
Current Applications of Absolute Bacterial Quantification in Microbiome Studies and Decision-Making Regarding Different Biological Questions


Abstract
:1. Introduction
2. Importance of Absolute Quantification for Biological Questions
3. Brief Description of Advanced Absolute Quantification Methods
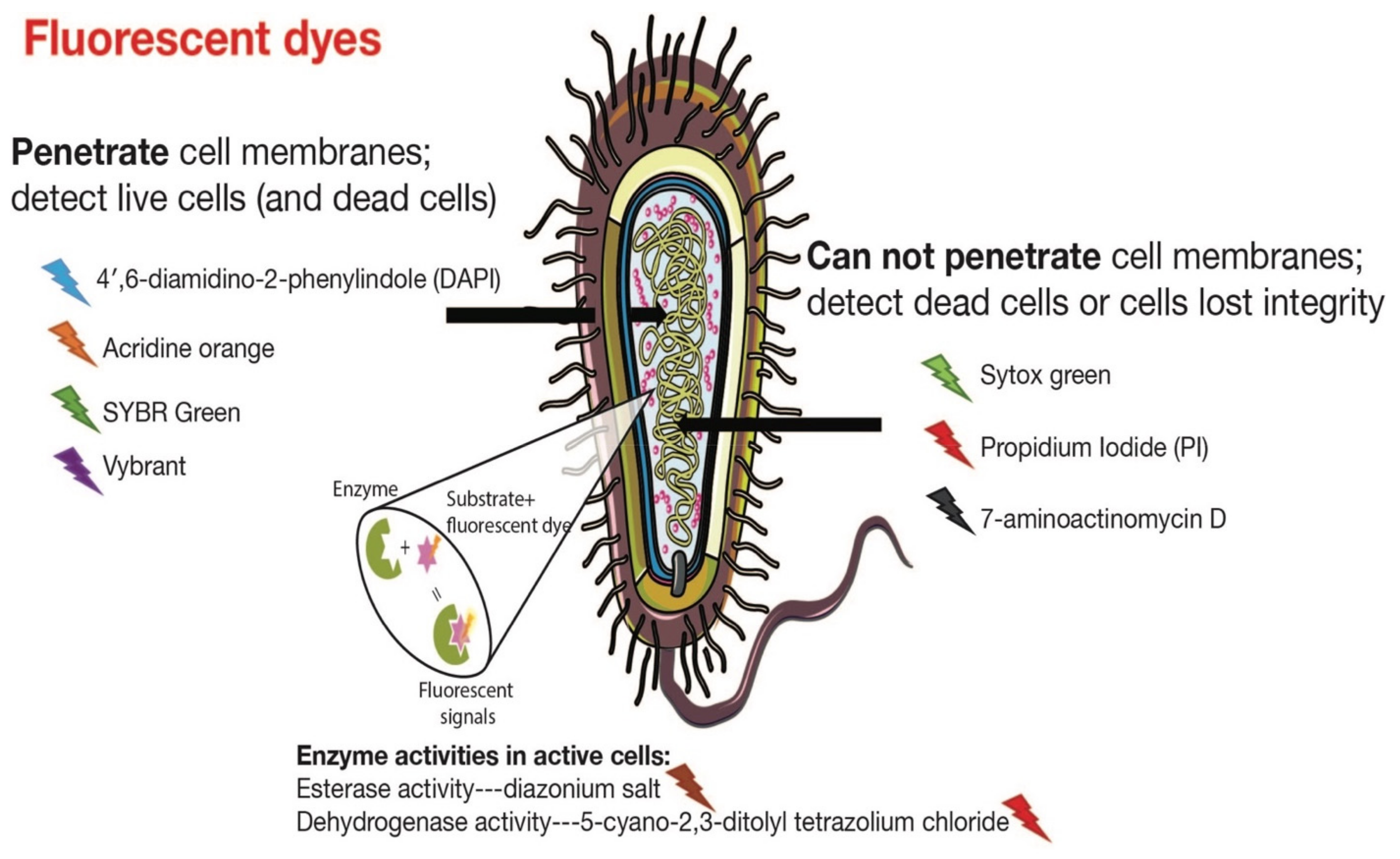
3.1. Fluorescence Spectroscopy
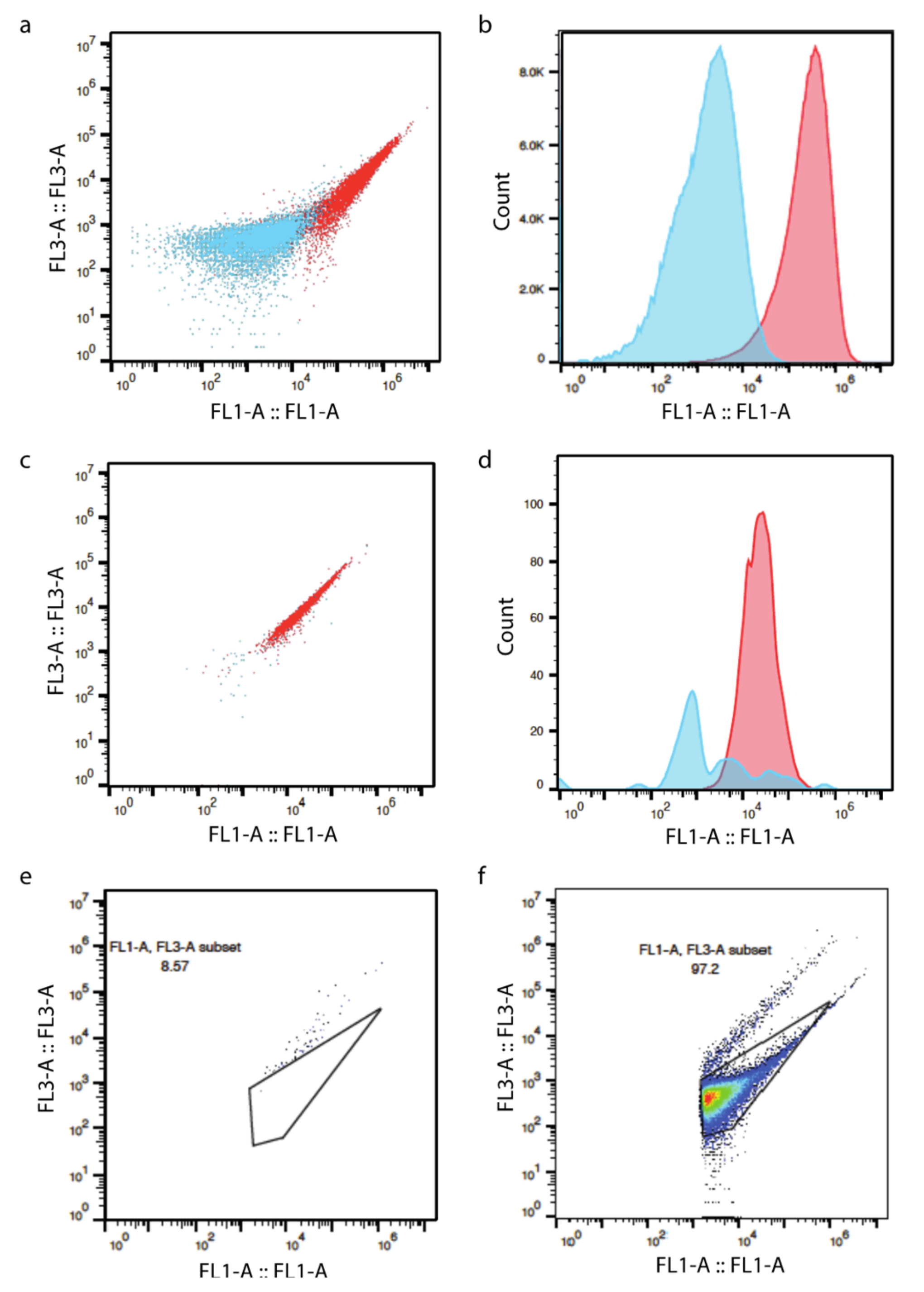
3.2. Flow Cytometry
3.3. Spike-In with Reference Markers
3.4. 16S qPCR and qRT-PCR Quantification
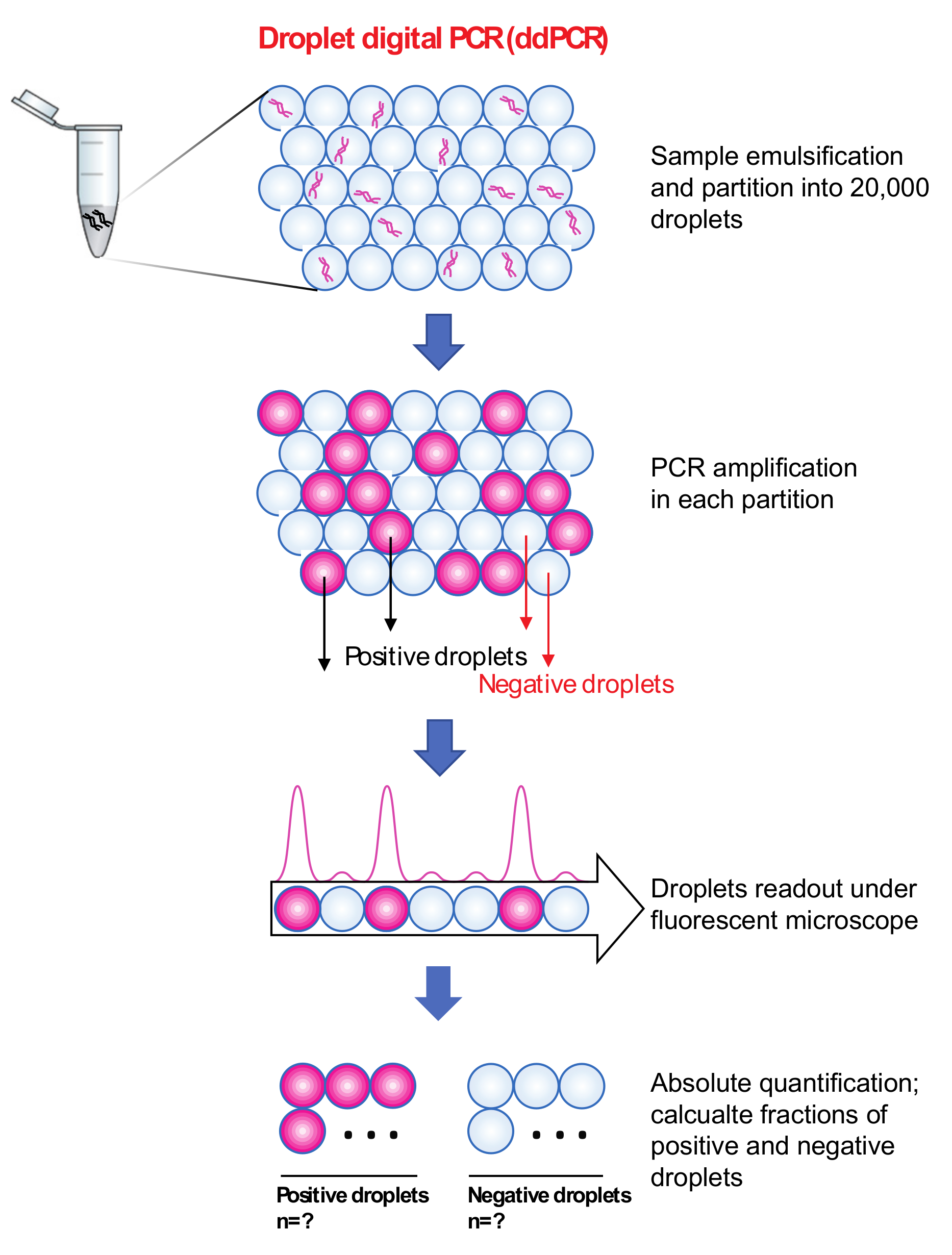
3.5. Droplet Digital PCR (ddPCR)
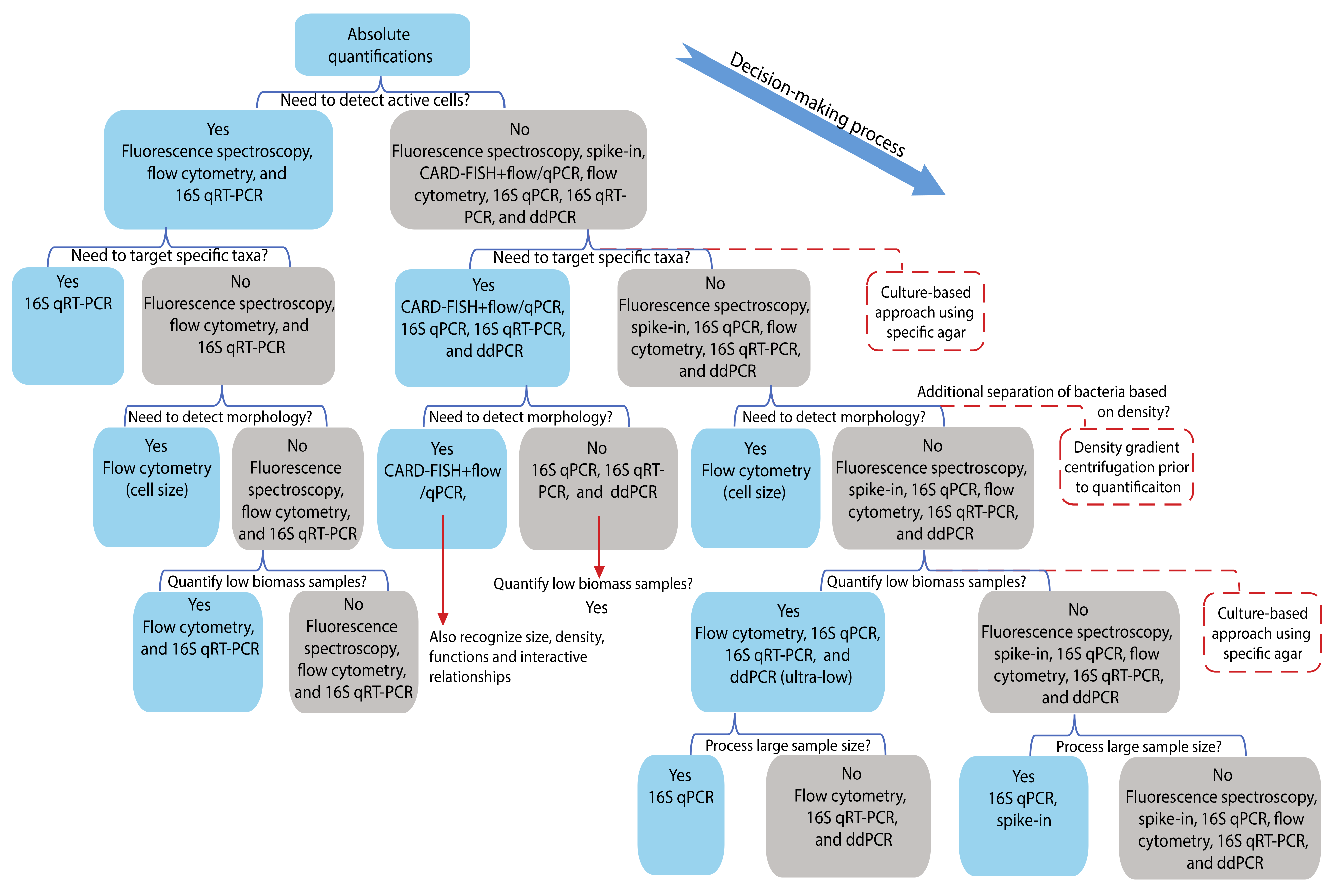
4. Decision-Making Regarding Different Biological Questions
4.1. Differentiation between Active and Dead Cells
4.2. Absolute Quantification of Specific Taxa of Interest
4.3. Absolute Quantification of Low Biomass Bacterial Samples
4.4. Rapid Quantification for a Large Number of Samples
4.5. Absolute Quantification of Bacteria Based on Other Features
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, Z.; Qu, Y.; Li, S.; Feng, K.; Wang, S.; Cai, W.; Liang, Y.; Li, H.; Xu, M.; Yin, H. Soil bacterial quantification approaches coupling with relative abundances reflecting the changes of taxa. Sci. Rep. 2017, 7, 4837. [Google Scholar] [CrossRef]
- Smets, W.; Leff, J.W.; Bradford, M.A.; McCulley, R.L.; Lebeer, S.; Fierer, N. A method for simultaneous measurement of soil bacterial abundances and community composition via 16S rRNA gene sequencing. Soil Biol. Biochem. 2016, 96, 145–151. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Tsai, T.; Wei, X.; Zuo, B.; Davis, E.; Rehberger, T.; Hernandez, S.; Jochems, E.J.; Maxwell, C.V.; Zhao, J. Effect of Lactylate and Bacillus subtilis on growth performance, peripheral blood cell profile, and gut microbiota of nursery pigs. Microorganisms 2021, 9, 803. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7, 109. [Google Scholar] [CrossRef] [Green Version]
- Harmsen, H.J.; Pouwels, S.D.; Funke, A.; Bos, N.A.; Dijkstra, G. Crohn’s disease patients have more IgG-binding fecal bacteria than controls. Clin. Vaccine Immunol. 2012, 19, 515–521. [Google Scholar] [CrossRef]
- Longo, A.V.; Zamudio, K.R. Temperature variation, bacterial diversity, and fungal infection dynamics in the amphibian skin. Mol. Ecol. 2017, 26, 4787–4797. [Google Scholar] [CrossRef]
- Stämmler, F.; Gläsner, J.; Hiergeist, A.; Holler, E.; Weber, D.; Oefner, P.J.; Gessner, A.; Spang, R. Adjusting microbiome profiles for differences in microbial load by spike-in bacteria. Microbiome 2016, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Morton, J.T.; Sanders, J.; Quinn, R.A.; McDonald, D.; Gonzalez, A.; Vázquez-Baeza, Y.; Navas-Molina, J.A.; Song, S.J.; Metcalf, J.L.; Hyde, E.R.; et al. Balance trees reveal microbial niche differentiation. MSystems 2017, 2, e00162-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peralta-Sánchez, J.M.; Martín-Platero, A.M.; Wegener-Parfrey, L.; Martínez-Bueno, M.; Rodríguez-Ruano, S.; Navas-Molina, J.A.; Vázquez-Baeza, Y.; Martín-Gálvez, D.; Martín-Vivaldi, M.; Ibáñez-Álamo, J.D.; et al. Bacterial density rather than diversity correlates with hatching success across different avian species. FEMS Microbiol. Ecol. 2018, 94, fiy022. [Google Scholar] [CrossRef] [Green Version]
- Tourlousse, D.M.; Yoshiike, S.; Ohashi, A.; Matsukura, S.; Noda, N.; Sekiguchi, Y. Synthetic spike-in standards for high-throughput 16S rRNA gene amplicon sequencing. Nucleic Acids Res. 2017, 45, e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandeputte, D.; Kathagen, G.; D’hoe, K.; Vieira-Silva, S.; Valles-Colomer, M.; Sabino, J.; Wang, J.; Tito, R.Y.; De Commer, L.; Darzi, Y.; et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 2017, 551, 507–511. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008, 453, 620. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-de-Castañeda, R.; Vela, A.I.; Lobato, E.; Briones, V.; Moreno, J. Prevalence of potentially pathogenic culturable bacteria on eggshells and in cloacae of female Pied Flycatchers in a temperate habitat in central Spain. J. Field Ornithol. 2011, 82, 215–224. [Google Scholar] [CrossRef]
- Hansen, C.M.; Meixell, B.W.; Van Hemert, C.; Hare, R.F.; Hueffer, K. Microbial infections are associated with embryo mortality in Arctic-nesting geese. Appl. Environ. Microbiol. 2015, 81, 5583–5592. [Google Scholar] [CrossRef] [Green Version]
- Soler, J.; Peralta-Sánchez, J.; Martín-Platero, A.; Martín-Vivaldi, M.; Martínez-Bueno, M.; Møller, A. The evolution of size of the uropygial gland: Mutualistic feather mites and uropygial secretion reduce bacterial loads of eggshells and hatching failures of European birds. J. Evol. Biol. 2012, 25, 1779–1791. [Google Scholar] [CrossRef]
- Møller, A.P.; Czirjak, G.Á.; Heeb, P. Feather micro-organisms and uropygial antimicrobial defences in a colonial passerine bird. Funct. Ecol. 2009, 23, 1097–1102. [Google Scholar] [CrossRef]
- Finnegan, S.; Droser, M.L. Relative and absolute abundance of trilobites and rhynchonelliform brachiopods across the Lower/Middle Ordovician boundary, eastern Basin and Range. Paleobiology 2005, 31, 480–502. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 1–22. [Google Scholar]
- Pearson, K. Mathematical contributions to the theory of evolution.—On a form of spurious correlation which may arise when indices are used in the measurement of organs. Proc. R. Soc. Lond. 1897, 60, 489–498. [Google Scholar]
- Lovell, D.; Taylor, J.; Zwart, A.; Helliwell, C. Caution! Compositions! Can Constraints on Omics Data Lead Analyses Astray; CSIRO 1: Canberra, Australia, 2010; pp. 1–44. [Google Scholar]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, S.; Xu, Z.Z.; Peddada, S.; Amir, A.; Bittinger, K.; Gonzalez, A.; Lozupone, C.; Zaneveld, J.R.; Vázquez-Baeza, Y.; Birmingham, A.; et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 2017, 5, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matchado, M.S.; Lauber, M.; Reitmeier, S.; Kacprowski, T.; Baumbach, J.; Haller, D.; List, M. Network analysis methods for studying microbial communities: A mini review. Comput. Struct. Biotechnol. J. 2021, 19, 2687–2698. [Google Scholar] [CrossRef]
- Yang, H.; Huang, X.; Fang, S.; Xin, W.; Huang, L.; Chen, C. Uncovering the composition of microbial community structure and metagenomics among three gut locations in pigs with distinct fatness. Sci. Rep. 2016, 6, 27427. [Google Scholar] [CrossRef] [Green Version]
- Luhung, I.; Uchida, A.; Lim, S.B.; Gaultier, N.E.; Kee, C.; Lau, K.J.; Gusareva, E.S.; Heinle, C.E.; Wong, A.; Premkrishnan, B.N.; et al. Experimental parameters defining ultra-low biomass bioaerosol analysis. NPJ Biofilms Microbiomes 2021, 7, 1–11. [Google Scholar] [CrossRef]
- Mahnert, A.; Vaishampayan, P.; Probst, A.J.; Auerbach, A.; Moissl-Eichinger, C.; Venkateswaran, K.; Berg, G. Cleanroom maintenance significantly reduces abundance but not diversity of indoor microbiomes. PLoS ONE 2015, 10, e0134848. [Google Scholar] [CrossRef] [PubMed]
- Minich, J.J.; Zhu, Q.; Janssen, S.; Hendrickson, R.; Amir, A.; Vetter, R.; Hyde, J.; Doty, M.M.; Stillwell, K.; Benardini, J. KatharoSeq Enables High-Throughput Microbiome Analysis from Low-Biomass Samples. mSystems 2018, 3, e00218-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, B.; Button, D. Characterizing aquatic bacteria according to population, cell size, and apparent DNA content by flow cytometry. Cytom. Part. A 1989, 10, 70–76. [Google Scholar] [CrossRef]
- Saint-Ruf, C.; Cordier, C.; Mégret, J.; Matic, I. Reliable detection of dead microbial cells by using fluorescent hydrazides. Appl. Environ. Microbiol. 2010, 76, 1674–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzaev, M.; Li, X.; Park, C.; Leung, W.-Y.; Roberts, L. Comparison of Nucleic Acid Gel Stains Cell Permeability, Safety, and Sensitivity of Ethidium Bromide Alternatives; Biotium: San Francisco, CA, USA, 2017. [Google Scholar]
- Roth, B.L.; Poot, M.; Yue, S.T.; Millard, P.J. Bacterial viability and antibiotic susceptibility testing with SYTOX green nucleic acid stain. Appl. Environ. Microbiol. 1997, 63, 2421–2431. [Google Scholar] [CrossRef] [Green Version]
- Herzenberg, L.A.; Parks, D.; Sahaf, B.; Perez, O.; Roederer, M.; Herzenberg, L.A. The history and future of the fluorescence activated cell sorter and flow cytometry: A view from Stanford. Clin. Chem. 2002, 48, 1819–1827. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, J.-E. Flow cytometry versus fluorescence microscopy. In Fluorescence Microscopy and Fluorescent Probes; Springer: Berlin/Heidelberg, Germany, 1996; pp. 61–66. [Google Scholar]
- Ambriz-Aviña, V.; Contreras-Garduño, J.A.; Pedraza-Reyes, M. Applications of flow cytometry to characterize bacterial physiological responses. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Prest, E.; Hammes, F.; Kötzsch, S.; van Loosdrecht, M.C.; Vrouwenvelder, J.S. Monitoring microbiological changes in drinking water systems using a fast and reproducible flow cytometric method. Water Res. 2013, 47, 7131–7142. [Google Scholar] [CrossRef] [PubMed]
- Seeger, M.; Stiel, A.C.; Ntziachristos, V. In vitro optoacoustic flow cytometry with light scattering referencing. Sci. Rep. 2021, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hammes, F.; Boon, N.; Chami, M.; Egli, T. Isolation and characterization of low nucleic acid (LNA)-content bacteria. Isme J. 2009, 3, 889–902. [Google Scholar] [CrossRef]
- Hammes, F.A.; Egli, T. New method for assimilable organic carbon determination using flow-cytometric enumeration and a natural microbial consortium as inoculum. Environ. Sci. Technol. 2005, 39, 3289–3294. [Google Scholar] [CrossRef]
- Van der Waaij, L.; Mesander, G.; Limburg, P.; Van der Waaij, D. Direct flow cytometry of anaerobic bacteria in human feces. Cytom. Part. A 1994, 16, 270–279. [Google Scholar] [CrossRef]
- Yang, L.; Lou, J.; Wang, H.; Wu, L.; Xu, J. Use of an improved high-throughput absolute abundance quantification method to characterize soil bacterial community and dynamics. Sci. Total Environ. 2018, 633, 360–371. [Google Scholar] [CrossRef]
- Kubista, M.; Andrade, J.M.; Bengtsson, M.; Forootan, A.; Jonák, J.; Lind, K.; Sindelka, R.; Sjöback, R.; Sjögreen, B.; Strömbom, L. The real-time polymerase chain reaction. Mol. Asp. Med. 2006, 27, 95–125. [Google Scholar] [CrossRef] [PubMed]
- Oldham, A.L.; Duncan, K.E. Similar gene estimates from circular and linear standards in quantitative PCR analyses using the prokaryotic 16S rRNA gene as a model. PLoS ONE 2012, 7, e51931. [Google Scholar] [CrossRef] [PubMed]
- Brankatschk, R.; Bodenhausen, N.; Zeyer, J.; Bürgmann, H. Simple absolute quantification method correcting for quantitative PCR efficiency variations for microbial community samples. Appl. Environ. Microbiol. 2012, 78, 4481–4489. [Google Scholar] [CrossRef] [Green Version]
- Callegari, M.; Crotti, E.; Fusi, M.; Marasco, R.; Gonella, E.; De Noni, I.; Romano, D.; Borin, S.; Tsiamis, G.; Cherif, A.; et al. Compartmentalization of bacterial and fungal microbiomes in the gut of adult honeybees. NPJ Biofilms Microbiomes 2021, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jian, C.; Luukkonen, P.; Yki-Järvinen, H.; Salonen, A.; Korpela, K. Quantitative PCR provides a simple and accessible method for quantitative microbiota profiling. PLoS ONE 2020, 15, e0227285. [Google Scholar] [CrossRef] [Green Version]
- Price, A.; Álvarez, L.A.; Whitby, C.; Larsen, J. How Many Microorganisms Are Present? Quantitative Reverse Transcription PCR (qRT-PCR); Springer: Berlin/Heidelberg, Germany, 2011; pp. 77–84. [Google Scholar]
- Ma, D.; Shanks, R.M.Q.; Davis, C.M., 3rd; Craft, D.W.; Wood, T.K.; Hamlin, B.R.; Urish, K.L. Viable bacteria persist on antibiotic spacers following two-stage revision for periprosthetic joint infection. J. Orthop Res. 2018, 36, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolan, A.; Burgess, C.M.; Barry, T.B.; Fanning, S.; Duffy, G. A novel quantitative reverse-transcription PCR (qRT-PCR) for the enumeration of total bacteria, using meat micro-flora as a model. J. Microbiol. Methods 2009, 77, 1–7. [Google Scholar] [CrossRef]
- Bui, X.T.; Wolff, A.; Madsen, M.; Bang, D.D. Reverse transcriptase real-time PCR for detection and quantification of viable Campylobacter jejuni directly from poultry faecal samples. Res. Microbiol. 2012, 163, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.; Behrens, S. Seasonal Dynamics of the Activated Sludge Microbiome in Sequencing Batch Reactors, Assessed Using 16S rRNA Transcript Amplicon Sequencing. Appl. Environ. Microbiol. 2020, 86, e00597-20. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.; Gibson, B.; Williams, A.; Alusta, P.; Buzatu, D.A.; Lee, Y.-J.; LiPuma, J.J.; Hussong, D.; Marasa, B.; Cerniglia, C.E. A comparison of culture-based, real-time PCR, droplet digital PCR and flow cytometric methods for the detection of Burkholderia cepacia complex in nuclease-free water and antiseptics. J. Ind. Microbiol. Biotechnol. 2020, 47, 475–484. [Google Scholar] [CrossRef]
- Gutiérrez-Aguirre, I.; Rački, N.; Dreo, T.; Ravnikar, M. Droplet digital PCR for absolute quantification of pathogens. In Plant Pathology; Humana Press: New York, NY, USA, 2015; pp. 331–347. [Google Scholar]
- Gobert, G.; Cotillard, A.; Fourmestraux, C.; Pruvost, L.; Miguet, J.; Boyer, M. Droplet digital PCR improves absolute quantification of viable lactic acid bacteria in faecal samples. J. Microbiol. Methods 2018, 148, 64–73. [Google Scholar] [CrossRef]
- Sze, M.A.; Abbasi, M.; Hogg, J.C.; Sin, D.D. A comparison between droplet digital and quantitative PCR in the analysis of bacterial 16S load in lung tissue samples from control and COPD GOLD 2. PLoS ONE 2014, 9, e110351. [Google Scholar] [CrossRef]
- Kim, T.G.; Jeong, S.Y.; Cho, K.S. Comparison of droplet digital PCR and quantitative real-time PCR for examining population dynamics of bacteria in soil. Appl. Microbiol. Biotechnol. 2014, 98, 6105–6113. [Google Scholar] [CrossRef] [PubMed]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, I.; Lindström, S.; Källgren, M.; Strålin, K.; Mölling, P. 16S rDNA droplet digital PCR for monitoring bacterial DNAemia in bloodstream infections. PLoS ONE 2019, 14, e0224656. [Google Scholar] [CrossRef] [PubMed]
- Berney, M.; Hammes, F.; Bosshard, F.; Weilenmann, H.-U.; Egli, T. Assessment and interpretation of bacterial viability by using the LIVE/DEAD BacLight Kit in combination with flow cytometry. Appl. Environ. Microbiol. 2007, 73, 3283–3290. [Google Scholar] [CrossRef] [Green Version]
- Vives-Rego, J.; Lebaron, P.; Nebe-von Caron, G. Current and future applications of flow cytometry in aquatic microbiology. FEMS Microbiol. Rev. 2000, 24, 429–448. [Google Scholar] [CrossRef] [PubMed]
- Auty, M.; Gardiner, G.; McBrearty, S.; O’sullivan, E.; Mulvihill, D.; Collins, J.; Fitzgerald, G.; Stanton, C.; Ross, R. Direct in situ viability assessment of bacteria in probiotic dairy products using viability staining in conjunction with confocal scanning laser microscopy. Appl. Environ. Microbiol. 2001, 67, 420–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barka, T.; Anderson, P.J. Histochemical methods for acid phosphatase using hexazonium pararosanilin as coupler. J. Histochem. Cytochem. 1962, 10. [Google Scholar] [CrossRef] [Green Version]
- Sieracki, M.E.; Cucci, T.L.; Nicinski, J. Flow cytometric analysis of 5-cyano-2, 3-ditolyl tetrazolium chloride activity of marine bacterioplankton in dilution cultures. Appl. Environ. Microbiol. 1999, 65, 2409–2417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Props, R.; Kerckhof, F.-M.; Rubbens, P.; De Vrieze, J.; Sanabria, E.H.; Waegeman, W.; Monsieurs, P.; Hammes, F.; Boon, N. Absolute quantification of microbial taxon abundances. ISME J. 2017, 11, 584–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, K.; Tsuji, H.; Asahara, T.; Matsumoto, K.; Takada, T.; Nomoto, K. Establishment of an analytical system for the human fecal microbiota, based on reverse transcription-quantitative PCR targeting of multicopy rRNA molecules. Appl. Environ. Microbiol. 2009, 75, 1961–1969. [Google Scholar] [CrossRef] [Green Version]
- Boyer, M.; Combrisson, J. Analytical opportunities of quantitative polymerase chain reaction in dairy microbiology. Int. Dairy J. 2013, 30, 45–52. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, Y.; Yan, Y.; Xue, H. Rapid and Sensitive Quantification of Anammox Bacteria by Flow Cytometric Analysis Based on Catalyzed Reporter Deposition Fluorescence In Situ Hybridization. Environ. Sci. Technol. 2019, 53, 6895–6905. [Google Scholar] [CrossRef]
- Piwosz, K.; Mukherjee, I.; Salcher, M.M.; Grujčić, V.; Šimek, K. CARD-FISH in the Sequencing Era: Opening a New Universe of Protistan Ecology. Front. Microbiol. 2021, 12, 397. [Google Scholar] [CrossRef] [PubMed]
- Piwosz, K.; Shabarova, T.; Pernthaler, J.; Posch, T.; Šimek, K.; Porcal, P.; Salcher, M.M. Bacterial and Eukaryotic Small-Subunit Amplicon Data Do Not Provide a Quantitative Picture of Microbial Communities, but They Are Reliable in the Context of Ecological Interpretations. mSphere 2020, 5, e00052-20. [Google Scholar] [CrossRef] [Green Version]
- Fadeev, E.; Cardozo-Mino, M.G.; Rapp, J.Z.; Bienhold, C.; Salter, I.; Salman-Carvalho, V.; Molari, M.; Tegetmeyer, H.E.; Buttigieg, P.L.; Boetius, A. Comparison of two 16S rRNA primers (V3–V4 and V4–V5) for studies of arctic microbial communities. Front. Microbiol. 2021, 12, 283. [Google Scholar] [CrossRef]
- Kuo, J.-T.; Chang, L.-L.; Yen, C.-Y.; Tsai, T.-H.; Chang, Y.-C.; Huang, Y.-T.; Chung, Y.-C. Development of Fluorescence In Situ Hybridization as a Rapid, Accurate Method for Detecting Coliforms in Water Samples. Biosensors 2021, 11, 8. [Google Scholar] [CrossRef]
- Jimenez, L.; Jashari, T.; Vasquez, J.; Zapata, S.; Bochis, J.; Kulko, M.; Ellman, V.; Gardner, M.; Choe, T. Real-time PCR detection of Burkholderia cepacia in pharmaceutical products contaminated with low levels of bacterial contamination. PDA J. Pharm. Sci. Technol. 2018, 72, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Furlan, J.P.R.; Pitondo-Silva, A.; Braz, V.S.; Gallo, I.F.L.; Stehling, E.G. Evaluation of different molecular and phenotypic methods for identification of environmental Burkholderia cepacia complex. World J. Microbiol. Biotechnol. 2019, 35, 39. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Fiskal, A.; Han, X.; Dubois, N.; Bernasconi, S.M.; Lever, M.A. Improving the accuracy of flow cytometric quantification of microbial populations in sediments: Importance of cell staining procedures. Front. Microbiol. 2019, 10, 720. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tandon, R.; Ancheta, C.; Arroyo, P.; Gilbert, J.A.; Stephens, B.; Kelley, S.T. Quantitative profiling of built environment bacterial and fungal communities reveals dynamic material dependent growth patterns and microbial interactions. Indoor Air 2021, 31, 188–205. [Google Scholar] [CrossRef] [PubMed]
- Luksamijarulkul, P.; Pipitsangjan, S. Microbial air quality and bacterial surface contamination in ambulances during patient services. Oman Med. J. 2015, 30, 104. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.-F.; Chen, C.-M.; Li, X.-Y.; Chen, J.-J.; Wang, Y.-G.; Ouyang, S.; Ji, T.-X.; Xia, Y.; Guo, X.-G. Development of a droplet digital PCR method for detection of Streptococcus agalactiae. BMC Microbiol. 2020, 20, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, S.F.; Smith, B.J.; Hein, R.; Roller, B.R.; Schmidt, T.M. rrnDB: Improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 2015, 43, D593–D598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Howe, S.; Wei, X.; Deng, F.; Tsai, T.; Chai, J.; Xiao, Y.; Yang, H.; Maxwell, C.V.; Li, Y. Comprehensive Cultivation of the Swine Gut Microbiome Reveals High Bacterial Diversity and Guides Bacterial Isolation in Pigs. mSystems 2021, 6, e0047721. [Google Scholar]
- Carson, C.F.; Inglis, T.J. Air sampling to assess potential generation of aerosolized viable bacteria during flow cytometric analysis of unfixed bacterial suspensions. Gates Open Res. 2017, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, J.; Yang, L.; Wang, H.; Wu, L.; Xu, J. Assessing soil bacterial community and dynamics by integrated high-throughput absolute abundance quantification. PeerJ 2018, 6, e4514. [Google Scholar] [CrossRef] [PubMed]
- Longnecker, K.; Sherr, B.F.; Sherr, E. Activity and phylogenetic diversity of bacterial cells with high and low nucleic acid content and electron transport system activity in an upwelling ecosystem. Appl. Environ. Microbiol. 2005, 71, 7737–7749. [Google Scholar] [CrossRef] [Green Version]
- Salcher, M.M.; Pernthaler, J.; Posch, T. Seasonal bloom dynamics and ecophysiology of the freshwater sister clade of SAR11 bacteria “that rule the waves” (LD12). ISME J. 2011, 5, 1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, Y.; Kim, C.; Nagata, T. Vertical and seasonal variations of bacterioplankton subgroups with different nucleic acid contents: Possible regulation by phosphorus. Appl. Environ. Microbiol. 2005, 71, 5828–5836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasol, J.M.; Del Giorgio, P.A. Using flow cytometry for counting natural planktonic bacteria and understanding the structure of planktonic bacterial communities. Sci. Mar. 2000, 64, 197–224. [Google Scholar] [CrossRef] [Green Version]
- Porter, J.; Edwards, C.; Morgan, J.; Pickup, R. Rapid, automated separation of specific bacteria from lake water and sewage by flow cytometry and cell sorting. Appl. Environ. Microbiol. 1993, 59, 3327–3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Chen, W.; Zhang, H.; Peng, L.; Liu, L.; Han, Z.; Wan, N.; Li, L.; Song, L. A flow cytometer based protocol for quantitative analysis of bloom-forming cyanobacteria (Microcystis) in lake sediments. J. Environ. Sci. 2012, 24, 1709–1716. [Google Scholar] [CrossRef]
- Liu, J.; Hao, Z.; Ma, L.; Ji, Y.; Bartlam, M.; Wang, Y. Spatio-temporal variations of high and low nucleic acid content bacteria in an exorheic river. PLoS ONE 2016, 11, e0153678. [Google Scholar] [CrossRef] [PubMed]
- Palesse, S.; Colombet, J.; Ram, A.P.; Sime-Ngando, T. Linking host prokaryotic physiology to viral lifestyle dynamics in a temperate freshwater lake (Lake Pavin, France). Microb. Ecol. 2014, 68, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Read, D.S.; Gweon, H.S.; Bowes, M.J.; Newbold, L.K.; Field, D.; Bailey, M.J.; Griffiths, R.I. Catchment-scale biogeography of riverine bacterioplankton. ISME J. 2015, 9, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, C.E.; Bochdansky, A.B. A simple separation method for downstream biochemical analysis of aquatic microbes. J. Microbiol. Methods 2015, 111, 78–86. [Google Scholar] [CrossRef]
- Graham, J. Biological Centrifugation; Garland Science: New York, NY, USA, 2020. [Google Scholar]
- Mortimer, M.; Petersen, E.J.; Buchholz, B.A.; Holden, P.A. Separation of bacteria, protozoa and carbon nanotubes by density gradient centrifugation. Nanomaterials 2016, 6, 181. [Google Scholar] [CrossRef]
- Hinzke, T.; Kleiner, M.; Meister, M.; Schlüter, R.; Hentschker, C.; Pané-Farré, J.; Hildebrandt, P.; Felbeck, H.; Sievert, S.M.; Bonn, F. Bacterial symbiont subpopulations have different roles in a deep-sea symbiosis. eLife 2021, 10, e58371. [Google Scholar] [CrossRef] [PubMed]
- Heinrichs, M.E.; De Corte, D.; Engelen, B.; Pan, D. An Advanced Protocol for the Quantification of Marine Sediment Viruses via Flow Cytometry. Viruses 2021, 13, 102. [Google Scholar] [CrossRef]
- Blaud, A.; Maïder, A.; Clark, I.M. Quantification of Ammonia Oxidizing Bacterial Abundances in Environmental Samples by Quantitative-PCR. In The Plant Microbiome; Springer: Berlin/Heidelberg, Germany, 2021; pp. 135–146. [Google Scholar]
- Lei, L.; Gu, J.; Wang, X.; Song, Z.; Yu, J.; Wang, J.; Dai, X.; Zhao, W. Effects of phosphogypsum and medical stone on nitrogen transformation, nitrogen functional genes, and bacterial community during aerobic composting. Sci. Total Environ. 2021, 753, 141746. [Google Scholar] [CrossRef]
- Kubota, K. CARD-FISH for environmental microorganisms: Technical advancement and future applications. Microbes Environ. 2013, 28, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Matturro, B.; Rossetti, S.; Leitão, P. CAtalyzed reporter deposition fluorescence in situ hybridization (CARD-FISH) for complex environmental samples. In Fluorescence In-Situ Hybridization (FISH) for Microbial Cells; Springer: New York, NY, USA, 2021; pp. 129–140. [Google Scholar]
- Stern, R.; Kraberg, A.; Bresnan, E.; Kooistra, W.H.; Lovejoy, C.; Montresor, M.; Morán, X.A.G.; Not, F.; Salas, R.; Siano, R. Molecular analyses of protists in long-term observation programmes—current status and future perspectives. J. Plankton Res. 2018, 40, 519–536. [Google Scholar] [CrossRef]
- Priest, T.; Fuchs, B.; Amann, R.; Reich, M. Diversity and biomass dynamics of unicellular marine fungi during a spring phytoplankton bloom. Environ. Microbiol. 2021, 23, 448–463. [Google Scholar] [CrossRef] [PubMed]
- Šimek, K.; Grujčić, V.; Mukherjee, I.; Kasalický, V.; Nedoma, J.; Posch, T.; Mehrshad, M.; Salcher, M.M. Cascading effects in freshwater microbial food webs by predatory Cercozoa, Katablepharidacea and ciliates feeding on aplastidic bacterivorous cryptophytes. FEMS Microbiol. Ecol. 2020, 96, fiaa121. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Absolute Quantification Method | Major Applications (Published) | Advantages | Limitations/Concerns | References |
|---|---|---|---|---|
| Fluorescence spectroscopy | Aquatic, soil, food and beverage, and air | High affinity; multiple dye selection to distinguish both live and dead cells | Fail to stain dead cells with complete DNA degradation; some dyes bind both DNA and RNA | Gordon et al., 2017, Guzaev et al., 2017, Saint-Ruf et al., 2010, Sieracki et al., 1999, Auty et al., 2001, |
| CARD-FISH + flow cytometry/qPCR | Aquatic | Direct quantification of specific taxa; detects both live and dead cells; provides insights for function, morphology, and ecology among taxa | Large population of cells are required for rare taxa detection; possibility of unspecific probe binding; Sample fixation may cause operation and efficiency biases; background noise | Hinzke et al., 2021, Kuo et al., 2021, Piwosz et al., 2021, Priest et al., 2021, Neuenschwander et al., 2015, Kubota et al., 2013 |
| Flow cytometry | Feces, aquatic, and soil | Rapid; single cell enumeration; flexible parameters based on physiological characteristics; capability to differentiate live and dead cells | Background noise exclusion may be required; gating strategy; dilution may be required; not ideal for complex systems/heterogeneous samples | Luhung et al., 2021, Heinrichs et al., 2021, Xu et al., 2021, Zhu et al., 2019, Deng et a., 2019, Vandeputte et al., 2017, Prest et al., 2013, Berney et al., 2007, Longnecker et al., 2005, Salcher et al., 2011 |
| Spike-in with internal reference | Soil, sludge, and feces | Rapid; easy incorporation into high throughput sequencing; high sensitivity; easy handling | Internal reference, spiking amount, and spiking time point can greatly affect the accuracy; 16S rRNA copy number calibration possibly needed. | Yang et al., 2018, Tourlousse et al., 2017, Smets et al., 2016, Lou et al., 2018, Stämmler et al., 2016 |
| 16S qPCR | Feces, clinical (lung), soil, plant, air, and aquatic | Directly quantifies specific taxa; cost-effective and easy handling; high sensitivity; compatible with low biomass samples | 16S rRNA copy number calibration may be needed; PCR-related biases exist; standard curves are required | Luhung et al., 2021, Callegari et al., 2021, Blaud et al., 2021, Lei et al., 2021, Jian et al., 2020, Vandeputte et al., 2017, Stoddard et al., 2015, Sze et al., 2014, Brankatschk et al., 2012 |
| 16S qRT-PCR | Clinical (joint infection), food safety, feces, sludge, water remediation, and soil | High resolution and sensitivity; directly quantifies specific taxa; detects active cells; compatible with low biomass samples | More of an approximation for protein synthesis than overall cell count; unstable RNA/RNA degradation; 16S rRNA copy number calibration may be needed | Ma et al., 2018, Johnston and Behrens, 2020, Bui et al., 2012, Boyer and Combrisson, 2013, Kim et al., 2014, Stoddard et al., 2015, Matsuda et al., 2009 |
| ddPCR | Clinical (lung, bloodstream infection), air, feces, and soil | Applicable to low concentrations of DNA; directly quantify specific taxa; high throughput capabilities, and no standard curve needed; compatible with low biomass samples | Dilutions are required for high concentrated template; may require a large number of replicates | Luhung et al., 2021, Ahn et al., 2020, Zeng et al., 2020, Sze et al., 2014, Kim et al., 2014, Ziegler et al., 2019, Gobert et al., 2018 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Howe, S.; Deng, F.; Zhao, J. Current Applications of Absolute Bacterial Quantification in Microbiome Studies and Decision-Making Regarding Different Biological Questions. Microorganisms 2021, 9, 1797. https://doi.org/10.3390/microorganisms9091797
Wang X, Howe S, Deng F, Zhao J. Current Applications of Absolute Bacterial Quantification in Microbiome Studies and Decision-Making Regarding Different Biological Questions. Microorganisms. 2021; 9(9):1797. https://doi.org/10.3390/microorganisms9091797
Chicago/Turabian StyleWang, Xiaofan, Samantha Howe, Feilong Deng, and Jiangchao Zhao. 2021. "Current Applications of Absolute Bacterial Quantification in Microbiome Studies and Decision-Making Regarding Different Biological Questions" Microorganisms 9, no. 9: 1797. https://doi.org/10.3390/microorganisms9091797