
Impact of Marine Aquaculture on the Microbiome Associated with Nearby Holobionts: The Case of Patella caerulea Living in Proximity of Sea Bream Aquaculture Cages

 , , , , ,
, , , , ,
Abstract
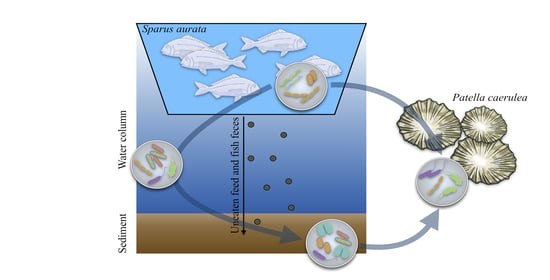
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Site Description, Samples Collection, and Environmental Data
2.2. Microbial DNA Extraction, 16S rRNA Gene Amplification, and Sequencing
2.3. Bioinformatics and Statistics
3. Results
3.1. Diversity and Compositional Structure of the Marine Microbial Communities at Aquaculture and Control Site in the Licata Harbor
3.2. Changes in the Limpet Microbiome and Surrounding Metacommunities at the Aquaculture Site in the Licata Harbor
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grigorakis, K.; Rigos, G. Aquaculture effects on environmental and public welfare—The case of Mediterranean mariculture. Chemosphere 2011, 85, 899–919. [Google Scholar] [CrossRef] [PubMed]
- Rosa, R.; Marques, A.; Nunes, M.L. Impact of climate change in Mediterranean aquaculture. Rev. Aquac. 2012, 4, 163–177. [Google Scholar] [CrossRef]
- FAO. The State of World Fisheries and Aquaculture 2018—Meeting the Sustainable Development Goals; FAO: Rome, Italy, 2018; Available online: http://www.fao.org/documents/card/en/c/I9540EN/ (accessed on 7 January 2021).
- Diego-McGlone, M.L.S.; Azanza, R.V.; Villanoy, C.L.; Jacinto, G.S. Eutrophic waters, algal bloom and fish kill in fish farming areas in Bolinao, Pangasinan, Philippines. Mar. Pollut. Bull. 2008, 57, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Tičina, V.; Katavić, I.; Grubišić, L. Marine Aquaculture Impacts on Marine Biota in Oligotrophic Environments of the Mediterranean Sea—A Review. Front. Mar. Sci. 2020, 7. [Google Scholar] [CrossRef]
- Moncada, C.; Hassenrück, C.; Gärdes, A.; Conaco, C. Microbial community composition of sediments influenced by intensive mariculture activity. FEMS Microbiol. Ecol. 2019, 95. [Google Scholar] [CrossRef]
- Kalantzi, I.; Rico, A.; Mylona, K.; Pergantis, S.A.; Tsapakis, M. Fish farming, metals and antibiotics in the eastern Mediterranean Sea: Is there a threat to sediment wildlife? Sci. Total. Environ. 2021, 764, 142843. [Google Scholar] [CrossRef] [PubMed]
- Holmer, M.; Duarte, C.M.; Heilskov, A.; Olesen, B.; Terrados, J. Biogeochemical conditions in sediments enriched by organic matter from net-pen fish farms in the Bolinao area, Philippines. Mar. Pollut. Bull. 2003, 46, 1470–1479. [Google Scholar] [CrossRef]
- Karakassis, I.; Tsapakis, M.; Hatziyanni, E.; Papadopoulou, K.-N.; Plaiti, W. Impact of cage farming of fish on the seabed in three Mediterranean coastal areas. ICES J. Mar. Sci. 2000, 57, 1462–1471. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Lázaro, C.; Belando, M.D.; Marín-Guirao, L.; Navarrete-Mier, F.; Marín, A. Relationship between sedimentation rates and benthic impact on Maërl beds derived from fish farming in the Mediterranean. Mar. Environ. Res. 2011, 71, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavicchioli, R.; Ripple, W.J.; Timmis, K.N.; Azam, F.; Bakken, L.R.; Baylis, M.; Behrenfeld, M.J.; Boetius, A.; Boyd, P.W.; Classen, A.T.; et al. Scientists’ warning to humanity: Microorganisms and climate change. Nat. Rev. Genet. 2019, 17, 569–586. [Google Scholar] [CrossRef] [Green Version]
- Luna, G.; Corinaldesi, C.; Dell’Anno, A.; Pusceddu, A.; Danovaro, R. Impact of aquaculture on benthic virus–prokaryote interactions in the Mediterranean Sea. Water Res. 2013, 47, 1156–1168. [Google Scholar] [CrossRef]
- Hornick, K.M.; Buschmann, A.H. Insights into the diversity and metabolic function of bacterial communities in sediments from Chilean salmon aquaculture sites. Ann. Microbiol. 2018, 68, 63–77. [Google Scholar] [CrossRef]
- Ape, F.; Manini, E.; Quero, G.M.; Luna, G.M.; Sarà, G.; Vecchio, P.; Brignoli, P.; Ansferri, S.; Mirto, S. Biostimulation of in situ microbial degradation processes in organically-enriched sediments mitigates the impact of aquaculture. Chemosphere 2019, 226, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Portillo, E.; Villamor, A.; Fernandez-Gonzalez, V.; Antón, J.; Sanchez-Jerez, P. Exploring changes in bacterial communities to assess the influence of fish farming on marine sediments. Aquaculture 2019, 506, 459–464. [Google Scholar] [CrossRef]
- Shi, R.; Xu, S.; Qi, Z.; Zhu, Q.; Huang, H.; Weber, F. Influence of suspended mariculture on vertical distribution profiles of bacteria in sediment from Daya Bay, Southern China. Mar. Pollut. Bull. 2019, 146, 816–826. [Google Scholar] [CrossRef]
- Zhang, K.; Zheng, X.; He, Z.; Yang, T.; Shu, L.; Xiao, F.; Wu, Y.; Wang, B.; Li, Z.; Chen, P.; et al. Fish growth enhances microbial sulfur cycling in aquaculture pond sediments. Microb. Biotechnol. 2020, 13, 1597–1610. [Google Scholar] [CrossRef]
- Haro-Moreno, J.M.; Coutinho, F.H.; Zaragoza-Solas, A.; Picazo, A.; Almagro-Moreno, S.; López-Pérez, M. Dysbiosis in marine aquaculture revealed through microbiome analysis: Reverse ecology for environmental sustainability. FEMS Microbiol. Ecol. 2020, 96. [Google Scholar] [CrossRef]
- Bordenstein, S.R.; Theis, K.R. Host Biology in Light of the Microbiome: Ten Principles of Holobionts and Hologenomes. PLoS Biol. 2015, 13, e1002226. [Google Scholar] [CrossRef] [Green Version]
- Leibold, M.A.; Holyoak, M.; Mouquet, N.; Amarasekare, P.; Chase, J.M.; Hoopes, M.F.; Holt, R.D.; Shurin, J.B.; Law, R.; Tilman, D.; et al. The metacommunity concept: A framework for multi-scale community ecology. Ecol. Lett. 2004, 7, 601–613. [Google Scholar] [CrossRef]
- Adair, K.L.; Douglas, A.E. Making a microbiome: The many determinants of host-associated microbial community composition. Curr. Opin. Microbiol. 2017, 35, 23–29. [Google Scholar] [CrossRef]
- Cleary, D.F.R.; Swierts, T.; Coelho, F.J.R.C.; Polónia, A.R.M.; Huang, Y.M.; Ferreira, M.R.S.; Putchakarn, S.; Carvalheiro, L.; Van Der Ent, E.; Ueng, J.-P.; et al. The sponge microbiome within the greater coral reef microbial metacommunity. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Trevathan-Tackett, S.M.; Sherman, C.D.; Huggett, M.J.; Campbell, A.H.; Laverock, B.; Hurtado-McCormick, V.; Seymour, J.R.; Firl, A.; Messer, L.F.; Ainsworth, T.D.; et al. A horizon scan of priorities for coastal marine microbiome research. Nat. Ecol. Evol. 2019, 3, 1509–1520. [Google Scholar] [CrossRef]
- Baquiran, J.I.P.; Conaco, C. Sponge-microbe partnerships are stable under eutrophication pressure from mariculture. Mar. Pollut. Bull. 2018, 136, 125–134. [Google Scholar] [CrossRef]
- Della Santina, P.; Chelazzi, G. Temporal organization of foraging in two Mediterranean limpets, Patella rustica L. and P. coerulea L. J. Exp. Mar. Biol. Ecol. 1991, 153, 75–85. [Google Scholar] [CrossRef]
- Reguera, P.; Couceiro, L.; Fernández, N. A review of the empirical literature on the use of limpets Patella spp. (Mollusca: Gastropoda) as bioindicators of environmental quality. Ecotoxicol. Environ. Saf. 2018, 148, 593–600. [Google Scholar] [CrossRef]
- Viñas, L.; Pérez-Fernández, B.; Soriano, J.A.; López, M.; Bargiela, J.; Alves, I. Limpet (Patella sp.) as a biomonitor for organic pollutants. A proxy for mussel? Mar. Pollut. Bull. 2018, 133, 271–280. [Google Scholar] [CrossRef]
- Coleman, R.A.; Underwood, A.J.; Benedetti-Cecchi, L.; Åberg, P.; Arenas, F.; Arrontes, J.; Castro, J.; Hartnoll, R.G.; Jenkins, S.R.; Paula, J.; et al. A continental scale evaluation of the role of limpet grazing on rocky shores. Oecologia 2006, 147, 556–564. [Google Scholar] [CrossRef]
- Mente, E.; Nikouli, E.; Antonopoulou, E.; Martin, S.A.M.; Kormas, K.A. Core versus diet-associated and postprandial bacterial communities of the rainbow trout (Oncorhynchus mykiss) midgut and faeces. Biol. Open 2018, 7, bio034397. [Google Scholar] [CrossRef] [Green Version]
- Mehrbach, C.; Culberson, C.H.; Hawley, J.E.; Pytkowicx, R.M. Measurement of the apparent dissociation constants of carbonic acid in seawater at atmospheric pressure1. Limnol. Oceanogr. 1973, 18, 897–907. [Google Scholar] [CrossRef]
- Dickson, A.; Millero, F. A comparison of the equilibrium constants for the dissociation of carbonic acid in seawater media. Deep. Sea Res. Part A Oceanogr. Res. Pap. 1987, 34, 1733–1743. [Google Scholar] [CrossRef]
- Dickson, A.G. Thermodynamics of the dissociation of boric acid in synthetic seawater from 273.15 to 318.15 K. Deep. Sea Res. Part A Oceanogr. Res. Pap. 1990, 37, 755–766. [Google Scholar] [CrossRef]
- Musella, M.; Wathsala, R.; Tavella, T.; Rampelli, S.; Barone, M.; Palladino, G.; Biagi, E.; Brigidi, P.; Turroni, S.; Franzellitti, S.; et al. Tissue-scale microbiota of the Mediterranean mussel (Mytilus galloprovincialis) and its relationship with the environment. Sci. Total. Environ. 2020, 717, 137209. [Google Scholar] [CrossRef]
- Quero, G.M.; Perini, L.; Pesole, G.; Manzari, C.; Lionetti, C.; Bastianini, M.; Marini, M.; Luna, G.M. Seasonal rather than spatial variability drives planktonic and benthic bacterial diversity in a microtidal lagoon and the adjacent open sea. Mol. Ecol. 2017, 26, 5961–5973. [Google Scholar] [CrossRef]
- Turroni, S.; Fiori, J.; Rampelli, S.; Schnorr, S.L.; Consolandi, C.; Barone, M.; Biagi, E.; Fanelli, F.; Mezzullo, M.; Crittenden, A.N.; et al. Fecal metabolome of the Hadza hunter-gatherers: A host-microbiome integrative view. Sci. Rep. 2016, 6, 32826. [Google Scholar] [CrossRef]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, F.; Biagi, E.; Rampelli, S.; Fiori, J.; Zama, D.; Soverini, M.; Barone, M.; Leardini, D.; Muratore, E.; Prete, A.; et al. Enteral Nutrition in Pediatric Patients Undergoing Hematopoietic SCT Promotes the Recovery of Gut Microbiome Homeostasis. Nutrients 2019, 11, 2958. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Culhane, A.C.; Thioulouse, J.; Perrière, G.; Higgins, D.G. MADE4: An R package for multivariate analysis of gene expression data. Bioinformatics 2005, 21, 2789–2790. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, N.E.; Ferry, M. ggtern: Ternary Diagrams Using ggplot2. J. Stat. Softw. 2018, 87, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Pohlert, T. The pairwise multiple comparison of mean ranks package (PMCMR). R Package 2014, 27, 10. [Google Scholar]
- Meyer, D.; Zeileis, A.; Hornik, K. vcd: Visualizing Categorical Data. R Package Version 1.4-8. 2020. Available online: https://cran.r-project.org/(accessed on 15 January 2021).
- Yoon, J.-H.; Lee, C.-H.; Kang, S.-J.; Oh, T.-K. Psychrobacter celer sp. nov., isolated from sea water of the South Sea in Korea. Int. J. Syst. Evol. Microbiol. 2005, 55, 1885–1890. [Google Scholar] [CrossRef] [Green Version]
- Infante-Dominguez, C.; Lawson, P.A.; Johnson, C.N.; Sánchez-Porro, C.; Ventosa, A. Fodinicurvata halophila sp. nov., a moderately halophilic bacterium from a marine saltern. Int. J. Syst. Evol. Microbiol. 2015, 65, 766–771. [Google Scholar] [CrossRef]
- Ferreira, C.; Soares, A.R.; Lamosa, P.; Santos, M.A.; Da Costa, M.S. Comparison of the compatible solute pool of two slightly halophilic planctomycetes species, Gimesia maris and Rubinisphaera brasiliensis. Extremophiles 2016, 20, 811–820. [Google Scholar] [CrossRef]
- Kallscheuer, N.; Jogler, M.; Wiegand, S.; Peeters, S.H.; Heuer, A.; Boedeker, C.; Jetten, M.S.M.; Rohde, M.; Jogler, C. Rubinisphaera italica sp. nov. isolated from a hydrothermal area in the Tyrrhenian Sea close to the volcanic island Panarea. Antonie Leeuwenhoek 2019, 113, 1727–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Yosef, D.Z.; Ben-Dov, E.; Kushmaro, A. Amorphus coralli gen. nov., sp. nov., a marine bacterium isolated from coral mucus, belonging to the order Rhizobiales. Int. J. Syst. Evol. Microbiol. 2008, 58, 2704–2709. [Google Scholar] [CrossRef] [Green Version]
- Fraser, C.M.; Gocayne, J.D.; White, O.; Adams, M.D.; Clayton, R.A.; Fleischmann, R.D.; Bult, C.J.; Kerlavage, A.R.; Sutton, G.; Kelley, J.M.; et al. The Minimal Gene Complement of Mycoplasma genitalium. Science 1995, 270, 397–404. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.-H.; Yu, M.; Wang, H.; Austin, B. Vibrio atypicus sp. nov., isolated from the digestive tract of the Chinese prawn (Penaeus chinensis O’sbeck). Int. J. Syst. Evol. Microbiol. 2010, 60, 2517–2523. [Google Scholar] [CrossRef]
- Abdel-Aziz, M.; Eissa, A.E.; Hanna, M.; Okada, M.A. Identifying some pathogenic Vibrio/Photobacterium species during mass mortalities of cultured Gilthead seabream (Sparus aurata) and European seabass (Dicentrarchus labrax) from some Egyptian coastal provinces. Int. J. Veter-Sci. Med. 2013, 1, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Sohn, H.; Kim, J.; Jin, C.; Lee, J. Identification of Vibrio species isolated from cultured olive flounder (Paralichthys olivaceus) in Jeju Island, South Korea. Fish. Aquat. Sci. 2019, 22, 14. [Google Scholar] [CrossRef]
- Kuebutornye, F.K.A.; Abarike, E.D.; Lu, Y.; Hlordzi, V.; Sakyi, M.E.; Afriyie, G.; Wang, Z.; Li, Y.; Xie, C.X. Mechanisms and the role of probiotic Bacillus in mitigating fish pathogens in aquaculture. Fish Physiol. Biochem. 2020, 46, 819–841. [Google Scholar] [CrossRef]
- Liquete, C.; Piroddi, C.; Drakou, E.G.; Gurney, L.; Katsanevakis, S.; Charef, A.; Egoh, B. Current Status and Future Prospects for the Assessment of Marine and Coastal Ecosystem Services: A Systematic Review. PLoS ONE 2013, 8, e67737. [Google Scholar] [CrossRef] [Green Version]
- Dudek, M.; Adams, J.; Swain, M.; Hegarty, M.; Huws, S.; Gallagher, J. Metaphylogenomic and Potential Functionality of the Limpet Patella pellucida’s Gastrointestinal Tract Microbiome. Int. J. Mol. Sci. 2014, 15, 18819–18839. [Google Scholar] [CrossRef] [Green Version]
- Pita, L.; Rix, L.; Slaby, B.M.; Franke, A.; Hentschel, U. The sponge holobiont in a changing ocean: From microbes to ecosystems. Microbiome 2018, 6, 1–18. [Google Scholar] [CrossRef]
- Wilkins, L.G.E.; Leray, M.; O’Dea, A.; Yuen, B.; Peixoto, R.S.; Pereira, T.J.; Bik, H.M.; Coil, D.A.; Duffy, J.E.; Herre, E.A.; et al. Host-associated microbiomes drive structure and function of marine ecosystems. PLoS Biol. 2019, 17, e3000533. [Google Scholar] [CrossRef] [Green Version]
- Biagi, E.; Caroselli, E.; Barone, M.; Pezzimenti, M.; Teixido, N.; Soverini, M.; Rampelli, S.; Turroni, S.; Gambi, M.C.; Brigidi, P.; et al. Patterns in microbiome composition differ with ocean acidification in anatomic compartments of the Mediterranean coral Astroides calycularis living at CO2 vents. Sci. Total. Environ. 2020, 724, 138048. [Google Scholar] [CrossRef]
- Sizikov, S.; Burgsdorf, I.; Handley, K.M.; Lahyani, M.; Haber, M.; Steindler, L. Characterization of sponge-associated Verrucomicrobia: Microcompartment-based sugar utilization and enhanced toxin–antitoxin modules as features of host-associated Opitutales. Environ. Microbiol. 2020, 22, 4669–4688. [Google Scholar] [CrossRef] [PubMed]
- Cabello-Yeves, P.J.; Ghai, R.; Mehrshad, M.; Picazo, A.; Camacho, A.; Rodriguez-Valera, F. Reconstruction of Diverse Verrucomicrobial Genomes from Metagenome Datasets of Freshwater Reservoirs. Front. Microbiol. 2017, 8, 2131. [Google Scholar] [CrossRef] [Green Version]
- Sichert, A.; Corzett, C.H.; Schechter, M.S.; Unfried, F.; Markert, S.; Becher, D.; Fernandez-Guerra, A.; Liebeke, M.; Schweder, T.; Polz, M.F.; et al. Verrucomicrobia use hundreds of enzymes to digest the algal polysaccharide fucoidan. Nat. Microbiol. 2020, 5, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- King, G.M.; Judd, C.; Kuske, C.R.; Smith, C. Analysis of Stomach and Gut Microbiomes of the Eastern Oyster (Crassostrea virginica) from Coastal Louisiana, USA. PLoS ONE 2012, 7, e51475. [Google Scholar] [CrossRef] [Green Version]
- Cornall, A.M.; Rose, A.; Streten, C.; McGuinness, K.; Parry, D.L.; Gibb, K. Molecular screening of microbial communities for candidate indicators of multiple metal impacts in marine sediments from northern Australia. Environ. Toxicol. Chem. 2016, 35, 468–484. [Google Scholar] [CrossRef] [PubMed]
- Baltar, F.; Gutiérrez-Rodríguez, A.; Meyer, M.; Skudelny, I.; Sander, S.; Thomson, B.; Nodder, S.; Middag, R.; Morales, S.E. Specific Effect of Trace Metals on Marine Heterotrophic Microbial Activity and Diversity: Key Role of Iron and Zinc and Hydrocarbon-Degrading Bacteria. Front. Microbiol. 2018, 9, 3190. [Google Scholar] [CrossRef]
- Ontañon, O.M.; Landi, C.; Carleo, A.; Gagliardi, A.; Bianchi, L.; González, P.S.; Agostini, E.; Bini, L. What makes A. guillouiae SFC 500-1A able to co-metabolize phenol and Cr(VI)? A proteomic approach. J. Hazard. Mater. 2018, 354, 215–224. [Google Scholar] [CrossRef]
- Lamin, H.; Alami, S.; Bouhnik, O.; Bennis, M.; Benkritly, S.; Abdelmoumen, H.; Bedmar, E.J.; Idrissi, M.M. Identification of the endosymbionts from Sulla spinosissima growing in a lead mine tailings in Eastern Morocco as Mesorhizobium camelthorni sv. Aridi. J. Appl. Microbiol. 2020, 10. [Google Scholar] [CrossRef]
- Duarte, L.N.; Coelho, F.J.R.C.; Oliveira, V.; Cleary, D.F.R.; Martins, P.; Gomes, N.C.M. Characterization of bacterioplankton communities from a hatchery recirculating aquaculture system (RAS) for juvenile sole (Solea senegalensis) production. PLoS ONE 2019, 14, e0211209. [Google Scholar] [CrossRef]
- Choi, A.; Cho, H.; Kim, B.; Kim, H.; Jung, R.; Lee, W.; Hyun, J. Effects of finfish aquaculture on biogeochemistry and bacterial communities associated with sulfur cycles in highly sulfidic sediments. Aquac. Environ. Interact. 2018, 10, 413–427. [Google Scholar] [CrossRef]
- Sharifah, E.N.; Eguchi, M. Mixed cultures of the phytoplankton Nannochloropsis oculata and the marine bacterium Sulfitobacter sp. RO3 inhibit the growth of virulent strains of the major fish pathogen Vibrio anguillarum. Aquac. Sci. 2012, 60, 39–45. [Google Scholar] [CrossRef]
- Feldman, S.H.; Wimsatt, J.; Marchang, R.E.; Johnson, A.J.; Brown, W.; Mitchell, J.C.; Sleeman, J.M. A Novel Mycoplasma Detected in Association with Upper Respiratory Disease Syndrome in Free-Ranging Eastern Box Turtles (Terrapene Carolina Carolina) in Virginia. J. Wildl. Dis. 2006, 42, 279–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, M.L. The Microbiome of the Eastern Oyster, Crassostrea Virginica (Gmelin, 1791): Temporal and Spatial Variation, Environmental Influences, and Its Impact on Host Physiology. Ph.D. Thesis, University of Connecticut (UCONN), Storrs, Connecticut, 2016. [Google Scholar]
- Aceves, A.K.; Johnson, P.; Bullard, S.A.; LaFrentz, S.; Arias, C.R. Description and characterization of the digestive gland microbiome in the freshwater mussel Villosa nebulosa (Bivalvia: Unionidae). J. Molluscan Stud. 2018, 84, 240–246. [Google Scholar] [CrossRef] [Green Version]
- Pierce, M.L.; Ward, J.E. Microbial Ecology of the Bivalvia, with an Emphasis on the Family Ostreidae. J. Shellfish. Res. 2018, 37, 793–806. [Google Scholar] [CrossRef]
- Pierce, M.L.; Ward, J.E. Gut Microbiomes of the Eastern Oyster (Crassostrea virginica) and the Blue Mussel (Mytilus edulis): Temporal Variation and the Influence of Marine Aggregate-Associated Microbial Communities. mSphere 2019, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Mathai, P.P.; Magnone, P.; Dunn, H.M.; Sadowsky, M.J. Water and sediment act as reservoirs for microbial taxa associated with invasive dreissenid mussels. Sci. Total. Environ. 2020, 703, 134915. [Google Scholar] [CrossRef]
- Marzocchi, U.; Bonaglia, S.; Zaiko, A.; Quero, G.M.; Vybernaite-Lubiene, I.; Politi, T.; Samuiloviene, A.; Zilius, M.; Bartoli, M.; Cardini, U. Zebra Mussel Holobionts Fix and Recycle Nitrogen in Lagoon Sediments. Front. Microbiol. 2021, 11. [Google Scholar] [CrossRef]
- Fraune, S.; Zimmer, M. Host-specificity of environmentally transmitted Mycoplasma-like isopod symbionts. Environ. Microbiol. 2008, 10, 2497–2504. [Google Scholar] [CrossRef]
- Holm, J.B.; Heidelberg, K.B. Microbiomes of Muricea californica and M. fruticosa: Comparative Analyses of Two Co-occurring Eastern Pacific Octocorals. Front. Microbiol. 2016, 7, 917. [Google Scholar] [CrossRef] [PubMed]
- Montalto, V.; Rinaldi, A.; Ape, F.; Mangano, M.; Gristina, M.; Sarà, G.; Mirto, S. Functional role of biofouling linked to aquaculture facilities in Mediterranean enclosed locations. Aquac. Environ. Interact. 2020, 12, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Minich, J.J.; Poore, G.D.; Jantawongsri, K.; Johnston, C.; Bowie, K.; Bowman, J.; Knight, R.; Nowak, B.; Allen, E.E. Microbial Ecology of Atlantic Salmon (Salmo salar) Hatcheries: Impacts of the Built Environment on Fish Mucosal Microbiota. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef]
- Floris, R.; Manca, S.; Fois, N. Microbial ecology of intestinal tract of gilthead sea bream (Sparus aurata Linnaeus, 1758) from two coastal lagoons of Sardinia (Italy). Trans. Water Bullet. 2013, 7, 4–12. [Google Scholar] [CrossRef]
- Estruch, G.; Collado, M.C.; Peñaranda, D.S.; Vidal, A.T.; Cerdá, M.J.; Martínez, G.P.; Martinezllorens, S. Impact of Fishmeal Replacement in Diets for Gilthead Sea Bream (Sparus aurata) on the Gastrointestinal Microbiota Determined by Pyrosequencing the 16S rRNA Gene. PLoS ONE 2015, 10, e0136389. [Google Scholar] [CrossRef] [PubMed]
- Legrand, T.P.; Wynne, J.W.; Weyrich, L.S.; Oxley, A.P. A microbial sea of possibilities: Current knowledge and prospects for an improved understanding of the fish microbiome. Rev. Aquac. 2019, 12, 1101–1134. [Google Scholar] [CrossRef]
- Floris, R.; Scanu, G.; Fois, N.; Rizzo, C.; Malavenda, R.; Spanò, N.; Giudice, A.L. Intestinal bacterial flora of Mediterranean gilthead sea bream (Sparus aurataLinnaeus) as a novel source of natural surface active compounds. Aquac. Res. 2018, 49, 1262–1273. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palladino, G.; Rampelli, S.; Scicchitano, D.; Musella, M.; Quero, G.M.; Prada, F.; Mancuso, A.; Seyfarth, A.M.; Turroni, S.; Candela, M.; et al. Impact of Marine Aquaculture on the Microbiome Associated with Nearby Holobionts: The Case of Patella caerulea Living in Proximity of Sea Bream Aquaculture Cages. Microorganisms 2021, 9, 455. https://doi.org/10.3390/microorganisms9020455
Palladino G, Rampelli S, Scicchitano D, Musella M, Quero GM, Prada F, Mancuso A, Seyfarth AM, Turroni S, Candela M, et al. Impact of Marine Aquaculture on the Microbiome Associated with Nearby Holobionts: The Case of Patella caerulea Living in Proximity of Sea Bream Aquaculture Cages. Microorganisms. 2021; 9(2):455. https://doi.org/10.3390/microorganisms9020455
Chicago/Turabian StylePalladino, Giorgia, Simone Rampelli, Daniel Scicchitano, Margherita Musella, Grazia Marina Quero, Fiorella Prada, Arianna Mancuso, Anne Mette Seyfarth, Silvia Turroni, Marco Candela, and et al. 2021. "Impact of Marine Aquaculture on the Microbiome Associated with Nearby Holobionts: The Case of Patella caerulea Living in Proximity of Sea Bream Aquaculture Cages" Microorganisms 9, no. 2: 455. https://doi.org/10.3390/microorganisms9020455