni, V.; Tognolini, M.; Bertoni, S.; Barocelli, E. 1,8-Naphthyridines VIII. Novel 5-aminoimidazo[1,2-a][1,8]naphthyridine-6-carboxamide and 5-amino[1,2,4]triazolo[4,3-a][1,8] naphthyridine-6-carboxamide derivatives showing potent analgesic or anti-inflammatory activity, respectively, and completely devoid of acute gastrolesivity. Eur. J. Med. Chem. 2010, 45, 352–366. [Google Scholar] [CrossRef]
- Ferrarini, P.L.; Badawneh, M.; Franconi, F.; Manera, C.; Miceli, M.; Mori, C.; Saccomanni, G. Synthesis and antiplatelet activity of some 2,7-di(N-cycloamino)-3-phenyl-1,8-naphthyridine derivatives. Farmaco 2001, 56, 311–318. [Google Scholar] [CrossRef]
- Santilli, A.A.; Scotese, A.C.; Bauer, R.F.; Bell, S.C. 2-Oxo-1,8-naphthyridine-3-carboxylic acid derivatives with potent gastric antisecretory properties. J. Med. Chem. 1987, 30, 2270–2277. [Google Scholar] [CrossRef]
- Ferrarini, P.L.; Mori, C.; Tellini, N. Synthesis and local anesthetic activity of (E)- and (Z)-diethylaminoethyliminothers of 1,8-naphtyridine. Farmaco 1990, 45, 385–389. [Google Scholar]
- Leonard, J.T.; Gangadhar, R.; Gnanasam, S.K.; Ramachandran, S.; Saravanan, M.; Sridhar, S.K. Synthesis and pharmacological activities of 1,8-naphthyridine derivatives. Biol. Pharm. Bull. 2002, 25, 798–802. [Google Scholar] [CrossRef]
- Ferrarini, P.L.; Mori, C.; Calderone, V.; Calzolari, L.; Nieri, P.; Martinotti, E.; Saccomanni, G. Synthesis of 1,8-naphthyridine derivatives: Potential antihypertensive agents – Part VIII. Eur. J. Med. Chem. 1999, 34, 505–513. [Google Scholar] [CrossRef]
- Ferrarini, P.L.; Mori, C.; Badawneh, M.; Calderone, V.; Calzolari, L.; Loffredo, T.; Martinotti, E.; Saccomanni, G. Synthesis of l,8-naphthyridine derivatives: Potential antihypertensive agents – Part VII. Eur. J. Med. Chem. 1998, 33, 383–397. [Google Scholar] [CrossRef]
- Ferrarini, P.L.; Mori, C.; Badawneh, M.; Calderone, V.; Greco, R.; Manera, C.; Martinelli, A.; Nieri, P.; Saccomanni, G. Synthesis and β-blocking activity of (R,S)-(E)-oximeethers of 2,3-dihydro-1,8-naphthyridine and 2,3-dihydrothiopyrano[2,3-b]pyridine: Potential antihypertensive agents – Part IX. Eur. J. Med. Chem. 2000, 35, 815. [Google Scholar] [CrossRef]
- Graf, H.; Franz, L.; Sauter, H.; Ammermann, E.; Pommer, E-H. Substituted 1,8-naphthyridine derivatives and fungicides containing them. U.S. Patent 4,801,592, 31 January 1989. [Google Scholar]
- Saupe, T.; Schaefer, P.; Meyer, N.; Wuerzer, B.; Westphalen, K.O. Substituted 1,8-naphthyridines, their preparation and their use as antidotes. U.S. Patent 5,258,356, 2 November 1993. [Google Scholar]
- Cotrel, C.; Guyon, C.; Roussel, G.; Taurand, G. Anxiolytic amides derived from certain 1,8-naphthyridine-2-amines. U.S. Patent 4,753,933, 28 June 1988. [Google Scholar]
- Litvinov, V.P.; Roman, S.V.; Dyachenko, V.D. Naphthyridines. Structure, physicochemical properties and general methods of synthesis. Russ. Chem. Rev. 2000, 69, 201–220. [Google Scholar] [CrossRef]
- Smart, B.E. Fluorine substituent effects (on bioactivity). J. Fluorine Chem. 2001, 109, 3–11. [Google Scholar] [CrossRef]
- Druzhinin, S.V.; Balenkova, E.S.; Nenajdenko, V.G. Recent advances in the chemistry of α,β-unsatured trifluoromethylketones. Tetrahedron 2007, 63, 7753–7808. [Google Scholar]
- Eichler, E.; Rooney, C.S.; Williams, H.W.R. 1,8-Naphthyridines. Part I. Synthesis of some trifluoromethyl-1,8-naphthyridines derivatives. J. Heterocycl. Chem. 1976, 13, 41–42. [Google Scholar] [CrossRef]
- Naik, T.R.R.; Naik, H.S.B.; Raghavendra, M.; Naik, S.G.K. Synthesis of thieno[2,3-b]benzo[1,8]naphthyridine-2-carboxylic acids under microwave irradiation and interaction with DNA studies. ARKIVOC 2006, xv, 84–94. [Google Scholar]
- Chen, S.; Chen, R.; He, M.; Pang, R.; Tan, Z.; Yang, M. Design, synthesis, and biological evaluation of novel quinoline derivatives as HIV–1 Tat–TAR interaction inhibitors. Bioorg. Med. Chem. 2009, 17, 1948–1956. [Google Scholar] [CrossRef]
- Pizzio, L.; Romanelli, G.; Vázquez, P.; Autino, J.; Blanco, M.; Cáceres, C. Keggin heteropolyacid-based catalysts for the preparation of substituted ethyl β-arylaminocrotonates, intermediates in the synthesis of 4-quinolones. Appl. Catal. A 2006, 308, 1531–1560. [Google Scholar]
- Hauser, C.R.; Reynolds, G.A. Reactions of β-keto esters with aromatic amines. Syntheses of 2- and 4-hydroxyquinoline derivatives. J. Am. Chem. Soc. 1948, 70, 2402–2404. [Google Scholar]
- Manske, R.H.F.; Kulka, M. The Skraup synthesis of quinolines. Org. React. 1953, 7, 59. [Google Scholar]
- Bonacorso, H.G.; Righi, F.J.; Rodrigues, C.A.; Cechinel, C.A.; Costa, M.B.; Wastowski, A.D.; Martins, M.A.P.; Zanatta, N. New efficient approach for the synthesis of 2-alkyl(aryl)substituted 4H-pyrido[1,2-a]pyrimidin-4-ones. J. Heterocycl. Chem. 2006, 43, 229–233. [Google Scholar] [CrossRef]
- Brown, E.V. 1,8-Naphthyridines. I. Derivatives of 2- and 4-methyl-1,8-naphthyridines. J. Org. Chem. 1965, 30, 1607–1610. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Lourega, R.V.; Wastowski, A.D.; Flores, A.F.C.; Zanatta, N.; Martins, M.A.P. β-Alkoxyvinyl trichloromethyl ketones as N-heterocyclic acylating agent. A new access to 5H-thiazolo[3,2-a]pyrimidin-5-ones. Tetrahedron Lett. 2002, 43, 9315–9318. [Google Scholar]
- Bonacorso, H.G.; Lourega, R.V.; Deon, E.D.; Zanatta, N.; Martins, M.A.P. The first synthesis of dihydro-3H-pyrido[2,3-b][1,4]diazepinols and a new alternative approach for diazepinone analogues. Tetrahedron Lett. 2007, 48, 4835–4838. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Lourega, R.V.; Righi, F.J.; Deon, E.D.; Zanatta, N.; Martins, M.A.P. Preparation of new 2-amino- and 2,3-diamino-pyridine trifluoroacetyl enamine derivatives and their application to the synthesis of trifluoromethyl-containing 3H-pyrido[2,3-b][1,4]diazepinols. J. Heterocycl. Chem. 2008, 45, 1679–1686. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Lourega, R.V.; Porte, L.M.F.; Deon, E.D.; Flores, A.F.C.; Zanatta, N.; Martins, M.A.P. Regiospecific synthesis of 3H-pyrido[2,3-b][1,4]diazepin-4(5H)-ones via haloform reaction with the isolation of N3-[3-oxo-4,4,4-trichloroalk-1-en-1-yl]-2,3-diaminopyridine intermediates. J. Heterocycl. Chem. 2009, 46, 603–609. [Google Scholar] [CrossRef]
- Gerus, I.I.; Gorbunova, M.G.; Kukhar, V.P. Ethoxyvinyl polyfluoroalkyl ketones – versatile synthones in fluoroorganic chemistry. J. Fluorine Chem. 1994, 69, 195–198. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Duarte, S.H.G.; Zanatta, N.; Martins, M.A.P. Regiospecific Synthesis of 3-alkyl-2-aryl-4-trifluoromethylbenzo[h]quinolines by intramolecular cyclization of N-(2-alkyl-1-aryl-3-oxo-4,4,4-trifluorobut-1-en-1-yl)-1-naphthylamines. Synthesis 2002, 1037–1042. [Google Scholar]
- Bonacorso, H.G.; Drekener, R.L.; Rodríguez, I.R.; Vezzosi, R.P.; Costa, M.B.; Martins, M.A.P.; Zanatta, N. Synthesis of new fluorine-containing dihydrobenzo[c]acridines from trifluoroacetyl dihydronaphthalene and substituted anilines. J. Fluorine Chem. 2005, 126, 1384–1389. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Moraes, T.S.; Zanatta, N.; Martins, M.A.P.; Flores, A.F.C. Synthesis of new trifluoromethyl-containing cycloalka[b]quinolines derived from alkoxycycloalkenes. ARKIVOC 2008, xvi, 75–83. [Google Scholar]
- Bonacorso, H.G.; Moraes, T.S.; Zanatta, N.; Martins, M.A.P. Synthesis of new fluorine-containing 1,2,3,4-tetrahydroacridines. Synth. Commun. 2009, 39, 3677–3686. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Bittencourt, S.R.T.; Lourega, R.V.; Flores, A.F.C.; Zanatta, N.; Martins, M.A.P. A Convenient synthetic method for fully conjugated 3-alkyl- and 3-aryl-5-trifluoromethyl-1-methyl-1,2-thiazine 1-oxide from β-alkoxyvinyl trifluoromethyl ketones. Synthesis 2000, 1431–1434. [Google Scholar]
- Bonacorso, H.G.; Wentz, A.P.; Bittencourt, S.T.R.; Marques, L.M.L.; Zanatta, N.; Martins, M.A.P. Synthesis of some N-[1-alkyl(aryl)-3-oxo-4,4,4-trichloro(trifluoro)-1-buten-1-yl]-o-aminophenols and o-phenylenediamines as potential anticancer agents. Synth. Commum. 2002, 32, 335–341. [Google Scholar] [CrossRef]
- Bonacorso, H.G.; Andrighetto, R.; Zanatta, N.; Martins, M.A.P. The unexpected cyclization routes of N,N’-bis(oxotrifluoroalkenyl)-1,3-phenylenediamines in polyphosphoric acid medium. Tetrahedron Lett. 2010, 51, 3752–3755. [Google Scholar] [CrossRef]
- Effenberger, F.; Maier, R.; Schonwalder, K.H.; Ziegler, T. Enolether, XIII. Die acylierung von enolethern mit reaktiven carbonsäure-chloriden. Chem. Ber. 1982, 115, 2766–2782. [Google Scholar] [CrossRef]
- Kamitori, Y.; Hojo, M.; Masuda, R.; Fujitani, T.; Kobuchi, T.; Nishigaki, T. A new convenient synthetic method for 3-allyl-1,1,1-trifluoroacetylacetone and its derivatives. Synthesis 1986, 340–342. [Google Scholar]
- Hojo, M.; Masuda, R.; Okada, E. A useful one-step synthesis of β-trihaloacetylvinyl ethers and trihaloacetylketene acetals. Synthesis 1986, 1013–1014. [Google Scholar]
- Colla, A.; Martins, M.A.P.; Clar, G.; Krimmer, S.; Fischer, P. Trihaloacetylated enol ethers - general synthetic procedure and heterocyclic ring closure reactions with hydroxylamine. Synthesis 1991, 483–486. [Google Scholar]
- Ezell, E.L.; Thummel, R.P.; Martin, G.E. Correlation of resonances of strongly coupled spin systems via responses due to strong coupling in homonuclear two-dimensional J-resolved spectra: Total assignment of the 1H-NMR spectrum of 2-(2'-pyridyl)-1,8-naphthyridine. J. Heterocycl.Chem. 1984, 21, 817–823. [Google Scholar] [CrossRef]
- Flores, A.F.C.; Siqueira, G.M.; Freitag, R.; Zanatta, N.; Martins, M.A.P. Síntese de 2-trialoacetil-cicloexanonas e -pentanonas: Um estudo comparativo dos rendimentos de reação de enoléteres, cetais e enaminas frente à trialometilacetilantes. Quim. Nova 1994, 17, 298–300. [Google Scholar]
- Bonacorso, H.G.; Costa, M.B.; Moura, S.; Pizzuti, L.; Martins, M.A.P.; Zanatta, N.; Flores, A.F.C. Synthesis, 17O NMR spectroscopy and structure of 2-trifluoroacetyl-1-methoxycycloalkenes. J. Fluorine Chem. 2005, 126, 1396–1402. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 2b, 2k, 3a-m, 4c, 4f, 4g, 4i, 4j are available from the authors.
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bonacorso, H.G.; Andrighetto, R.; Krüger, N.; Zanatta, N.; Martins, M.A.P. General Pathway for a Convenient One-Pot Synthesis of Trifluoromethyl-Containing 2-amino-7-alkyl(aryl/heteroaryl)-1,8-naphthyridines and Fused Cycloalkane Analogues. Molecules 2011, 16, 2817-2832. https://doi.org/10.3390/molecules16042817
Bonacorso HG, Andrighetto R, Krüger N, Zanatta N, Martins MAP. General Pathway for a Convenient One-Pot Synthesis of Trifluoromethyl-Containing 2-amino-7-alkyl(aryl/heteroaryl)-1,8-naphthyridines and Fused Cycloalkane Analogues. Molecules. 2011; 16(4):2817-2832. https://doi.org/10.3390/molecules16042817
Chicago/Turabian StyleBonacorso, Helio G., Rosália Andrighetto, Nícolas Krüger, Nilo Zanatta, and Marcos A. P. Martins. 2011. "General Pathway for a Convenient One-Pot Synthesis of Trifluoromethyl-Containing 2-amino-7-alkyl(aryl/heteroaryl)-1,8-naphthyridines and Fused Cycloalkane Analogues" Molecules 16, no. 4: 2817-2832. https://doi.org/10.3390/molecules16042817