
Microbial Diversity and Community Structure of Wastewater-Driven Microalgal Biofilms
 , , and
, , and
Abstract
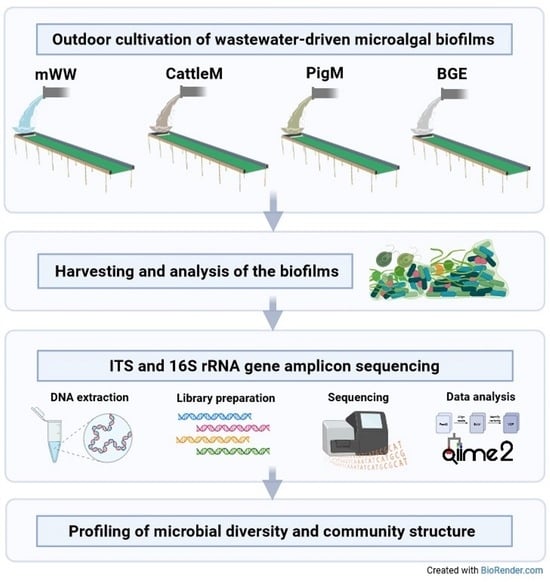
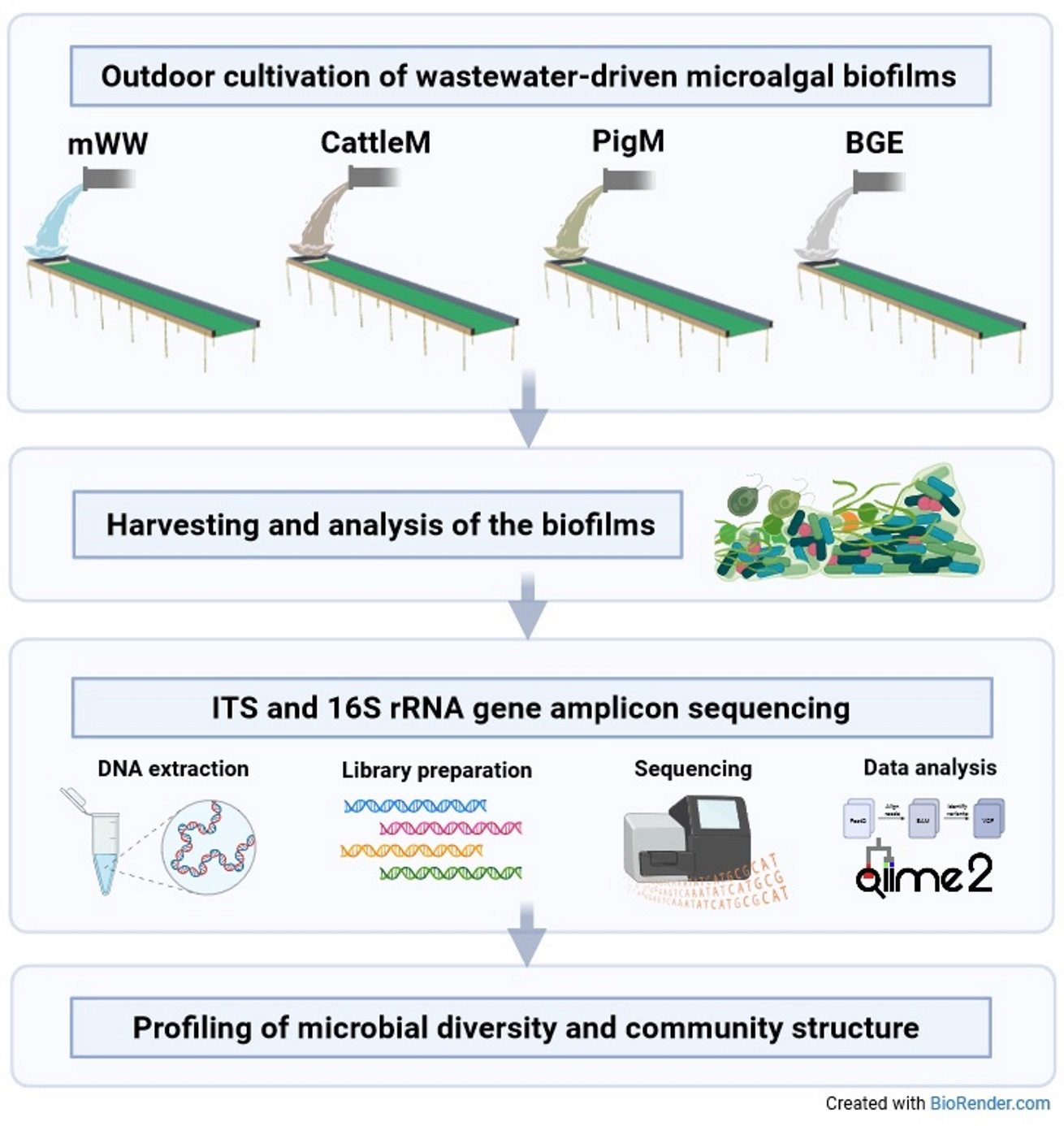
:
1. Introduction
2. Materials and Methods
2.1. Sample Sources, Sample Collection, and DNA Isolation
2.2. Amplicon Library Preparation and Sequencing
2.3. Bioinformatic Processing
3. Results
3.1. Physiochemical Properties of the Wastewater
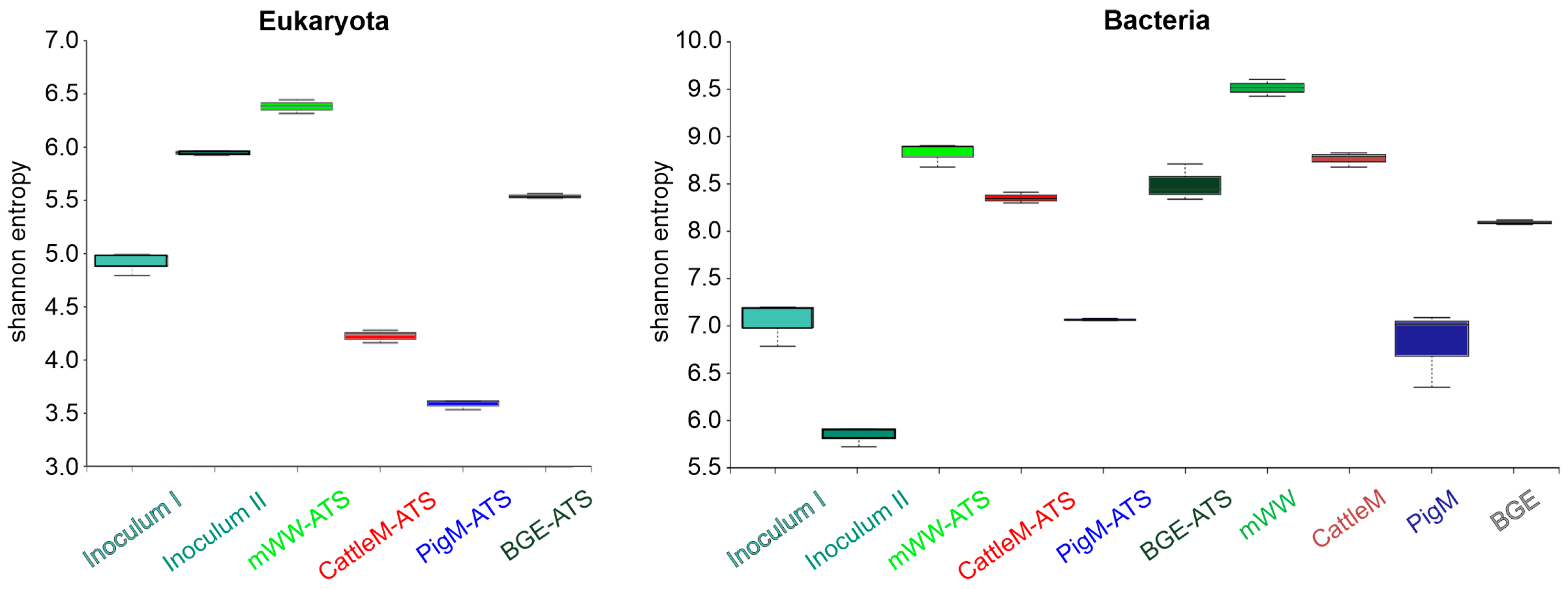
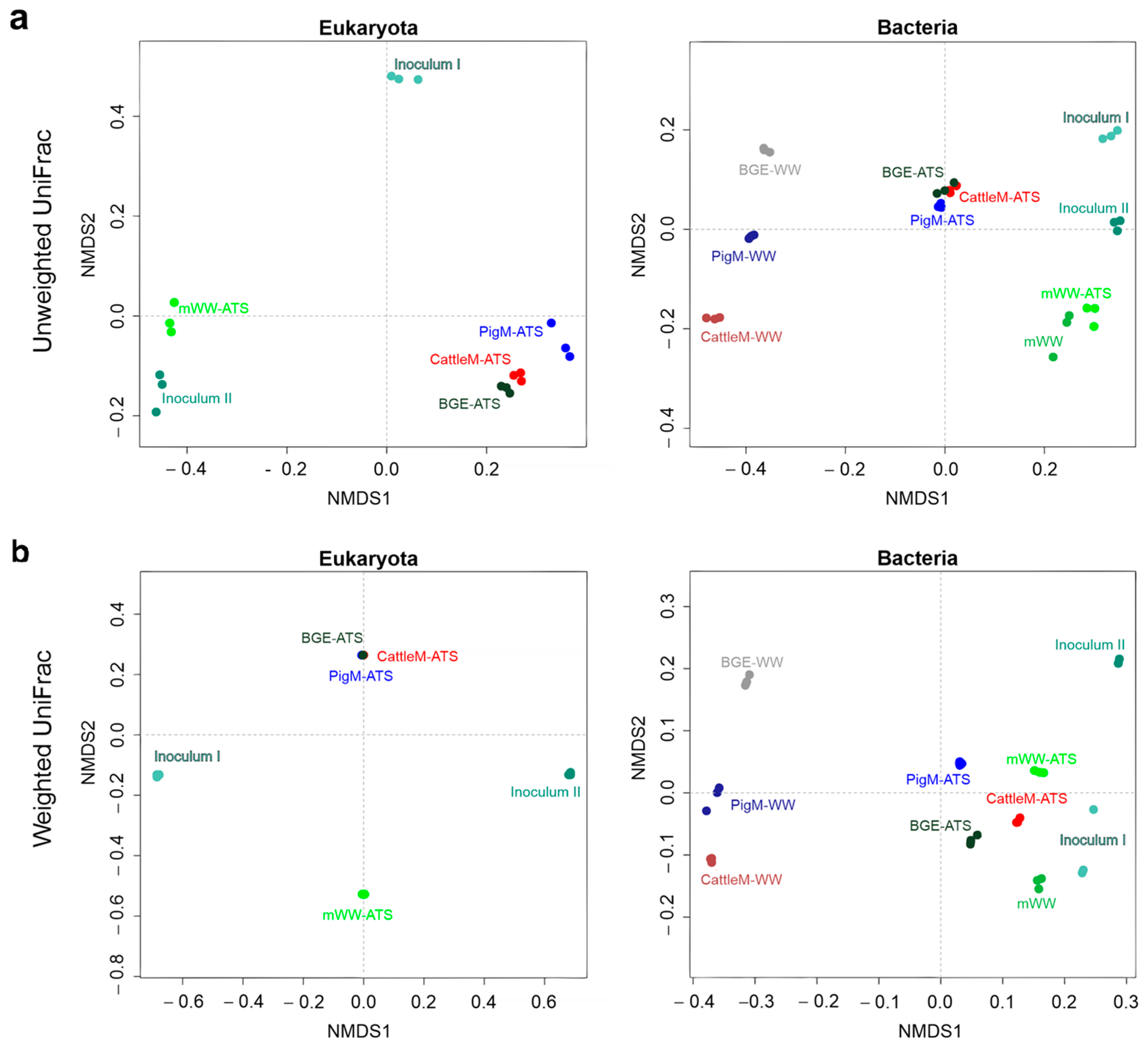
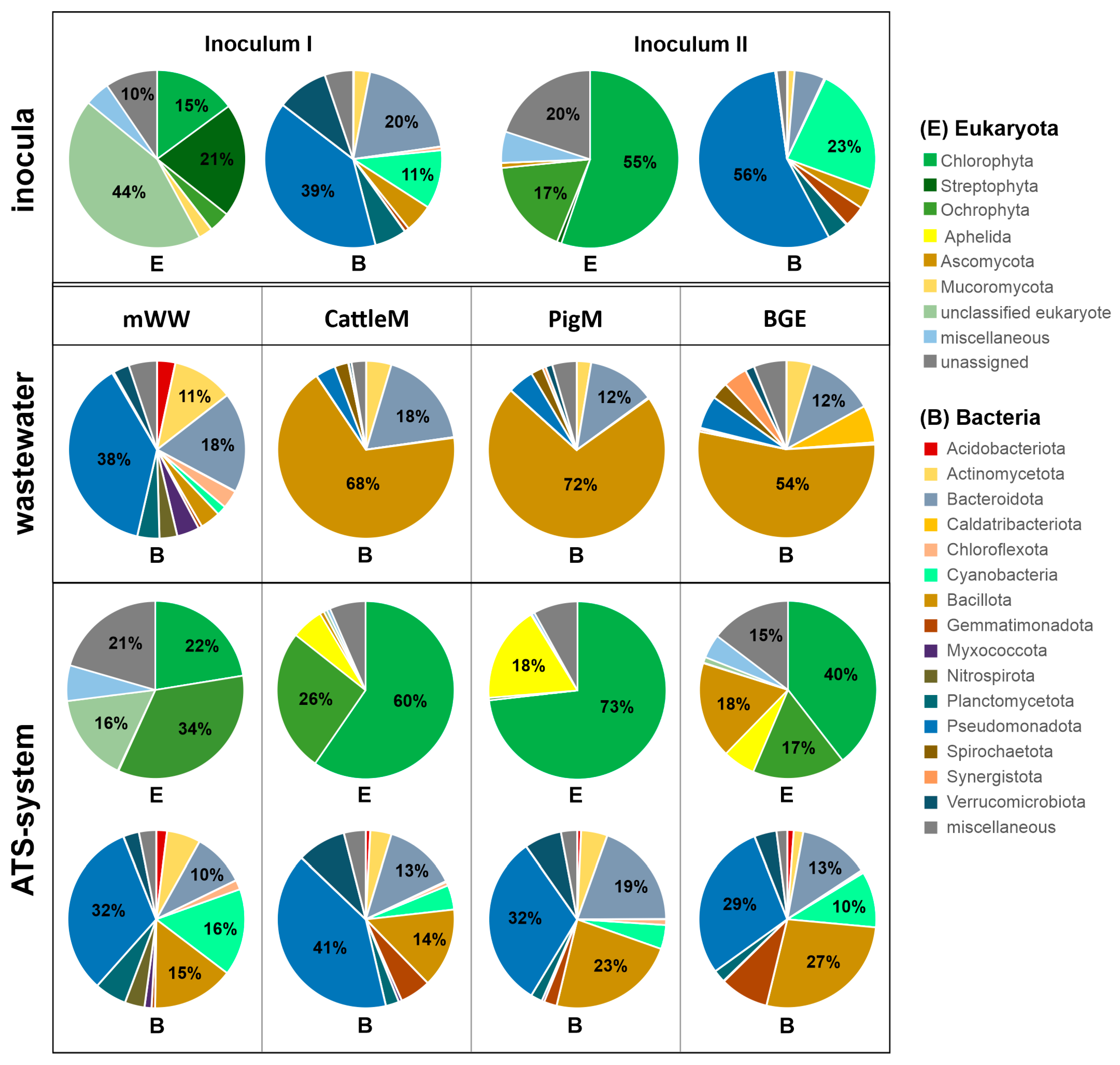
3.2. Biodiversity of the Different Wastewater-Driven Microbiomes
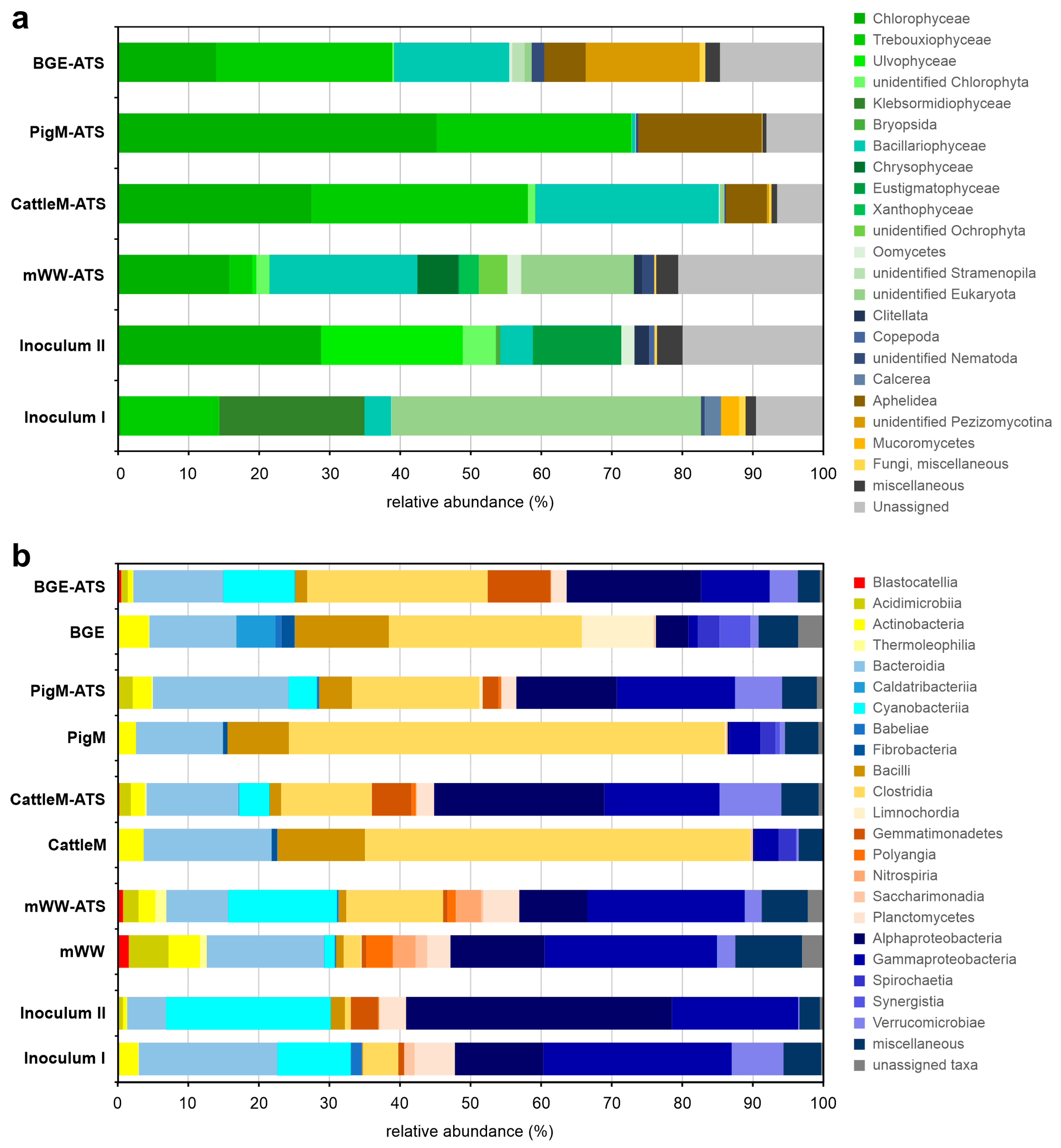
3.3. Taxonomic Profiling of the Communities of ATS Biofilms
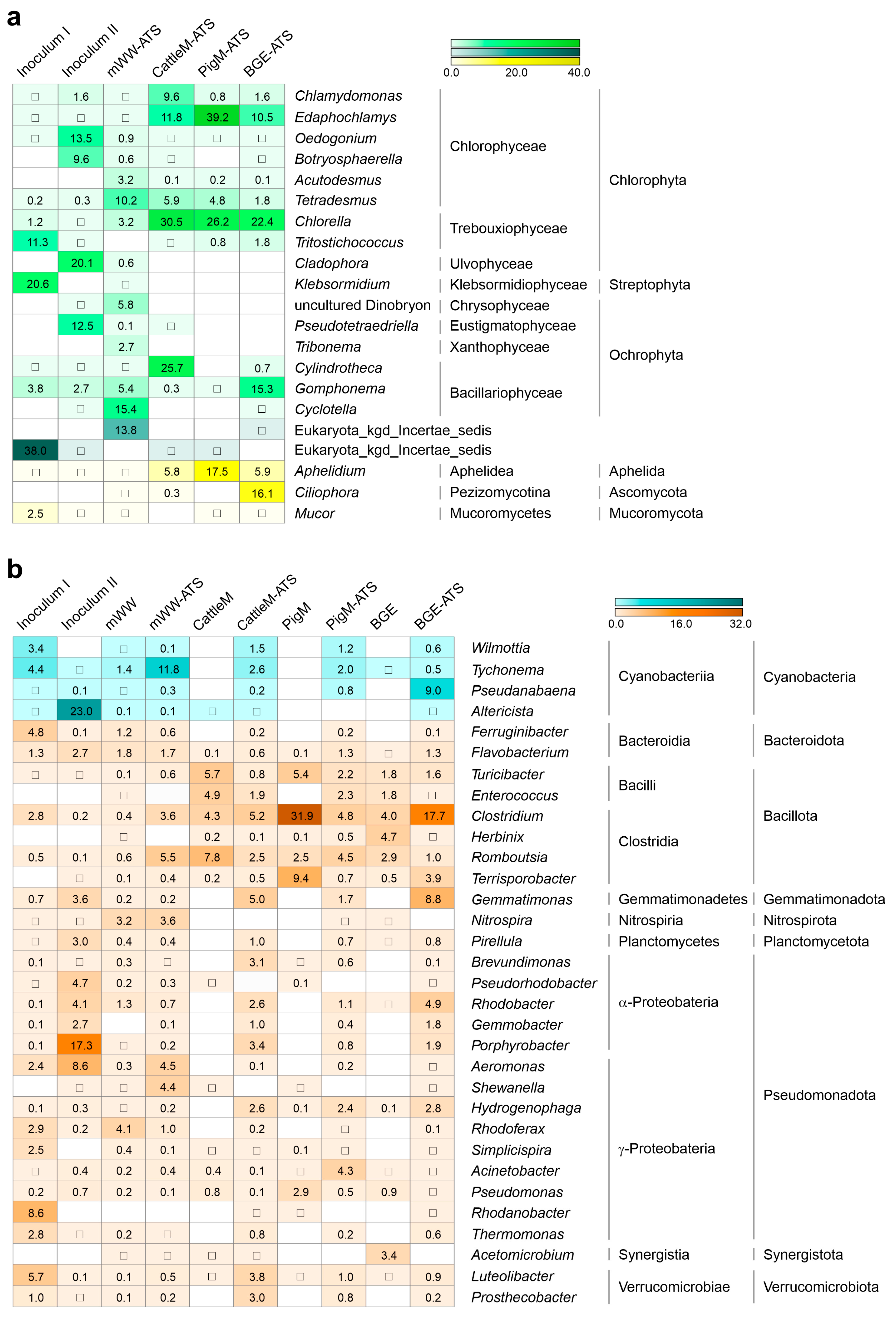
3.4. Microbial Taxa Forming the Wastewater-Driven Biofilm Structures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WWAP (United Nations World Water Assessment Programme). The United Nations World WATER Development Report. Wastewater: The Untapped Resource; UNESCO: Paris, France, 2017. [Google Scholar]
- Oruganti, R.K.; Katam, K.; Show, P.L.; Gadhamshetty, V.; Upadhyayula, V.K.K.; Bhattacharyya, D. A comprehensive review on the use of algal-bacterial systems for wastewater treatment with emphasis on nutrient and micropollutant removal. Bioengineered 2022, 13, 10412–10453. [Google Scholar] [CrossRef] [PubMed]
- Acién, F.G.; Gómez-Serrano, C.; Morales-Amaral, M.D.M.; Fernández-Sevilla, J.M.; Molina-Grima, E. Wastewater treatment using microalgae: How realistic a contribution might it be to significant urban wastewater treatment? Appl. Microbiol. Biotechnol. 2016, 100, 9013–9022. [Google Scholar] [CrossRef] [PubMed]
- Georgianna, D.R.; Mayfield, S.P. Exploiting diversity and synthetic biology for the production of algal biofuels. Nature 2012, 488, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.J.; Janssen, M.; Südfeld, C.; D’Adamo, S.; Wijffels, R.H. Hypes, hopes, and the way forward for microalgal biotechnology. Trends Biotechnol. 2023, 41, 452–471. [Google Scholar] [CrossRef] [PubMed]
- Klassen, V.; Blifernez-Klassen, O.; Wobbe, L.; Schlüter, A.; Kruse, O.; Mussgnug, J.H. Efficiency and biotechnological aspects of biogas production from microalgal substrates. J. Biotechnol. 2016, 234, 7–26. [Google Scholar] [CrossRef] [PubMed]
- Formighieri, C. Solar-to-Fuel Conversion in Algae and Cyanobacteria, 1st ed.; Springer International Publishing: Cham, Switzerland, 2015; ISBN 978-3-319-16730-5. [Google Scholar]
- Reinecke, D.; Bischoff, L.S.; Klassen, V.; Blifernez-Klassen, O.; Grimm, P.; Kruse, O.; Klose, H.; Schurr, U. Nutrient recovery from wastewaters by algal biofilm for fertilizer production part 1: Case study on the techno-economical aspects at pilot-scale. Sep. Purif. Technol. 2023, 305, 122471. [Google Scholar] [CrossRef]
- Hallmann, A. Algal transgenics and biotechnology. Transgenic Plant J. 2007, 1, 81–98. [Google Scholar]
- Vale, F.; Sousa, C.A.; Sousa, H.; Simões, L.C.; McBain, A.J.; Simões, M. Bacteria and microalgae associations in periphyton—Mechanisms and biotechnological opportunities. FEMS Microbiol. Rev. 2023, 47, fuad047. [Google Scholar] [CrossRef]
- Blifernez-Klassen, O.; Klassen, V.; Wibberg, D.; Cebeci, E.; Henke, C.; Rückert, C.; Chaudhari, S.; Rupp, O.; Blom, J.; Winkler, A.; et al. Phytoplankton consortia as a blueprint for mutually beneficial eukaryote-bacteria ecosystems based on the biocoenosis of Botryococcus consortia. Sci. Rep. 2021, 11, 1726. [Google Scholar] [CrossRef]
- Seymour, J.R.; Amin, S.A.; Raina, J.B.; Stocker, R. Zooming in on the phycosphere: The ecological interface for phytoplankton-bacteria relationships. Nat. Microbiol. 2017, 2, 17065. [Google Scholar] [CrossRef]
- Ramanan, R.; Kim, B.-H.; Cho, D.-H.; Oh, H.-M.; Kim, H.-S. Algae–bacteria interactions: Evolution, ecology and emerging applications. Biotechnol. Adv. 2016, 34, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Wijffels, R.H.; Smidt, H.; Sipkema, D. The effect of the algal microbiome on industrial production of microalgae. Microb. Biotechnol. 2018, 11, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Klassen, V.; Blifernez-Klassen, O.; Bax, J.; Kruse, O. Wastewater-borne microalga Chlamydomonas sp.: A robust chassis for efficient biomass and biomethane production applying low-N cultivation strategy. Bioresour. Technol. 2020, 315, 123825. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.B.; Smith, A.G. Exploring mutualistic interactions between microalgae and bacteria in the omics age. Curr. Opin. Plant Biol. 2015, 26, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Hmelo, L.R.; Van Tol, H.M.; Durham, B.P.; Carlson, L.T.; Heal, K.R.; Morales, R.L.; Berthiaume, C.T.; Parker, M.S.; Djunaedi, B.; et al. Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 2015, 522, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Hejna, M.; Kapuścińska, D.; Aksmann, A. Pharmaceuticals in the aquatic environment: A review on eco-toxicology and the remediation potential of algae. Int. J. Environ. Res. Public Health 2022, 19, 7717. [Google Scholar] [CrossRef] [PubMed]
- Abinandan, S.; Shanthakumar, S. Challenges and opportunities in application of microalgae (Chlorophyta) for wastewater treatment: A review. Renew. Sustain. Energy Rev. 2015, 52, 123–132. [Google Scholar] [CrossRef]
- Gonçalves, A.L.; Pires, J.C.M.; Simões, M. A review on the use of microalgal consortia for wastewater treatment. Algal Res. 2017, 24, 403–415. [Google Scholar] [CrossRef]
- Bui, X.-T.; Chiemchaisri, C.; Fujioka, T.; Varjani, S. Water and Wastewater Treatment Technologies; Springer: Singapore, 2019; ISBN 9811332584. [Google Scholar]
- Whitton, R.; Ometto, F.; Pidou, M.; Jarvis, P.; Villa, R.; Jefferson, B. Microalgae for municipal wastewater nutrient remediation: Mechanisms, reactors and outlook for tertiary treatment. Environ. Technol. Rev. 2015, 4, 133–148. [Google Scholar] [CrossRef]
- Valigore, J.M.; Gostomski, P.A.; Wareham, D.G.; O’Sullivan, A.D. Effects of hydraulic and solids retention times on productivity and settleability of microbial (microalgal-bacterial) biomass grown on primary treated wastewater as a biofuel feedstock. Water Res. 2012, 46, 2957–2964. [Google Scholar] [CrossRef]
- Saini, S.; Tewari, S.; Dwivedi, J.; Sharma, V. Biofilm Mediated Wastewater Treatment: A Comprehensive Review. Mater. Adv. 2023, 4, 1415–1443. [Google Scholar] [CrossRef]
- Adey, W.H. The microcosm: A new tool for reef research. Coral Reefs 1983, 1, 193–201. [Google Scholar] [CrossRef]
- Craggs, R.L. Wastewater treatment by algal turf scrubbing. Water Sci. Technol. 2001, 44, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Adey, W.H.; Laughinghouse, H.D., IV; Miller, J.B.; Hayek, L.-A.C.; Thompson, J.G.; Bertman, S.; Hampel, K.; Puvanendran, S. Algal turf scrubber (ATS) floways on the Great Wicomico River, Chesapeake Bay: Productivity, algal community structure, substrate and chemistry1. J. Phycol. 2013, 49, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hong, Y. Microalgae Biofilm and Bacteria Symbiosis in Nutrient Removal and Carbon Fixation from Wastewater: A Review. Curr. Pollut. Rep. 2022, 8, 128–146. [Google Scholar] [CrossRef]
- Lamprecht, O.; Wagner, B.; Derlon, N.; Tlili, A. Synthetic periphyton as a model system to understand species dynamics in complex microbial freshwater communities. NPJ Biofilms Microbiomes 2022, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- D’Aiuto, P.E.; Patt, J.M.; Albano, J.P.; Shatters, R.G.; Evens, T.J. Algal turf scrubbers: Periphyton production and nutrient recovery on a South Florida citrus farm. Ecol. Eng. 2015, 75, 404–412. [Google Scholar] [CrossRef]
- DIN EN ISO 11885 2009–09; Water Quality—Determination of Selected Elements by Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) (ISO 11885:2007); German version EN ISO 11885:2009. Deutsches Institut für Normung: Berlin, Germany, 2009.
- DIN 38406-E5-2 1983–10; German Standard Methods for the Examination of Water, Waste Water and Sludge; Cations (Group E); Determination of Ammonia-Nitrogen (E 5). Deutsches Institut für Normung: Berlin, Germany, 1983.
- VDLUFA Methodenbuch. Verband Dtsch. Landwirtsch. Untersuchungs- und Forschungsanstalten e.V. 1985. Available online: https://www.vdlufa.de/9methodenbuch/ (accessed on 1 October 2023).
- Klassen, V.; Blifernez-Klassen, O.; Hoekzema, Y.; Mussgnug, J.H.; Kruse, O. A novel one-stage cultivation/fermentation strategy for improved biogas production with microalgal biomass. J. Biotechnol. 2015, 215, 44–51. [Google Scholar] [CrossRef]
- Zhou, J.; Bruns, M.A.; Tiedje, J.M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 1996, 62, 316–322. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. Guide Methods Appl. 1990, 18, 315–322. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Joshi Nikhil, F.J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files, Version 1.33. 2011. Available online: https://github.com/najoshi/sickle (accessed on 1 October 2023).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://github.com/s-andrews/FastQC (accessed on 1 October 2023).
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Oren, A.; Garrity, G.M. Valid publication of the names of forty-two phyla of prokaryotes. Int. J. Syst. Evol. Microbiol. 2021, 71, 005056. [Google Scholar] [CrossRef] [PubMed]
- Ortiz Tena, F.; Bickel, V.; Steinweg, C.; Posten, C. Continuous microalgae cultivation for wastewater treatment—Development of a process strategy during day and night. Sci. Total Environ. 2024, 912, 169082. [Google Scholar] [CrossRef] [PubMed]
- Carles, L.; Wullschleger, S.; Joss, A.; Eggen, R.I.L.; Schirmer, K.; Schuwirth, N.; Stamm, C.; Tlili, A. Impact of wastewater on the microbial diversity of periphyton and its tolerance to micropollutants in an engineered flow-through channel system. Water Res. 2021, 203, 117486. [Google Scholar] [CrossRef] [PubMed]
- Dueholm, M.K.D.; Nierychlo, M.; Andersen, K.S.; Rudkjøbing, V.; Knutsson, S.; Arriaga, S.; Bakke, R.; Boon, N.; Bux, F.; Christensson, M.; et al. MiDAS 4: A global catalogue of full-length 16S rRNA gene sequences and taxonomy for studies of bacterial communities in wastewater treatment plants. Nat. Commun. 2022, 13, 1908. [Google Scholar] [CrossRef] [PubMed]
- Pushpakumara, B.L.D.U.; Tandon, K.; Willis, A.; Verbruggen, H. Unravelling microalgal-bacterial interactions in aquatic ecosystems through 16S rRNA gene-based co-occurrence networks. Sci. Rep. 2023, 13, 2743. [Google Scholar] [CrossRef]
- Hassa, J.; Klang, J.; Benndorf, D.; Pohl, M.; Hülsemann, B.; Mächtig, T.; Effenberger, M.; Pühler, A.; Schlüter, A.; Theuerl, S. Indicative marker microbiome structures deduced from the taxonomic inventory of 67 full-scale anaerobic digesters of 49 agricultural biogas plants. Microorganisms 2021, 9, 1457. [Google Scholar] [CrossRef]
- Li, J.; Rui, J.; Yao, M.; Zhang, S.; Yan, X.; Wang, Y.; Yan, Z.; Li, X. Substrate type and free ammonia determine bacterial community structure in full-scale mesophilic anaerobic digesters treating cattle or swine manure. Front. Microbiol. 2015, 6, 1337. [Google Scholar] [CrossRef]
- Liu, J.; Pemberton, B.; Scales, P.J.; Martin, G.J.O. Ammonia tolerance of filamentous algae Oedogonium, Spirogyra, Tribonema and Cladophora, and its implications on wastewater treatment processes. Algal Res. 2023, 72, 103126. [Google Scholar] [CrossRef]
- Averina, S.; Polyakova, E.; Senatskaya, E.; Pinevich, A. A new cyanobacterial genus Altericista and three species, A. lacusladogae sp. nov., A. violacea sp. nov., and A. variichlora sp. nov., described using a polyphasic approach. J. Phycol. 2021, 57, 1517–1529. [Google Scholar] [CrossRef]
- Seto, K.; Matsuzawa, T.; Kuno, H.; Kagami, M. Morphology, Ultrastructure, and Molecular Phylogeny of Aphelidium collabens sp. nov. (Aphelida), a Parasitoid of a Green Alga Coccomyxa sp. Protist 2020, 171, 125728. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Guo, L.-D. Endophytic fungi VI. Ciliophora quercus sp. nov. from China. Nova Hedwig. 2007, 85, 403–406. [Google Scholar] [CrossRef]
- Fito, J.; Alemu, K. Microalgae–bacteria consortium treatment technology for municipal wastewater management. Nanotechnol. Environ. Eng. 2018, 4, 4. [Google Scholar] [CrossRef]
- Croft, M.T.; Warren, M.J.; Smith, A.G. Algae need their vitamins. Eukaryot. Cell 2006, 5, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Hoeger, A.-L.; Jehmlich, N.; Kipping, L.; Griehl, C.; Noll, M. Associated bacterial microbiome responds opportunistic once algal host Scenedesmus vacuolatus is attacked by endoparasite Amoeboaphelidium protococcarum. Sci. Rep. 2022, 12, 13187. [Google Scholar] [CrossRef] [PubMed]
- Goecke, F.; Thiel, V.; Wiese, J.; Labes, A.; Imhoff, J.F. Algae as an important environment for bacteria—Phylogenetic relationships among new bacterial species isolated from algae. Phycologia 2013, 52, 14–24. [Google Scholar] [CrossRef]
- Krohn-Molt, I.; Alawi, M.; Förstner, K.U.; Wiegandt, A.; Burkhardt, L.; Indenbirken, D.; Thieß, M.; Grundhoff, A.; Kehr, J.; Tholey, A.; et al. Insights into Microalga and bacteria interactions of selected phycosphere biofilms using metagenomic, transcriptomic, and proteomic approaches. Front. Microbiol. 2017, 8, 1941. [Google Scholar] [CrossRef]
- Akbari, A.; Wang, Z.; He, P.; Wang, D.; Lee, J.; Han, I.L.; Li, G.; Gu, A.Z. Unrevealed roles of polyphosphate-accumulating microorganisms. Microb. Biotechnol. 2021, 14, 82–87. [Google Scholar] [CrossRef]
- Koblížek, M. Ecology of aerobic anoxygenic phototrophs in aquatic environments. FEMS Microbiol. Rev. 2015, 39, 854–870. [Google Scholar] [CrossRef]
- Debabov, V.G. Acetogens: Biochemistry, Bioenergetics, Genetics, and Biotechnological Potential. Microbiology 2021, 90, 273–297. [Google Scholar] [CrossRef]
- Gerritsen, J.; Hornung, B.; Ritari, J.; Paulin, L.; Rijkers, G.T.; Schaap, P.J.; de Vos, W.M.; Smidt, H. A comparative and functional genomics analysis of the genus Romboutsia provides insight into adaptation to an intestinal lifestyle. bioRxiv 2019, 845511. [Google Scholar] [CrossRef]
- Daims, H.; Wagner, M. Nitrospira. Trends Microbiol. 2018, 26, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sekiguchi, Y.; Hanada, S.; Hugenholtz, P.; Kim, H.; Kamagata, Y.; Nakamura, K. Gemmatimonas aurantiaca gen. nov., sp. nov., a gram-negative, aerobic, polyphosphate-accumulating microorganism, the first cultured representative of the new bacterial phylum Gemmatimonadetes phyl. nov. Int. J. Syst. Evol. Microbiol. 2003, 53, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Coyne, K.J.; Wang, Y.; Johnson, G. Algicidal Bacteria: A Review of Current Knowledge and Applications to Control Harmful Algal Blooms. Front. Microbiol. 2022, 13, 871177. [Google Scholar] [CrossRef] [PubMed]
- Meyer, N.; Bigalke, A.; Kaulfuß, A.; Pohnert, G. Strategies and ecological roles of algicidal bacteria. FEMS Microbiol. Rev. 2017, 41, 880–899. [Google Scholar] [CrossRef] [PubMed]
- Nishu, S.D.; Kang, Y.; Han, I.; Jung, T.Y.; Lee, T.K. Nutritional status regulates algicidal activity of Aeromonas sp. L23 against cyanobacteria and green algae. PLoS ONE 2019, 14, e0213370. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, Y.; Song, T.; Li, L. Isolation and characterization of a newly isolated algicidal Aeromonas sp. J. Pure Appl. Microbiol. 2014, 8, 1379–1385. [Google Scholar]
- Tiedje, J.M. Shewanella—The environmentally versatile genome. Nat. Biotechnol. 2002, 20, 1093–1094. [Google Scholar] [CrossRef]
- Pokrzywinski, K.L.; Place, A.R.; Warner, M.E.; Coyne, K.J. Investigation of the algicidal exudate produced by Shewanella sp. IRI-160 and its effect on dinoflagellates. Harmful Algae 2012, 19, 23–29. [Google Scholar] [CrossRef]
- Zhou, J.; Lyu, Y.; Richlen, M.L.; Anderson, D.M.; Cai, Z. Quorum Sensing Is a Language of Chemical Signals and Plays an Ecological Role in Algal-Bacterial Interactions. Crit. Rev. Plant Sci. 2016, 35, 81–105. [Google Scholar] [CrossRef]
- Rajamani, S.; Bauer, W.D.; Robinson, J.B.; Farrow, J.M.; Pesci, E.C.; Teplitski, M.; Gao, M.; Sayre, R.T.; Phillips, D.A. The Vitamin Riboflavin and Its Derivative Lumichrome Activate the LasR Bacterial Quorum-Sensing Receptor. Mol. Plant-Microbe Interact. 2008, 21, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Amaral-Zettler, L.A.; Zettler, E.R.; Mincer, T.J. Ecology of the plastisphere. Nat. Rev. Microbiol. 2020, 18, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Celler, K.; Hödl, I.; Simone, A.; Battin, T.J.; Picioreanu, C. A mass-spring model unveils the morphogenesis of phototrophic Diatoma biofilms. Sci. Rep. 2014, 4, 3649. [Google Scholar] [CrossRef] [PubMed]
- Najdek, M.; Blažina, M.; Djakovac, T.; Kraus, R. The role of the diatom Cylindrotheca closterium in a mucilage event in the northern Adriatic Sea: Coupling with high salinity water intrusions. J. Plankton Res. 2005, 27, 851–862. [Google Scholar] [CrossRef]
- Masitah, E.D.; ISLAMY, R.A. Checklist of Freshwater Periphytic Diatoms in the Midstream of Brantas River, East Java. Biodiversitas J. Biol. Divers. 2023, 24, 3269–3281. [Google Scholar] [CrossRef]
- Vidal, L.; Ballot, A.; Azevedo, S.; Padisák, J.; Welker, M. Introduction to cyanobacteria. In Toxic Cyanobacteria in Water, 2nd ed.; Chorus, I., Welker, M., Eds.; CRC Press: Boca Raton, FL, USA, 2021; pp. 163–211. ISBN 978-1-003-08144-9. [Google Scholar]
- Liu, J.; Pemberton, B.; Lewis, J.; Scales, P.J.; Martin, G.J.O. Wastewater treatment using filamentous algae—A review. Bioresour. Technol. 2020, 298, 122556. [Google Scholar] [CrossRef]
- Gao, Y.; Jiang, X.; Lin, D.; Chen, Y.; Tong, Z. The Starvation Resistance and Biofilm Formation of Enterococcus faecalisin Coexistence with Candida albicans, Streptococcus gordonii, Actinomyces viscosus, or Lactobacillus acidophilus. J. Endod. 2016, 42, 1233–1238. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | mWW | PigM | CattleM | BGE | ||
|---|---|---|---|---|---|---|
| pH | 6.7 | 7.6 | 8.0 | 7.2 | ||
| COD | mg O2 | mg L−1 | <15 | 1847.6 | 2638.6 | 5265.3 |
| Ammonium | NH4-N | mg L−1 | 0.8 | 173.0 | 50.0 | 158.5 |
| Nitrate | NO3-N | mg L−1 | 1.6 | 25.4 | 8.8 | 11.6 |
| Total Nitrogen | N | mg L−1 | 3.9 | 207.5 | 111.0 | 250.5 |
| Total Sulphur | S | mg L−1 | 15.5 | 9.5 | 28.0 | |
| Total Phosphorous | P | mg L−1 | 1.6 | 98.0 | 49.0 | 92.5 |
| Orthophosphate | PO4-P | mg L−1 | 1.4 | 23.2 | 12.7 | 28.3 |
| Potassium | K | mg L−1 | 16.0 | 164.5 | 145.0 | 353.0 |
| Calcium | Ca | mg L−1 | 40.0 | 41.0 | 82.5 | 142.0 |
| Magnesium | Mg | mg L−1 | 4.0 | 62.0 | 27.5 | 42.0 |
| TS | % w/w | <0.1 | 0.21 | 0.28 | 0.47 | |
| VS | % w/w | <0.1 | 0.15 | 0.21 | 0.34 | |
| N:P molar ratio | 5.4 | 4.7 | 5.0 | 6.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blifernez-Klassen, O.; Hassa, J.; Reinecke, D.L.; Busche, T.; Klassen, V.; Kruse, O. Microbial Diversity and Community Structure of Wastewater-Driven Microalgal Biofilms. Microorganisms 2023, 11, 2994. https://doi.org/10.3390/microorganisms11122994
Blifernez-Klassen O, Hassa J, Reinecke DL, Busche T, Klassen V, Kruse O. Microbial Diversity and Community Structure of Wastewater-Driven Microalgal Biofilms. Microorganisms. 2023; 11(12):2994. https://doi.org/10.3390/microorganisms11122994
Chicago/Turabian StyleBlifernez-Klassen, Olga, Julia Hassa, Diana L. Reinecke, Tobias Busche, Viktor Klassen, and Olaf Kruse. 2023. "Microbial Diversity and Community Structure of Wastewater-Driven Microalgal Biofilms" Microorganisms 11, no. 12: 2994. https://doi.org/10.3390/microorganisms11122994