Profiling the Oral Microbiome and Plasma Biochemistry of Obese Hyperglycemic Subjects in Qatar


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Plasma Biochemistry
2.3. 16S rRNA Sequencing
2.4. Bioinformatics
2.5. Statistical Analysis
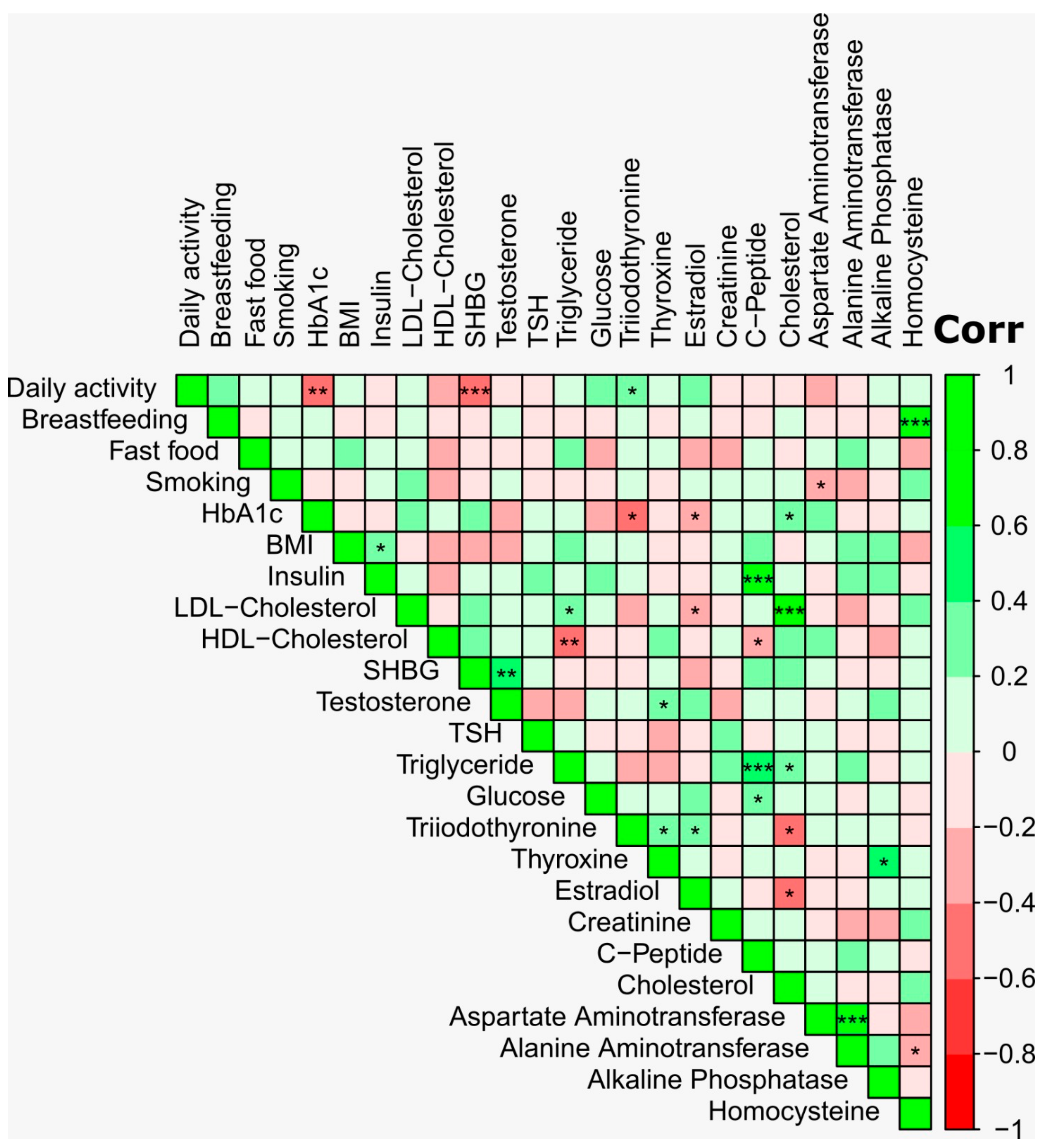
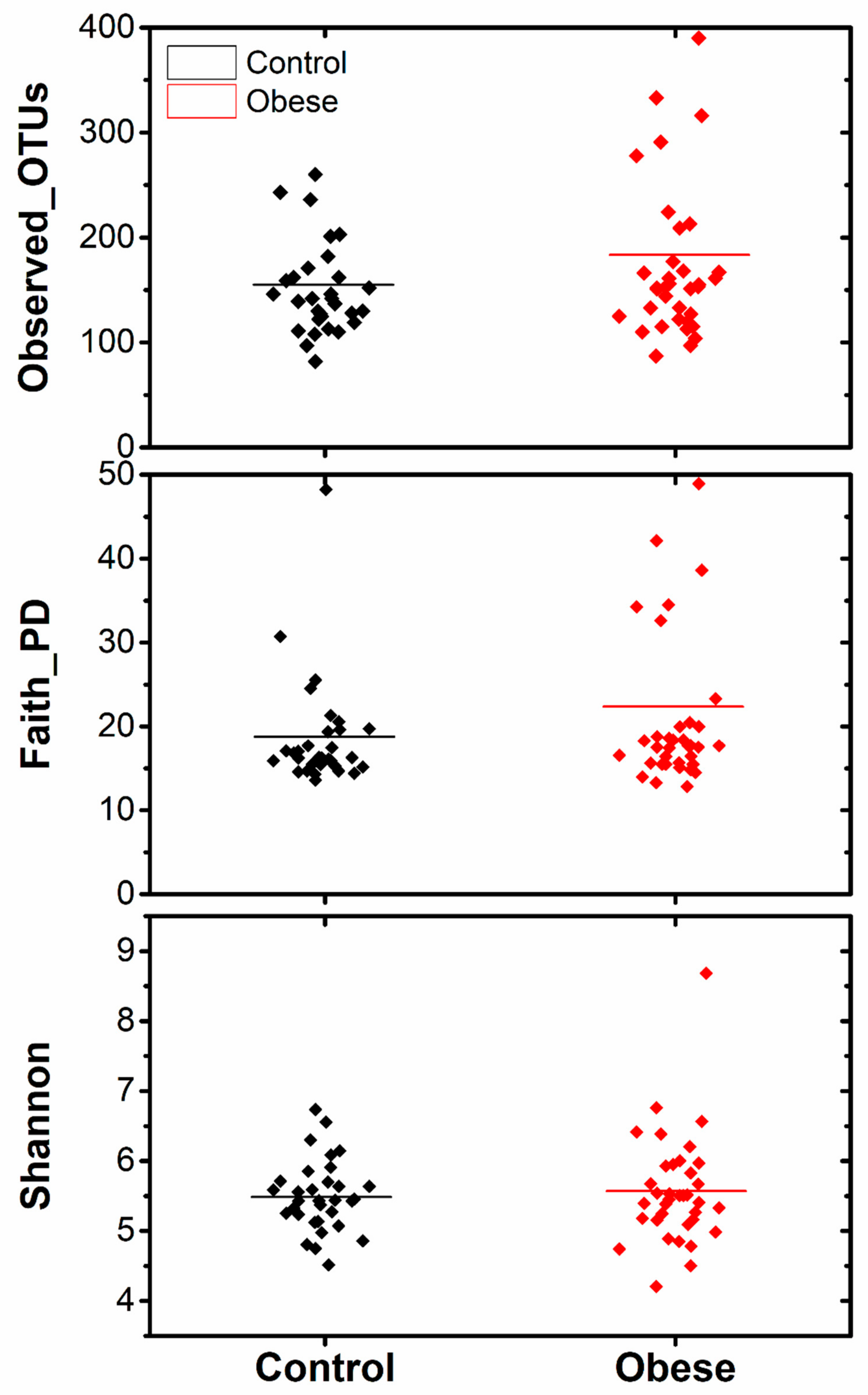
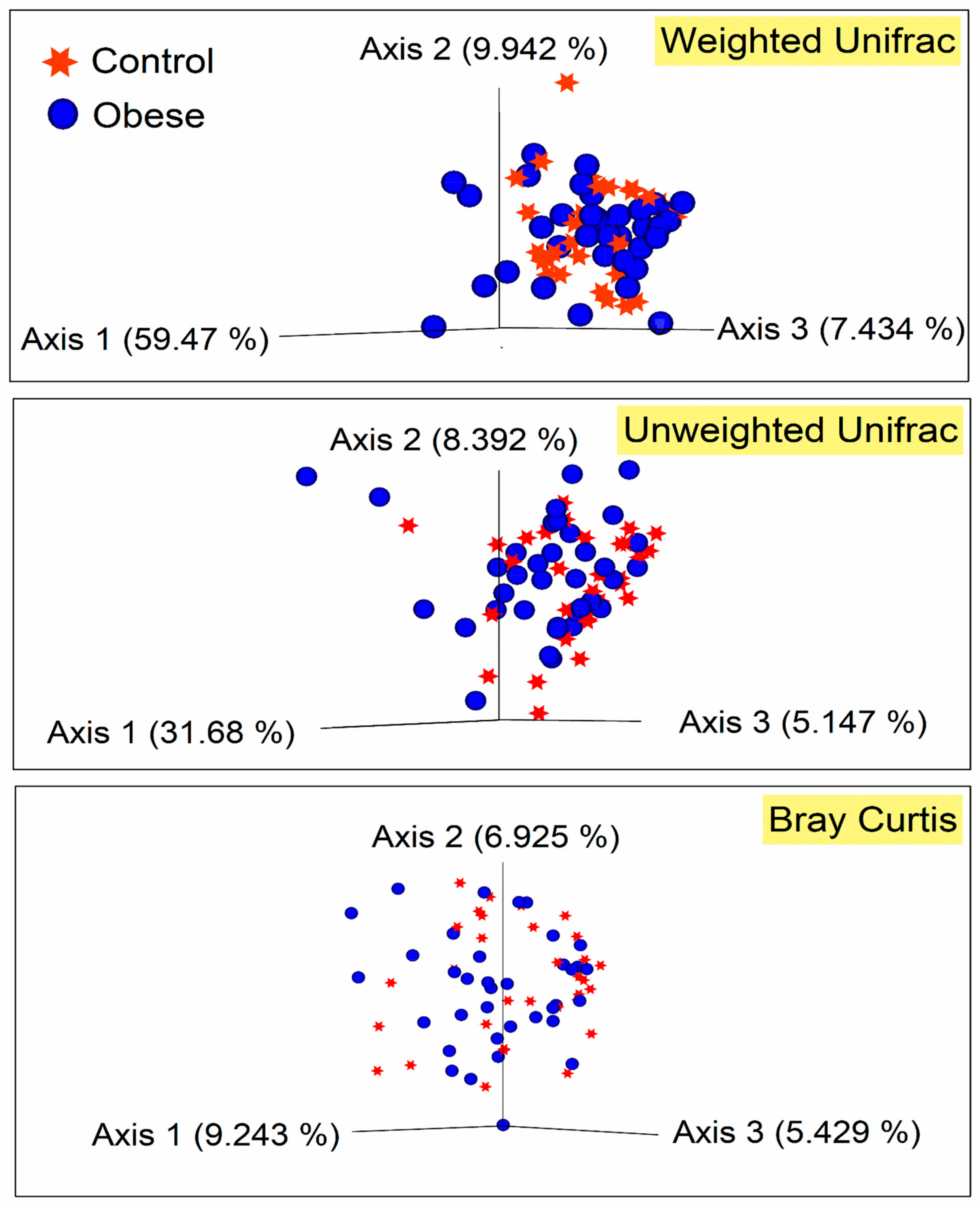
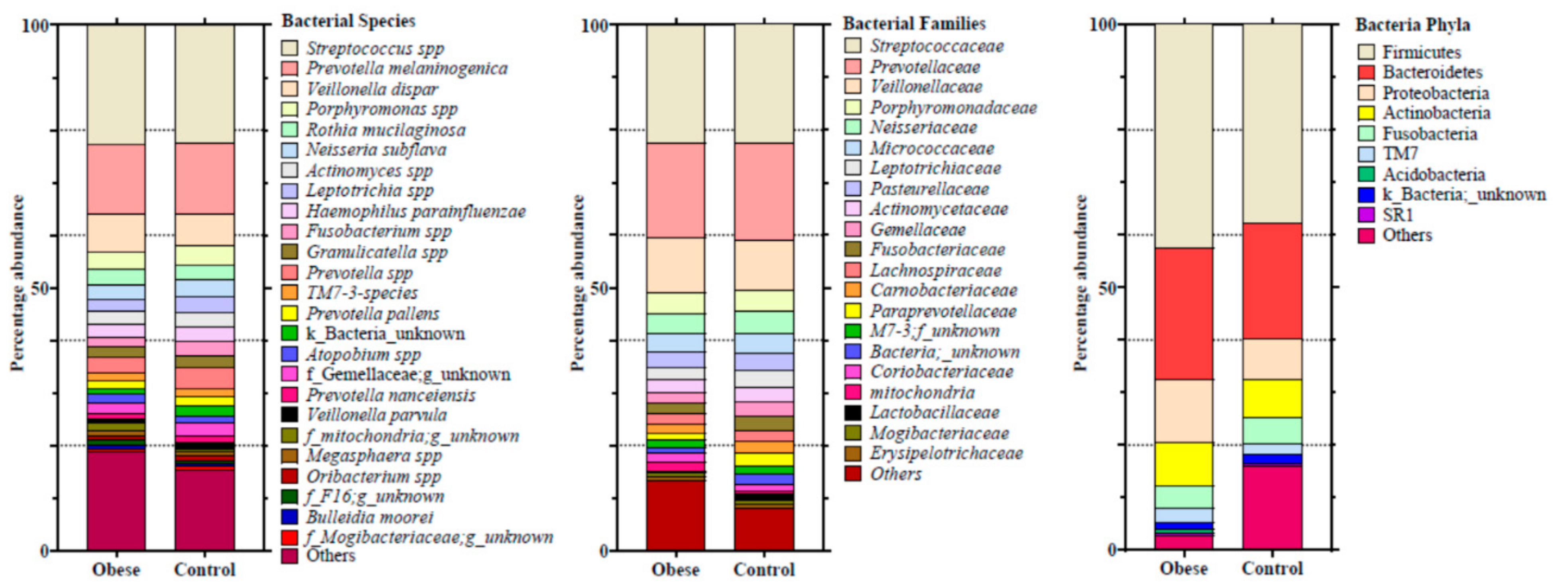
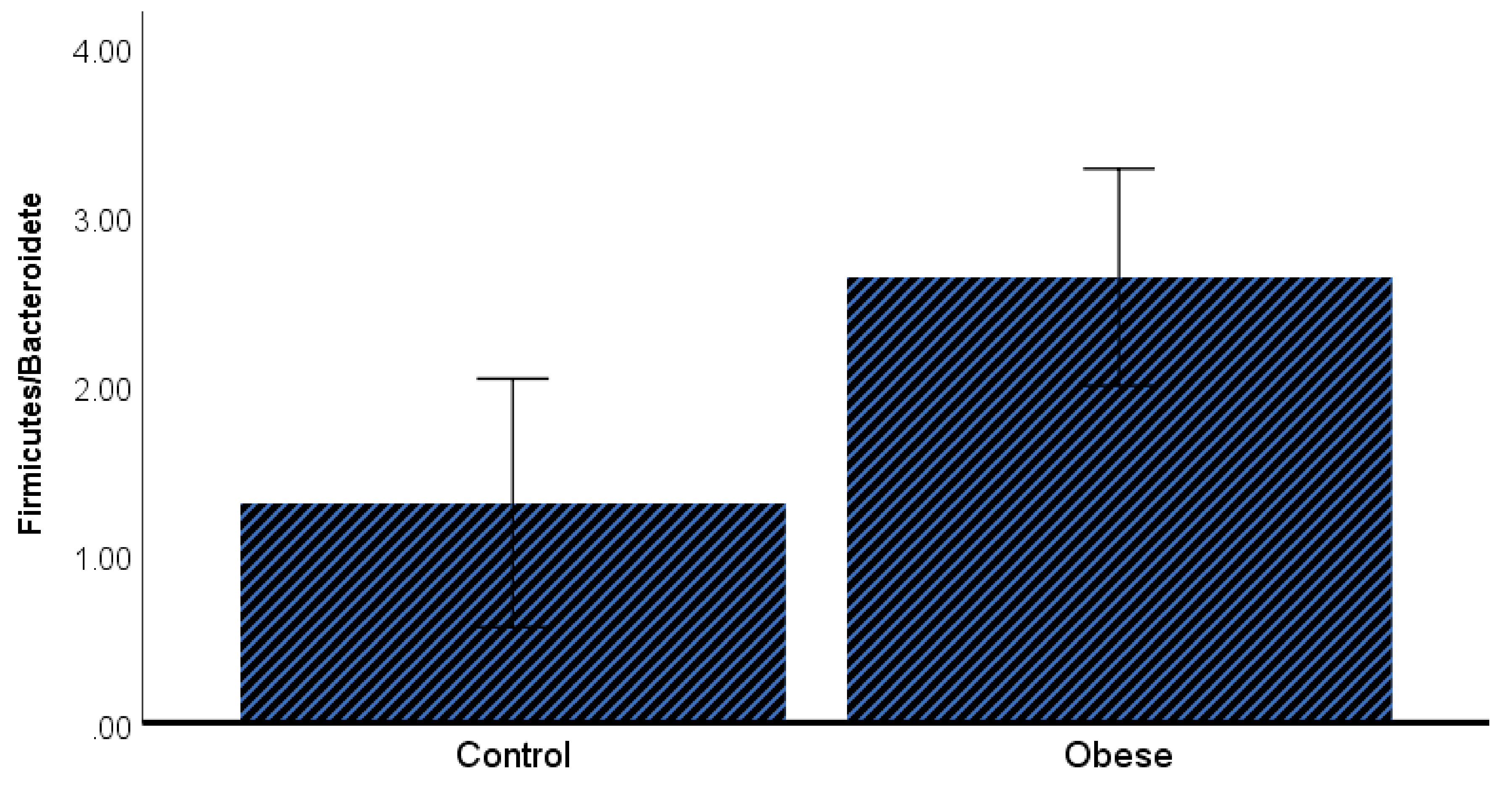
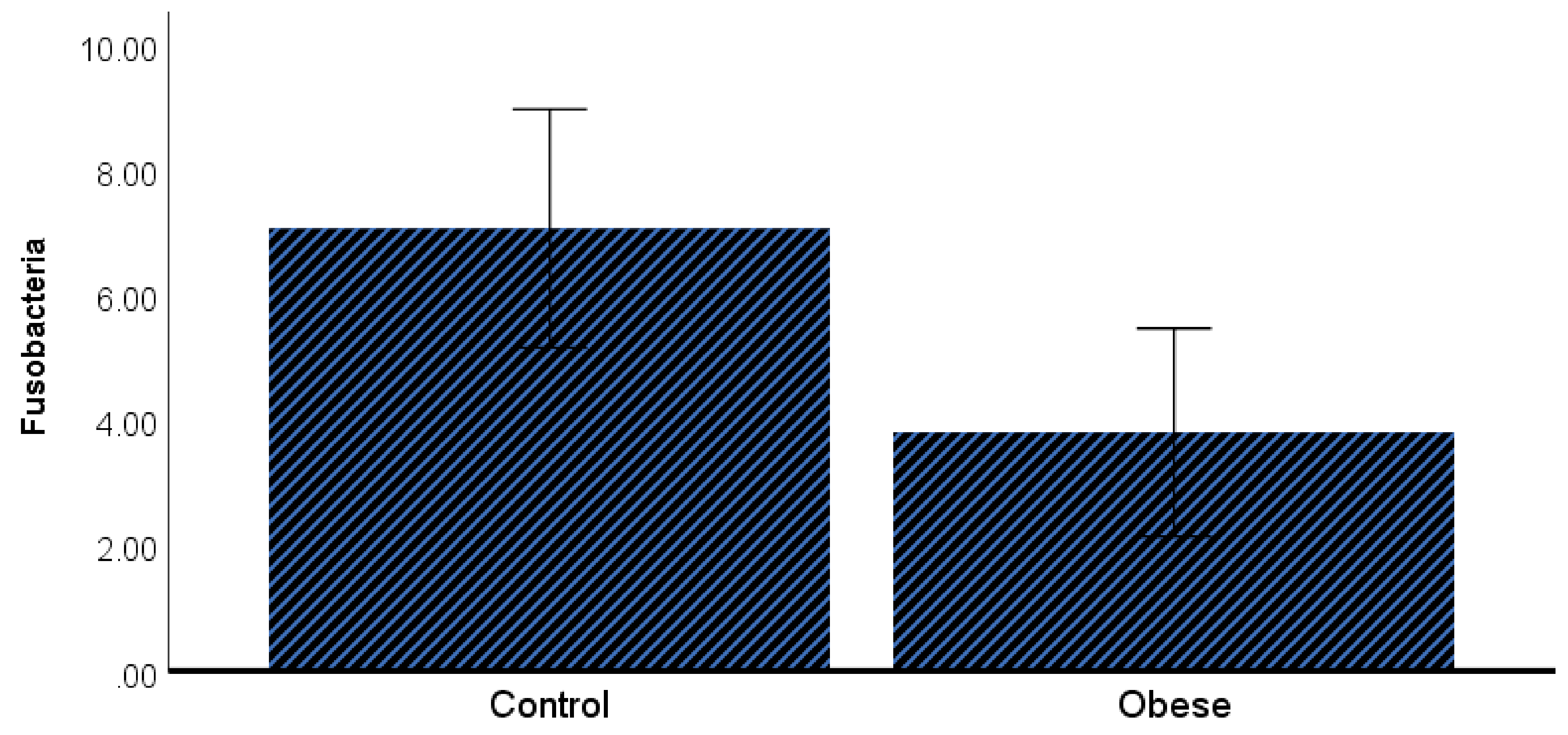
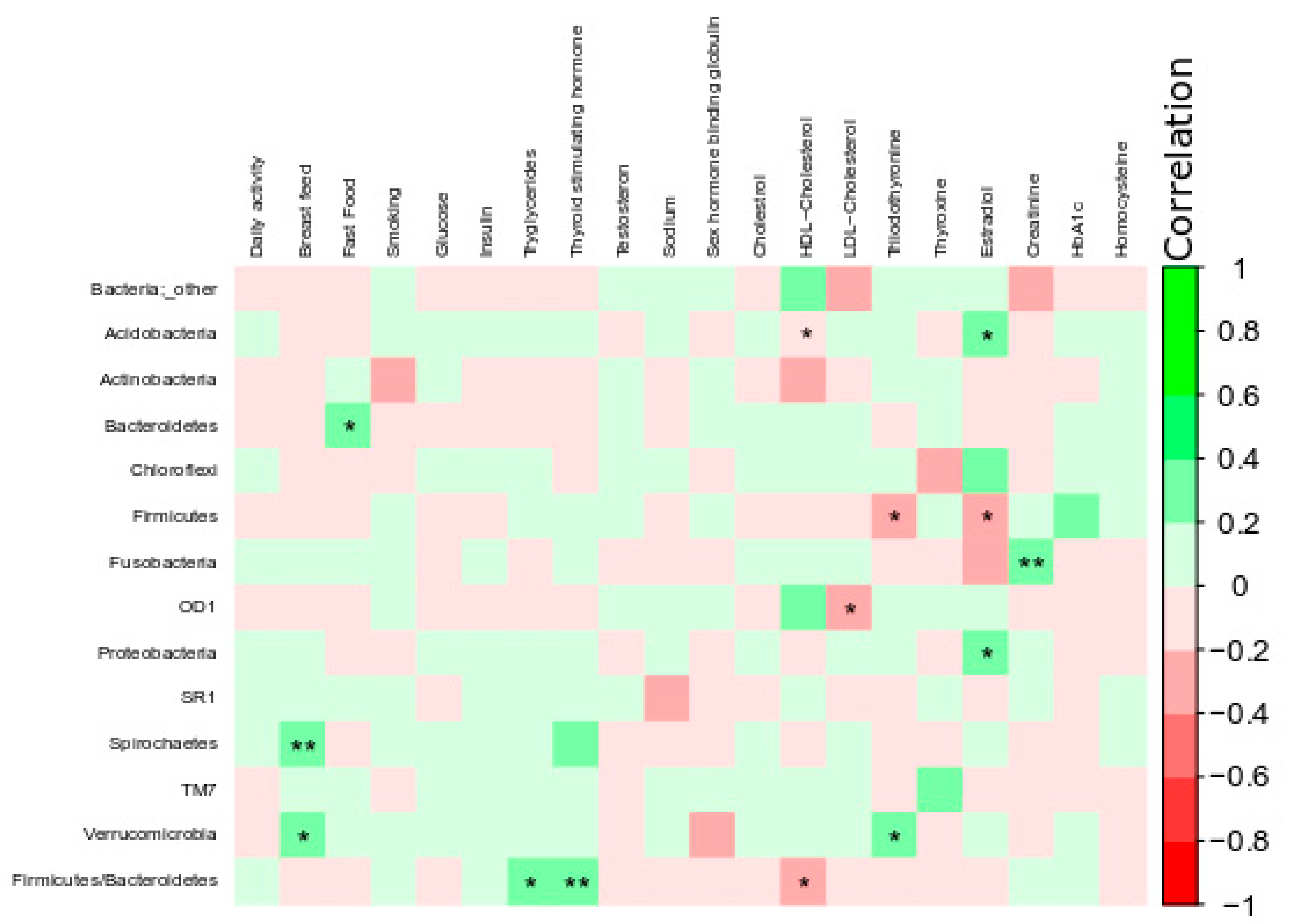
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fuchsberger, C.; Flannick, J.; Teslovich, T.M.; Mahajan, A.; Agarwala, V.; Gaulton, K.J.; Ma, C.; Fontanillas, P.; Moutsianas, L.; McCarthy, D.J. The genetic architecture of type 2 diabetes. Nature 2016, 536, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colberg, S.R.; Sigal, R.J.; Yardley, J.E.; Riddell, M.C.; Dunstan, D.W.; Dempsey, P.C.; Horton, E.S.; Castorino, K.; Tate, D.F. Physical activity/exercise and diabetes: A position statement of the American Diabetes Association. Diabetes Care 2016, 39, 2065–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabák, A.G.; Herder, C.; Rathmann, W.; Brunner, E.J.; Kivimäki, M. Prediabetes: A high-risk state for developing diabetes. Lancet 2012, 379, 2279. [Google Scholar] [CrossRef] [Green Version]
- Collaboration, E.R.F. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [CrossRef] [Green Version]
- Sohail, M.U.; Yassine, H.M.; Sohail, A.; Al Thani, A.A. Impact of Physical Exercise on Gut Microbiome, Inflammation, and the Pathobiology of Metabolic Disorders. Rev. Diabet. Stud. RDS 2019, 15, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.-Z.; Du, W.-T.; Xu, Y.-L.; Cheng, S.-Z.; Liu, Z.-J. Gut microbiome-based medical methodologies for early-stage disease prevention. Microb. Pathog. 2017, 105, 122–130. [Google Scholar] [CrossRef]
- Wang, J.; Jia, H. Metagenome-wide association studies: Fine-mining the microbiome. Nat. Rev. Microbiol. 2016, 14, 508. [Google Scholar] [CrossRef]
- Demmer, R.T.; Breskin, A.; Rosenbaum, M.; Zuk, A.; LeDuc, C.; Leibel, R.; Paster, B.; Desvarieux, M.; Jacobs, D.R., Jr.; Papapanou, P.N. The subgingival microbiome, systemic inflammation and insulin resistance: The oral infections, glucose intolerance and insulin resistance study. J. Clin. Periodontol. 2017, 44, 255–265. [Google Scholar] [CrossRef]
- Ding, S.; Chi, M.M.; Scull, B.P.; Rigby, R.; Schwerbrock, N.M.; Magness, S.; Jobin, C.; Lund, P.K. High-fat diet: Bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE 2010, 5, e12191. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027. [Google Scholar] [CrossRef]
- Allin, K.H.; Tremaroli, V.; Caesar, R.; Jensen, B.A.; Damgaard, M.T.; Bahl, M.I.; Licht, T.R.; Hansen, T.H.; Nielsen, T.; Dantoft, T.M. Aberrant intestinal microbiota in individuals with prediabetes. Diabetologia 2018, 61, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.-R.; Xu, Y.-S.; Ji, M.-M.; Zhang, L.; Li, D.; Lang, Q.; Zhang, L.; Ji, G.; Liu, B.-C. Association of the oral microbiome with the progression of impaired fasting glucose in a Chinese elderly population. J. Oral Microbiol. 2019, 11, 1605789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y. The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat. Med. 2015, 21, 895. [Google Scholar] [CrossRef] [PubMed]
- Blekkenhorst, L.C.; Bondonno, N.P.; Liu, A.H.; Ward, N.C.; Prince, R.L.; Lewis, J.R.; Devine, A.; Croft, K.D.; Hodgson, J.M.; Bondonno, C.P. Nitrate, the oral microbiome, and cardiovascular health: A systematic literature review of human and animal studies. Am. J. Clin. Nutr. 2018, 107, 504–522. [Google Scholar] [CrossRef] [Green Version]
- Michaud, D.S.; Izard, J.; Rubin, Z.; Johansson, I.; Weiderpass, E.; Tjønneland, A.; Olsen, A.; Overvad, K.; Boutron-Ruault, M.C.; Clavel-Chapelon, F. Lifestyle, dietary factors, and antibody levels to oral bacteria in cancer-free participants of a European cohort study. Cancer Causes Control 2013, 24, 1901–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhatalo, A.; Blackwell, J.R.; L’Heureux, J.E.; Williams, D.W.; Smith, A.; van der Giezen, M.; Winyard, P.G.; Kelly, J.; Jones, A.M. Nitrate-responsive oral microbiome modulates nitric oxide homeostasis and blood pressure in humans. Free Radic. Biol. Med. 2018, 124, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Zarco, M.; Vess, T.; Ginsburg, G. The oral microbiome in health and disease and the potential impact on personalized dental medicine. Oral Dis. 2012, 18, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Cai, Q.; Steinwandel, M.; Hargreaves, M.K.; Bordenstein, S.R.; Blot, W.J.; Zheng, W.; Shu, X.O. Association of oral microbiome with type 2 diabetes risk. J. Periodontal Res. 2017, 52, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480. [Google Scholar] [CrossRef] [Green Version]
- Zupancic, M.L.; Cantarel, B.L.; Liu, Z.; Drabek, E.F.; Ryan, K.A.; Cirimotich, S.; Jones, C.; Knight, R.; Walters, W.A.; Knights, D. Analysis of the gut microbiota in the old order Amish and its relation to the metabolic syndrome. PLoS ONE 2012, 7, e43052. [Google Scholar] [CrossRef]
- Al Kuwari, H.; Al Thani, A.; Al Marri, A.; Al Kaabi, A.; Abderrahim, H.; Afifi, N.; Qafoud, F.; Chan, Q.; Tzoulaki, I.; Downey, P. The Qatar Biobank: Background and methods. BMC Public Health 2015, 15, 1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Dabhani, K.; Tsilidis, K.; Murphy, N.; Ward, H.; Elliott, P.; Riboli, E.; Gunter, M.; Tzoulaki, I. Prevalence of vitamin D deficiency and association with metabolic syndrome in a Qatari population. Nutr. Diabetes 2017, 7, e263. [Google Scholar] [CrossRef] [PubMed]
- Thani, A.A.; Fthenou, E.; Paparrodopoulos, S.; Marri, A.A.; Shi, Z.; Qafoud, F.; Afifi, N. Qatar Biobank Cohort Study: Study Design and First Results. Am. J. Epidemiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Anwar, H.; Suchodolski, J.S.; Ullah, M.I.; Hussain, G.; Shabbir, M.Z.; Mustafa, I.; Sohail, M.U. Shiitake Culinary-Medicinal Mushroom, Lentinus edodes (Agaricomycetes), Supplementation Alters Gut Microbiome and Corrects Dyslipidemia in Rats. Int. J. Med. Mushrooms 2019, 21. [Google Scholar] [CrossRef] [PubMed]
- Sohail, M.U.; Hume, M.E. Evaluation of Antimicrobial Action of Chitosan and Acetic Acid on Broiler Cecal Bacterial Profiles in Anaerobic Cultures Inoculated with Salmonella Typhimurium. J. Appl. Poult. Res. 2018, 28, 176–183. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581. [Google Scholar] [CrossRef] [Green Version]
- Wajid, B.; Sohail, M.U.; Ekti, A.R.; Serpedin, E. The A, C, G, and T of Genome Assembly. BioMed Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. Peer J. Preprints 2018, 6, e27295v2. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Suchodolski, J.S.; Foster, M.L.; Sohail, M.U.; Leutenegger, C.; Queen, E.V.; Steiner, J.M.; Marks, S.L. The fecal microbiome in cats with diarrhea. PLoS ONE 2015, 10, e0127378. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Larsen, N.; Vogensen, F.K.; Van Den Berg, F.W.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef] [PubMed]
- Brant, R. Hypothesis Testing: Two-Sample Inference-Estimation of Sample Size and Power for Comparing Two Means, Fundamentals in Biostatistics. 2013. Available online: https://www.stat.ubc.ca/~rollin/stats/ssize/n2.html (accessed on 17 September 2019).
- Al Hayek, A.A.; Robert, A.A.; Alshammari, G.; Hakami, H.; Al Dawish, M.A. Assessment of hypogonadism in men with type 2 diabetes: A cross-sectional study from Saudi Arabia. Clin. Med. Insights Endocrinol. Diabetes 2017, 10, 1179551417710209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asare-Anane, H.; Ofori, E.; Agyemang, Y.; Oppong, S.; Tagoe, E.; Bani, S.; Ateku, R.; Bawa, T. Obesity and testosterone levels in Ghanaian men with type 2 diabetes. Clin. Diabetes 2014, 32, 61–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.R.; Clegg, D.J.; Prossnitz, E.R.; Barton, M. Obesity, insulin resistance and diabetes: Sex differences and role of oestrogen receptors. Acta Physiol. 2011, 203, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Wallace, I.R.; McKinley, M.C.; Bell, P.M.; Hunter, S.J. Sex hormone binding globulin and insulin resistance. Clin. Endocrinol. 2013, 78, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afsar, B.; Karaca, H. The relationship between insulin, insulin resistance, parathyroid hormone, cortisol, testosterone, and thyroid function tests in the presence of nephrolithiasis: A comprehensive analysis. Cent. Eur. J. Urol. 2014, 67, 58. [Google Scholar]
- Chaker, L.; Ligthart, S.; Korevaar, T.I.; Hofman, A.; Franco, O.H.; Peeters, R.P.; Dehghan, A. Thyroid function and risk of type 2 diabetes: A population-based prospective cohort study. BMC Med. 2016, 14, 150. [Google Scholar] [CrossRef] [Green Version]
- Paun, A.; Danska, J.S. Modulation of type 1 and type 2 diabetes risk by the intestinal microbiome. Pediatric Diabetes 2016, 17, 469–477. [Google Scholar] [CrossRef]
- Keying, X.; Gui, Q.; Yang, Y. Study on the changes in intestinal phylum firmicutes in elderly patients with type 2 diabetes. Chin. J. Geriatr. 2017, 36, 195–198. [Google Scholar]
- Koliada, A.; Syzenko, G.; Moseiko, V.; Budovska, L.; Puchkov, K.; Perederiy, V.; Gavalko, Y.; Dorofeyev, A.; Romanenko, M.; Tkach, S. Association between body mass index and Firmicutes/Bacteroidetes ratio in an adult Ukrainian population. BMC Microbiol. 2017, 17, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anbalagan, R.; Srikanth, P.; Mani, M.; Barani, R.; Seshadri, K.G.; Janarthanan, R. Next generation sequencing of oral microbiota in Type 2 diabetes mellitus prior to and after neem stick usage and correlation with serum monocyte chemoattractant-1. Diabetes Res. Clin. Pract. 2017, 130, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Xiao, E.; Mattos, M.; Vieira, G.H.A.; Chen, S.; Corrêa, J.D.; Wu, Y.; Albiero, M.L.; Bittinger, K.; Graves, D.T. Diabetes enhances IL-17 expression and alters the oral microbiome to increase its pathogenicity. Cell Host Microbe 2017, 22, 120–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, W.; Penney, N.C.; Cronin, O.; Garcia-Perez, I.; Molloy, M.G.; Holmes, E.; Shanahan, F.; Cotter, P.D.; O’Sullivan, O. The microbiome of professional athletes differs from that of more sedentary subjects in composition and particularly at the functional metabolic level. Gut 2018, 67, 625–633. [Google Scholar] [CrossRef]
- Hu, J.; Iragavarapu, S.; Nadkarni, G.N.; Huang, R.; Erazo, M.; Bao, X.; Verghese, D.; Coca, S.; Ahmed, M.K.; Peter, I. Location-specific oral microbiome possesses features associated with CKD. Kidney Int. Rep. 2018, 3, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Zaura, E.; Keijser, B.J.; Huse, S.M.; Crielaard, W. Defining the healthy” core microbiome” of oral microbial communities. BMC Microbiol. 2009, 9, 259. [Google Scholar] [CrossRef] [Green Version]
- Goodson, J.; Groppo, D.; Halem, S.; Carpino, E. Is obesity an oral bacterial disease? J. Dent. Res. 2009, 88, 519–523. [Google Scholar] [CrossRef]
- Si, J.; Lee, C.; Ko, G. Oral microbiota: Microbial biomarkers of metabolic syndrome independent of host genetic factors. Front. Cell. Infect. Microbiol. 2017, 7, 516. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Parameter | Study Group | Corrected p-Value | |
|---|---|---|---|
| Obese | Control | ||
| BMI | 35.65 ± 4.92 | 22.73 ± 1.52 | < 0.001 |
| Hemoglobin A1c (HbA1c) (%) | 5.68 ± 0.51 | 5.14 ± 0.76 | < 0.001 |
| Fasting glucose (mmol/L) | 5.96 ± 0.42 | 4.80 ± 0.35 | < 0.001 |
| Insulin (umol/L) | 16.56 ± 7.95 | 6.42 ± 1.86 | < 0.001 |
| Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) | 4.3 ± 1.7 | 1.3 ± 0.3 | < 0.001 |
| Triglyceride (mmol/L) | 1.74 ± 1.12 | 1.18 ± 0.74 | 0.051 |
| Total cholesterol (mmol/L) | 5.09 ± 0.97 | 5.32 ± 1.03 | 0.382 |
| HDL cholesterol (mmol/L) | 1.11 ± 0.28 | 1.32 ± 0.43 | 0.772 |
| LDL cholesterol (mmol/L) | 3.23 ± 0.89 | 3.47 ± 0.86 | 0.215 |
| Thyroid-stimulating hormone (mIU/L) | 1.91 ± 1.38 | 1.95 ± 1.48 | 0.890 |
| Triiodothyronine (pmol/L) | 4.21 ± 0.70 | 3.99 ± 0.64 | 0.213 |
| Thyroxine (pmol/L) | 12.62 ± 1.41 | 13.01 ± 1.48 | 0.208 |
| Testosterone (nmol/L) | 13.38 ± 3.67 | 22.74 ± 7.35 | 0.005 |
| Sex hormone-binding globulin (nmol/L) | 26.20 ± 12.01 | 37.00 ± 16.28 | 0.034 |
| Estradiol (pmol/L) | 111.43 ± 42.26 | 92.27 ± 36.89 | 0.030 |
| Creatinine (µmol/L) | 72.05 ± 8.72 | 77.53 ± 9.19 | 0.027 |
| C-peptide (ng/mL) | 3.04 ± 0.96 | 1.51 ± 0.50 | 0.048 |
| Aspartate aminotransferase (U/L) | 24.78 ± 9.84 | 21.61 ± 8.74 | 0.120 |
| Alanine aminotransferase (U/L) | 39.68 ± 18.97 | 24.75 ± 13.86 | 0.004 |
| Alkaline phosphatase (U/L) | 73.92 ± 14.35 | 68.36 ± 12.83 | 0.111 |
| Homocysteine (umol/L) | 9.65 ± 2.54 | 10.53 ± 2.85 | 0.216 |
| * Daily activity | 2.39 ± 1.61 | 3.00 ± 1.66 | 0.195 |
| * Breastfed in infancy | 1.12 ± 0.33 | 1.00 ± 0.00 | 0.137 |
| * Fast food consumption | 2.53 ± 1.42 | 3.22 ± 1.55 | 0.212 |
| * Smoking | 1.19 ± 1.24 | 1.51 ± 1.36 | 0.343 |
| * Antibiotic usage in last one year | 2.39 ± 1.04 | 1.49 ± 0.50 | 0.204 |
| Bacterium | Predictor | Adjusted R Square | Std. Error of the Estimate | p-Value |
|---|---|---|---|---|
| Acidobacteria | Estradiol | 0.067 | 1.7 | 0.023 |
| Estradiol | 0.13 | 1.7 | 0.005 | |
| HDL cholesterol | 0.024 | |||
| Firmicutes | Estradiol | 0.097 | 9.7 | 0.008 |
| Estradiol | 0.141 | 9.4 | 0.002 | |
| HDL cholesterol | 0.046 | |||
| Estradiol | 0.186 | 9.2 | 0.003 | |
| HDL cholesterol | 0.028 | |||
| Triiodothyronine | 0.043 | |||
| Fusobacteria | Creatinine | 0.103 | 3.6 | 0.006 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sohail, M.U.; Elrayess, M.A.; Al Thani, A.A.; Al-Asmakh, M.; Yassine, H.M. Profiling the Oral Microbiome and Plasma Biochemistry of Obese Hyperglycemic Subjects in Qatar. Microorganisms 2019, 7, 645. https://doi.org/10.3390/microorganisms7120645
Sohail MU, Elrayess MA, Al Thani AA, Al-Asmakh M, Yassine HM. Profiling the Oral Microbiome and Plasma Biochemistry of Obese Hyperglycemic Subjects in Qatar. Microorganisms. 2019; 7(12):645. https://doi.org/10.3390/microorganisms7120645
Chicago/Turabian StyleSohail, Muhammad U., Mohamed A. Elrayess, Asma A. Al Thani, Maha Al-Asmakh, and Hadi M. Yassine. 2019. "Profiling the Oral Microbiome and Plasma Biochemistry of Obese Hyperglycemic Subjects in Qatar" Microorganisms 7, no. 12: 645. https://doi.org/10.3390/microorganisms7120645