Induction and Genome Analysis of HY01, a Newly Reported Prophage from an Emerging Shrimp Pathogen Vibrio campbellii

 ,
, 
 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial strain and Culture Conditions
2.2. Prophage Induction Using Mitomycin C, Heat Treatment, and UV Radiation
2.3. Preparation of Concentrated Phage
2.4. Transmission Electron Microscopy (TEM)
2.5. Phage DNA Extraction
2.6. Phage DNA Assembly and Analysis
2.7. Phylogenetic Analysis
2.8. Accession Numbers of Phage HY01 Genome
2.9. Statistical Analysis
3. Results
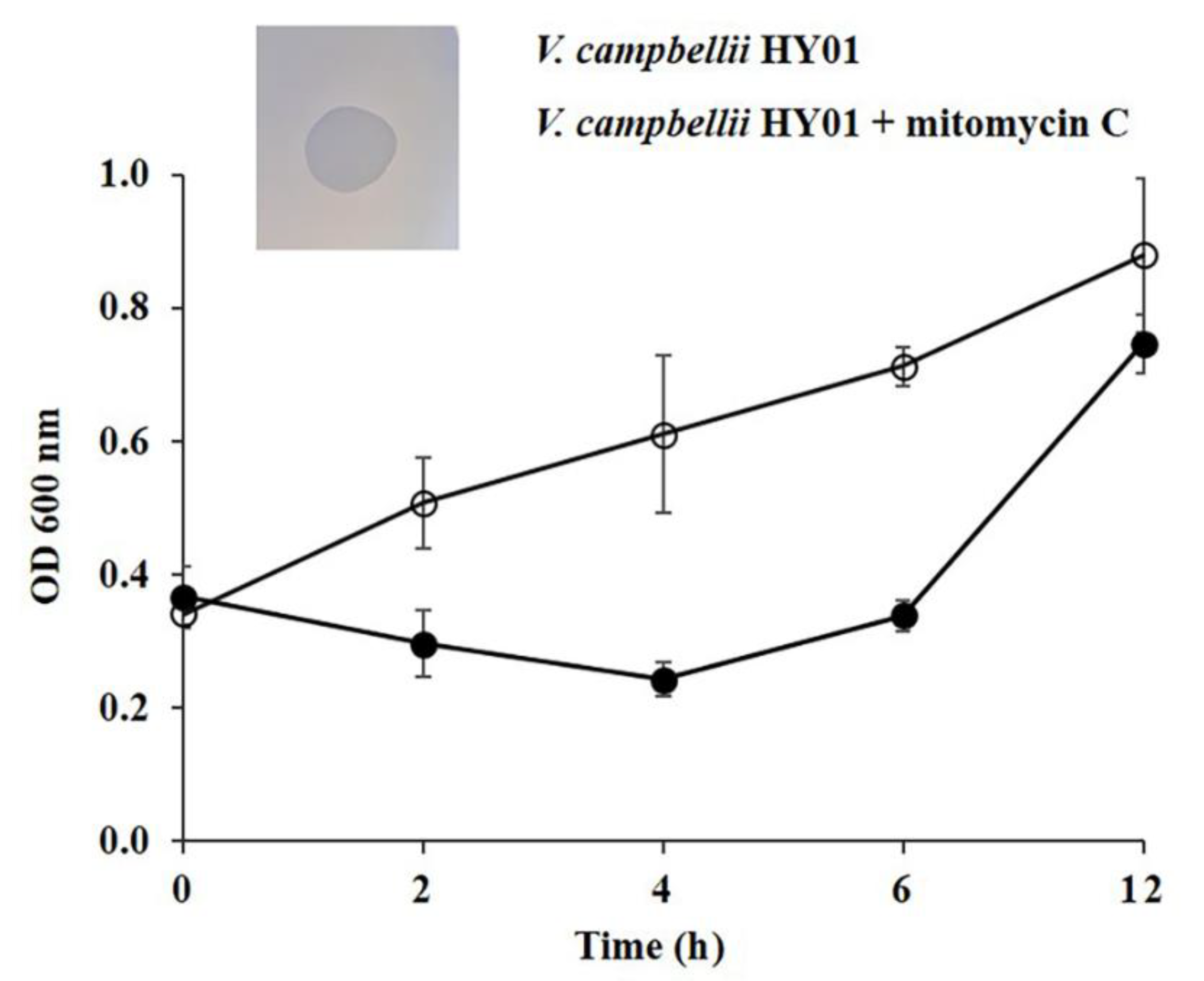
3.1. Induction of Prophage HY01
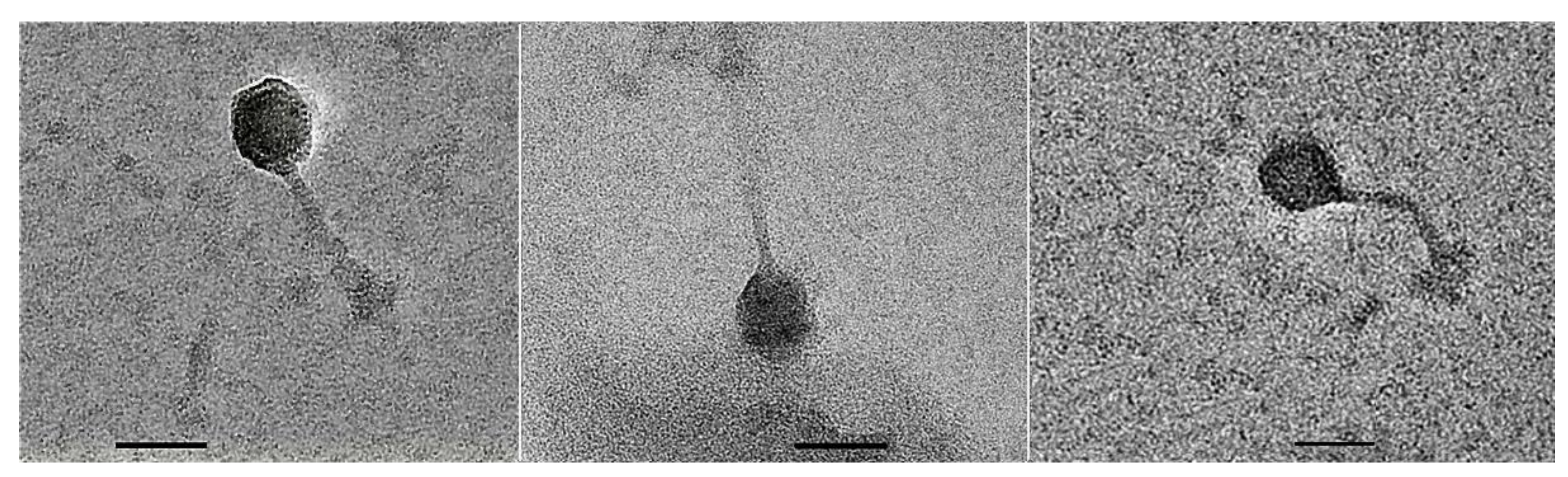
3.2. Transmission Electron Micrographs of Phage Particle
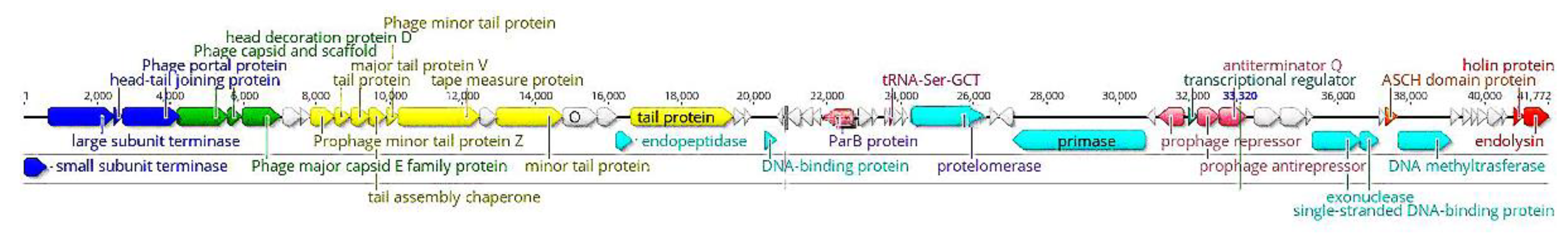
3.3. Phage HY01 Genome and Comparative Genomic Analysis
3.4. ORFs Analysis and Comparative Proteomic Analysis of Phage HY01
3.4.1. DNA Packaging
3.4.2. Head Structural Components and Assembly
3.4.3. Tail Structural Components and Assembly
3.4.4. DNA Replication, Modulation, and Repair
3.4.5. Lysogeny Control
3.4.6. The Lysis Module
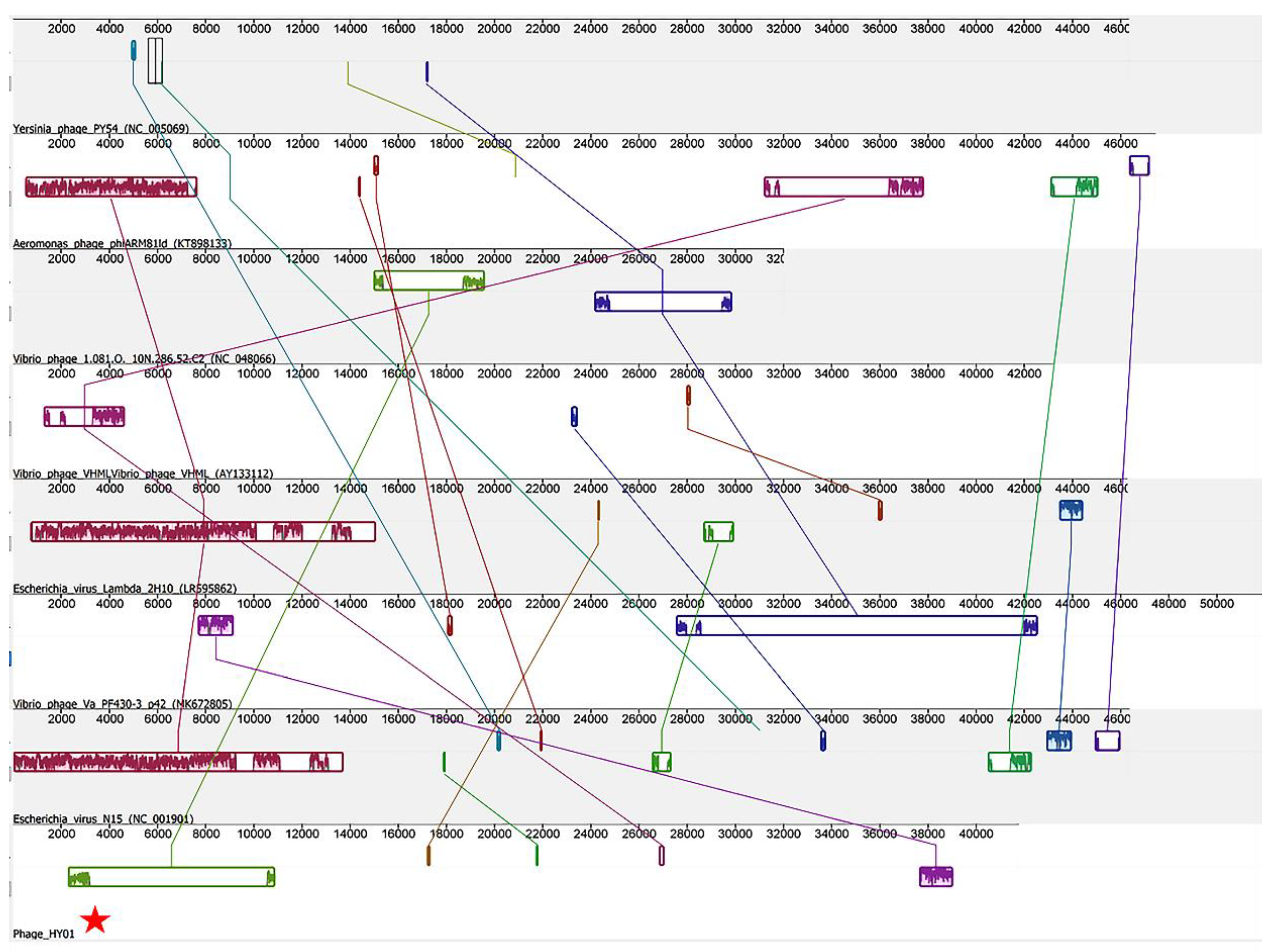
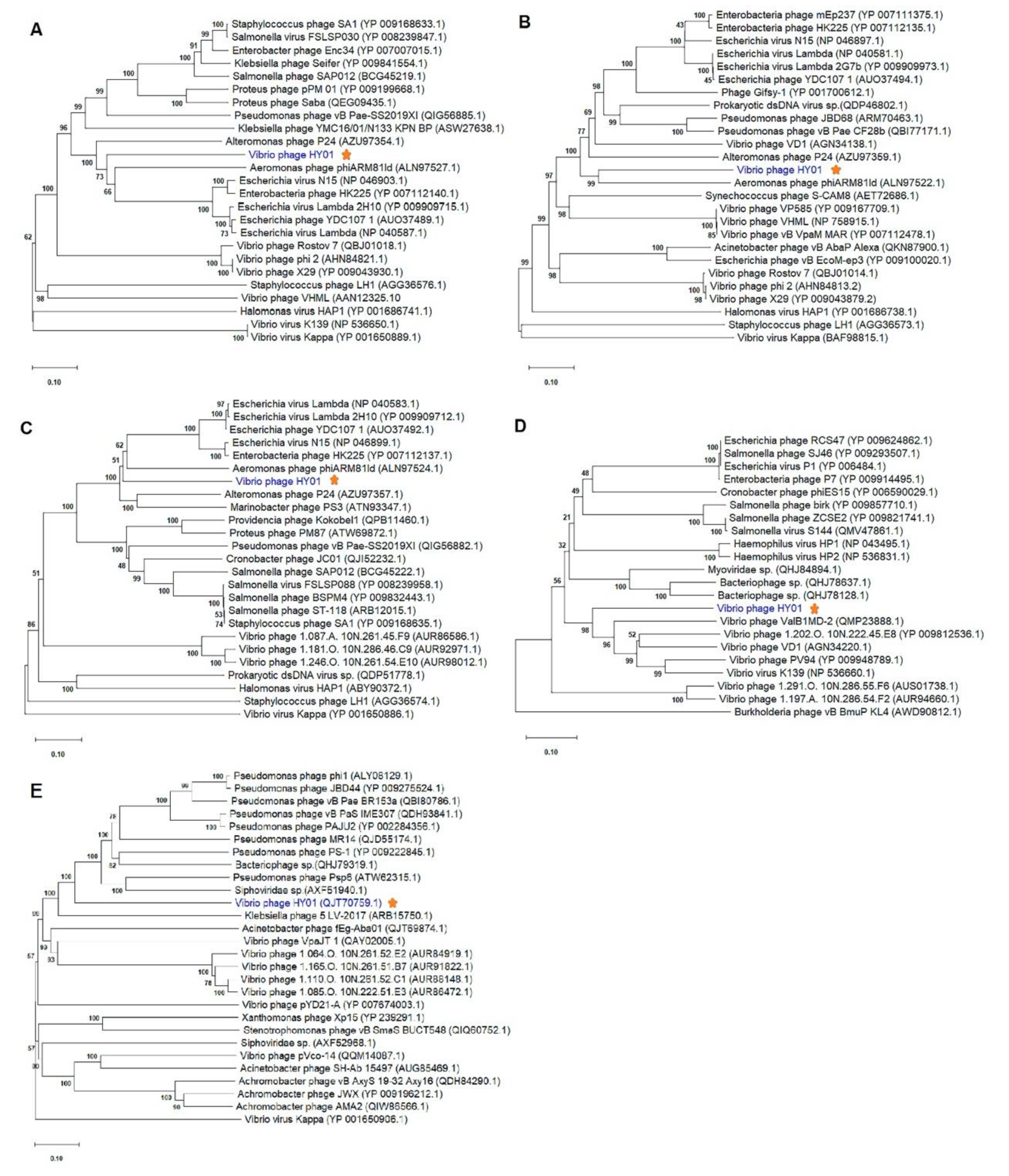
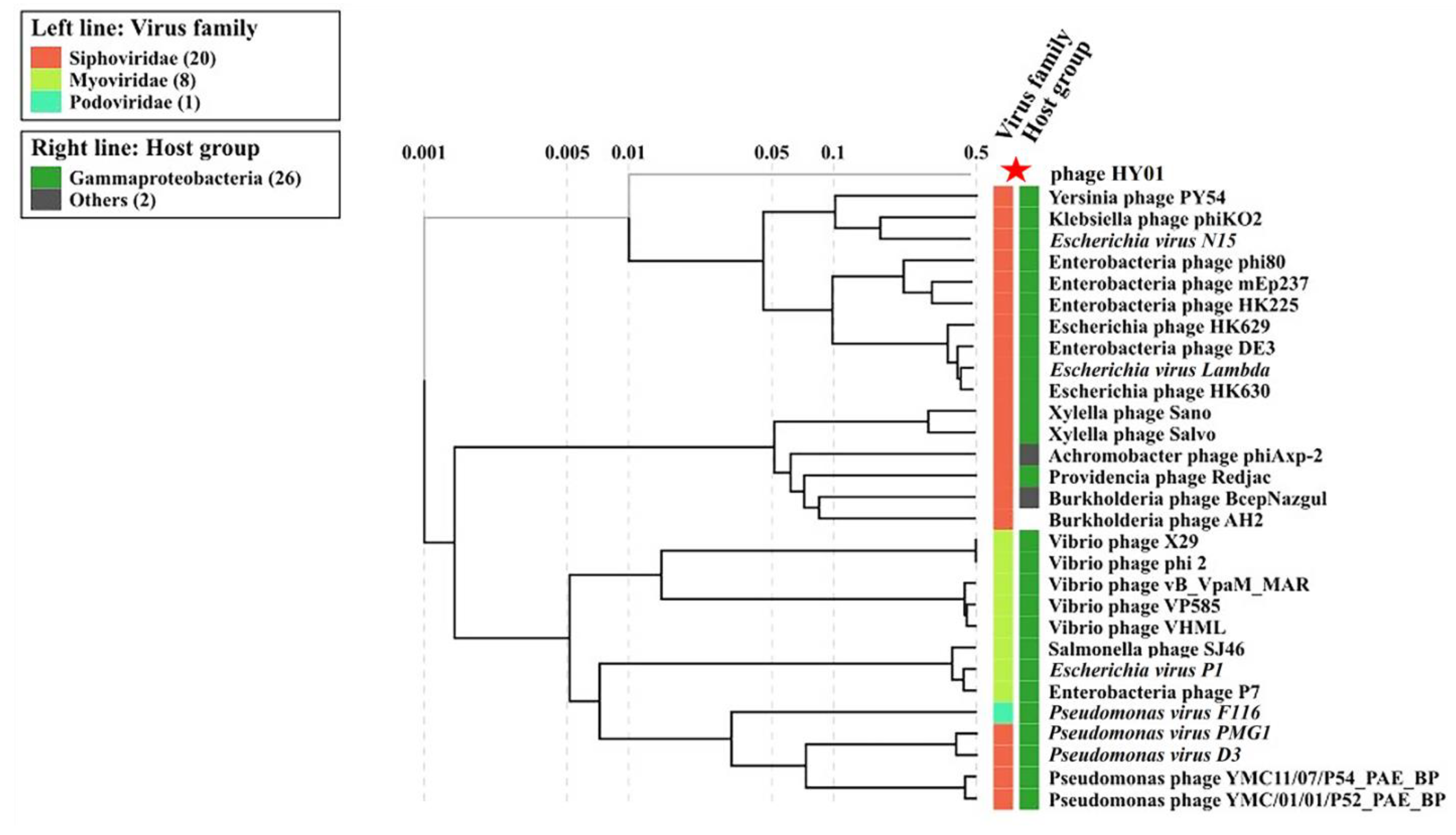
3.5. Phylogenetic Analysis of Phage HY01 with Other Related Phages
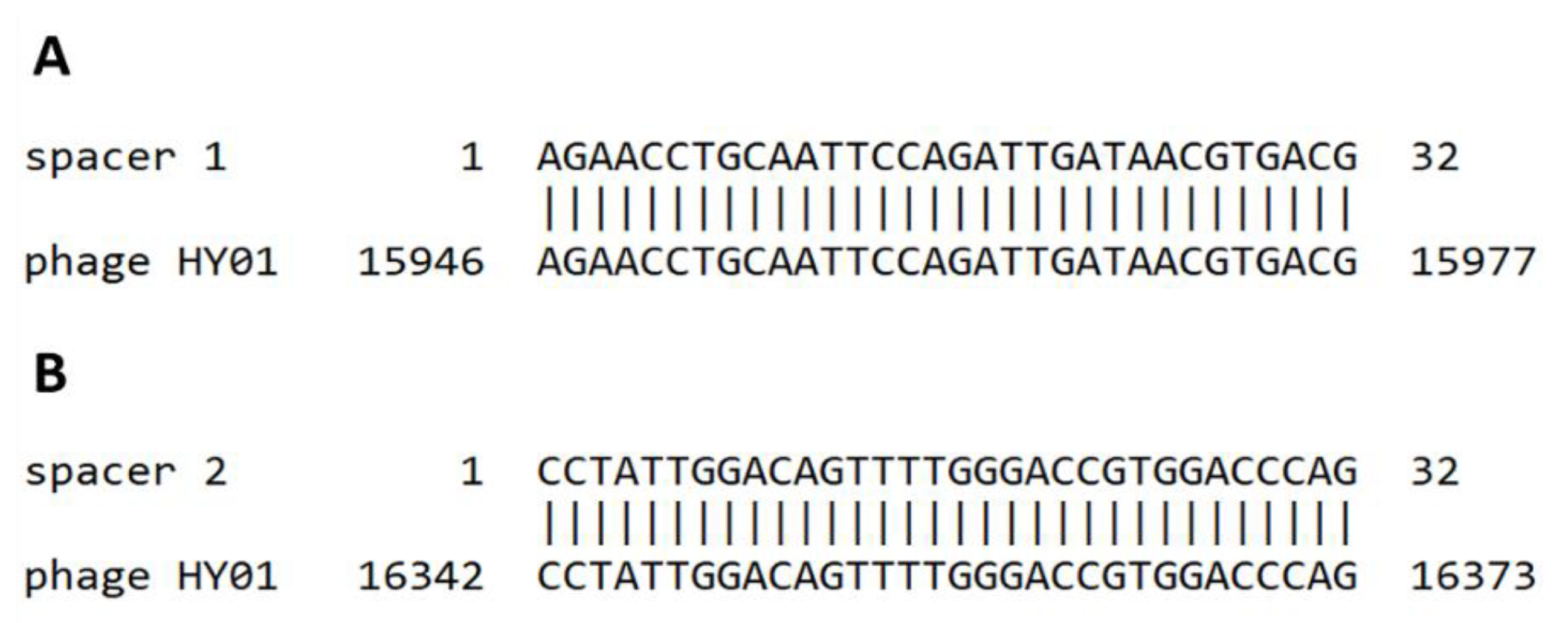
3.6. CRISPR Spacer Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thompson, F.L.; Iida, T.; Swings, J. Biodiversity of Vibrios. Microbiol. Mol. Bio. Rev. 2004, 68, 403. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; Wang, Z.; Malanoski, A.P.; O’Grady, E.A.; Wimpee, C.F.; Vuddhakul, V.; Alves, N., Jr.; Thompson, F.L.; Gomez-Gil, B.; Vora, G.J. Comparative genomic analyses identify the Vibrio harveyi genome sequenced strains BAA-1116 and HY01 as Vibrio campbellii. Environ. Microbiol. Rep. 2010, 2, 81–89. [Google Scholar] [CrossRef]
- Rattanama, P.; Srinitiwarawong, K.; Thompson, J.R.; Pomwised, R.; Supamattaya, K.; Vuddhakul, V. Shrimp pathogenicity, hemolysis, and the presence of hemolysin and TTSS genes in Vibrio harveyi isolated from Thailand. Dis. Aquat. 2009, 86, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.V.; Winstanley, C.; Fothergill, J.L.; James, C.E. The role of temperate bacteriophages in bacterial infection. FEMS Microbiol. Lett. 2016, 363, fnw015. [Google Scholar] [CrossRef] [Green Version]
- Faruque, S.M. Role of Phages in the Epidemiology of Cholera. In Cholera Outbreaks; Nair, G.B., Takeda, Y., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 165–180. [Google Scholar]
- Faruque, S.M.; Mekalanos, J.J. Phage-bacterial interactions in the evolution of toxigenic Vibrio cholerae. Virulence 2012, 3, 556–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, D.; Kauffman, K.; Hussain, F.; Kalatzis, P.; Rørbo, N.; Polz, M.F.; Middelboe, M. Widespread distribution of prophage-encoded virulence factors in marine Vibrio communities. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vidgen, M.; Carson, J.; Higgins, M.; Owens, L. Changes to the phenotypic profile of Vibrio harveyi when infected with the Vibrio harveyi myovirus-like (VHML) bacteriophage. J. Appl. Microbiol. 2006, 100, 481–487. [Google Scholar] [CrossRef]
- Bochow, S.; Elliman, J.; Owens, L. Bacteriophage adenine methyltransferase: A life cycle regulator? Modelled using Vibrio harveyi myovirus like. J. Appl. Microbiol. 2012, 113, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Waddell, T.E.; Franklin, K.; Mazzocco, A.; Kropinski, A.M.; Johnson, R.P. Generalized Transduction by Lytic Bacteriophages. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 293–303. [Google Scholar]
- Choi, S.; Dunams, D.; Jiang, S. Transfer of cholera toxin genes from O1 to non-O1/O139 strains by vibriophages from California coastal waters. J. Appl. Microbiol. 2010, 108, 1015–1022. [Google Scholar] [CrossRef]
- Crothers-Stomps, C.; Høj, L.; Bourne, D.; Hall, M.; Owens, L. Isolation of lytic bacteriophage against Vibrio harveyi. J. Appl. Microbiol. 2010, 108, 1744–1750. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-P.; Gong, T.; Jost, G.; Liu, W.-H.; Ye, D.-Z.; Luo, Z.-H. Isolation and characterization of five lytic bacteriophages infecting a Vibrio strain closely related to Vibrio owensii. FEMS Microbiol. Lett. 2013, 348, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Lal, T.M.; Sano, M.; Ransangan, J. Genome characterization of a novel vibriophage VpKK5 (Siphoviridae) specific to fish pathogenic strain of Vibrio parahaemolyticus. J. Basic Microbiol. 2016, 56, 872–888. [Google Scholar] [CrossRef]
- Sangseedum, C.; Vuddhakul, V.; Mittraparp-arthorn, P. Isolation and Host Range of Vibrio campbellii Bacteriophages Isolated from Cockles. In Proceedings of the National and International Graduate Research Conference; Poj Sarasin Building, Khon Kaen University: Khon Kaen, Thailand, 2017; pp. 241–247. [Google Scholar]
- Luo, Z.H.; Yu, Y.P.; Jost, G.; Liu, W.H.; Huang, X.L.; Gu, L. Characterization of two bacteriophages for specific treatment of biofilm formed by a Vibrio sp. isolated from an abalone farm. Aquac. Res. 2016, 47, 3964–3972. [Google Scholar] [CrossRef]
- Lorenz, N.; Reiger, M.; Toro-Nahuelpan, M.; Brachmann, A.; Poettinger, L.; Plener, L.; Lassak, J.; Jung, K. Identification and initial characterization of prophages in Vibrio campbellii. PLoS ONE 2016, 11, e0156010. [Google Scholar] [CrossRef]
- Jäckel, C.; Hertwig, S.; Scholz, H.C.; Nöckler, K.; Reetz, J.; Hammerl, J.A. Prevalence, Host Range, and Comparative Genomic Analysis of Temperate Ochrobactrum Phages. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clokie, M.R.; Kropinski, A. Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2009; pp. 69–81. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, D.; Marcu, A.; Liang, Y.; Wishart, D.S. PHAST, PHASTER and PHASTEST: Tools for finding prophage in bacterial genomes. Brief. Bioinform. 2019, 20, 1560–1567. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Paez-Espino, D.; Roux, S.; Chen, I.-M.A.; Palaniappan, K.; Ratner, A.; Chu, K.; Huntemann, M.; Reddy, T.B.K.; Pons, J.C.; Llabrés, M. IMG/VR v. 2.0: An integrated data management and analysis system for cultivated and environmental viral genomes. Nucleic Acids Res. 2019, 47, D678–D686. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Jiang, S.; Paul, J. Significance of lysogeny in the marine environment: Studies with isolates and a model of lysogenic phage production. Microb. Ecol. 1998, 35, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.-W. Frequency of morphological phage descriptions in the year 2000. Arch. Virol. 2001, 146, 843–857. [Google Scholar] [CrossRef]
- Bailly-Bechet, M.; Vergassola, M.; Rocha, E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007, 17, 1486–1495. [Google Scholar] [CrossRef] [Green Version]
- Botstein, D. A theory of modular evolution for bacteriophages. Ann. N. Y. Acad. Sci. 1980, 354, 484. [Google Scholar] [CrossRef]
- Veesler, D.; Cambillau, C. A common evolutionary origin for tailed-bacteriophage functional modules and bacterial machineries. Microbiol. Mol. Biol. 2011, 75, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Guo, P. High resolution structure of hexameric herpesvirus DNA-packaging motor elucidates revolving mechanism and ends 20-year fervent debate. Protein Cell 2020, 11, 311–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcaid, M.; Bergeron, A.; Poisson, G. The evolution of the tape measure protein: Units, duplications and losses. BMC Bioinform. 2011, 12, S10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pell, L.G.; Cumby, N.; Clark, T.E.; Tuite, A.; Battaile, K.P.; Edwards, A.M.; Chirgadze, N.Y.; Davidson, A.R.; Maxwell, K.L. A conserved spiral structure for highly diverged phage tail assembly chaperones. J. Mol. Biol. 2013, 425, 2436–2449. [Google Scholar] [CrossRef] [PubMed]
- Lan, S.-F.; Huang, C.-H.; Chang, C.-H.; Liao, W.-C.; Lin, I.H.; Jian, W.-N.; Wu, Y.-G.; Chen, S.-Y.; Wong, H.-C. Characterization of a new plasmid-like prophage in a pandemic Vibrio parahaemolyticus O3:K6 strain. Appl. Environ. Microbiol. 2009, 75, 2659–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, V.M.; Oldfield, L.M.; Hatfull, G.F. An Unusual Phage Repressor Encoded by Mycobacteriophage BPs. PLoS ONE 2015, 10, e0137187. [Google Scholar] [CrossRef] [Green Version]
- Grigoriev, P.S.; Łobocka, M.B. Determinants of segregational stability of the linear plasmid-prophage N15 of Escherichia coli. Mol. Microbiol. 2001, 42, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Łobocka, M.B.; Rose, D.J.; Plunkett, G.; Rusin, M.; Samojedny, A.; Lehnherr, H.; Yarmolinsky, M.B.; Blattner, F.R. Genome of Bacteriophage P1. J. Bacteriol. 2004, 186, 7032. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Deaton, J.; Young, R. Sizing the holin lesion with an endolysin-β-galactosidase fusion. J. Bacteriol. 2003, 185, 779–787. [Google Scholar] [CrossRef] [Green Version]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar]
- Wang, X.; Wood, T.K. Cryptic prophages as targets for drug development. Drug Resist. Updat. 2016, 27, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Loessner, M.; Goeppl, S.; Busse, M. Comparative inducibility of bacteriophage in naturally lysogenic and lysogenized strains of Listeria spp. by UV light and Mitomycin C. Lett. Appl. Microbiol. 1991, 12, 196–199. [Google Scholar] [CrossRef]
- Byrne, M.; Kropinski, A.M. The genome of the Pseudomonas aeruginosa generalized transducing bacteriophage F116. Gene 2005, 346, 187–194. [Google Scholar] [CrossRef]
- Williamson, S.J.; Paul, J. Environmental factors that influence the transition from lysogenic to lytic existence in the ϕHSIC/Listonella pelagia marine phage–host system. Microb. Ecol. 2006, 52, 217–225. [Google Scholar] [CrossRef]
- Łoś, J.M.; Łoś, M.; Węgrzyn, G.; Węgrzyn, A. Differential efficiency of induction of various lambdoid prophages responsible for production of Shiga toxins in response to different induction agents. Microb. Pathog. 2009, 47, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.M. Preferential Orientation Preferential Orientation of Natural Lambdoid Prophages and Bacterial Chromosome Organization. Theor. Popul. Biol. 2002, 61, 503–507. [Google Scholar] [CrossRef]
- Rasmussen, K.K.; Frandsen, K.E.H.; Boeri Erba, E.; Pedersen, M.; Varming, A.K.; Hammer, K.; Kilstrup, M.; Thulstrup, P.W.; Blackledge, M.; Jensen, M.R.; et al. Structural and dynamics studies of a truncated variant of CI repressor from bacteriophage TP901-1. Sci. Rep. 2016, 6, 29574. [Google Scholar] [CrossRef]
- Young, R.; Wang, N.; Roof, W.D. Phages will out: Strategies of host cell lysis. Trends Microbiol. 2000, 8, 120–128. [Google Scholar] [CrossRef]
- Garneau, J.E.; Dupuis, M.-È.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadán, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Kalatzis, P.G.; Rørbo, N.I.; Castillo, D.; Mauritzen, J.J.; Jørgensen, J.; Kokkari, C.; Zhang, F.; Katharios, P.; Middelboe, M. Stumbling across the Same Phage: Comparative Genomics of Widespread Temperate Phages Infecting the Fish Pathogen Vibrio anguillarum. Viruses 2017, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.L.; Waldor, M.K. Bacteriophage control of bacterial virulence. Infect. Immun. 2002, 70, 3985–3993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.; Wang, L.; Tian, H.; Wei, D.; Liu, Y. Pathogenicity and genomic characterization of Vibrio parahaemolyticus strain PB1937 causing shrimp acute hepatopancreatic necrosis disease in China. Ann. Microbiol. 2018, 68, 175–184. [Google Scholar] [CrossRef]
- Hoikkala, V.; Almeida, G.M.; Laanto, E.; Sundberg, L.-R. Aquaculture as a source of empirical evidence for coevolution between CRISPR-Cas and phage. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180100. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Region Length | Completeness * | Score | # Total Proteins | Region Position | Most Common Phage | GC % |
|---|---|---|---|---|---|---|---|
| 1 | 41.7Kb | intact | 110 | 60 | 1–41,772 | PHAGE_Entero_N15_NC_001901(9) | 47.45% |
| ORF | Start | Stop | Strand | Length (bp) | BLASTp Best Hit (Gene) [Taxa] | Coverage (%) | E value | Identity (%) | Accession Blast Hit |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 603 | + | 603 | small subunit terminase [Aeromonas phage phiARM81ld] | 82 | 2e −31 | 40.48 | ALN97521.1 |
| 2 | 683 | 2467 | + | 1785 | large subunit terminase [Aeromonas phage phiARM81ld] | 99 | 3e−120 | 38.47 | ALN97522.1 |
| 3 | 2464 | 2673 | + | 210 | head–tail joining protein [Escherichia virus Lambda] | 95 | 1e−11 | 48.48 | NP_040582.1 |
| 4 | 2673 | 4229 | + | 1557 | Phage portal protein [Escherichia virus Lambda_2H10] | 95 | 0.0 | 55.98 | VUD36612.1 |
| 5 | 4222 | 5574 | + | 1353 | Phage capsid and scaffold [Escherichia virus Lambda] | 94 | 3e−121 | 45.90 | VUF53141.1 |
| 6 | 5588 | 5929 | + | 342 | Head decoration protein D [Aeromonas phage phiARM81ld] | 72 | 2e−12 | 40.96 | ALN97526.1 |
| 7 | 5969 | 7006 | + | 1038 | phage major capsid E family protein [Escherichia phage YDC107_1] | 98 | 6e−89 | 42.52 | AUO37489.1 |
| 8 | 7071 | 7562 | + | 492 | - | - | - | - | - |
| 9 | 7571 | 7861 | + | 291 | - | - | - | - | - |
| 10 | 7858 | 8490 | + | 633 | Prophage minor tail protein Z [Escherichia virus N15] | 93 | 1.00e−44 | 41.62 | NP_046906.1 |
| 11 | 8480 | 8929 | + | 450 | tail protein [Enterobacteria phage mEp043 c-1] | 95 | 4e−09 | 26.53 | YP_007111512.1 |
| 12 | 8932 | 9429 | + | 498 | major tail protein V [Enterobacteria phage HK225] | 91 | 1e−47 | 51.66 | YP_007112145.1 |
| 13 | 9432 | 9914 | + | 483 | tail assembly chaperone [Enterobacteria phage phi80] | 90 | 8e−05 | 27.52 | YP_007947938.1 |
| 14 | 9938 | 10,270 | + | 333 | Phage minor tail protein [Escherichia phage mEp460_ev081] | 100 | 1e−05 | 35.14 | VUF53221.1 |
| 15 | 10,251 | 12,473 | + | 2223 | tail length tape-measure protein 1 [Pseudoalteromonas phage SL25] | 68 | 8e−35 | 27.32 | ASU03386.1 |
| 16 | 12,480 | 12,947 | + | 468 | hypothetical protein [Pseudomonas phage PS-1] | 74 | 1e−08 | 31.62 | YP_009222842.1 |
| 17 | 12,947 | 14,743 | + | 1797 | minor tail protein [Arthrobacter phage Ingrid] | 48 | 2e−16 | 27.87 | QFG08696.1 |
| 18 | 14,744 | 15,691 | + | 948 | - | - | - | - | - |
| 19 | 15,693 | 16,229 | + | 537 | hypothetical protein [Bacteriophage sp.] | 96 | 3e−28 | 40.70 | QHJ79317.1 |
| 20 | 16,229 | 16,642 | + | 414 | endopeptidase [Pseudomonas phage PS-1] | 80 | 7e−08 | 32.43 | YP_009222844.1 |
| 21 | 16,614 | 19,427 | + | 2814 | tail protein [Pseudomonas phage PS-1] | 98 | 4e−137 | 32.24 | YP_009222845.1 |
| 22 | 19,471 | 19,731 | + | 261 | hypothetical protein NVP1081O_260 [Vibrio phage 1.081.O._10N.286.52.C2] | 89 | 9e−12 | 42.31 | AUR85995.1 |
| 23 | 19,738 | 19,920 | + | 183 | - | - | - | - | - |
| 24 | 20,301 | 20,627 | + | 327 | putative DNA-binding protein [Vibrio phage VHML] | 73 | 5e−11 | 35.44 | NP_758895.1 |
| 25 | 20,660 | 20,815 | - | 156 | - | - | - | - | - |
| 26 | 20,893 | 21,252 | - | 360 | hypothetical protein CETO_181 [Vibrio phage Ceto] | 89 | 4e−06 | 32.43 | YP_009621244.1 |
| 27 | 21,272 | 21,562 | - | 291 | - | - | - | - | - |
| 28 | 21,583 | 21,813 | - | 231 | - | - | - | - | - |
| 29 | 21,824 | 22,786 | - | 963 | ParB protein [Escherichia virus N15] | 61 | 2e−51 | 46.70 | NP_046922.1 |
| 30 | 22,798 | 23,187 | + | 390 | - | - | - | - | - |
| 31 | 23,252 | 23,377 | + | 126 | - | - | - | - | - |
| 32 | 23,594 | 23,731 | + | 138 | - | - | - | - | - |
| 33 | 23,733 | 23,817 | + | 85 | tRNA-Ser-GCT | - | - | - | - |
| 34 | 23,846 | 24,037 | + | 192 | - | - | - | - | - |
| 35 | 24,037 | 24,198 | + | 162 | - | - | - | - | - |
| 36 | 24,247 | 26,307 | + | 2061 | protelomerase [Yersinia phage PY54] | 74 | 3e−68 | 34.21 | CAC88681.1 |
| 37 | 26,450 | 26,665 | + | 216 | - | - | - | - | - |
| 38 | 26,695 | 27,084 | - | 390 | - | - | - | - | - |
| 39 | 27,084 | 30,713 | - | 3630 | primase [Aeromonas phage phiARM81ld] | 84 | 0.0 | 36.02 | ALN97565.1 |
| 40 | 30,768 | 30,971 | - | 204 | - | - | - | - | - |
| 41 | 31,075 | 31,770 | - | 696 | prophage repressor [Enterobacterial phage mEp390] | 62 | 7e−22 | 41.50 | YP_007112454.1 |
| 42 | 31,912 | 32,139 | + | 228 | putative transcriptional regulator (cro analog) [Salmonella phage Fels-1] | 90 | 2e−04 | 36.23 | YP_001700550.1 |
| 43 | 32,159 | 32,758 | + | 600 | prophage antirepressor [Halomonas virus HAP1] | 78 | 2e−16 | 25.77 | YP_001686774.1 |
| 44 | 32,771 | 33,493 | + | 723 | antiterminator Q [Yersinia phage PY54] | 85 | 2e−12 | 26.42 | NP_892088.1 |
| 45 | 33,698 | 34,393 | + | 696 | - | - | - | - | - |
| 46 | 34,424 | 35,101 | + | 678 | hypothetical protein VH12019_00006 [Vibrio phage VH1_2019] | 34 | 2e−10 | 34.62 | QHJ74333.1 |
| 47 | 35,111 | 35,290 | + | 180 | - | - | - | - | - |
| 48 | 35,300 | 36,598 | + | 1299 | putative exonuclease [Vibrio phage 1.205.O._10N.222.51.A7] | 68 | 6e−116 | 54.73 | AUR95306.1 |
| 49 | 36,598 | 37,104 | + | 507 | single-stranded DNA-binding protein [Vibrio phage VD1] | 100 | 7e−82 | 73.21 | AGN34167.1 |
| 50 | 37,131 | 37,316 | + | 186 | - | - | - | - | - |
| 51 | 37,309 | 37,650 | + | 342 | ASCH domain protein [Vibrio phage 1.104.O._10N.286.49.A12] | 85 | 3e−06 | 35.00 | AUR87783.1 |
| 52 | 37,650 | 39,077 | + | 1428 | DNA methyltransferase [Vibrio phage Va_PF430-3_p42] | 97 | 0.0 | 66.40 | QCW19890.1 |
| 53 | 39,077 | 39,328 | + | 252 | - | - | - | - | - |
| 54 | 39,443 | 39,655 | + | 213 | - | - | - | - | - |
| 55 | 39,659 | 39,841 | + | 183 | hypothetical protein VPQG_00007 [Vibrio phage VBpm10] | 63 | 8e−11 | 68.42 | AGF90980.1 |
| 56 | 39,845 | 40,093 | + | 249 | - | - | - | - | - |
| 57 | 40,093 | 40,476 | + | 384 | hypothetical protein [Escherichia virus N15] | 66 | 2e−18 | 42.86 | NP_046953.1 |
| 58 | 40,493 | 40,621 | + | 129 | - | - | - | - | - |
| 59 | 40,835 | 41,116 | + | 282 | Bacteriophage holin HP1 family protein [Bacteriophage APSE-7] | 52 | 1e−07 | 46.94 | CAB3623628.1 |
| 60 | 41,116 | 41,772 | + | 657 | lysozyme-like domain protein [Vibrio phage 1.202.O._10N.222.45.E8] | 97 | 4e−61 | 50.00 | YP_009812536.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nuidate, T.; Kuaphiriyakul, A.; Surachat, K.; Mittraparp-arthorn, P. Induction and Genome Analysis of HY01, a Newly Reported Prophage from an Emerging Shrimp Pathogen Vibrio campbellii. Microorganisms 2021, 9, 400. https://doi.org/10.3390/microorganisms9020400
Nuidate T, Kuaphiriyakul A, Surachat K, Mittraparp-arthorn P. Induction and Genome Analysis of HY01, a Newly Reported Prophage from an Emerging Shrimp Pathogen Vibrio campbellii. Microorganisms. 2021; 9(2):400. https://doi.org/10.3390/microorganisms9020400
Chicago/Turabian StyleNuidate, Taiyeebah, Aphiwat Kuaphiriyakul, Komwit Surachat, and Pimonsri Mittraparp-arthorn. 2021. "Induction and Genome Analysis of HY01, a Newly Reported Prophage from an Emerging Shrimp Pathogen Vibrio campbellii" Microorganisms 9, no. 2: 400. https://doi.org/10.3390/microorganisms9020400