

Characterization of phage vB_EcoS-EE09 infecting E. coli DSM613 Isolated from Wastewater Treatment Plant Effluent and Comparative Proteomics of the Infected and Non-Infected Host
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Host Strain
2.2. Collection of Samples for Bacteriophage Isolation
2.3. Phage Enrichment and Isolation
2.4. Thermal and pH Stability Analysis
2.5. Host Range Analysis
2.6. Genome Sequencing and Analysis
2.7. One-Step Growth Curve Experiments
2.8. Transmission Electron Microscopy (TEM)
2.9. Protein Analysis
2.10. Analysis of the Proteomic Data
3. Results and Discussion
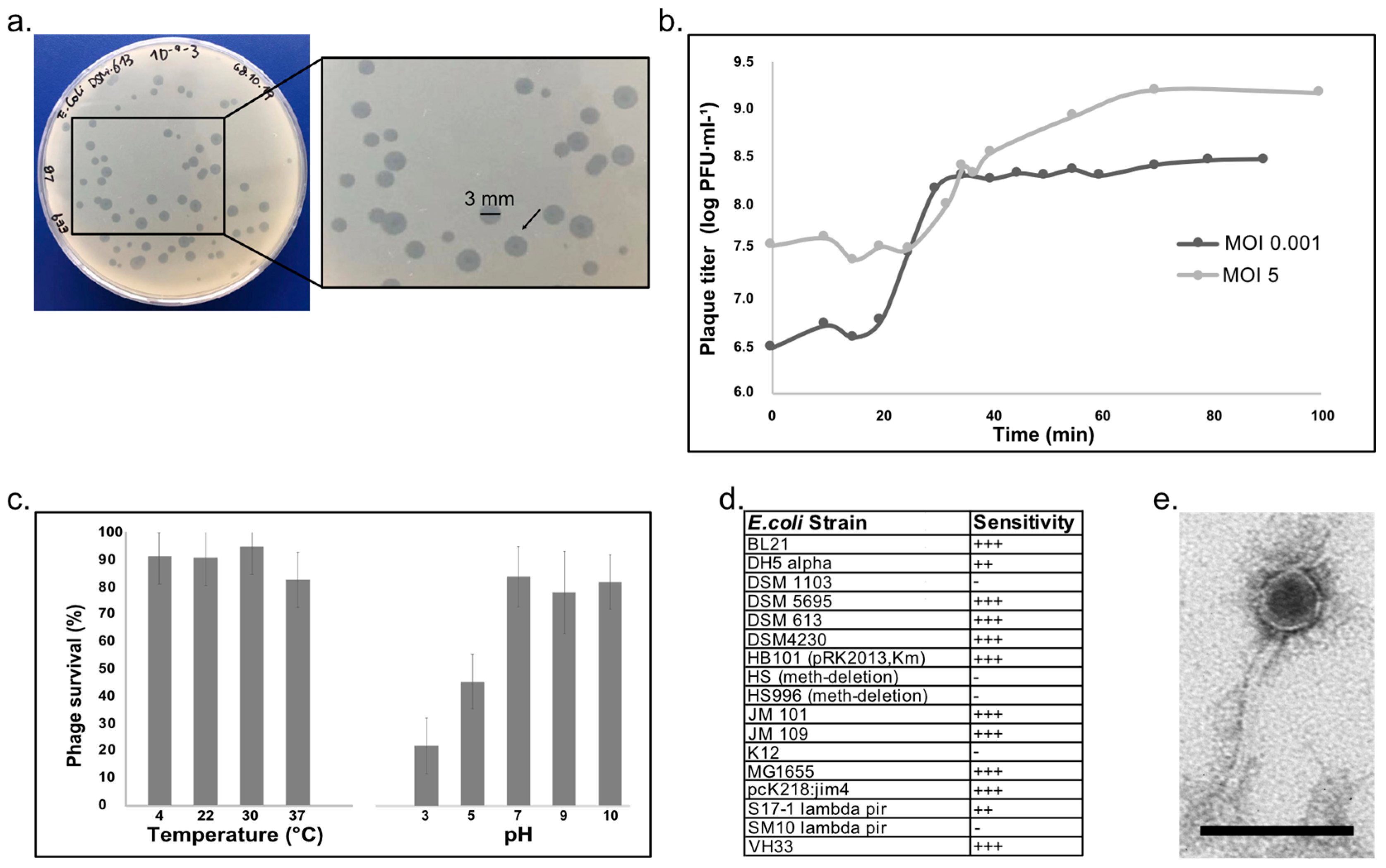
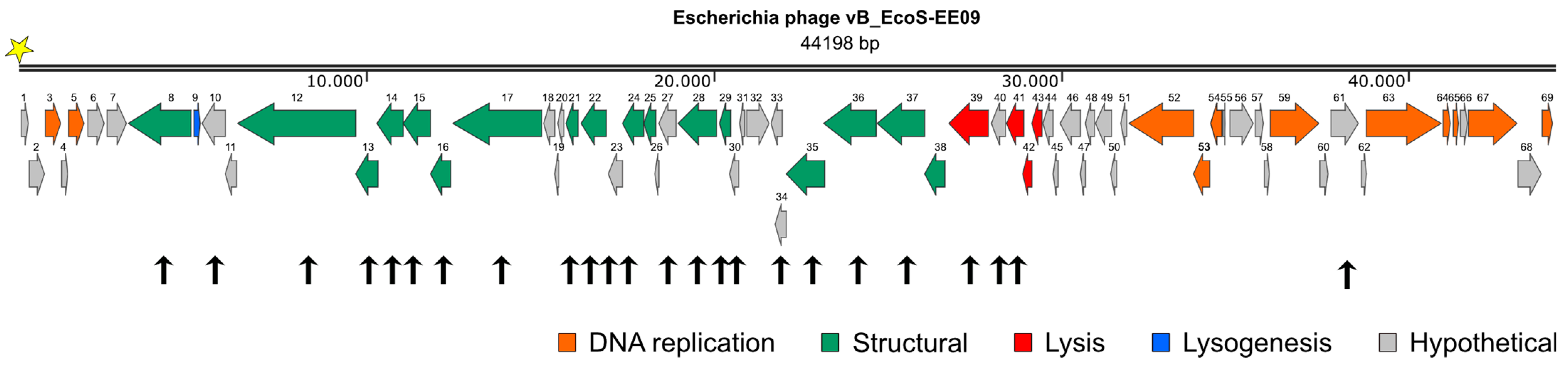
3.1. Characterization of Escherichia Phage vB_EcoS-EE09
3.2. Proteomic Analysis of Cell-Free Lysates of Escherichia Phage EE09
3.3. Detection of Escherichia Phage EE09 Proteins in Infected E. coli Cultures
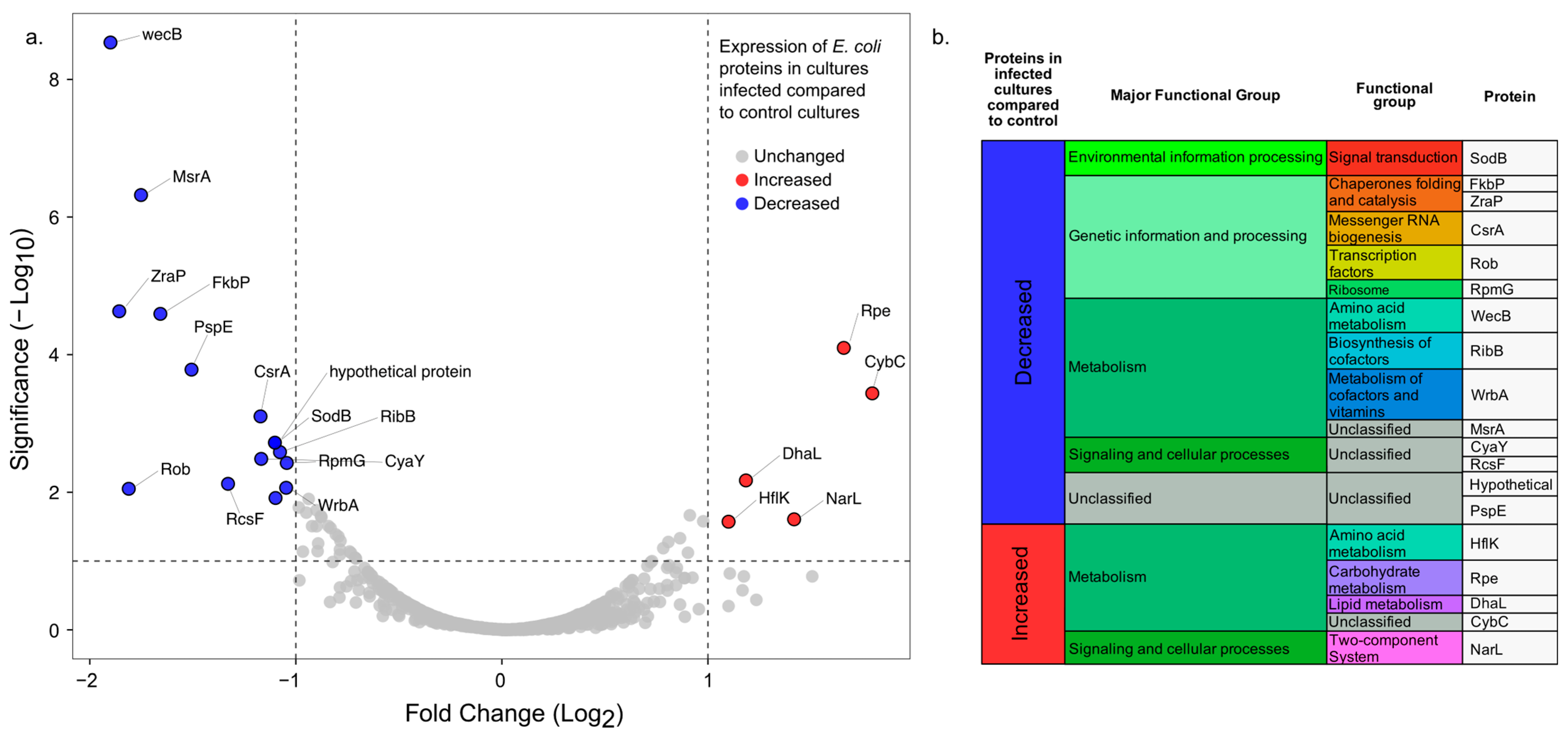
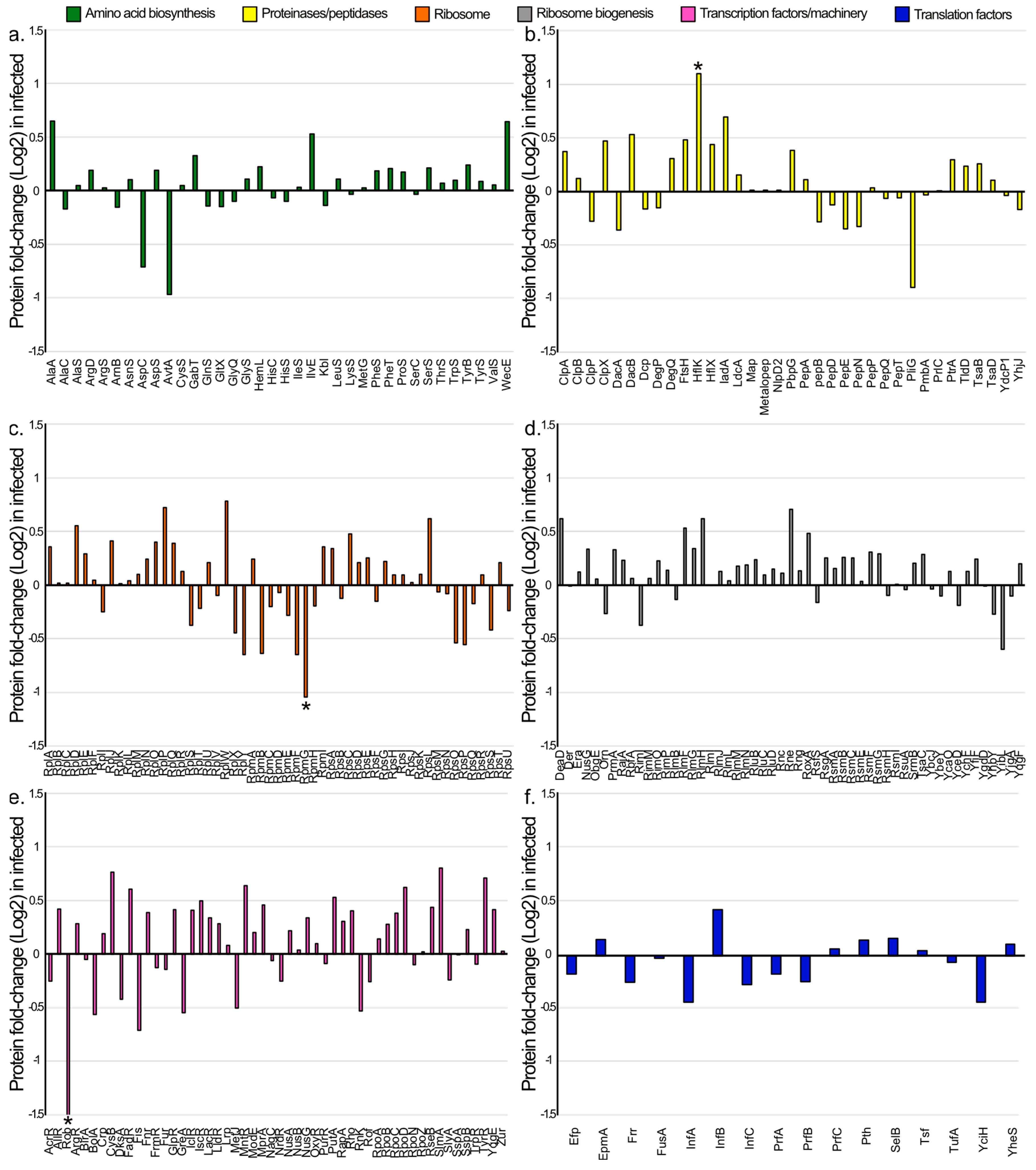
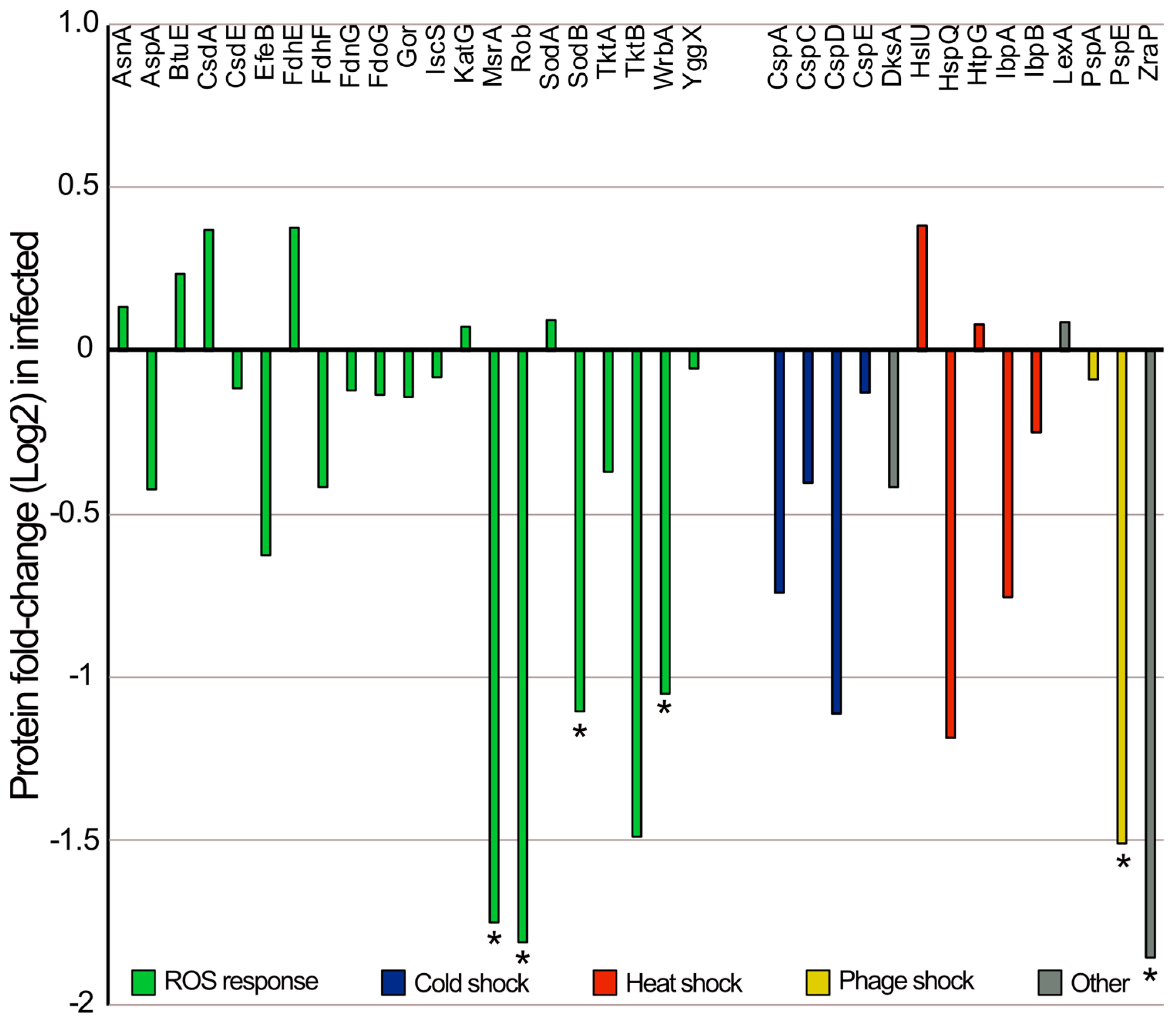
3.4. Comparative Proteomic Analysis between E. coli Infected with Phage EE09 and Non-Infected Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hendrix, R.W.; Smith, M.C.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary relationships among diverse bacteriophages and prophages: All the world’s a phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef]
- Zimmerman, A.E.; Howard-Varona, C.; Needham, D.M.; John, S.G.; Worden, A.Z.; Sullivan, M.B.; Waldbauer, J.R.; Coleman, M.L. Metabolic and biogeochemical consequences of viral infection in aquatic ecosystems. Nat. Rev. Microbio. 2020, 18, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B.; Brockhurst, M.A. Bacteria-phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol. Rev. 2014, 38, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Ankrah, N.Y.; May, A.L.; Middleton, J.L.; Jones, D.R.; Hadden, M.K.; Gooding, J.R.; LeCleir, G.R.; Wilhelm, S.W.; Campagna, S.R.; Buchan, A. Phage infection of an environmentally relevant marine bacterium alters host metabolism and lysate composition. ISME J. 2014, 8, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Maimaiti, Z.; Li, Z.; Xu, C.; Chen, J.; Chai, W. Global trends and hotspots of phage therapy for bacterial infection: A bibliometric visualized analysis from 2001 to 2021. Front. Microbiol. 2022, 13, 1067803. [Google Scholar] [CrossRef]
- Wu, B.; Wang, R.; Fane, A.G. The roles of bacteriophages in membrane-based water and wastewater treatment processes: A review. Water Res. 2017, 110, 120–132. [Google Scholar] [CrossRef]
- Ji, M.; Liu, Z.; Sun, K.; Li, Z.; Fan, X.; Li, Q. Bacteriophages in water pollution control: Advantages and limitations. Front. Environ. Sci. Eng. 2020, 15, 84. [Google Scholar] [CrossRef]
- Korf, I.H.E.; Meier-Kolthoff, J.P.; Adriaenssens, E.M.; Kropinski, A.M.; Nimtz, M.; Rohde, M.; van Raaij, M.J.; Wittmann, J. Still something to discover: Novel insights into Escherichia coli phage diversity and taxonomy. Viruses 2019, 11, 454. [Google Scholar] [CrossRef]
- Olsen, N.S.; Forero-Junco, L.; Kot, W.; Hansen, L.H. Exploring the remarkable diversity of culturable Escherichia coli phages in the Danish wastewater environment. Viruses 2020, 12, 986. [Google Scholar] [CrossRef]
- Smith, R.; O’Hara, M.; Hobman, J.L.; Millard, A.D. Draft genome sequences of 14 Escherichia coli phages isolated from cattle slurry. Genome Announc. 2015, 3. [Google Scholar] [CrossRef]
- Salmond, G.P.C.; Fineran, P.C. A century of the phage: Past, present and future. Nat. Rev. Microbio. 2015, 13, 777–786. [Google Scholar] [CrossRef]
- Chegini, Z.; Khoshbayan, A.; Vesal, S.; Moradabadi, A.; Hashemi, A.; Shariati, A. Bacteriophage therapy for inhibition of multi drug-resistant uropathogenic bacteria: A narrative review. Ann. Clin. Microbiol. Antimicrob. 2021, 20, 30. [Google Scholar] [CrossRef]
- Sarker, S.A.; Brssow, H. From bench to bed and back again: Phage therapy of childhood Escherichia coli diarrhea. Ann. N. Y. Acad. Sci. 2016, 1372, 42–52. [Google Scholar] [CrossRef]
- Anderson, C.L.; Sullivan, M.B.; Fernando, S.C. Dietary energy drives the dynamic response of bovine rumen viral communities. Microbiome 2017, 5, 155. [Google Scholar] [CrossRef]
- Willenbücher, K.; Wibberg, D.; Huang, L.; Conrady, M.; Ramm, P.; Gätcke, J.; Busche, T.; Brandt, C.; Szewzyk, U.; Schlüter, A.; et al. Phage genome diversity in a biogas-producing microbiome analyzed by Illumina and Nanopore GridION sequencing. Microorganisms 2022, 10, 368. [Google Scholar] [CrossRef]
- Roux, S.; Hallam, S.J.; Woyke, T.; Sullivan, M.B. Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. Elife 2015, 4, e08490. [Google Scholar] [CrossRef]
- Wright, B.W.; Logel, D.Y.; Mirzai, M.; Pascovici, D.; Molloy, M.P.; Jaschke, P.R. Proteomic and transcriptomic analysis of Microviridae φX174 infection reveals broad upregulation of host Escherichia coli membrane damage and heat shock responses. mSystems 2021, 6, e00046-21. [Google Scholar] [CrossRef] [PubMed]
- Brum, J.R.; Ignacio-Espinoza, J.C.; Kim, E.H.; Trubl, G.; Jones, R.M.; Roux, S.; VerBerkmoes, N.C.; Rich, V.I.; Sullivan, M.B. Illuminating structural proteins in viral “dark matter” with metaproteomics. Proc. Natl. Acad. Sci. USA 2016, 113, 2436–2441. [Google Scholar] [CrossRef] [PubMed]
- Howard-Varona, C.; Hargreaves, K.R.; Solonenko, N.E.; Markillie, L.M.; White, R.A., 3rd; Brewer, H.M.; Ansong, C.; Orr, G.; Adkins, J.N.; Sullivan, M.B. Multiple mechanisms drive phage infection efficiency in nearly identical hosts. ISME J. 2018, 12, 1605–1618. [Google Scholar] [CrossRef] [PubMed]
- Howard-Varona, C.; Lindback, M.M.; Bastien, G.E.; Solonenko, N.; Zayed, A.A.; Jang, H.; Andreopoulos, B.; Brewer, H.M.; Glavina del Rio, T.; Adkins, J.N.; et al. Phage-specific metabolic reprogramming of virocells. ISME J. 2020, 14, 881–895. [Google Scholar] [CrossRef] [PubMed]
- Bleriot, I.; Blasco, L.; Pacios, O.; Fernández-García, L.; López, M.; Ortiz-Cartagena, C.; Barrio-Pujante, A.; Fernández-Cuenca, F.; Pascual, Á.; Martínez-Martínez, L.; et al. Proteomic study of the interactions between phages and the bacterial host Klebsiella pneumoniae. Microbiol. Spectr. 2023, 11, e03974-22. [Google Scholar] [CrossRef] [PubMed]
- Van Tewest, R.K.; Andrew, M. Bacteriophage enrichment from water and soil. Methods Mol. Biol. 2009, 501, 15–21. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages by double agar overlay plaque assay. Methods Mol. Biol. 2009, 501, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.C.; Hair, B.B.; Wienclaw, T.M.; Murdock, M.H.; Hatch, J.B.; Trent, A.T.; White, T.D.; Haskell, K.J.; Berges, B.K. Isolation and host range of bacteriophage with lytic activity against methicillin-resistant Staphylococcus aureus and potential use as a fomite decontaminant. PLoS ONE 2015, 10, e0131714. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, V.; Bunk, B.; Meier-Kolthoff, J.P.; Spröer, C.; Poehlein, A.; Dogs, M.; Nguyen, M.; Petersen, J.; Daniel, R.; Overmann, J.; et al. Cobaviruses—A new globally distributed phage group infecting Rhodobacteraceae in marine ecosystems. ISME J. 2019, 13, 1404–1421. [Google Scholar] [CrossRef]
- Nikolenko, S.I.; Korobeynikov, A.I.; Alekseyev, M.A. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genom. 2013, 14 (Suppl. S1), S7. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Garneau, J.R.; Depardieu, F.; Fortier, L.-C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef] [PubMed]
- Ecale Zhou, C.L.; Malfatti, S.; Kimbrel, J.; Philipson, C.; McNair, K.; Hamilton, T.; Edwards, R.; Souza, B. MultiPhATE: Bioinformatics pipeline for functional annotation of phage isolates. Bioinformatics 2019, 35, 4402–4404. [Google Scholar] [CrossRef] [PubMed]
- McNair, K.; Zhou, C.; Dinsdale, E.A.; Souza, B.; Edwards, R.A.; Hancock, J. PHANOTATE: A novel approach to gene identification in phage genomes. Bioinformatics 2019, 35, 4537–4542. [Google Scholar] [CrossRef] [PubMed]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, 491–498. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- McNair, K.; Bailey, B.A.; Edwards, R.A. PHACTS, a computational approach to classifying the lifestyle of phages. Bioinformatics 2012, 28, 614–618. [Google Scholar] [CrossRef]
- Shang, J.; Tang, X.; Sun, Y. PhaTYP: Predicting the lifestyle for bacteriophages using BERT. Brief. Bioinform. 2023, 24, bbac487. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A novel tool to calculate the intergenomic similarities of prokaryote-infecting viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Moraru, C. VirClust-A tool for hierarchical clustering, core protein detection and annotation of (prokaryotic) viruses. Viruses 2023, 15, 1007. [Google Scholar] [CrossRef] [PubMed]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kropinski, A.M. Practical Advice on the One-Step Growth Curve. In Bacteriophages: Methods and Protocols, Volume 3; Clokie, M.R.J., Kropinski, A.M., Lavigne, R., Eds.; Springer: New York, NY, USA, 2018; pp. 41–47. [Google Scholar]
- Ackermann, H.W. Basic phage electron microscopy. Methods Mol. Biol. 2009, 501, 113–126. [Google Scholar] [CrossRef]
- Eziuzor, S.C.; Correa, F.B.; Peng, S.; Schultz, J.; Kleinsteuber, S.; da Rocha, U.N.; Adrian, L.; Vogt, C. Structure and functional capacity of a benzene-mineralizing, nitrate-reducing microbial community. J. Appl. Microbiol. 2022, 132, 2795–2811. [Google Scholar] [CrossRef]
- Schopper, S.; Kahraman, A.; Leuenberger, P.; Feng, Y.; Piazza, I.; Müller, O.; Boersema, P.J.; Picotti, P. Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry. Nat. Protoc. 2017, 12, 2391–2410. [Google Scholar] [CrossRef]
- Klaes, S.; Madan, S.; Deobald, D.; Cooper, M.; Adrian, L. GroEL-Proteotyping of bacterial communities using tandem mass spectrometry. Int. J. Mol. Sci. 2023, 24, 15692. [Google Scholar] [CrossRef]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 2020, 10, 20560. [Google Scholar] [CrossRef]
- Abedon, S.T. Lysis from without. Bacteriophage 2011, 1, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Mudgal, P.; Breidt, F.; Lubkin, S.R.; Sandeep, K.P. Quantifying the Significance of Phage Attack on Starter Cultures: A Mechanistic Model for Population Dynamics of Phage and Their Hosts Isolated from Fermenting Sauerkraut. Appl. Environ. Microbiol. 2006, 72, 3908–3915. [Google Scholar] [CrossRef]
- Silpe, J.E.; Bassler, B.L. A host-produced quorum-sensing autoinducer controls a phage lysis-lysogeny decision. Cell 2019, 176, 268–280.e13. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.; Brister, J.R. How to name and classify your phage: An informal guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Feiss, M.; Widner, W.; Miller, G.; Johnson, G.; Christiansen, S. Structure of the bacteriophage lambda cohesive end site: Location of the sites of terminase binding (cosB) and nicking (cosN). Gene 1983, 24, 207–218. [Google Scholar] [CrossRef]
- Latka, A.; Maciejewska, B.; Majkowska-Skrobek, G.; Briers, Y.; Drulis-Kawa, Z. Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl. Microbiol. Biotechnol. 2017, 101, 3103–3119. [Google Scholar] [CrossRef]
- Lemay, M.-L.; Otto, A.; Maaß, S.; Plate, K.; Becher, D.; Moineau, S. Investigating Lactococcus lactis MG1363 response to phage p2 infection at the proteome level*[S]. Mol. Cell Proteom. 2019, 18, 704–714. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Nagar, N.; Ecker, N.; Loewenthal, G.; Avram, O.; Ben-Meir, D.; Biran, D.; Ron, E.; Pupko, T. Harnessing Machine Learning To Unravel Protein Degradation in Escherichia coli. mSystems 2021, 6, e01296-20. [Google Scholar] [CrossRef]
- Noble, J.A.; Innis, M.A.; Koonin, E.V.; Rudd, K.E.; Banuett, F.; Herskowitz, I. The Escherichia coli hflA locus encodes a putative GTP-binding protein and two membrane proteins, one of which contains a protease-like domain. Proc. Natl. Acad. Sci. USA 1993, 90, 10866–10870. [Google Scholar] [CrossRef]
- Bandyopadhyay, K.; Parua, P.K.; Datta, A.B.; Parrack, P. Escherichia coli HflK and HflC can individually inhibit the HflB (FtsH)-mediated proteolysis of λCII in vitro. Arch. Biochem. Biophys. 2010, 501, 239–243. [Google Scholar] [CrossRef]
- Erez, Z.; Steinberger-Levy, I.; Shamir, M.; Doron, S.; Stokar-Avihail, A.; Peleg, Y.; Melamed, S.; Leavitt, A.; Savidor, A.; Albeck, S.; et al. Communication between viruses guides lysis-lysogeny decisions. Nature 2017, 541, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, T.; Yu, M.; Chen, Y.L.; Jin, M. The life cycle transitions of temperate phages: Regulating factors and potential ecological implications. Viruses 2022, 14, 1904. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Mohr, I. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 2011, 9, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Dkhili, S.; Ribeiro, M.; Ghariani, S.; Yahia, H.B.; Hillion, M.; Poeta, P.; Slama, K.B.; Hébraud, M.; Igrejas, G. Bacteriophages as antimicrobial agents? proteomic insights on three novel lytic bacteriophages infecting ESBL-producing Escherichia coli. Omics 2021, 25, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Andrade, S.L.A.; Patridge, E.V.; Ferry, J.G.; Einsle, O. Crystal structure of the NADH:quinone oxidoreductase WrbA from Escherichia coli. J. Bacteriol. 2007, 189, 9101–9107. [Google Scholar] [CrossRef]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- St John, G.; Brot, N.; Ruan, J.; Erdjument-Bromage, H.; Tempst, P.; Weissbach, H.; Nathan, C. Peptide methionine sulfoxide reductase from Escherichia coli and Mycobacterium tuberculosis protects bacteria against oxidative damage from reactive nitrogen intermediates. Proc. Natl. Acad. Sci. USA 2001, 98, 9901–9906. [Google Scholar] [CrossRef]
- Wilkinson, H.C.; Dalby, P.A. The Two-Species Model of transketolase explains donor substrate-binding, inhibition and heat-activation. Sci. Rep. 2020, 10, 4148. [Google Scholar] [CrossRef]
- Duval, V.; Lister, I.M. MarA, SoxS and Rob of Escherichia coli—Global regulators of multidrug resistance, virulence and stress response. Int. J. Biotechnol. Wellness Ind. 2013, 2, 101–124. [Google Scholar] [CrossRef]
- Dong, T.G.; Dong, S.; Catalano, C.; Moore, R.; Liang, X.; Mekalanos, J.J. Generation of reactive oxygen species by lethal attacks from competing microbes. Proc. Natl. Acad. Sci. USA 2015, 112, 2181–2186. [Google Scholar] [CrossRef] [PubMed]
- van der Weel, L.; As, K.S.; Dekker, W.J.C.; van den Eijnden, L.; van Helmond, W.; Schiphorst, C.; Hagen, W.R.; Hagedoorn, P.-L. ZraP, the most prominent zinc protein under zinc stress conditions has no direct role in in-vivo zinc tolerance in Escherichia coli. J. Inorg. Biochem. 2019, 192, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Adams, H.; Teertstra, W.; Demmers, J.; Boesten, R.; Tommassen, J. Interactions between phage-shock proteins in Escherichia coli. J. Bacteriol. 2003, 185, 1174–1180. [Google Scholar] [CrossRef] [PubMed]
- Shimuta, T.R.; Nakano, K.; Yamaguchi, Y.; Ozaki, S.; Fujimitsu, K.; Matsunaga, C.; Noguchi, K.; Emoto, A.; Katayama, T. Novel heat shock protein HspQ stimulates the degradation of mutant DnaA protein in Escherichia coli. Genes Cells 2004, 9, 1151–1166. [Google Scholar] [CrossRef]
- Abdelwahed, E.K.; Hussein, N.A.; Moustafa, A.; Moneib, N.A.; Aziz, R.K. Gene networks and pathways involved in Escherichia coli response to multiple stressors. Microorganisms 2022, 10, 1793. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2021, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CDS vB_EcoS-EE09 | Annotation | Protein Length (Amino Acids) | Protein Mass | Number of Detected Peptides | Coverage (%) |
|---|---|---|---|---|---|
| CDS8 | Tail fiber protein | 606 | 65.6 kDa | 14 | 34 |
| CDS10 | Hypothetical protein | 224 | 23.3 kDa | 2 | 12 |
| CDS12 | Tail tip, host specificity protein J | 1139 | 125.8 kDa | 25 | 31 |
| CDS13 | Tail tip assembly protein | 205 | 21.3 kDa | 1 | 9 |
| CDS14 | Phage tail assembly protein | 246 | 27.5 kDa | 1 | 4 |
| CDS15 | Phage minor tail protein | 262 | 28.8 kDa | 8 | 44 |
| CDS16 | Tail protein | 199 | 21.8 kDa | 3 | 21 |
| CDS17 | Tail tape measure protein H | 854 | 90.9 kDa | 31 | 45 |
| CDS21 | Tape measure chaperone | 103 | 11.0 kDa | 2 | 19 |
| CDS22 | Major tail protein | 241 | 25.6 kDa | 3 | 16 |
| CDS23 | Hypothetical protein | 140 | 15.0 kDa | 2 | 18 |
| CDS24 | Putative tail protein | 117 | 12.9 kDa | 1 | 6 |
| CDS27 | Hypothetical protein | 166 | 17.8 kDa | 1 | 9 |
| CDS28 | Major capsid protein | 366 | 38.4 kDa | 8 | 28 |
| CDS29 | Capsid and scaffold protein | 99 | 10.7 kDa | 8 | 80 |
| CDS30 | Hypothetical protein | 88 | 9.8 kDa | 5 | 44 |
| CDS34 | Hypothetical protein | 102 | 11.4 Kda | 1 | 12 |
| CDS35 | Phage capsid and scaffold | 367 | 41.0 Kda | 3 | 11 |
| CDS36 | Minor tail protein | 506 | 55.2 kDa | 15 | 36 |
| CDS37 | Phage terminase | 461 | 52.2 kDa | 1 | 3 |
| CDS39 | Calcineurin-like phoshoesterase | 380 | 42.8 kDa | 3 | 11 |
| CDS40 | Hypothetical protein | 137 | 13.6 kDa | 3 | 28 |
| CDS41 | Putative phage lysin | 163 | 18.2 kDa | 5 | 41 |
| CDS61 | Hypothetical protein | 766 | 30.7 kDa | 2 | 11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrero-Canosa, J.; Wang, L.; Oyugi, A.; Klaes, S.; Fischer, P.; Adrian, L.; Szewzyk, U.; Cooper, M. Characterization of phage vB_EcoS-EE09 infecting E. coli DSM613 Isolated from Wastewater Treatment Plant Effluent and Comparative Proteomics of the Infected and Non-Infected Host. Microorganisms 2023, 11, 2688. https://doi.org/10.3390/microorganisms11112688
Barrero-Canosa J, Wang L, Oyugi A, Klaes S, Fischer P, Adrian L, Szewzyk U, Cooper M. Characterization of phage vB_EcoS-EE09 infecting E. coli DSM613 Isolated from Wastewater Treatment Plant Effluent and Comparative Proteomics of the Infected and Non-Infected Host. Microorganisms. 2023; 11(11):2688. https://doi.org/10.3390/microorganisms11112688
Chicago/Turabian StyleBarrero-Canosa, Jimena, Luyao Wang, Angelah Oyugi, Simon Klaes, Pascal Fischer, Lorenz Adrian, Ulrich Szewzyk, and Myriel Cooper. 2023. "Characterization of phage vB_EcoS-EE09 infecting E. coli DSM613 Isolated from Wastewater Treatment Plant Effluent and Comparative Proteomics of the Infected and Non-Infected Host" Microorganisms 11, no. 11: 2688. https://doi.org/10.3390/microorganisms11112688