Effect of Washing, Waxing and Low-Temperature Storage on the Postharvest Microbiome of Apple

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Harvesting of Apples, Treatments and Experimental Design
2.2. Library Preparation and Sequencing
2.3. Bioinformatic and Statistical Analyses
3. Results
3.1. Sequencing Results
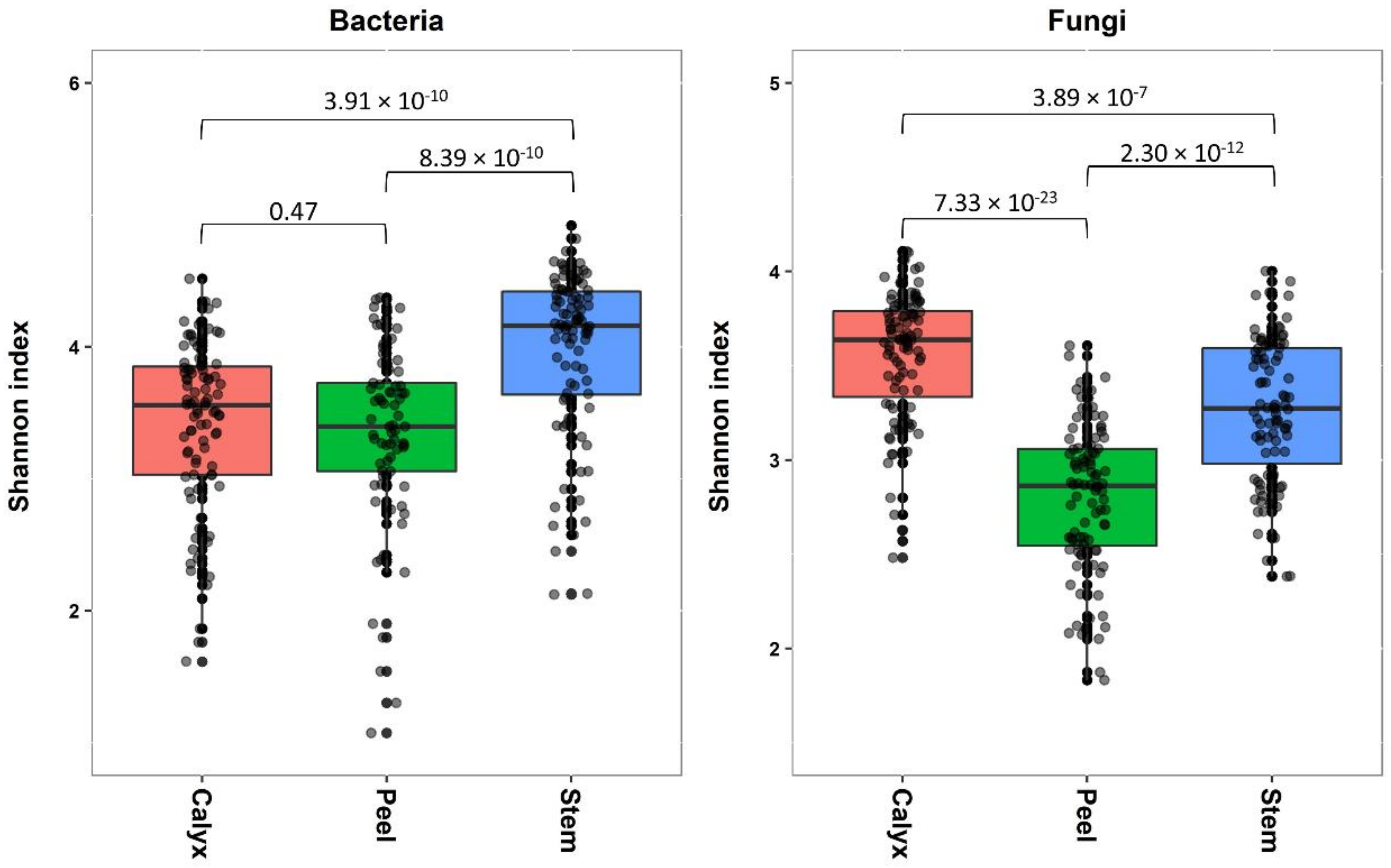
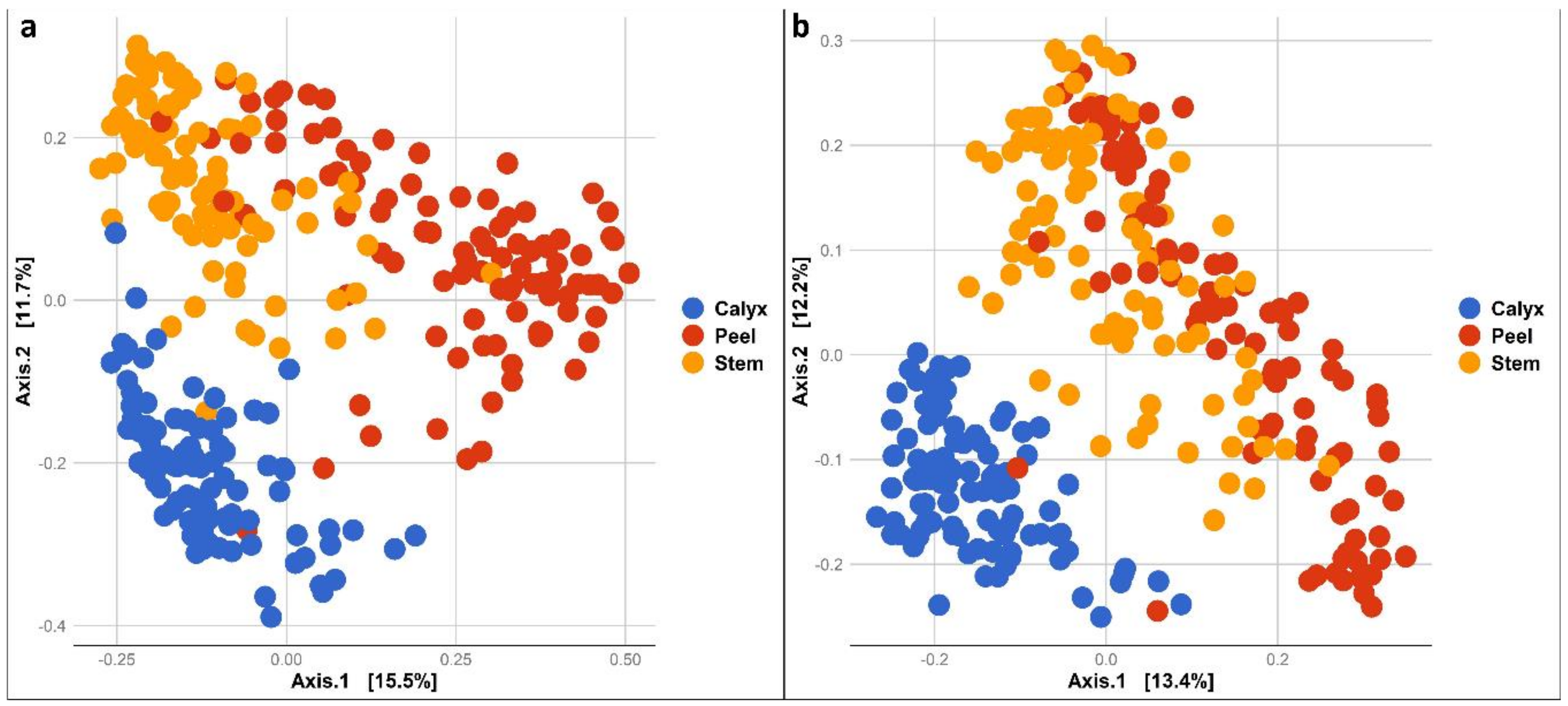
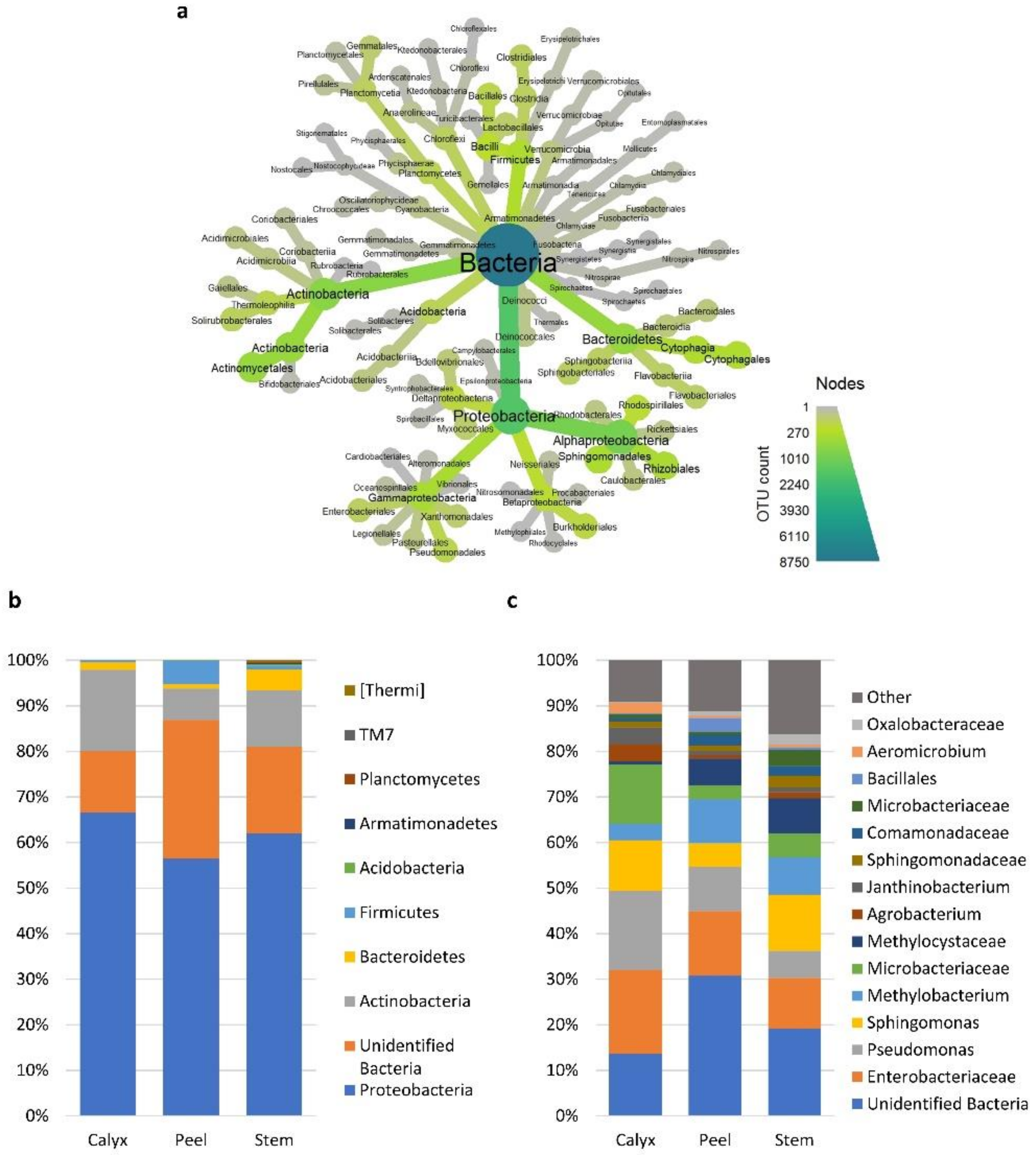
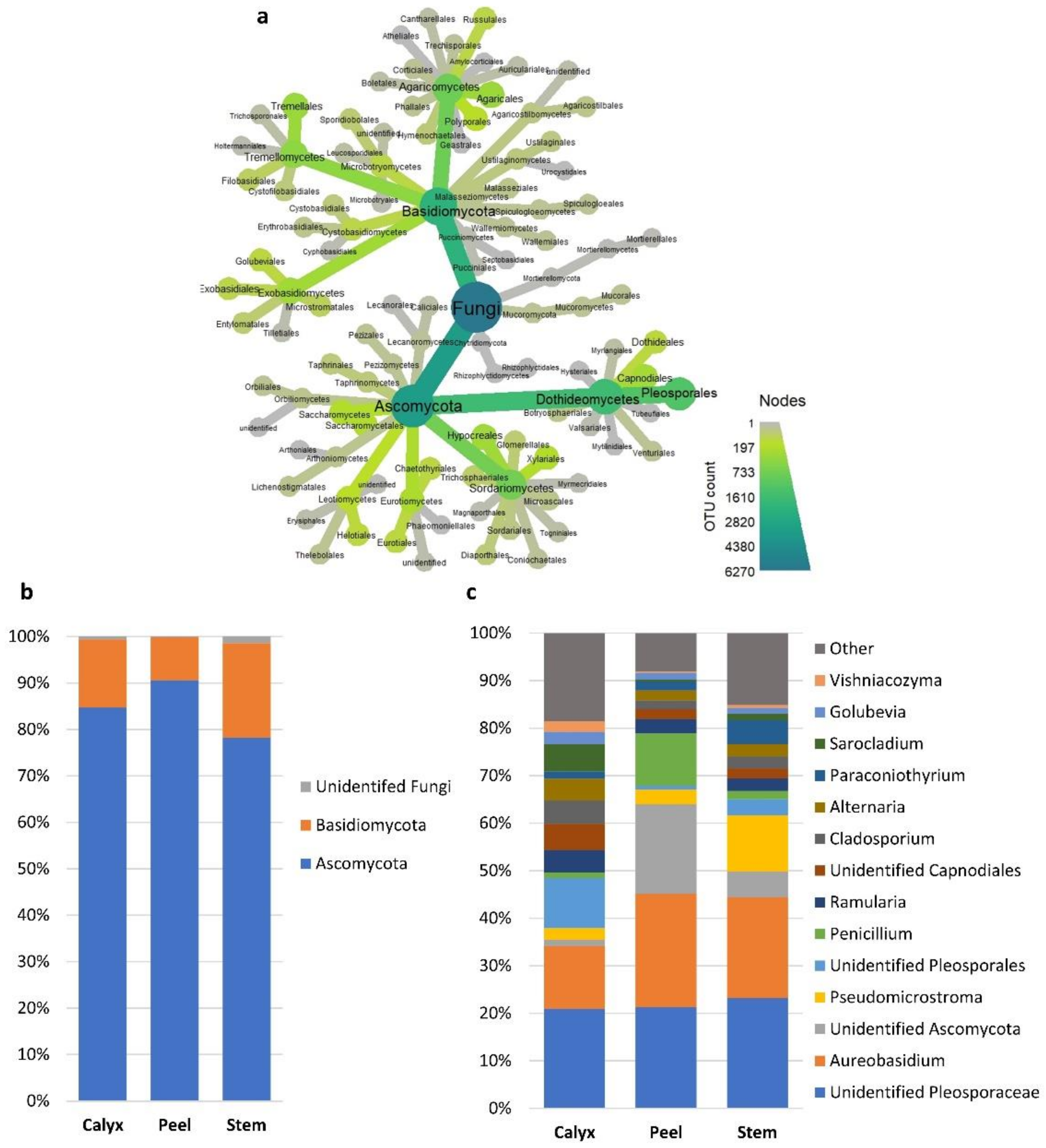
3.2. Bacterial and Fungal Communities Associated with Apple Calyx-End, Stem-End, and Peel Tissues
3.3. Effect of Washing and Waxing and Time of Storage on Apple Peel, Stem-End, and Calyx-End Tissues
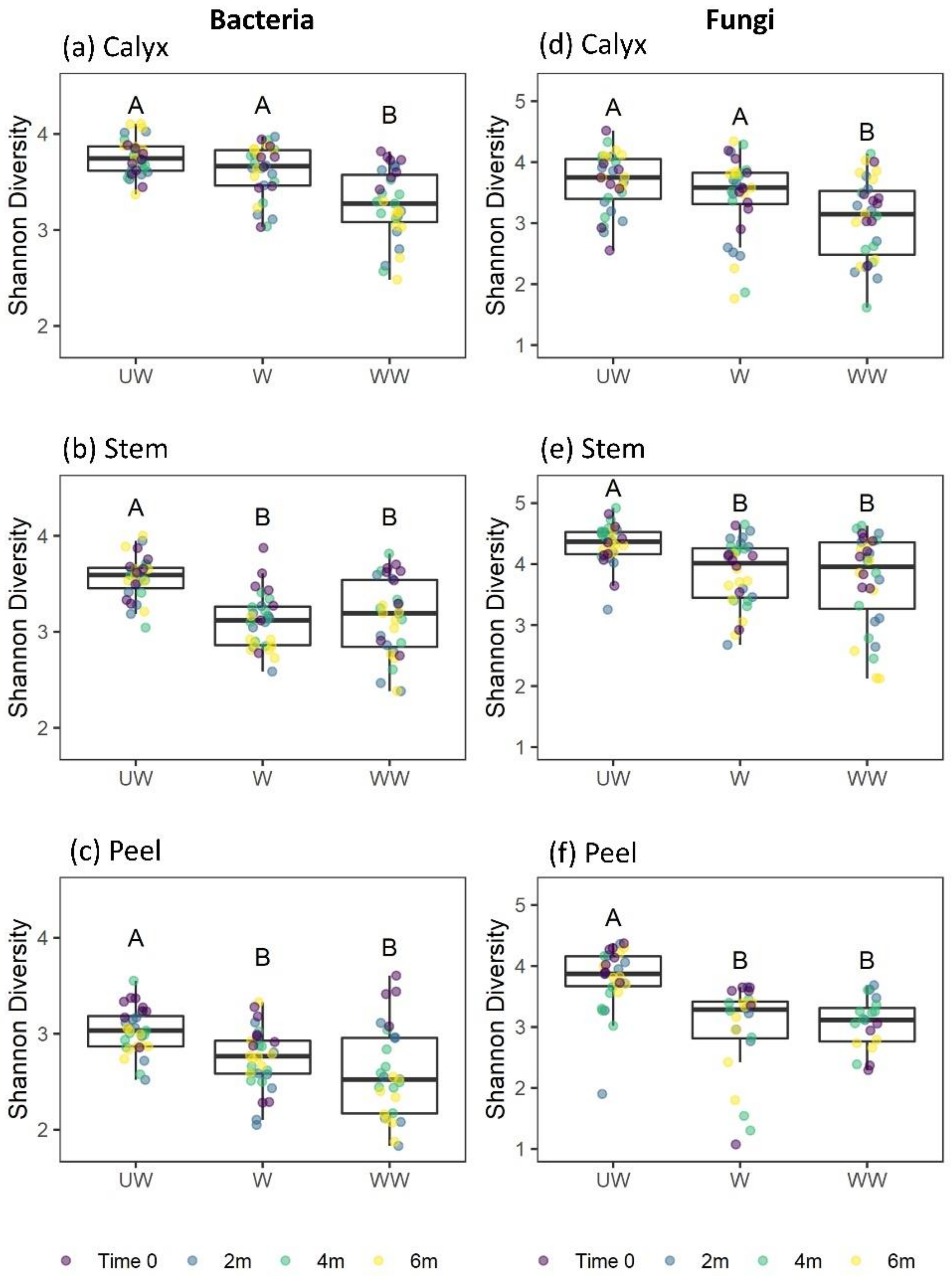
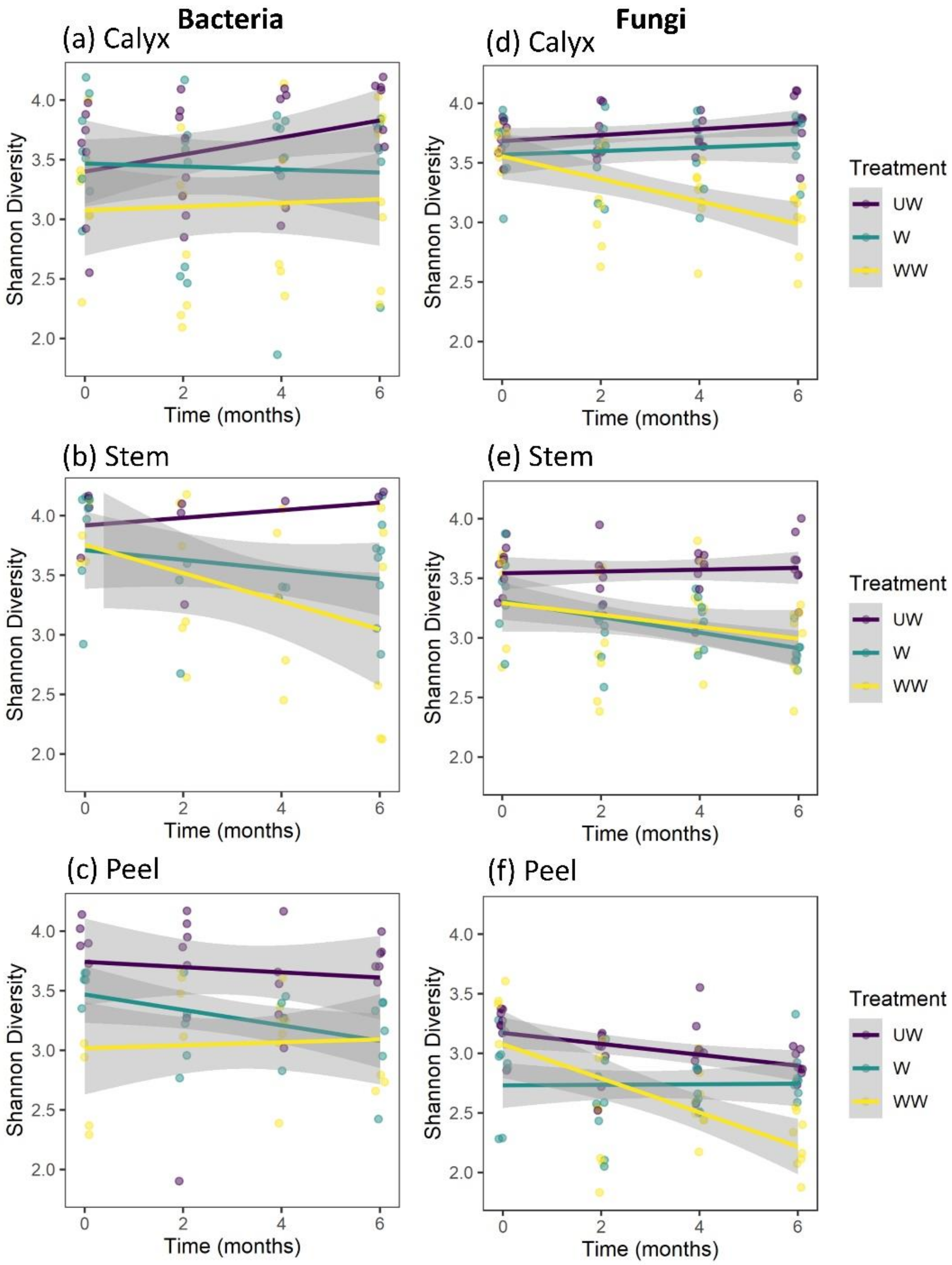
3.3.1. Shannon Diversity
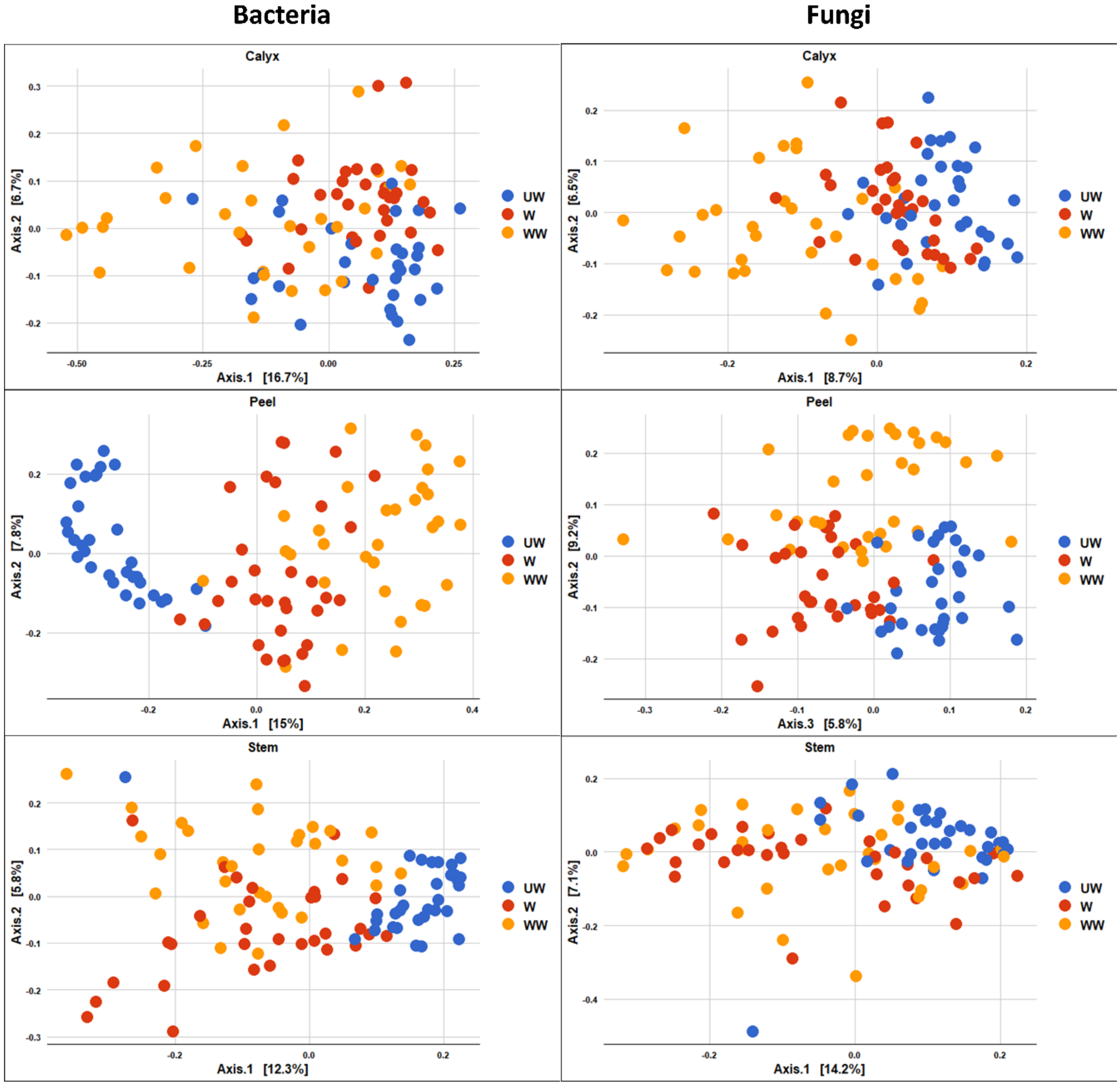
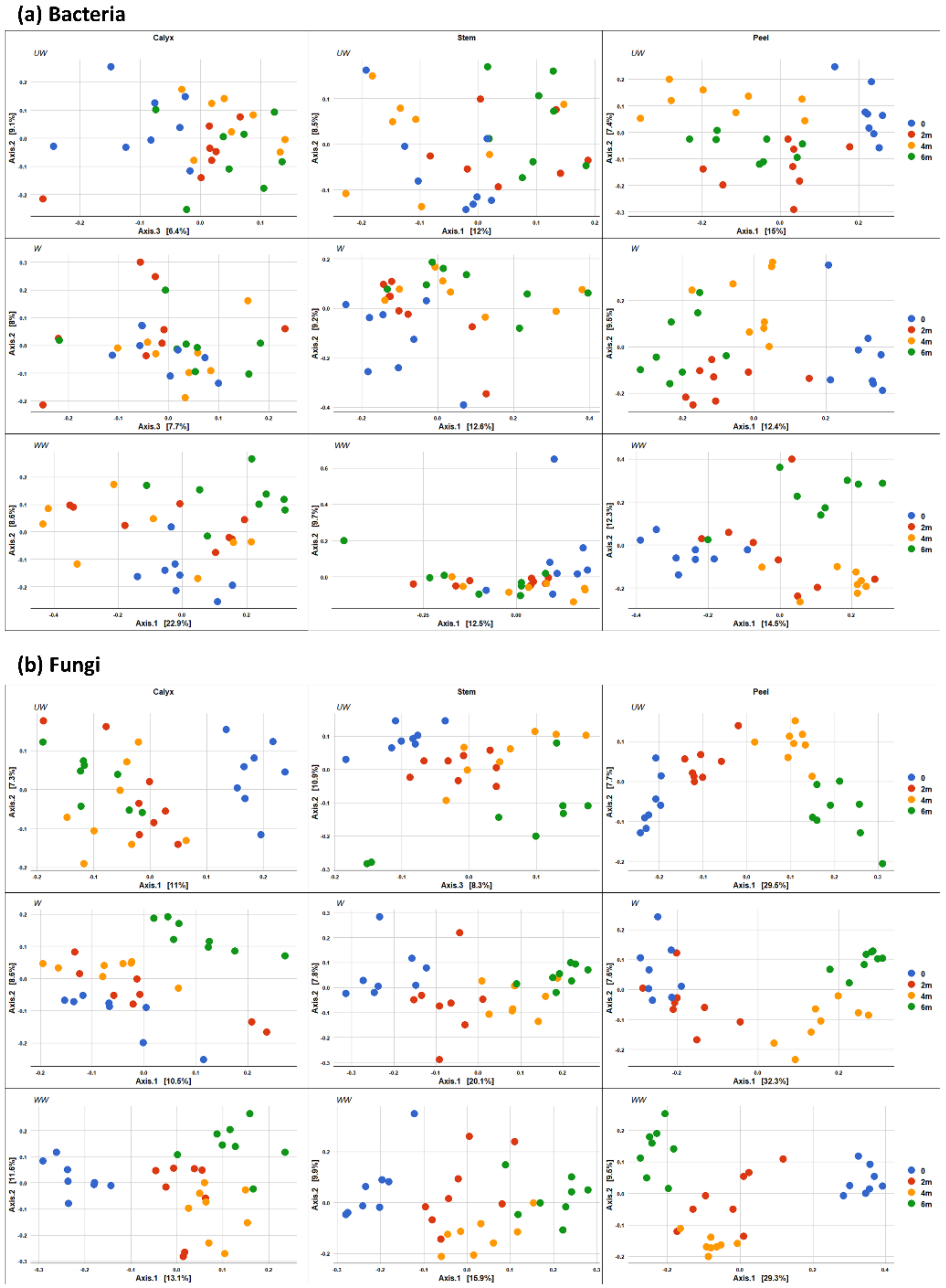
3.3.2. Community Composition
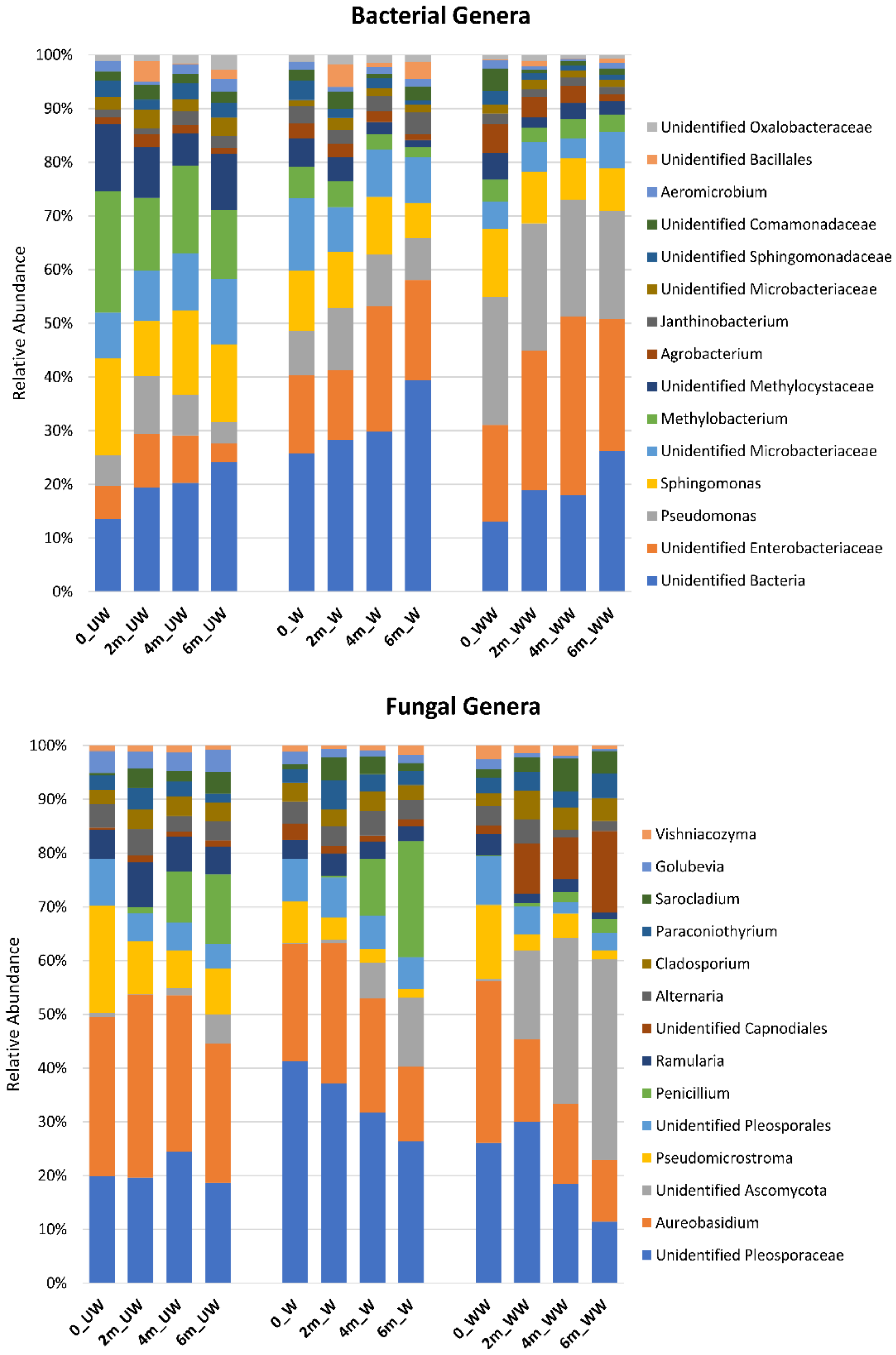
3.4. Effect of Postharvest Treatments and Storage Time on the Relative Abundance of Microbial Taxa
4. Discussion
4.1. Tissue
4.2. Treatment
4.3. Time
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cryan, J.F.; Dinan, T.G. Mind-Altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Raina, T.K.; Kumar, A.; Singh, J.; Prasad, R. Plant microbiome: A reservoir of novel genes and metabolites. Plant Gene 2019, 18, 100177. [Google Scholar] [CrossRef]
- Hammer, T.J.; Sanders, J.G.; Fierer, N. Not all animals need a microbiome. FEMS Microbiol. Lett. 2019, 366. [Google Scholar] [CrossRef] [Green Version]
- Proctor, L.; LoTempio, J.; Marquitz, A.; Daschner, P.; Xi, D.; Flores, R.; Brown, L.; Ranallo, R.; Maruvada, P.; Regan, K.; et al. A review of 10 years of human microbiome research activities at the US National Institutes of Health, Fiscal Years 2007–2016. Microbiome 2019, 7, 31. [Google Scholar] [CrossRef]
- Compant, S.; Samad, A.; Faist, H.; Sessitsch, A. A review on the plant microbiome: Ecology, functions, and emerging trends in microbial application. J. Adv. Res. 2019, 19, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.-K.; Pineda, A.; Hannula, S.E.; Kielak, A.M.; Setyarini, S.N.; Bezemer, T.M. Steering root microbiomes of a commercial horticultural crop with plant-soil feedbacks. Appl. Soil Ecol. 2020, 150, 103468. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Cacciola, S.O.; Mosca, S.; Zappia, R.; Schena, L. Analysis of the Fungal Diversity in Citrus Leaves with Greasy Spot Disease Symptoms. Microb. Ecol. 2017, 73, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Abdelfattah, A.; Sanzani, S.M.; Wisniewski, M.; Berg, G.; Cacciola, S.O.; Schena, L. Revealing Cues for Fungal Interplay in the Plant-Air Interface in Vineyards. Front. Plant Sci. 2019, 10. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Li Destri Nicosia, M.G.; Cacciola, S.O.; Droby, S.; Schena, L. Metabarcoding Analysis of Fungal Diversity in the Phyllosphere and Carposphere of Olive (Olea europaea). PLoS ONE 2015, 10, e0131069. [Google Scholar] [CrossRef] [Green Version]
- Abdelfattah, A.; Wisniewski, M.; Droby, S.; Schena, L. Spatial and compositional variation in the fungal communities of organic and conventionally grown apple fruit at the consumer point-of-purchase. Hortic. Res. 2016, 3, 16047. [Google Scholar] [CrossRef] [Green Version]
- Abdelfattah, A.; Malacrinò, A.; Wisniewski, M.; Cacciola, S.O.; Schena, L. Metabarcoding: A powerful tool to investigate microbial communities and shape future plant protection strategies. Biol. Control 2018, 120, 1–10. [Google Scholar] [CrossRef]
- Abdelfattah, A.; Wisniewski, M.; Li Destri Nicosia, M.G.; Cacciola, S.O.; Schena, L. Metagenomic Analysis of Fungal Diversity on Strawberry Plants and the Effect of Management Practices on the Fungal Community Structure of Aerial Organs. PLoS ONE 2016, 11, e0160470. [Google Scholar] [CrossRef]
- Berg, G.; Erlacher, A.; Grube, M. The Edible Plant Microbiome: Importance and Health Issues. In Principles of Plant-Microbe Interactions: Microbes for Sustainable Agriculture; Lugtenberg, B., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 419–426. [Google Scholar] [CrossRef]
- Wassermann, B.; Müller, H.; Berg, G. An apple a day: which bacteria do we eat with organic and conventional apples? Front. Microbiol. 2019, 10, 1629. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Chung, T.; Chen, Y.; Macarisin, D.; LaBorde, L.; Kovac, J. The occurrence of Listeria monocytogenes is associated with built environment microbiota in three tree fruit processing facilities. Microbiome 2019, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Abdelfattah, A.; Norelli, J.; Burchard, E.; Schena, L.; Droby, S.; Wisniewski, M. Apple endophytic microbiota of different rootstock/scion combinations suggests a genotype-specific influence. Microbiome 2018, 6, 18. [Google Scholar] [CrossRef]
- Droby, S.; Wisniewski, M. The fruit microbiome: A new frontier for postharvest biocontrol and postharvest biology. Postharvest Biol. Technol. 2018, 140, 107–112. [Google Scholar] [CrossRef]
- Wisniewski, M.; Droby, S.; Norelli, J.; Liu, J.; Schena, L. Alternative management technologies for postharvest disease control: The journey from simplicity to complexity. Postharvest Biol. Technol. 2016, 122, 3–10. [Google Scholar] [CrossRef]
- Wisniewski, M.; Droby, S. The postharvest microbiome: The other half of sustainability. Biol. Control 2019, 137, 104025. [Google Scholar] [CrossRef]
- Macarisin, D.; Sheth, I.; Hur, M.; Wooten, A.; Kwon, H.J.; Gao, Z.; De Jesus, A.; Jurick, W.; Chen, Y. Survival of outbreak, food, and environmental strains of Listeria monocytogenes on whole apples as affected by cultivar and wax coating. Sci. Rep. 2019, 9, 12170. [Google Scholar] [CrossRef]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, D.S.; Yourstone, S.; Mieczkowski, P.; Jones, C.D.; Dangl, J.L. Practical innovations for high-throughput amplicon sequencing. Nat. Methods 2013, 10, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Toju, H.; Tanabe, A.S.; Yamamoto, S.; Sato, H. High-Coverage ITS Primers for the DNA-Based Identification of Ascomycetes and Basidiomycetes in Environmental Samples. PLoS ONE 2012, 7, e40863. [Google Scholar] [CrossRef] [Green Version]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Academic Press Inc.: San Diego, CA, USA, 1990; Volume 18, pp. 315–322. [Google Scholar]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abarenkov, K.; Henrik Nilsson, R.; Larsson, K.-H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; Pennanen, T.; et al. The UNITE database for molecular identification of fungi—recent updates and future perspectives. New Phytol. 2010, 186, 281–285. [Google Scholar] [CrossRef]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Martinez Arbizu, P. PairwiseAdonis: Pairwise Multilevel Comparison Using Adonis. R Package Version 0.4. 2020. Available online: https://github.com/pmartinezarbizu/pairwiseAdonis (accessed on 14 March 2020).
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Hothorn, T.; Bretz, F.; Westfall, P. Simultaneous Inference in General Parametric Models. Biom. J. 2008, 50, 346–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Package ‘Vegan’. Community Ecology, Package. Version 2.5-6. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 23 June 2020).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Foster, Z.S.L.; Sharpton, T.J.; Grünwald, N.J. Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput. Biol. 2017, 13, e1005404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Wassermann, B.; Kusstatscher, P.; Berg, G. Microbiome Response to Hot Water Treatment and Potential Synergy with Biological Control on Stored Apples. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, E.; Olsson, C.; He, J.; Stark, T.; Sadowska, Z.; Molin, G.; Ahrné, S.; Alsanius, B.; Håkansson, Å. Effects of household washing on bacterial load and removal of Escherichia coli from lettuce and “ready-to-eat” salads. Food Sci. Nutr. 2017, 5, 1215–1220. [Google Scholar] [CrossRef]
- Shen, Y.; Nie, J.; Dong, Y.; Kuang, L.; Li, Y.; Zhang, J. Compositional shifts in the surface fungal communities of apple fruits during cold storage. Postharvest Biol. Technol. 2018, 144, 55–62. [Google Scholar] [CrossRef]
- Cui, Z.; Huntley, R.B.; Zeng, Q.; Steven, B. Temporal and spatial dynamics in the apple flower microbiome in the presence of the phytopathogen Erwinia amylovora. bioRxiv 2020, 19, 956078. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria | Fungi | |||
|---|---|---|---|---|
| F Value | Pr (>F) | F Value | Pr (>F) | |
| Treatment | 26.973 | 2.56 × 10−11 | 56.58 | <2 × 10−16 |
| Tissue | 35.98 | 1.96 × 10−14 | 165.926 | <2 × 10−16 |
| Time | 1.831 | 0.1773 | 20.982 | 7.16 × 10−6 |
| Treatment × Tissue | 2.068 | 0.0855 | 3.995 | 0.00365 |
| Treatment × Time | 1.407 | 0.2469 | 12.049 | 9.85 × 10−6 |
| Tissue × Time | 1.455 | 0.2353 | 2.417 | 0.09116 |
| Treatment × Tissue × Time | 0.726 | 0.5751 | 3.609 | 0.00696 |
| Calyx | Stem | Peel | |||||
|---|---|---|---|---|---|---|---|
| F Value | Pr (>F) | F Value | Pr (>F) | F Value | Pr (>F) | ||
| Bacteria | Treatment | 7.564 | 0.000925 | 10.577 | 7.49 × 10−5 | 16.051 | 2.02 × 10−6 |
| Time | 0.231 | 0.631866 | 3.574 | 0.0619 | 1.036 | 0.312 | |
| Treatment × Time | 0.608 | 0.546641 | 2.189 | 0.1179 | 0.325 | 0.724 | |
| Fungi | Treatment | 30.518 | 8.07 × 10−11 | 22.535 | 1.25 × 10−8 | 17.097 | 5.60 × 10−7 |
| Time | 1.977 | 0.16317 | 6.58 | 0.012 | 14.15 | 0.000307 | |
| Treatment × Time | 10.119 | 1.10 × 10−4 | 2.575 | 0.0819 | 7.898 | 0.00071 | |
| Bacteria | Fungi | |||
|---|---|---|---|---|
| R2 | Pr (>F) | R2 | Pr (>F) | |
| Tissue | 0.19293 | 0.001 | 0.16309 | 0.001 |
| Treatment | 0.06442 | 0.001 | 0.04962 | 0.001 |
| Time | 0.0382 | 0.001 | 0.09419 | 0.001 |
| Tissue × Treatment | 0.04249 | 0.001 | 0.02634 | 0.001 |
| Treatment × Time | 0.02803 | 0.001 | 0.03742 | 0.001 |
| Tissue × Time | 0.0275 | 0.001 | 0.03841 | 0.001 |
| Tissue × Treatment × Time | 0.03766 | 0.002 | 0.04455 | 0.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelfattah, A.; Whitehead, S.R.; Macarisin, D.; Liu, J.; Burchard, E.; Freilich, S.; Dardick, C.; Droby, S.; Wisniewski, M. Effect of Washing, Waxing and Low-Temperature Storage on the Postharvest Microbiome of Apple. Microorganisms 2020, 8, 944. https://doi.org/10.3390/microorganisms8060944
Abdelfattah A, Whitehead SR, Macarisin D, Liu J, Burchard E, Freilich S, Dardick C, Droby S, Wisniewski M. Effect of Washing, Waxing and Low-Temperature Storage on the Postharvest Microbiome of Apple. Microorganisms. 2020; 8(6):944. https://doi.org/10.3390/microorganisms8060944
Chicago/Turabian StyleAbdelfattah, Ahmed, Susan R. Whitehead, Dumitru Macarisin, Jia Liu, Erik Burchard, Shiri Freilich, Christopher Dardick, Samir Droby, and Michael Wisniewski. 2020. "Effect of Washing, Waxing and Low-Temperature Storage on the Postharvest Microbiome of Apple" Microorganisms 8, no. 6: 944. https://doi.org/10.3390/microorganisms8060944