

Regulatory Functions of Hypoxia in Host–Parasite Interactions: A Focus on Enteric, Tissue, and Blood Protozoa
Abstract
:1. Introduction
2. Hypoxia and Hypoxia-Inducible Factor
Mechanisms of Tissue Hypoxia: HIF, Prolyl-Hydroxylases (PHDs), and Metabolism
3. Protozoan Parasites and Hypoxia
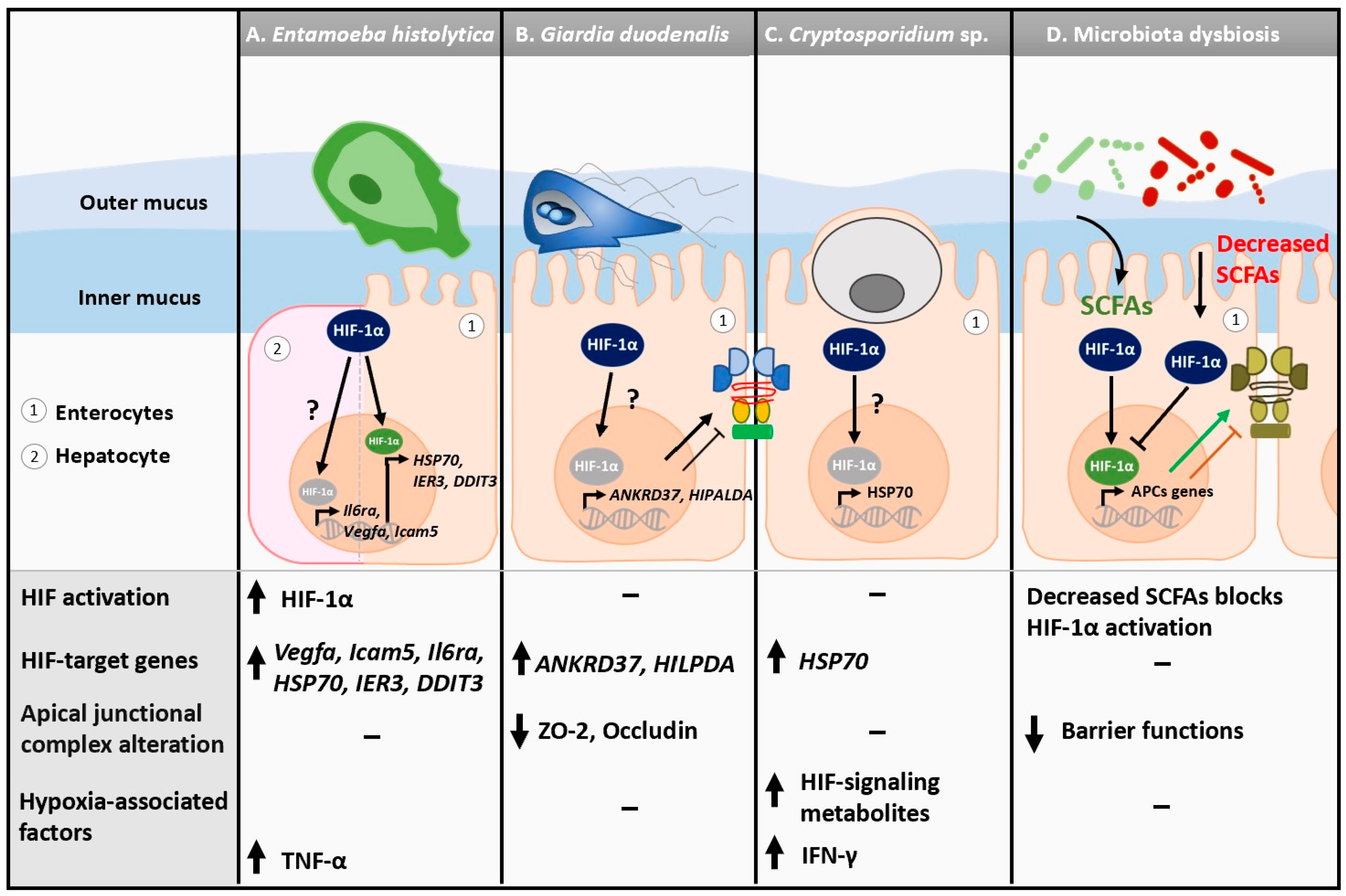
3.1. Enteric Protozoan Parasites: Oxygen Metabolism and Host Hypoxic Response
3.1.1. Hypoxia in the Gastrointestinal Tract
3.1.2. Entamoeba histolytica
3.1.3. Giardia Duodenalis
3.1.4. Cryptosporidium spp.
3.1.5. Hypoxia and Gut Epithelial Barrier Functions during Protozoan Infections
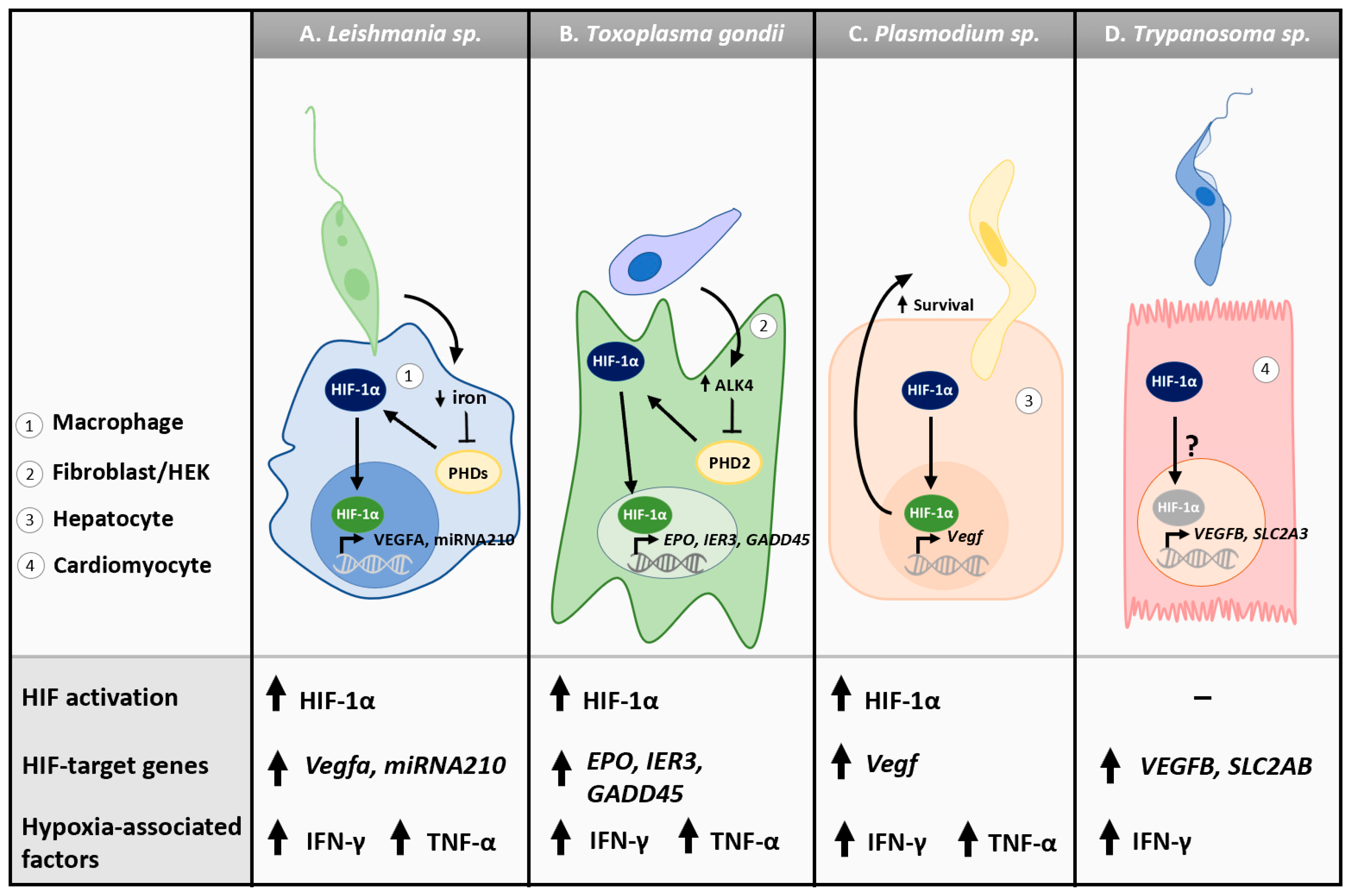
3.2. Tissue and Blood Parasites: Oxygen Metabolism and Host Hypoxic Responses
3.2.1. Tissue Hypoxia
3.2.2. Leishmania spp.
3.2.3. Toxoplasma gondii
3.2.4. Plasmodium spp.
3.2.5. Trypanosoma spp.
4. Host Immune Response and Markers of Hypoxia during Protozoan Infections
4.1. IL-1β and NLRP3
4.2. TNFα
4.3. INF-γ
4.4. IL-33
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zheng, L.; Kelly, C.J.; Colgan, S.P. Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C350–C360. [Google Scholar] [CrossRef] [Green Version]
- Schaible, B.; Schaffer, K.; Taylor, C.T. Hypoxia, innate immunity and infection in the lung. Respir. Physiol. Neurobiol. 2010, 174, 235–243. [Google Scholar] [CrossRef] [Green Version]
- McKeown, S.R. Defining normoxia, physoxia and hypoxia in tumours—Implications for treatment response. Br. J. Radiol. 2014, 87, 20130676. [Google Scholar] [CrossRef] [Green Version]
- He, G.; Shankar, R.A.; Chzhan, M.; Samouilov, A.; Kuppusamy, P.; Zweier, J.L. Noninvasive measurement of anatomic structure and intraluminal oxygenation in the gastrointestinal tract of living mice with spatial and spectral EPR imaging. Proc. Natl. Acad. Sci. USA 1999, 96, 4586–4591. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, W.G.; Lowndes, R.H.; Young, H.L. Intraoperative tissue oximetry in the human gastrointestinal tract. Am. J. Surg. 1990, 159, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, T.; Paul, T. Oxygen Conformance in Hepatocytes. Am. J. Physiol. 1993, 265 Pt 1, L395–L402. [Google Scholar] [CrossRef] [PubMed]
- Budinger, G.R.; Chandel, N.; Shao, Z.H.; Li, C.Q.; Melmed, A.; Becker, L.B.; Schumacker, P.T. Cellular energy utilization and supply during hypoxia in embryonic cardiac myocytes. Am. J. Physiol.-Lung Cell Mol. Physiol. 1996, 270, L44–L53. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [Green Version]
- Singhal, R.; Shah, Y.M. Oxygen battle in the gut: Hypoxia and hypoxia-inducible factors in metabolic and inflammatory responses in the intestine. J. Biol. Chem. 2020, 295, 10493–10505. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential Roles of Hypoxia-Inducible Factor 1α (HIF-1α) and HIF-2α in Hypoxic Gene Regulation. Mol. Cell Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaśkiewicz, M.; Moszyńska, A.; Króliczewski, J.; Cabaj, A.; Bartoszewska, S.; Charzyńska, A.; Gebert, M.; Dąbrowski, M.; Collawn, J.F.; Bartoszewski, R. The transition from HIF-1 to HIF-2 during prolonged hypoxia results from reactivation of PHDs and HIF1A mRNA instability. Cell Mol. Biol. Lett. 2022, 27, 109. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Lemos, R.; Liu, X.; Powis, G. The Hypoxia-Associated Factor Switches Cells from HIF-1α- to HIF-2α-Dependent Signaling Promoting Stem Cell Characteristics, Aggressive Tumor Growth and Invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular Characterization and Chromosomal Localization of a Third α- Class Hypoxia Inducible Factor Subunit, HIF3α. Gene Expr. 1998, 7, 205–213. [Google Scholar] [PubMed]
- Hara, S.; Hamada, J.; Kobayashi, C.; Kondo, Y.; Imura, N. Expression and Characterization of Hypoxia-Inducible Factor (HIF)-3α in Human Kidney: Suppression of HIF-Mediated Gene Expression by HIF-3α. Biochem. Biophys. Res. Commun. 2001, 287, 808–813. [Google Scholar] [CrossRef]
- Li, Q.F.; Wang, X.R.; Yang, Y.W.; Lin, H. Hypoxia upregulates hypoxia inducible factor (HIF)-3α expression in lung epithelial cells: Characterization and comparison with HIF-1α. Cell Res. 2006, 16, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Safran, M.; Kaelin, W.G., Jr. HIF hydroxylation and the mammalian oxygen-sensing pathway. J. Clin. Investig. 2003, 111, 779–783. [Google Scholar] [CrossRef] [Green Version]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-alpha to the von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine Hydroxylation of the HIF Transactivation Domain: A Hypoxic Switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Corrado, C.; Fontana, S. Hypoxia and HIF Signaling: One Axis with Divergent Effects. Int. J. Mol. Sci. 2020, 21, 5611. [Google Scholar] [CrossRef] [PubMed]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 838205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L.; Jiang, B.-H.; Leung, S.W.; Passantino, R.; Concordet, J.-P.; Maire, P.; Giallongo, A. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase A Gene Promoters Contain Essential Binding Sites for Hypoxia-inducible Factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Bartoszewski, R.; Moszyńska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Króliczewski, J.; Dąbrowski, M.; et al. Primary endothelial Cell-Specific regulation of Hypoxia-Inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef]
- Augustin, H.G.; Koh, G.Y.; Thurston, G.; Alitalo, K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat. Rev. Mol. Cell Biol. 2009, 10, 165–177. [Google Scholar] [CrossRef]
- Gabai, V.L.; Meng, L.; Kim, G.; Mills, T.A.; Benjamin, I.J.; Sherman, M.Y. Heat Shock Transcription Factor Hsf1 Is Involved in Tumor Progression via Regulation of Hypoxia-Inducible Factor 1 and RNA-Binding Protein HuR. Mol. Cell Biol. 2012, 32, 929–940. [Google Scholar] [CrossRef] [Green Version]
- Kierans, S.J.; Taylor, C.T. Regulation of glycolysis by the hypoxia-inducible factor (HIF): Implications for cellular physiology. J. Physiol. 2020, 599, 23–37. [Google Scholar] [CrossRef]
- Shepherd, A.P. Metabolic control of intestinal oxygenation and blood flow. Fed. Proc. 1982, 41, 2084–2089. [Google Scholar]
- Matheson, P.J.; Wilson, M.A.; Garrison, R. Regulation of Intestinal Blood Flow. J. Surg. Res. 2000, 93, 182–196. [Google Scholar] [CrossRef] [Green Version]
- Fisher, E.M.; Khan, M.; Salisbury, R.; Kuppusamy, P. Noninvasive Monitoring of Small Intestinal Oxygen in a Rat Model of Chronic Mesenteric Ischemia. Cell Biochem. Biophys. 2013, 67, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albenberg, L.; Esipova, T.V.; Judge, C.P.; Bittinger, K.; Chen, J.; Laughlin, A.; Grunberg, S.; Baldassano, R.N.; Lewis, J.D.; Li, H.; et al. Correlation Between Intraluminal Oxygen Gradient and Radial Partitioning of Intestinal Microbiota. Gastroenterology 2014, 147, 1055–1063.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konjar, Š.; Pavšič, M.; Veldhoen, M. Regulation of Oxygen Homeostasis at the Intestinal Epithelial Barrier Site. Int. J. Mol. Sci. 2021, 22, 9170. [Google Scholar] [CrossRef]
- Taylor, C.T.; Colgan, S.P. Hypoxia and gastrointestinal disease. J. Mol. Med. 2007, 85, 1295–1300. [Google Scholar] [CrossRef]
- Colgan, S.P.; Taylor, C. Hypoxia: An alarm signal during intestinal inflammation. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 281–287. [Google Scholar] [CrossRef] [Green Version]
- Bertout, J.A.; Patel, S.A.; Simon, M.C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 2008, 8, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Ruan, K.; Song, G.; Ouyang, G. Role of hypoxia in the hallmarks of human cancer. J. Cell Biochem. 2009, 107, 1053–1062. [Google Scholar] [CrossRef]
- Shi, R.; Liao, C.; Zhang, Q. Hypoxia-Driven Effects in Cancer: Characterization, Mechanisms, and Therapeutic Implications. Cells 2021, 10, 678. [Google Scholar] [CrossRef]
- Sebestyén, A.; Kopper, L.; Dankó, T.; Tímár, J. Hypoxia Signaling in Cancer: From Basics to Clinical Practice. Pathol. Oncol. Res. 2021, 27, 1609802. [Google Scholar] [CrossRef] [PubMed]
- Freiberg, R.A.; Hammond, E.M.; Dorie, M.J.; Welford, S.M.; Giaccia, A.J. DNA Damage during Reoxygenation Elicits a Chk2-Dependent Checkpoint Response. Mol. Cell Biol. 2006, 26, 1598–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, S.A.; Fines, K.; Ng, J.; Traboulsi, D.; Lee, J.; Ihara, E.; Li, Y.; Willmore, W.G.; Chung, D.; Scully, M.M.; et al. Hypoxia-Inducible Factor Signaling Provides Protection in Clostridium difficile-Induced Intestinal Injury. Gastroenterology 2010, 139, 259–269.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karhausen, J.; Furuta, G.T.; Tomaszewski, J.E.; Johnson, R.S.; Colgan, S.P.; Haase, V.H. Epithelial Hypoxia-Inducible Factor-1 Is Protective in Murine Experimental Colitis. J. Clin. Investig. 2004, 114, 1098–1106. [Google Scholar] [CrossRef] [Green Version]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and Inflammation Hypoxia-Induced Inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Stauffer, W.; Ravdin, J.I. Entamoeba histolytica: An update. Curr. Opin. Infect. Dis. 2003, 16, 479–485. [Google Scholar] [CrossRef]
- Stanley, S.L. Amoebiasis. Lancet 2003, 361, 1025–1034. [Google Scholar] [CrossRef]
- Pineda, E.; Perdomo, D. Entamoeba histolytica under Oxidative Stress: What Countermeasure Mechanisms Are in Place? Cells 2017, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Santos, F.; Nequiz, M.; Hernández-Cuevas, N.A.; Hernández, K.; Pineda, E.; Encalada, R.; Guillén, N.; Luis-García, E.; Saralegui, A.; Saavedra, E.; et al. Maintenance of intracellular hypoxia and adequate heat shock response are essential requirements for pathogenicity and virulence of Entamoeba histolytica. Cell Microbiol. 2015, 17, 1037–1051. [Google Scholar] [CrossRef]
- Ghadirian, E.; Denis, M. In vivo activation of macrophages by IFN-γ to kill Entamoeba histolytica trophozoites in vitro. Parasite Immunol. 1992, 14, 397–404. [Google Scholar] [CrossRef]
- Lin, J.Y.; Chadee, K. Macrophage cytotoxicity against Entamoeba histolytica trophozoites is mediated by nitric oxide from L-arginine. J. Immunol. 1992, 148, 3999–4005. [Google Scholar] [CrossRef] [PubMed]
- Vicente, J.B.; Ehrenkaufer, G.M.; Saraiva, L.M.; Teixeira, M.; Singh, U. Entamoeba histolytica modulates a complex repertoire of novel genes in response to oxidative and nitrosative stresses: Implications for amebic pathogenesis. Cell Microbiol. 2009, 11, 51–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbar, A.; Chatterjee, N.S.; Sen, P.; Debnath, A.; Pal, A.; Bera, T.; Das, P. Genes induced by a high-oxygen environment in Entamoeba histolytica. Mol. Biochem. Parasitol. 2004, 133, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Arias, D.G.; Gutierrez, C.E.; Iglesias, A.A.; Guerrero, S.A. Thioredoxin-linked metabolism in Entamoeba histolytica. Free Radic. Biol. Med. 2007, 42, 1496–1505. [Google Scholar] [CrossRef]
- Groneberg, M.; Hoenow, S.; Marggraff, C.; Fehling, H.; Metwally, N.G.; Hansen, C.; Bruchhaus, I.; Tiegs, G.; Sellau, J.; Lotter, H. HIF-1α modulates sex-specific Th17/Treg responses during hepatic amoebiasis. J. Hepatol. 2021, 76, 160–173. [Google Scholar] [CrossRef]
- Zhang, Z.; Stanley, S.L. Stereotypic and specific elements of the human colonic response to Entamoeba histolytica and Shigella flexneri. Cell Microbiol. 2004, 6, 535–554. [Google Scholar] [CrossRef] [Green Version]
- Verma, R.N.; Malik, Z.; Subbarao, N.; Singh, G.P.; Sinha, D.N. Entamoeba histolytica HM-1: IMSS gene expression profiling identifies key hub genes, potential biomarkers, and pathways in Amoebiasis infection: A systematic network meta-analysis. Biosci. Rep. 2022, 42, BSR20220191. [Google Scholar] [CrossRef]
- Roxstrom-Lindquist, K.; Ringqvist, E.; Palm, D.; Svard, S. Giardia lamblia-Induced Changes in Gene Expression in Differentiated Caco-2 Human Intestinal Epithelial Cells. Infect. Immun. 2005, 73, 8204–8208. [Google Scholar] [CrossRef] [Green Version]
- Rojas, L.; Grüttner, J.; Ma’ayeh, S.; Xu, F.; Svärd, S.G. Dual RNA Sequencing Reveals Key Events When Different Giardia Life Cycle Stages Interact with Human Intestinal Epithelial Cells In Vitro. Front. Cell Infect. Microbiol. 2022, 12, 862211. [Google Scholar] [CrossRef]
- Anatskaya, O.V.; Sidorenko, N.V.; Matveev, I.V.; Kropotov, A.V.; Vinogradov, A.E. Remodeling of rat cardiomyocytes after neonatal cryptosporidiosis. II. Deformation, excessive polyploidization, and HIF-1α overexpression. Cell Tissue Biol. 2012, 6, 472–484. [Google Scholar] [CrossRef]
- Deng, M.; Lancto, C.A.; Abrahamsen, M.S. Cryptosporidium parvum regulation of human epithelial cell gene expression. Int. J. Parasitol. 2004, 34, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Degrossoli, A.; Bosetto, M.C.; Lima, C.B.C.; Giorgio, S. Expression of hypoxia-inducible factor 1α in mononuclear phagocytes infected with Leishmania amazonensis. Immunol. Lett. 2007, 114, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Degrossoli, A.; Arrais-Silva, W.W.; Colhone, M.C.; Gadelha, F.R.; Joazeiro, P.P.; Giorgio, S. The Influence of Low Oxygen on Macrophage Response to Leishmania Infection. Scand. J. Immunol. 2011, 74, 165–175. [Google Scholar] [CrossRef]
- Araújo, A.P.; Arrais-Silva, W.W.; Giorgio, S. Acta Histochemica Infection by Leishmania amazonensis in Mice: A Potential Model for Chronic Hypoxia. Acta Histochem. 2012, 114, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kumar, A.; Das, S.; Kumar, A.; Abhishek, K.; Verma, S.; Mandal, A.; Singh, R.K.; Das, P. Leishmania donovani Activates Hypoxia Inducible Factor-1α and miR-210 for Survival in Macrophages by Downregulation of NF-κB Mediated Pro-inflammatory Immune Response. Front. Microbiol. 2018, 9, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettadapura, M.; Roys, H.; Bowlin, A.; Venugopal, G.; Washam, C.L.; Fry, L.; Murdock, S.; Wanjala, H.; Byrum, S.D.; Weinkopff, T. HIF-α Activation Impacts Macrophage Function during Murine Leishmania major Infection. Pathogens 2021, 10, 1584. [Google Scholar] [CrossRef] [PubMed]
- Spear, W.; Chan, D.; Coppens, I.; Johnson, R.S.; Giaccia, A.; Blader, I.J. The host cell transcription factor hypoxia-inducible factor 1 is required for Toxoplasma gondii growth and survival at physiological oxygen levels. Cell Microbiol. 2005, 8, 339–352. [Google Scholar] [CrossRef]
- Liu, Y.; Zou, X.; Ou, M.; Ye, X.; Zhang, B.; Wu, T.; Dong, S.; Chen, X.; Liu, H.; Zheng, Z.; et al. Toxoplasma gondii Cathepsin C1 inhibits NF-κB signalling through the positive regulation of the HIF-1α/EPO axis. Acta Trop. 2019, 195, 35–43. [Google Scholar] [CrossRef]
- Syn, G.; Anderson, D.; Blackwell, J.M.; Jamieson, S.E. Toxoplasma gondii Infection Is Associated with Mitochondrial Dysfunction In-Vitro. Front. Cell Infect. Microbiol. 2017, 7, 512. [Google Scholar] [CrossRef] [Green Version]
- Blader, I.J.; Manger, I.D.; Boothroyd, J.C. Microarray Analysis Reveals Previously Unknown Changes in Toxoplasma gondii-infected Human Cells. J. Biol. Chem. 2001, 276, 24223–24231. [Google Scholar] [CrossRef] [Green Version]
- Park, M.-K.; Ko, E.-J.; Jeon, K.-Y.; Kim, H.; Jo, J.-O.; Baek, K.-W.; Kang, Y.-J.; Choi, Y.H.; Hong, Y.; Ock, M.S.; et al. Induction of Angiogenesis by Malarial Infection through Hypoxia Dependent Manner. Korean J. Parasitol. 2019, 57, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Venturini, G.; Alvim, J.M.; Padilha, K.; Toepfer, C.N.; Gorham, J.M.; Wasson, L.K.; Biagi, D.; Schenkman, S.; Carvalho, V.M.; Salgueiro, J.S.; et al. Cardiomyocyte infection by Trypanosoma cruzi promotes innate immune response and glycolysis activation. Front. Cell Infect. Microbiol. 2023, 13, 1098457. [Google Scholar] [CrossRef]
- Ivanov, A.I. Giardia and giardiasis. Bulg. J. Vet. Med. 2010, 13, 65–80. [Google Scholar]
- Cotton, J.A.; Beatty, J.K.; Buret, A.G. Host parasite interactions and pathophysiology in Giardia infections. Int. J. Parasitol. 2011, 41, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Halliez, M.C.M.; Buret, A.G. Extra-intestinal and long term consequences of Giardia duodenalis infections. World J. Gastroenterol. 2013, 19, 8974–8985. [Google Scholar] [CrossRef]
- Allain, T.; Amat, C.B.; Motta, J.-P.; Manko, A.; Buret, A.G. Interactions of Giardia sp. with the intestinal barrier: Epithelium, mucus, and microbiota. Tissue Barriers 2017, 5, e1274354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ankarklev, J.; Jerlström-Hultqvist, J.; Ringqvist, E.; Troell, K.; Svärd, S.G. Behind the smile: Cell biology and disease mechanisms of Giardia species. Nat. Rev. Microbiol. 2010, 8, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, D.; Harris, J.C.; Maroulis, S.; Biagini, G.A.; Wadley, R.B.; Turner, M.P.; Edwards, M.R. The microaerophilic flagellate Giardia intestinalis: Oxygen and its reaction products collapse membrane potential and cause cytotoxicity. Microbiology 2000, 146, 3109–3118. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Collins, L.J. Reconstruction of Sugar Metabolic Pathways of Giardia lamblia. Int. J. Proteom. 2012, 2012, 980829. [Google Scholar] [CrossRef] [Green Version]
- Lindmark, D.G. Energy metabolism of the anaerobic protozoon Giardia lamblia. Mol. Biochem. Parasitol. 1980, 1, 1–12. [Google Scholar] [CrossRef]
- Brown, D.M.; Upcroft, J.A.; Upcroft, P. Free radical detoxification in Giardia duodenalis. Mol. Biochem. Parasitol. 1995, 72, 47–56. [Google Scholar] [CrossRef]
- Mastronicola, D.; Giuffrè, A.; Testa, F.; Mura, A.; Forte, E.; Bordi, E.; Pucillo, L.P.; Fiori, P.L.; Sarti, P. Giardia intestinalis escapes oxidative stress by colonizing the small intestine: A molecular hypothesis. IUBMB Life 2011, 63, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Di Matteo, A.; Scandurra, F.M.; Testa, F.; Forte, E.; Sarti, P.; Brunori, M.; Giuffrè, A. The O2-scavenging Flavodiiron Protein in the Human Parasite Giardia intestinalis. J. Biol. Chem. 2008, 283, 4061–4068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townson, S.M.; Hanson, G.R.; Upcroft, J.A.; Upcroft, P. A purified ferredoxin from Giardia duodenalis. JBIC J. Biol. Inorg. Chem. 1994, 220, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Paget, T.A.; Macechko, P.T.; Jarroll, E.L. Metabolic Changes in Giardia intestinalis during Differentiation. J. Parasitol. 1998, 84, 222–226. [Google Scholar] [CrossRef]
- Holthaus, D.; Kraft, M.R.; Krug, S.M.; Wolf, S.; Müller, A.; Betancourt, E.D.; Schorr, M.; Holland, G.; Knauf, F.; Schulzke, J.-D.; et al. Dissection of Barrier Dysfunction in Organoid-Derived Human Intestinal Epithelia Induced by Giardia duodenalis. Gastroenterology 2022, 162, 844–858. [Google Scholar] [CrossRef]
- Ma’Ayeh, S.Y.; Knörr, L.; Sköld, K.; Garnham, A.; Ansell, B.; Jex, A.; Svärd, S.G. Responses of the Differentiated Intestinal Epithelial Cell Line Caco-2 to Infection with the Giardia intestinalis GS Isolate. Front. Cell Infect. Microbiol. 2018, 8, 244. [Google Scholar] [CrossRef] [Green Version]
- Povero, D.; Johnson, S.M.; Liu, J. Hypoxia, hypoxia-inducible gene 2 (HIG2)/HILPDA, and intracellular lipolysis in cancer. Cancer Lett. 2020, 493, 71–79. [Google Scholar] [CrossRef]
- Kotloff, K.L.; Nataro, J.P.; Blackwelder, W.C.; Nasrin, D.; Farag, T.H.; Panchalingam, S.; Wu, Y.; Sow, S.O.; Sur, D.; Breiman, R.F.; et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): A prospective, case-control study. Lancet 2013, 382, 209–222. [Google Scholar] [CrossRef]
- Bouzid, M.; Hunter, P.R.; Chalmers, R.M.; Tyler, K.M. Cryptosporidium Pathogenicity and Virulence. Clin. Microbiol. Rev. 2013, 26, 115–134. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Widmer, G.; Wang, Y.; Ozaki, L.S.; Alves, J.M.; Serrano, M.G.; Puiu, D.; Manque, P.; Akiyoshi, D.; Mackey, A.J.; et al. The genome of Cryptosporidium hominis. Nature 2004, 431, 1107–1112. [Google Scholar] [CrossRef] [Green Version]
- Barta, J.R.; Thompson, R.A. What is Cryptosporidium? Reappraising its biology and phylogenetic affinities. Trends Parasitol. 2006, 22, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Bilung, L.M.; Tahar, A.S.; Yunos, N.E.; Apun, K.; Lim, Y.A.-L.; Nillian, E.; Hashim, H.F. Detection of Cryptosporidium and Cyclospora Oocysts from Environmental Water for Drinking and Recreational Activities in Sarawak, Malaysia. BioMed Res. Int. 2017, 2017, 4636420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Cao, L.; Chang, Y.; Fu, Y.; Wang, Y.; Zhang, K.; Zhang, S.; Zhang, L. Microbiome-Metabolomics Analysis of the Impacts of Cryptosporidium muris Infection in BALB/C Mice. Microbiol. Spectr. 2022, 11, e02175-22. [Google Scholar] [CrossRef] [PubMed]
- Glover, L.E.; Colgan, S.P. Epithelial Barrier Regulation by Hypoxia-Inducible Factor. Ann. Am. Thorac. Soc. 2017, 14, S233–S236. [Google Scholar] [CrossRef]
- Luo, P.-L.; Wang, Y.-J.; Yang, Y.-Y.; Yang, J.-J. Hypoxia-induced hyperpermeability of rat glomerular endothelial cells involves HIF-2α mediated changes in the expression of occludin and ZO-1. Braz. J. Med. Biol. Res. 2018, 51, 51. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, J.J.; McCaffrey, G.; Quigley, C.E.; Finch, J.; DeMarco, K.M.; Nametz, N.; Davis, T.P. Oxidative Stress Increases Blood-Brain Barrier Permeability and Induces Alterations in Occludin during Hypoxia-Reoxygenation. J. Cereb. Blood Flow Metab. 2010, 30, 1625–1636. [Google Scholar] [CrossRef] [Green Version]
- Dowdell, A.S.; Cartwright, I.M.; Goldberg, M.S.; Kostelecky, R.; Ross, T.; Welch, N.; Glover, L.E.; Colgan, S.P. The HIF target ATG9A is essential for epithelial barrier function and tight junction biogenesis. Mol. Biol. Cell 2020, 31, 2249–2258. [Google Scholar] [CrossRef]
- Saeedi, B.J.; Kao, D.J.; Kitzenberg, D.A.; Dobrinskikh, E.; Schwisow, K.D.; Masterson, J.C.; Kendrick, A.A.; Kelly, C.J.; Bayless, A.J.; Kominsky, D.J.; et al. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol. Biol. Cell 2015, 26, 2252–2262. [Google Scholar] [CrossRef] [Green Version]
- Leroy, A.; Lauwaet, T.; De Bruyne, G.; Cornelissen, M.; Mareel, M. Entamoeba histolytica disturbs the tight junction complex in human enteric T84 cell layers. FASEB J. 2000, 14, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Kissoon-Singh, V.; Moreau, F.; Trusevych, E.; Chadee, K. Entamoeba histolytica Exacerbates Epithelial Tight Junction Permeability and Proinflammatory Responses in Muc2 Mice. Am. J. Pathol. 2013, 182, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, Z.; Hellman, L.; Svärd, S.G. Cleavage specificity of recombinant Giardia intestinalis cysteine proteases: Degradation of immunoglobulins and defensins. Mol. Biochem. Parasitol. 2018, 227, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Allain, T.; Fekete, E.; Buret, A.G. Giardia Cysteine Proteases: The Teeth behind the Smile. Trends Parasitol. 2019, 35, 636–648. [Google Scholar] [CrossRef]
- Allain, T.; Buret, A.G. Chapter Five—Pathogenesis and Post-Infectious Complications in Giardiasis. In Giardia and Giardiasis, Part B; Ortega-Pierres, M.G.B.T.-A., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 107, pp. 173–199. [Google Scholar] [CrossRef]
- Souza, J.B.; Tsantarlis, K.; Tonelli, R.R. Oxygen-dependent regulation of permeability in low resistance intestinal epithelial cells infected with Giardia lamblia. Exp. Parasitol. 2022, 240, 108329. [Google Scholar] [CrossRef]
- Ankri, S. Entamoeba histolytica—Gut Microbiota Interaction: More Than Meets the Eye. Microorganisms 2021, 9, 581. [Google Scholar] [CrossRef]
- Karpe, A.V.; Hutton, M.L.; Mileto, S.J.; James, M.L.; Evans, C.; Shah, R.M.; Ghodke, A.B.; Hillyer, K.E.; Metcalfe, S.S.; Liu, J.-W.; et al. Cryptosporidiosis Modulates the Gut Microbiome and Metabolism in a Murine Infection Model. Metabolites 2021, 11, 380. [Google Scholar] [CrossRef] [PubMed]
- Charania, R.; Wade, B.E.; McNair, N.N.; Mead, J.R. Changes in the Microbiome of Cryptosporidium-Infected Mice Correlate to Differences in Susceptibility and Infection Levels. Microorganisms 2020, 8, 879. [Google Scholar] [CrossRef]
- Fekete, E.; Allain, T.; Siddiq, A.; Sosnowski, O.; Buret, A.G. Giardia spp. and the Gut Microbiota: Dangerous Liaisons. Front. Microbiol. 2021, 11, 618106. [Google Scholar] [CrossRef]
- Gerbaba, T.K.; Gupta, P.; Rioux, K.; Hansen, D.; Buret, A.G. Giardia duodenalis-induced alterations of commensal bacteria kill Caenorhabditis elegans: A new model to study microbial-microbial interactions in the gut. Am. J. Physiol.-Gastrointest Liver Physiol. 2015, 308, G550–G561. [Google Scholar] [CrossRef] [Green Version]
- Beatty, J.K.; Akierman, S.V.; Motta, J.-P.; Muise, S.; Workentine, M.L.; Harrison, J.J.; Bhargava, A.; Beck, P.L.; Rioux, K.P.; McKnight, G.W.; et al. Giardia duodenalis induces pathogenic dysbiosis of human intestinal microbiota biofilms. Int. J. Parasitol. 2017, 47, 311–326. [Google Scholar] [CrossRef]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, S.C.; Karl, J.P.; Weber, G.J. Effects of short-chain fatty acids on intestinal function in an enteroid model of hypoxia. Front. Physiol. 2022, 13, 1056233. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, P.; Pascual, J.; Xiao, G.; Lu, H. Effect of Hypoxia and Hyperoxia on Cerebral Blood Flow, Blood Oxygenation, and Oxidative Metabolism. J. Cereb. Blood Flow Metab. 2012, 32, 1909–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreau, A.; El Hafny-Rahbi, B.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestergaard, M.B.; Lindberg, U.; Aachmann-Andersen, N.J.; Lisbjerg, K.; Christensen, S.; Law, I.; Rasmussen, P.; Olsen, N.V.; Larsson, H.B.W. Acute hypoxia increases the cerebral metabolic rate—A magnetic resonance imaging study. J. Cereb. Blood Flow Metab. 2015, 36, 1046–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Kim, T.; Brumwell, A.N.; Driver, I.H.; Wei, Y.; Tan, V.; Jackson, J.R.; Xu, J.; Lee, D.-K.; Gotts, J.E.; et al. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat. Cell Biol. 2017, 19, 904–914. [Google Scholar] [CrossRef]
- Straseski, J.A.; Gibson, A.L.; Thomas-Virnig, C.L.; Allen-Hoffmann, B.L. Oxygen deprivation inhibits basal keratinocyte proliferation in a model of human skin and induces regio-specific changes in the distribution of epidermal adherens junction proteins, aquaporin-3, and glycogen. Wound Repair Regen. 2009, 17, 606–616. [Google Scholar] [CrossRef] [Green Version]
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 95–111. [Google Scholar] [CrossRef] [Green Version]
- Herwaldt, B.L. Leishmaniasis. Lancet 1999, 354, 1191–1199. [Google Scholar] [CrossRef] [Green Version]
- Schlein, Y. Leishmania and Sandflies: Interactions in the life cycle and transmission. Parasitol. Today 1993, 9, 255–258. [Google Scholar] [CrossRef]
- Kima, P.E. The amastigote forms of Leishmania are experts at exploiting host cell processes to establish infection and persist. Int. J. Parasitol. 2007, 37, 1087–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hellemond, J.J.; Van Der Meer, P.; Tielens, A.G.M. Leishmania infantum promastigotes have a poor capacity for anaerobic functioning and depend mainly on respiration for their energy generation. Parasitology 1997, 114, 351–360. [Google Scholar] [CrossRef]
- James, P.E.; Grinberg, O.Y.; Swartz, H.M. Superoxide production by phagocytosing macrophages in relation to the intracellular distribution of oxygen. J. Leukoc. Biol. 1998, 64, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Tovar, J.; Wilkinson, S.; Mottram, J.C.; Fairlamb, A.H. Evidence that trypanothione reductase is an essential enzyme in Leishmania by targeted replacement of the tryA gene locus. Mol. Microbiol. 1998, 29, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Nandal, A.; Ruiz, J.C.; Subramanian, P.; Ghimire-Rijal, S.; Sinnamon, R.A.; Stemmler, T.L.; Bruick, R.K.; Philpott, C.C. Activation of the HIF Prolyl Hydroxylase by the Iron Chaperones PCBP1 and PCBP2. Cell Metab. 2011, 14, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Mukhopadhyay, C.; Biswas, S.; Singh, V.K.; Mukhopadhyay, C.K. Intracellular Pathogen Leishmania donovani Activates Hypoxia Inducible Factor-1 by Dual Mechanism for Survival Advantage within Macrophage. PLoS ONE 2012, 7, e38489. [Google Scholar] [CrossRef]
- Pelegrini, M.D.; Pereira, J.B.; Costa, S.D.S.; Terreros, M.J.S.; Degrossoli, A.; Giorgio, S. Evaluation of hypoxia inducible factor targeting pharmacological drugs as antileishmanial agents. Asian Pac. J. Trop. Med. 2016, 9, 652–657. [Google Scholar] [CrossRef] [Green Version]
- Weinkopff, T.; Roys, H.; Bowlin, A.; Scott, P. Leishmania Infection Induces Macrophage Vascular Endothelial Growth Factor A Production in an ARNT/HIF-Dependent Manner. Infect. Immun. 2019, 87, e00088-19. [Google Scholar] [CrossRef] [Green Version]
- Bowlin, A.; Roys, H.; Wanjala, H.; Bettadapura, M.; Venugopal, G.; Surma, J.; Simon, M.C.; Weinkopff, T. Hypoxia-Inducible Factor Signaling in Macrophages Promotes Lymphangiogenesis in Leishmania major Infection. Infect. Immun. 2021, 89, e00124-21. [Google Scholar] [CrossRef]
- Alonso, D.; Serrano, E.; Bermejo, F.J.; Corral, R.S. HIF-1α-regulated MIF activation and Nox2-dependent ROS generation promote Leishmania amazonensis killing by macrophages under hypoxia. Cell Immunol. 2019, 335, 15–21. [Google Scholar] [CrossRef]
- Colhone, M.C.; Arrais-Silva, W.W.; Picoli, C.; Giorgio, S. Effect of hypoxia on macrophage infection by Leishmania amazonensis. J. Parasitol. 2004, 90, 510–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosseto, M.C.; Palma, P.V.B.; Covas, D.T.; Giorgio, S. Hypoxia modulates phenotype, inflammatory response, and leishmanial infection of human dendritic cells. Apmis 2010, 118, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Schatz, V.; Strüssmann, Y.; Mahnke, A.; Schley, G.; Waldner, M.; Ritter, U.; Wild, J.; Willam, C.; Dehne, N.; Brüne, B.; et al. Myeloid Cell-Derived HIF-1α Promotes Control of Leishmania major. J. Immunol. 2016, 197, 4034–4041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mota, C.A.; Lera-Nonose, D.S.S.L.; Brustolin, A.A.; Duarte, G.C.; dos Santos, M.C.M.; Lonardoni, M.V.C.; Silveira, T.G.V. Low expression of hypoxia-inducible factor-1α and differential expression of immune mediators during experimental infection with Leishmania (Viannia) spp. Cytokine 2022, 153, 155833. [Google Scholar] [CrossRef]
- Ben-Cheikh, A.; Bali, A.; Guerfali, F.Z.; Atri, C.; Attia, H.; Laouini, D. Hypoxia-Inducible Factor-1 Alpha Stabilization in Human Macrophages during Leishmania major Infection Is Impaired by Parasite Virulence. Korean J. Parasitol. 2022, 60, 317–325. [Google Scholar] [CrossRef]
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Robert-Gangneux, F.; Dardé, M.-L. Epidemiology of and Diagnostic Strategies for Toxoplasmosis. Clin. Microbiol. Rev. 2012, 25, 264–296. [Google Scholar] [CrossRef] [Green Version]
- Behnke, M.S.; Zhang, T.P.; Dubey, J.P.; Sibley, L.D. Toxoplasma gondii merozoite gene expression analysis with comparison to the life cycle discloses a unique expression state during enteric development. BMC Genom. 2014, 15, 350. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Abboud, M.I.; Chowdhury, R.; Tumber, A.; Hardy, A.P.; Lippl, K.; Lohans, C.T.; Pires, E.; Wickens, J.; McDonough, M.A.; et al. Biochemical and biophysical analyses of hypoxia sensing prolyl hydroxylases from Dictyostelium discoideum and Toxoplasma gondii. J. Biol. Chem. 2020, 295, 16545–16561. [Google Scholar] [CrossRef]
- Xu, Y.; Brown, K.M.; Wang, Z.A.; van der Wel, H.; Teygong, C.; Zhang, D.; Blader, I.J.; West, C.M. The Skp1 Protein from Toxoplasma Is Modified by a Cytoplasmic Prolyl 4-Hydroxylase Associated with Oxygen Sensing in the Social Amoeba Dictyostelium. J. Biol. Chem. 2012, 287, 25098–25110. [Google Scholar] [CrossRef] [Green Version]
- Seidi, A.; Muellner-Wong, L.S.; Rajendran, E.; Tjhin, E.T.; Dagley, L.F.; Aw, V.Y.; Faou, P.; Webb, A.I.; Tonkin, C.J.; Van Dooren, G.G. Elucidating the mitochondrial proteome of Toxoplasma gondii reveals the presence of a divergent cytochrome c oxidase. eLife 2018, 7, e38131. [Google Scholar] [CrossRef]
- Pino, P.; Foth, B.J.; Kwok, L.-Y.; Sheiner, L.; Schepers, R.; Soldati, T.; Soldati-Favre, D. Dual Targeting of Antioxidant and Metabolic Enzymes to the Mitochondrion and the Apicoplast of Toxoplasma gondii. PLoS Pathog. 2007, 3, e115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, L.Y.; Schlüter, D.; Clayton, C.; Soldati, D. The antioxidant systems in Toxoplasma gondii and the role of cytosolic catalase in defence against oxidative injury. Mol. Microbiol. 2004, 51, 47–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.M.; Suvorova, E.; Farrell, A.; McLain, A.; Dittmar, A.; Wiley, G.B.; Marth, G.; Gaffney, P.; Gubbels, M.-J.; White, M.; et al. Forward Genetic Screening Identifies a Small Molecule That Blocks Toxoplasma gondii Growth by Inhibiting Both Host- and Parasite-Encoded Kinases. PLoS Pathog. 2014, 10, e1004180. [Google Scholar] [CrossRef] [PubMed]
- Wiley, M.; Sweeney, K.R.; Chan, D.A.; Brown, K.M.; McMurtrey, C.; Howard, E.W.; Giaccia, A.J.; Blader, I.J. Toxoplasma gondii Activates Hypoxia-inducible Factor (HIF) by Stabilizing the HIF-1α Subunit via Type I Activin-like Receptor Kinase Receptor Signaling. J. Biol. Chem. 2010, 285, 26852–26860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lis, A.; Wiley, M.; Vaughan, J.; Gray, P.C.; Blader, I.J. The Activin Receptor, Activin-Like Kinase 4, Mediates Toxoplasma Gondii Activation of Hypoxia Inducible Factor-1. Front. Cell Infect. Microbiol. 2019, 9, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, M.T.; Teygong, C.; Wade, K.; Florimond, C.; Blader, J. Important Hypoxia-Inducible Transcription Factor 1 (HIF-1) Target Gene in Toxoplasma Gondii-Infected Cells. MBio 2015, 6, e00462-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gail, M.; Gross, U.; Bohne, W. Transcriptional profile of Toxoplasma gondii-infected human fibroblasts as revealed by gene-array hybridization. Mol. Genet. Genom. 2001, 265, 905–912. [Google Scholar] [CrossRef]
- Jagannathan, P.; Kakuru, A. Malaria in 2022: Increasing challenges, cautious optimism. Nat. Commun. 2022, 13, 12–14. [Google Scholar] [CrossRef]
- Sato, S. Plasmodium—A Brief Introduction to the Parasites Causing Human Malaria and Their Basic Biology. J. Physiol. Anthropol. 2021, 40, 1. [Google Scholar] [CrossRef]
- Egan, T.J. Recent advances in understanding the mechanism of hemozoin (malaria pigment) formation. J. Inorg. Biochem. 2008, 102, 1288–1299. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.G.; Matuschewski, K.; Zhang, M.; Rug, M. Plasmodium falciparum. Trends Parasitol. 2019, 35, 481–482. [Google Scholar] [CrossRef] [PubMed]
- Scheibel, L.W.; Ashton, S.H.; Trager, W. Plasmodium falciparum: Microaerophilic requirements in human red blood cells. Exp. Parasitol. 1979, 47, 410–418. [Google Scholar] [CrossRef]
- Krungkrai, J. The multiple roles of the mitochondrion of the malarial parasite. Parasitology 2004, 129, 511–524. [Google Scholar] [CrossRef]
- Murphy, A.D.; Doeller, J.E.; Hearn, B.; Lang-Unnasch, N. Plasmodium falciparum: Cyanide-Resistant Oxygen Consumption. Exp. Parasitol. 1997, 87, 112–120. [Google Scholar] [CrossRef]
- Vaidya, A.B.; Mather, M.W. Mitochondrial Evolution and Functions in Malaria Parasites. Annu. Rev. Microbiol. 2009, 63, 249–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrentino-Madamet, M.; Fall, B.; Benoit, N.; Camara, C.; Amalvict, R.; Fall, M.; Dionne, P.; Fall, K.B.; Nakoulima, A.; Diatta, B.; et al. Limited polymorphisms in k13 gene in Plasmodium falciparum isolates from Dakar, Senegal in 2012–2013. Malar. J. 2014, 13, 472. [Google Scholar] [CrossRef] [Green Version]
- Tsai, A.G.; Johnson, P.G.; Intaglietta, M. Oxygen Gradients in the Microcirculation. Physiol. Rev. 2003, 83, 933–963. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.; March, S.; Galstian, A.; Hanson, K.; Carvalho, T.; Mota, M.M.; Bhatia, S.N. Hypoxia promotes liver stage malaria infection in primary human hepatocytes in vitro. Dis. Model. Mech. 2013, 7, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Hempel, C.; Combes, V.; Hunt, N.H.; Kurtzhals, J.A.L.; Grau, G.E.R. CNS Hypoxia Is More Pronounced in Murine Cerebral than Noncerebral Malaria and Is Reversed by Erythropoietin. Am. J. Pathol. 2011, 179, 1939–1950. [Google Scholar] [CrossRef]
- Boeuf, P.; Tan, A.; Romagosa, C.; Radford, J.; Mwapasa, V.; Molyneux, M.E.; Meshnick, S.R.; Hunt, N.H.; Rogerson, S.J. Placental Hypoxia during Placental Malaria. J. Infect. Dis. 2008, 197, 757–765. [Google Scholar] [CrossRef]
- Medana, I.M.; Day, N.P.J.; Roberts, R.; Sachanonta, N.; Turley, H.; Pongponratn, E.; Hien, T.T.; White, N.J.; Turner, G.D.H. Induction of the vascular endothelial growth factor pathway in the brain of adults with fatal falciparum malaria is a non-specific response to severe disease. Histopathology 2010, 57, 282–294. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, M.C.; Andrews, N.W. Host cell invasion by Trypanosoma cruzi: A unique strategy that promotes persistence. FEMS Microbiol. Rev. 2012, 36, 734–747. [Google Scholar] [CrossRef] [Green Version]
- Macedo, A.; Pena, S. Genetic Variability of Trypanosoma cruzi: Implications for the Pathogenesis of Chagas Disease. Parasitol. Today 1998, 14, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Melo, R.D.F.P.; Guarneri, A.A.; Silber, A.M. The Influence of Environmental Cues on the Development of Trypanosoma cruzi in Triatominae Vector. Front. Cell Infect. Microbiol. 2020, 10, 27. [Google Scholar] [CrossRef] [Green Version]
- De-Simone, S.G.; Bourguignon, S.C.; Gonçalves, P.S.; Lechuga, G.C.; Provance, D.W. Metabolic Alteration of Trypanosoma cruzi during Differentiation of Epimastigote to Trypomastigote Forms. Pathogens 2022, 11, 268. [Google Scholar] [CrossRef]
- Saraiva, F.M.S.; Cosentino-Gomes, D.; Inacio, J.D.F.; Almeida-Amaral, E.E.; Louzada-Neto, O.; Rossini, A.; Nogueira, N.P.; Meyer-Fernandes, J.R.; Paes, M.C. Hypoxia Effects on Trypanosoma cruzi Epimastigotes Proliferation, Differentiation, and Energy Metabolism. Pathogens 2022, 11, 897. [Google Scholar] [CrossRef] [PubMed]
- Berná, L.; Chiribao, M.L.; Greif, G.; Rodriguez, M.; Alvarez-Valin, F.; Robello, C. Transcriptomic analysis reveals metabolic switches and surface remodeling as key processes for stage transition in Trypanosoma cruzi. PeerJ 2017, 5, e3017. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, R.L.S.; Barreto, R.F.S.M.; Polycarpo, C.R.; Gadelha, F.R.; Castro, S.L.; Oliveira, M.F. A comparative assessment of mitochondrial function in epimastigotes and bloodstream trypomastigotes of Trypanosoma cruzi. J. Bioenerg. Biomembr. 2011, 43, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Piacenza, L.; Peluffo, G.; Alvarez, M.N.; Martínez, A.; Radi, R. Trypanosoma cruzi Antioxidant Enzymes as Virulence Factors in Chagas Disease. Antioxid. Redox Signal. 2013, 19, 723–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, N.P.; Saraiva, F.M.S.; Sultano, P.E.; Cunha, P.R.B.B.; Laranja, G.A.T.; Justo, G.A.; Sabino, K.C.C.; Coelho, M.G.P.; Rossini, A.; Atella, G.C.; et al. Proliferation and Differentiation of Trypanosoma cruzi inside Its Vector Have a New Trigger: Redox Status. PLoS ONE 2015, 10, e0116712. [Google Scholar] [CrossRef] [Green Version]
- Eberhardt, N.; Sanmarco, L.M.; Bergero, G.; Theumer, M.G.; García, M.C.; Ponce, N.E.; Cano, R.C.; Aoki, M.P. Deficiency of CD73 activity promotes protective cardiac immunity against Trypanosoma cruzi infection but permissive environment in visceral adipose tissue. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1866, 165592. [Google Scholar] [CrossRef]
- Wallberg-Henriksson, J.R.Z.H.; Wallberg-Henriksson, J.H. GLUT4: A key player regulating glucose homeostasis? Insights from transgenic and knockout mice. Mol. Membr. Biol. 2001, 18, 205–211. [Google Scholar] [CrossRef]
- Adesse, D.; Iacobas, S.; Meirelles, M.d.N.; Tanowitz, H.B.; Spray, D.C.; Iacobas, D.A.; Garzoni, L.R. Transcriptomic Signatures of Alterations in a Myoblast Cell Line Infected with Four Distinct Strains of Trypanosoma cruzi. Am. J. Trop. Med. Hyg. 2010, 82, 846–854. [Google Scholar] [CrossRef] [PubMed]
- McGettrick, A.F.; Corcoran, S.E.; Barry, P.J.G.; McFarland, J.; Crès, C.; Curtis, A.M.; Franklin, E.; Corr, S.C.; Mok, K.H.; Cummins, E.P.; et al. Trypanosoma brucei metabolite indolepyruvate decreases HIF-1α and glycolysis in macrophages as a mechanism of innate immune evasion. Proc. Natl. Acad. Sci. USA 2016, 113, E7778–E7787. [Google Scholar] [CrossRef] [Green Version]
- Olivera, G.C.; Vetter, L.; Tesoriero, C.; Del Gallo, F.; Hedberg, G.; Basile, J.; Rottenberg, M.E. Role of T cells during the cerebral infection with Trypanosoma brucei. PLoS Negl. Trop. Dis. 2021, 15, e0009764. [Google Scholar] [CrossRef] [PubMed]
- Engwerda, C.R.; Ng, S.; Bunn, P.T. The Regulation of CD4+ T Cell Responses during Protozoan Infections. Front. Immunol. 2014, 5, 498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serradell, M.C.; Gargantini, P.R.; Saura, A.; Oms, S.R.; Rupil, L.L.; Berod, L.; Sparwasser, T.; Luján, H.D. Cytokines, Antibodies, and Histopathological Profiles during Giardia Infection and Variant-Specific Surface Protein-Based Vaccination. Infect. Immun. 2018, 86, e00773-17. [Google Scholar] [CrossRef] [Green Version]
- Malkov, M.I.; Lee, C.T.; Taylor, C.T. Regulation of the Hypoxia-Inducible Factor (HIF) by Pro-Inflammatory Cytokines. Cells 2021, 10, 2340. [Google Scholar] [CrossRef]
- Yang, S.; Yu, M.; Sun, L.; Xiao, W.; Yang, X.; Sun, L.; Zhang, C.; Ma, Y.; Yang, H.; Liu, Y.; et al. Interferon-γ-Induced Intestinal Epithelial Barrier Dysfunction by NF-κB/HIF-1α Pathway. J. Interf. Cytokine Res. 2014, 34, 195–203. [Google Scholar] [CrossRef]
- Hellwig-Burgel, T.; Rutkowski, K.; Metzen, E.; Fandrey, J.; Jelkmann, W. Interleukin-1β and Tumor Necrosis Factor-Stimulate DNA Binding of Hypoxia-Inducible Factor-1. Blood 1999, 94, 1561–1567. [Google Scholar] [CrossRef]
- Jung, Y.J.; Isaacs, J.S.; Lee, S.; Trepel, J.; Neckers, L. IL-1beta-Mediated up-Regulation of HIF-1alpha via an NFkap-paB/COX-2 Pathway Identifies HIF-1 as a Critical Link between Inflammation and Oncogenesis. FASEB J. 2003, 17, 2115–2117. [Google Scholar] [CrossRef] [Green Version]
- Sartori-Cintra, A.R.; de Mara, C.S.; Argolo, D.L.; Coimbra, I.B. Regulation of hypoxia-inducible factor-1α (HIF-1α) expression by interleukin-1β (IL-1β), insulin-like growth factors I (IGF-I) and II (IGF-II) in human osteoarthritic chondrocytes. Clinics 2012, 67, 35–40. [Google Scholar] [CrossRef]
- Qian, D.; Lin, H.-Y.; Wang, H.-M.; Zhang, X.; Liu, D.-L.; Li, Q.-L.; Zhu, C. Normoxic Induction of the Hypoxic-Inducible Factor-1α by Interleukin-1β Involves the Extracellular Signal-Regulated Kinase 1/2 Pathway in Normal Human Cytotrophoblast Cells1. Biol. Reprod. 2004, 70, 1822–1827. [Google Scholar] [CrossRef] [Green Version]
- Berra, E.; Pagès, G.; Pouysségur, J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev. 2000, 19, 139–145. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate Is a Danger Signal That Induces IL-1β via HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Shah, Y.M.; Ito, S.; Morimura, K.; Chen, C.; Yim, S.; Haase, V.H.; Gonzalez, F.J. Hypoxia-Inducible Factor Augments Experimental Colitis through an MIF–Dependent Inflammatory Signaling Cascade. Gastroenterology 2008, 134, 2036–2048.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann, D.C.; Wittig, I.; Brüne, B. TMEM126B deficiency reduces mitochondrial SDH oxidation by LPS, attenuating HIF-1α stabilization and IL-1β expression. Redox Biol. 2018, 20, 204–216. [Google Scholar] [CrossRef]
- Xu, J.; Zheng, Y.; Zhao, Y.; Zhang, Y.; Li, H.; Zhang, A.; Wang, X.; Wang, W.; Hou, Y.; Wang, J. Succinate/IL-1β Signaling Axis Promotes the Inflammatory Progression of Endothelial and Exacerbates Atherosclerosis. Front. Immunol. 2022, 13, 817572. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Geng, X.; Warren, J.; Cosky, E.E.P.; Kaura, S.; Stone, C.; Li, F.; Ding, Y. Hypoxia Inducible Factor-1α (HIF-1α) Mediates NLRP3 Inflammasome-Dependent-Pyroptotic and Apoptotic Cell Death Following Ischemic Stroke. Neuroscience 2020, 448, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Cosin-Roger, J.; Simmen, S.; Melhem, H.; Atrott, K.; Frey-Wagner, I.; Hausmann, M.; de Vallière, C.; Spalinger, M.R.; Spielmann, P.; Wenger, R.H.; et al. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat. Commun. 2017, 8, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, D.; Guan, S.; Wang, Z.; Ni, H.; Ding, D.; Xu, W.; Li, G. HIF-1α aggravated traumatic brain injury by NLRP3 inflammasome-mediated pyroptosis and activation of microglia. J. Chem. Neuroanat. 2021, 116, 101994. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, L.; Moreau, F.; Cornick, S.; Chadee, K. The NLRP3 Inflammasome Is a Pathogen Sensor for Invasive Entamoeba histolytica via Activation of α5β1 Integrin at the Macrophage-Amebae Intercellular Junction. PLoS Pathog. 2015, 11, e1004887. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Feng, M.; Zhao, Y.; Zhang, Y.; Zhou, R.; Zhou, H.; Pang, Z.; Tachibana, H.; Cheng, X. A Novel TLR4-Binding Domain of Peroxiredoxin from Entamoeba histolytica Triggers NLRP3 Inflammasome Activation in Macrophages. Front. Immunol. 2021, 12, 758451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yan, L.; Wang, L.; Seydel, K.B.; Li, E.; Ankri, S.; Mirelman, D.; Stanley, S.L. Entamoeba histolytica cysteine proteinases with interleukin-1 beta converting enzyme (ICE) activity cause intestinal inflammation and tissue damage in amoebiasis. Mol. Microbiol. 2000, 37, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Que, X.; Kim, S.-H.; Sajid, M.; Eckmann, L.; Dinarello, C.A.; McKerrow, J.H.; Reed, S.L. A Surface Amebic Cysteine Proteinase Inactivates Interleukin-18. Infect. Immun. 2003, 71, 1274–1280. [Google Scholar] [CrossRef] [Green Version]
- Manko-Prykhoda, A.; Allain, T.; Motta, J.-P.; Cotton, J.A.; Feener, T.; Oyeyemi, A.; Bindra, S.; Vallance, B.A.; Wallace, J.L.; Beck, P.; et al. Giardia spp. promote the production of antimicrobial peptides and attenuate disease severity induced by attaching and effacing enteropathogens via the induction of the NLRP3 inflammasome. Int. J. Parasitol. 2020, 50, 263–275. [Google Scholar] [CrossRef]
- Cotton, J.A.; Motta, J.-P.; Schenck, L.P.; Hirota, S.A.; Beck, P.L.; Buret, A.G. Giardia duodenalis Infection Reduces Granulocyte Infiltration in an in vivo Model of Bacterial Toxin-Induced Colitis and Attenuates Inflammation in Human Intestinal Tissue. PLoS ONE 2014, 9, e109087. [Google Scholar] [CrossRef]
- Cotton, J.A.; Bhargava, A.; Ferraz, J.G.; Yates, R.M.; Beck, P.L.; Buret, A.G. Giardia duodenalis Cathepsin B Proteases Degrade Intestinal Epithelial Interleukin-8 and Attenuate Interleukin-8-Induced Neutrophil Chemotaxis. Infect. Immun. 2014, 82, 2772–2787. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Ma’Ayeh, S.; Peirasmaki, D.; Lundström-Stadelmann, B.; Hellman, L.; Svärd, S.G. Secreted Giardia intestinalis cysteine proteases disrupt intestinal epithelial cell junctional complexes and degrade chemokines. Virulence 2018, 9, 879–894. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Yang, Y.; Fang, R.; Zhu, W.; Wu, J.; Li, X.; Patankar, J.V.; Li, W. Giardia duodenalis and Its Secreted PPIB Trigger Inflammasome Activation and Pyroptosis in Macrophages through TLR4-Induced ROS Signaling and A20-Mediated NLRP3 Deubiquitination. Cells 2021, 10, 3425. [Google Scholar] [CrossRef]
- Riestra, A.M.; Valderrama, J.A.; Patras, K.A.; Booth, S.D.; Quek, X.Y.; Tsai, C.-M.; Nizet, V. Trichomonas vaginalis Induces NLRP3 Inflammasome Activation and Pyroptotic Cell Death in Human Macrophages. J. Innate Immun. 2018, 11, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Cao, L.; Wang, X.; Dong, J.; Zhang, N.; Li, X.; Li, J.; Zhang, X.; Gong, P. Extracellular vesicles secreted by Giardia duodenalis regulate host cell innate immunity via TLR2 and NLRP3 inflammasome signaling pathways. PLoS Negl. Trop. Dis. 2021, 15, e0009304. [Google Scholar] [CrossRef]
- Zhao, P.; Cao, L.; Wang, X.; Li, J.; Dong, J.; Zhang, N.; Li, X.; Li, S.; Sun, M.; Zhang, X.; et al. Giardia duodenalis extracellular vesicles regulate the proinflammatory immune response in mouse macrophages in vitro via the MAPK, AKT and NF-κB pathways. Parasites Vectors 2021, 14, 358. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, Y.; Liu, M.; Qin, X.; Yu, X.; Zhao, H.; Li, X.; Li, W. COX-2 is required to mediate crosstalk of ROS-dependent activation of MAPK/NF-κB signaling with pro-inflammatory response and defense-related NO enhancement during challenge of macrophage-like cell line with Giardia duodenalis. PLoS Negl. Trop. Dis. 2022, 16, e0010402. [Google Scholar] [CrossRef]
- Gurung, P.; Karki, R.; Vogel, P.; Watanabe, M.; Bix, M.; Lamkanfi, M.; Kanneganti, T.-D. An NLRP3 inflammasome-triggered Th2-biased adaptive immune response promotes leishmaniasis. J. Clin. Investig. 2015, 125, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Santos, D.; Campos, T.M.; Saldanha, M.; Oliveira, S.C.; Nascimento, M.; Zamboni, D.; Machado, P.R.; Arruda, S.; Scott, P.; Carvalho, E.M.; et al. IL-1β Production by Intermediate Monocytes Is Associated with Immunopathology in Cutaneous Leishmaniasis. J. Investig. Dermatol. 2018, 138, 1107–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novais, F.O.; Carvalho, A.M.; Clark, M.L.; Carvalho, L.P.; Beiting, D.P.; Brodsky, I.E.; Carvalho, E.M.; Scott, P. CD8+ T cell cytotoxicity mediates pathology in the skin by inflammasome activation and IL-1β production. PLoS Pathog. 2017, 13, e1006196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charmoy, M.; Hurrell, B.P.; Romano, A.; Lee, S.H.; Ribeiro-Gomes, F.; Riteau, N.; Mayer-Barber, K.; Tacchini-Cottier, F.; Sacks, D.L. The Nlrp3 inflammasome, IL-1β, and neutrophil recruitment are required for susceptibility to a nonhealing strain of Leishmania major in C57BL/6 mice. Eur. J. Immunol. 2016, 46, 897–911. [Google Scholar] [CrossRef] [Green Version]
- Gov, L.; Schneider, C.A.; Lima, T.S.; Pandori, W.; Lodoen, M.B. NLRP3 and Potassium Efflux Drive Rapid IL-1β Release from Primary Human Monocytes during Toxoplasma gondii Infection. J. Immunol. 2017, 199, 2855–2864. [Google Scholar] [CrossRef] [Green Version]
- Pandori, W.J.; Lima, T.S.; Mallya, S.; Kao, T.H.; Gov, L.; Lodoen, M.B. Toxoplasma gondii activates a Syk-CARD9-NF-κB signaling axis and gasdermin D-independent release of IL-1β during infection of primary human monocytes. PLoS Pathog. 2019, 15, e1007923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, H.B.; de Salles, M.; Panatieri, R.H.; Boscardin, S.B.; Rodríguez-Málaga, S.M.; Álvarez, J.M.; Lima, M.R.D. IFN-γ-Induced Priming Maintains Long-Term Strain-Transcending Immunity against Blood-Stage Plasmodium chabaudi Malaria. J. Immunol. 2013, 191, 5160–5169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Pinho, R.T.; da Silva, W.S.; Côrtes, L.M.D.C.; Sousa, P.D.S.V.; Soares, R.O.D.A.; Alves, C.R. Production of MMP-9 and inflammatory cytokines by Trypanosoma cruzi-infected macrophages. Exp. Parasitol. 2014, 147, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Lamberet, A.; Rostan, O.; Dion, S.; Jan, A.; Guegan, H.; Manuel, C.; Samson, M.; Gangneux, J.-P.; Robert-Gangneux, F. IL-33/ST2 axis is involved in disease progression in the spleen during Leishmania donovani infection. Parasites Vectors 2020, 13, 320. [Google Scholar] [CrossRef] [PubMed]
- Rostan, O.; Gangneux, J.-P.; Piquet-Pellorce, C.; Manuel, C.; McKenzie, A.N.J.; Guiguen, C.; Samson, M.; Robert-Gangneux, F. The IL-33/ST2 Axis Is Associated with Human Visceral Leishmaniasis and Suppresses Th1 Responses in the Livers of BALB/c Mice Infected with Leishmania donovani. mBio 2013, 4, e00383-13. [Google Scholar] [CrossRef] [Green Version]
- Shio, M.T.; Eisenbarth, S.C.; Savaria, M.; Vinet, A.F.; Bellemare, M.-J.; Harder, K.W.; Sutterwala, F.S.; Bohle, D.S.; Descoteaux, A.; Flavell, R.A.; et al. Malarial Hemozoin Activates the NLRP3 Inflammasome through Lyn and Syk Kinases. PLoS Pathog. 2009, 5, e1000559. [Google Scholar] [CrossRef]
- Ouma, C.; Davenport, G.C.; Awandare, G.A.; Keller, C.C.; Were, T.; Otieno, M.F.; Vulule, J.M.; Martinson, J.; Ong’echa, J.M.; Ferrell, R.E.; et al. Polymorphic Variability in the Interleukin (IL)-1β Promoter Conditions Susceptibility to Severe Malarial Anemia and Functional Changes in IL-1β Production. J. Infect. Dis. 2008, 198, 1219–1226. [Google Scholar] [CrossRef]
- Guha, R.; Mathioudaki, A.; Doumbo, S.; Doumtabe, D.; Skinner, J.; Arora, G.; Siddiqui, S.; Li, S.; Kayentao, K.; Ongoiba, A.; et al. Plasmodium falciparum malaria drives epigenetic reprogramming of human monocytes toward a regulatory phenotype. PLoS Pathog. 2021, 17, e1009430. [Google Scholar] [CrossRef]
- Albina, J.E.; Mastrofrancesco, B.; Vessella, J.A.; Louis, C.A.; Henry, W.L., Jr.; Reichner, J.S. HIF-1 expression in healing wounds: HIF-1α induction in primary inflammatory cells by TNF-α. Am. J. Physiol. Cell Physiol. 2001, 281, C1971–C1977. [Google Scholar] [CrossRef]
- Haddad, J.J.; Land, S.C. A non-hypoxic, ROS-sensitive pathway mediates TNF-α-dependent regulation of HIF-1α. FEBS Lett. 2001, 505, 269–274. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.T. Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem. J. 2007, 409, 19–26. [Google Scholar] [CrossRef] [Green Version]
- van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1α by NF-κB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, H.; Suzuki, S.; Kawasaki, N.; Nakano, H.; Okazaki, T.; Chino, A.; Doi, T.; Saiki, I. Tumor Necrosis Factor-α-induced IKK Phosphorylation of NF-κB p65 on Serine 536 Is Mediated through the TRAF2, TRAF5, and TAK1 Signaling Pathway. J. Biol. Chem. 2003, 278, 36916–36923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remels, A.H.V.; Gosker, H.; Verhees, K.K.; Langen, R.C.J.; Schols, A.M.W.J. TNF-α-Induced NF-κB Activation Stimulates Skeletal Muscle Glycolytic Metabolism through Activation of HIF-1α. Endocrinology 2015, 156, 1770–1781. [Google Scholar] [CrossRef]
- Peterson, K.M.; Mondal, D.; Petri, W.A.; Duggal, P.; Haque, R.; Shu, J. Association between TNF-α and Entamoeba histolytica Diarrhea. Am. J. Trop. Med. Hyg. 2010, 82, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Helk, E.; Bernin, H.; Ernst, T.; Ittrich, H.; Jacobs, T.; Heeren, J.; Tacke, F.; Tannich, E.; Lotter, H. TNFα-Mediated Liver Destruction by Kupffer Cells and Ly6Chi Monocytes during Entamoeba histolytica Infection. PLoS Pathog. 2013, 9, e1003096. [Google Scholar] [CrossRef] [Green Version]
- Noor, Z.; Watanabe, K.; Abhyankar, M.M.; Burgess, S.L.; Buonomo, E.L.; Cowardin, C.A.; Petri, W.A. Role of Eosinophils and Tumor Necrosis Factor Alpha in Interleukin-25-Mediated Protection from Amebic Colitis. mBio 2017, 8, e02329-16. [Google Scholar] [CrossRef] [Green Version]
- Moonah, S.N.; Jiang, N.M.; Jr, W.A.P. Host Immune Response to Intestinal Amebiasis. PLoS Pathog. 2013, 9, e1003489. [Google Scholar] [CrossRef]
- Lin, J.Y.; Seguin, R.; Keller, K.; Chadee, K. Tumor necrosis factor alpha augments nitric oxide-dependent macrophage cytotoxicity against Entamoeba histolytica by enhanced expression of the nitric oxide synthase gene. Infect. Immun. 1994, 62, 1534–1541. [Google Scholar] [CrossRef] [Green Version]
- Blazquez, S.; Zimmer, C.; Guigon, G.; Olivo-Marin, J.-C.; Guillén, N.; Labruyère, E. Human Tumor Necrosis Factor is a Chemoattractant for the Parasite Entamoeba histolytica. Infect. Immun. 2006, 74, 1222–1232. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Li, E.; Shea-Donohue, T.; Singer, S.M. Tumour necrosis factor α contributes to protection against Giardia lamblia infection in mice. Parasite Immunol. 2007, 29, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Cruz, S.; Gomez-García, A.; Matadamas-Martínez, F.; Alvarado-Torres, J.A.; Meza-Cervantez, P.; Arriaga-Pizano, L.; Yépez-Mulia, L. Giardia lamblia: Identification of molecules that contribute to direct mast cell activation. Parasitol. Res. 2018, 117, 2555–2567. [Google Scholar] [CrossRef]
- Lean, I.-S.; McDonald, S.A.C.; Bajaj-Elliott, M.; Pollok, R.C.G.; Farthing, M.J.G.; McDonald, V. Interleukin-4 and Transforming Growth Factor β Have Opposing Regulatory Effects on Gamma Interferon-Mediated Inhibition of Cryptosporidium parvum Reproduction. Infect. Immun. 2003, 71, 4580–4585. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, S.; Mancassola, R.; Naciri, M.; Laurent, F. Cryptosporidium parvum-Specific Mucosal Immune Response in C57BL/6 Neonatal and Gamma Interferon-Deficient Mice: Role of Tumor Necrosis Factor Alpha in Protection. Infect. Immun. 2001, 69, 1635–1642. [Google Scholar] [CrossRef] [Green Version]
- Robinson, P.; Okhuysen, P.C.; Chappell, C.L.; Lewis, D.E.; Shahab, I.; Janecki, A.; White, A.C. Expression of Tumor Necrosis Factor Alpha and Interleukin 1β in Jejuna of Volunteers after Experimental Challenge with Cryptosporidium parvum Correlates with Exposure but Not with Symptoms. Infect. Immun. 2001, 69, 1172–1174. [Google Scholar] [CrossRef] [Green Version]
- de Sablet, T.; Potiron, L.; Marquis, M.; Bussière, F.I.; Lacroix-Lamandé, S.; Laurent, F. Cryptosporidium parvum increases intestinal permeability through interaction with epithelial cells and IL-1β and TNFα released by inflammatory monocytes. Cell Microbiol. 2016, 18, 1871–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lean, I.-S.; Lacroix-Lamandé, S.; Laurent, F.; McDonald, V. Role of Tumor Necrosis Factor Alpha in Development of Immunity against Cryptosporidium parvum Infection. Infect. Immun. 2006, 74, 4379–4382. [Google Scholar] [CrossRef] [Green Version]
- Green, S.J.; Crawford, R.M.; Hockmeyer, J.T.; Meltzer, M.S.; Nacy, C.A. Leishmania major amastigotes initiate the L-arginine-dependent killing mechanism in IFN-gamma-stimulated macrophages by induction of tumor necrosis factor-alpha. J. Immunol. 1990, 145, 4290–4297. [Google Scholar] [CrossRef] [PubMed]
- Nashleanas, M.; Kanaly, S.; Scott, P. Control of Leishmania major Infection in Mice Lacking TNF Receptors. J. Immunol. 1999, 160, 5506–5513. [Google Scholar] [CrossRef]
- Lucas, R.; Juillard, P.; Decoster, E.; Redard, M.; Burger, D.; Donati, Y.; Giroud, C.; Monso-Hinard, C.; De Kesel, T.; Buurman, W.A.; et al. Crucial role of tumor necrosis factor (TNF) receptor 2 and membrane-bound TNF in experimental cerebral malaria. Eur. J. Immunol. 1997, 27, 1719–1725. [Google Scholar] [CrossRef]
- Jacobs, P.; Radzioch, D.; Stevenson, M.M. A Th1-associated increase in tumor necrosis factor alpha expression in the spleen correlates with resistance to blood-stage malaria in mice. Infect. Immun. 1996, 64, 535–541. [Google Scholar] [CrossRef] [Green Version]
- Seixas, E.; Oliveira, P.; Nunes, J.F.M.; Coutinho, A. An experimental model for fatal malaria due to TNF-α-dependent hepatic damage. Parasitology 2008, 135, 683–690. [Google Scholar] [CrossRef]
- Grau, G.E.; Heremans, H.; Piguet, P.F.; Pointaire, P.; Lambert, P.H.; Billiau, A.; Vassalli, P. Monoclonal antibody against interferon gamma can prevent experimental cerebral malaria and its associated overproduction of tumor necrosis factor. Proc. Natl. Acad. Sci. USA 1989, 86, 5572–5574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schluter, D.; Kwok, L.-Y.; Lutjen, S.; Soltek, S.; Hoffmann, S.; Korner, H.; Deckert, M. Both Lymphotoxin-α and TNF Are Crucial for Control of Toxoplasma gondii in the Central Nervous System. J. Immunol. 2003, 170, 6172–6182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, E.C.S.; Garcia, I.; Vicentelli, M.H.; Vassalli, P.; Minoprio, P. Evidence for a protective role of tumor necrosis factor in the acute phase of Trypanosoma cruzi infection in mice. Infect. Immun. 1997, 65, 457–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalvão, F.; Almeida, G.M.; Silva, E.M.; Borges, V.M.; Vasconcellos, R.; Takiya, C.M.; Lopes, M.F.; Nunes, M.P.; DosReis, G.A. Apoptotic lymphocytes treated with IgG from Trypanosoma cruzi infection increase TNF-α secretion and reduce parasite replication in macrophages. Eur. J. Immunol. 2010, 40, 417–425. [Google Scholar] [CrossRef]
- Arnaiz, E.; Harris, A.L. Role of Hypoxia in the Interferon Response. Front. Immunol. 2022, 13, 821816. [Google Scholar] [CrossRef]
- Der, S.D.; Zhou, A.; Williams, B.R.G.; Silverman, R.H. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.-H.; Hsiao, H.-F.; Chen, T.-W.; Li, T.-K. Inflammatory interferon activates HIF-1α-mediated epithelial-to-mesenchymal transition via PI3K/AKT/mTOR pathway. J. Exp. Clin. Cancer Res. 2018, 37, 70. [Google Scholar] [CrossRef] [Green Version]
- Parra-Izquierdo, I.; Castaños-Mollor, I.; López, J.; Gómez, C.; Román, J.A.S.; Crespo, M.S.; García-Rodríguez, C. Lipopolysaccharide and interferon-γ team up to activate HIF-1α via STAT1 in normoxia and exhibit sex differences in human aortic valve interstitial cells. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 2168–2179. [Google Scholar] [CrossRef]
- Liu, H.; Li, M.; Wang, P.; Wang, F. Blockade of hypoxia-inducible factor-1α by YC-1 attenuates interferon-γ and tumor necrosis factor-α-induced intestinal epithelial barrier dysfunction. Cytokine 2011, 56, 581–588. [Google Scholar] [CrossRef]
- Wu, Y.; Meitzler, J.L.; Antony, S.; Juhasz, A.; Lu, J.; Jiang, G.; Liu, H.; Hollingshead, M.; Haines, D.C.; Butcher, D.; et al. Dual oxidase 2 and pancreatic adenocarcinoma: IFN-γ-mediated dual oxidase 2 overexpression results in H2O2-induced, ERK-associated up-regulation of HIF-1α and VEGF-A. Oncotarget 2016, 7, 68412–68433. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Elly, C.; Park, Y.; Liu, Y.-C. E3 Ubiquitin Ligase VHL Regulates Hypoxia-Inducible Factor-1α to Maintain Regulatory T Cell Stability and Suppressive Capacity. Immunity 2015, 42, 1062–1074. [Google Scholar] [CrossRef] [Green Version]
- Glover, L.E.; Colgan, S.P. Hypoxia and Metabolic Factors That Influence Inflammatory Bowel Disease Pathogenesis. Gastroenterology 2011, 140, 1748–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerber, S.A.; Yatsula, B.; Maier, C.L.; Sadler, T.J.; Whittaker, L.W.; Pober, J.S. Interferon-Gamma Induces Prolyl Hydroxylase (PHD)3 Through a STAT1-Dependent Mechanism in Human Endothelial Cells. Arter. Thromb. Vasc. Biol. 2009, 29, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Moraes, L.C.A.; França, E.L.; Pessoa, R.S.; Fagundes, D.L.G.; Gil Hernandes, M.; Ribeiro, V.P.; Gomes, M.A.; Honorio-França, A.C. The effect of IFN-γ and TGF-β in the functional activity of mononuclear cells in the presence of Entamoeba histolytica. Parasites Vectors 2015, 8, 413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellanos, C.; Ramos, C.; Ortiz-Ortiz, L. Effects of gamma interferon on syntheses of DNA and proteins by Entamoeba histolytica. Infect. Immun. 1989, 57, 2771–2775. [Google Scholar] [CrossRef] [Green Version]
- You, X.; Mead, J.R. Characterization of experimental Cryptosporidium parvum infection in IFN-γ knockout mice. Parasitology 1998, 117, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Flandin, J.-F.; Chano, F.; Descoteaux, A. RNA interference reveals a role for TLR2 and TLR3 in the recognition of Leishmania donovani promastigotes by interferon-γ-primed macrophages. Eur. J. Immunol. 2006, 36, 411–420. [Google Scholar] [CrossRef]
- Yap, B.G.S.; Sher, A. Effector Cells of Both Nonhemopoietic and Hemopoietic Origin are required for Interferon (IFN)- γ- and Tumor Necrosis Factor (TNF)- α -dependent Host Resistance to the Intracellular Pathogen, Toxoplasma gondii. J. Exp. Med. 1999, 189, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Orellana, M.A.; Schreiber, R.D.; Remington, J.S. Interferon-γ: The Major Mediator of Resistance against Toxoplasma gondii. Science 1988, 240, 516–518. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Gekker, G.; Hu, S.; Peterson, P.K. Human microglial cell defense against Toxoplasma gondii. The Role of Cytokines. J. Immunol. 1994, 152, 1246–1252. [Google Scholar] [CrossRef] [PubMed]
- Halonen, S.K.; Chiu, F.-C.; Weiss, L.M. Effect of Cytokines on Growth of Toxoplasma gondii in Murine Astrocytes. Infect. Immun. 1998, 66, 4989–4993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holscher, C.; Kohler, G.; Muller, U.; Mossmann, H.; Schaub, G.A.; Brombacher, F. Defective Nitric Oxide Effector Functions Lead to Extreme Susceptibility of Trypanosoma cruzi-Infected Mice Deficient in Gamma Interferon Receptor or Inducible Nitric Oxide Synthase. Infect. Immun. 1998, 66, 1208–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belnoue, E.; Potter, S.M.; Rosa, D.S.; Mauduit, M.; Grüner, A.C.; Kayibanda, M.; Mitchell, A.J.; Hunt, N.H.; Rénia, L. Control of pathogenic CD8+ T cell migration to the brain by IFN-γ during experimental cerebral malaria. Parasite Immunol. 2008, 30, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; He, C.; Wu, W.; Zhou, G.; Liu, F.; Cong, Y.; Liu, Z. Hypoxia inducible factor-1α-induced interleukin-33 expression in intestinal epithelia contributes to mucosal homeostasis in inflammatory bowel disease. Clin. Exp. Immunol. 2017, 187, 428–440. [Google Scholar] [CrossRef] [Green Version]
- Neumann, K.; Schiller, B.; Tiegs, G. NLRP3 Inflammasome and IL-33: Novel Players in Sterile Liver Inflammation. Int. J. Mol. Sci. 2018, 19, 2732. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an Interleukin-1-like Cytokine that Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Moussion, C.; Ortega, N.; Girard, J.-P. The IL-1-Like Cytokine IL-33 Is Constitutively Expressed in the Nucleus of Endothelial Cells and Epithelial Cells In Vivo: A Novel ‘Alarmin’? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef] [Green Version]
- Carriere, V.; Roussel, L.; Ortega, N.; Lacorre, D.-A.; Americh, L.; Aguilar, L.; Bouche, G.; Girard, J.-P. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 282–287. [Google Scholar] [CrossRef] [Green Version]
- Lunderius-Andersson, C.; Enoksson, M.; Nilsson, G. Mast Cells Respond to Cell Injury through the Recognition of IL-33. Front. Immunol. 2012, 3, 82. [Google Scholar] [CrossRef] [Green Version]
- Ryan, N.; Anderson, K.; Volpedo, G.; Varikuti, S.; Satoskar, M.; Satoskar, S.; Oghumu, S. The IL-33/ST2 Axis in Immune Responses against Parasitic Disease: Potential Therapeutic Applications. Front. Cell Infect. Microbiol. 2020, 10, 153. [Google Scholar] [CrossRef]
- Lechner, C.J.; Komander, K.; Hegewald, J.; Huang, X.; Gantin, R.G.; Soboslay, P.T.; Agossou, A.; Banla, M.; Köhler, C. Cytokine and chemokine responses to helminth and protozoan parasites and to fungus and mite allergens in neonates, children, adults, and the elderly. Immun. Ageing 2013, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotani, T.; Setiawan, J.; Konno, T.; Ihara, N.; Okamoto, S.; Saito, Y.; Murata, Y.; Noda, T.; Matozaki, T. Regulation of colonic epithelial cell homeostasis by mTORC1. Sci. Rep. 2020, 10, 13810. [Google Scholar] [CrossRef] [PubMed]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene 2014, 34, 2239–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef]
- Land, S.C.; Tee, A.R. Hypoxia-inducible Factor 1α Is Regulated by the Mammalian Target of Rapamycin (mTOR) via an mTOR Signaling Motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [Green Version]
- Xu, E.A.H.; Li, D.; Ma, J.; Zhao, Y.; Xu, L.; Tian, R.; Liu, Y.; Sun, L.; Su, J. The IL-33/ST2 axis affects tumor growth by regulating mitophagy in macrophages and reprogramming their polarization. Cancer Biol. Med. 2021, 18, 172–183. [Google Scholar] [CrossRef]
- Salmond, R.J.; Mirchandani, A.S.; Besnard, A.-G.; Bain, C.C.; Thomson, N.C.; Liew, F.Y. IL-33 induces innate lymphoid cell-mediated airway inflammation by activating mammalian target of rapamycin. J. Allergy Clin. Immunol. 2012, 130, 1159–1166.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nian, J.-B.; Zeng, M.; Zheng, J.; Zeng, L.-Y.; Fu, Z.; Huang, Q.-J.; Wei, X. Epithelial cells expressed IL-33 to promote degranulation of mast cells through inhibition on ST2/PI3K/mTOR-mediated autophagy in allergic rhinitis. Cell Cycle 2020, 19, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Liu, J.; Ma, R.; Hao, J.; Liang, Y.; Zhao, J.; Zhang, A.; Meng, H.; Lu, J. Interleukin-33: Metabolic checkpoints, metabolic processes, and epigenetic regulation in immune cells. Front. Immunol. 2022, 13, 900826. [Google Scholar] [CrossRef]
- Liu, J.; Wang, W.; Wang, L.; Chen, S.; Tian, B.; Huang, K.; Corrigan, C.J.; Ying, S.; Wang, W.; Wang, C. IL-33 Initiates Vascular Remodelling in Hypoxic Pulmonary Hypertension by up-Regulating HIF-1α and VEGF Expression in Vascular Endothelial Cells. Ebiomedicine 2018, 33, 196–210. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Li, D.; Zhang, X.; Wan, Q.; Zhang, W.; Zheng, M.; Zou, L.; Elly, C.; Lee, J.H.; Liu, Y.-C. E3 Ligase VHL Promotes Group 2 Innate Lymphoid Cell Maturation and Function via Glycolysis Inhibition and Induction of Interleukin-33 Receptor. Immunity 2018, 48, 258–270.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, S.; Dias, T.H. Hypoxia-Inducible Factor (HIF) as a Target for Novel Therapies in Rheumatoid Arthritis. Front. Pharmacol. 2016, 7, 184. [Google Scholar] [CrossRef] [Green Version]
- Uddin, J.; Leslie, J.L.; Burgess, S.L.; Oakland, N.; Thompson, B.; Abhyankar, M.; Revilla, J.; Frisbee, A.; Donlan, A.N.; Kumar, P.; et al. The IL-33-ILC2 pathway protects from amebic colitis. Mucosal Immunol. 2022, 15, 165–175. [Google Scholar] [CrossRef]
- Nakamura, R.; Yoshizawa, A.; Moriyasu, T.; Deloer, S.; Senba, M.; Kikuchi, M.; Koyasu, S.; Moro, K.; Hamano, S. Group 2 Innate Lymphoid Cells Exacerbate Amebic Liver Abscess in Mice. iScience 2020, 23, 101544. [Google Scholar] [CrossRef]
- Bedi, B.; McNair, N.N.; Mead, J.R. Dendritic cells play a role in host susceptibility to Cryptosporidium parvum infection. Immunol. Lett. 2014, 158, 42–51. [Google Scholar] [CrossRef]
- Wang, Y.; Gong, A.-Y.; Ma, S.; Chen, X.; Strauss-Soukup, J.K.; Chen, X.-M. Delivery of parasite Cdg7_Flc_0990 RNA transcript into intestinal epithelial cells during Cryptosporidium parvum infection suppresses host cell gene transcription through epigenetic mechanisms. Cell Microbiol. 2017, 19, e12760. [Google Scholar] [CrossRef] [Green Version]
- Still, K.M.; Batista, S.J.; O’brien, C.A.; Oyesola, O.O.; Früh, S.P.; Webb, L.M.; Smirnov, I.; Kovacs, M.A.; Cowan, M.N.; Hayes, N.W.; et al. Astrocytes promote a protective immune response to brain Toxoplasma gondii infection via IL-33-ST2 signaling. PLoS Pathog. 2020, 16, e1009027. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.A.; Roberts, F.; Nickdel, M.B.; Brombacher, F.; McKenzie, A.N.J.; Henriquez, F.L.; Alexander, J.; Roberts, C.W. IL-33 receptor (T1/ST2) signalling is necessary to prevent the development of encephalitis in mice infected with Toxoplasma gondii. Eur. J. Immunol. 2010, 40, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Ryffel, B.; Huang, F.; Robinet, P.; Panek, C.; Couillin, I.; Erard, F.; Piotet, J.; Le Bert, M.; Mackowiak, C.; Arias, M.T.; et al. Blockade of IL-33R/ST2 Signaling Attenuates Toxoplasma gondii Ileitis Depending on IL-22 Expression. Front. Immunol. 2019, 10, 702. [Google Scholar] [CrossRef] [PubMed]
- Besnard, A.-G.; Guabiraba, R.; Niedbala, W.; Palomo, J.; Reverchon, F.; Shaw, T.N.; Couper, K.N.; Ryffel, B.; Liew, F.Y. IL-33-Mediated Protection against Experimental Cerebral Malaria Is Linked to Induction of Type 2 Innate Lymphoid Cells, M2 Macrophages and Regulatory T Cells. PLoS Pathog. 2015, 11, e1004607. [Google Scholar] [CrossRef] [Green Version]
- Palomo, J.; Reverchon, F.; Piotet, J.; Besnard, A.-G.; Couturier-Maillard, A.; Maillet, I.; Tefit, M.; Erard, F.; Mazier, D.; Ryffel, B.; et al. Critical role of IL-33 receptor ST2 in experimental cerebral malaria development. Eur. J. Immunol. 2015, 45, 1354–1365. [Google Scholar] [CrossRef]
- Glineur, C.; Leleu, I.; Pied, S. The IL-33/ST2 Pathway in Cerebral Malaria. Int. J. Mol. Sci. 2022, 23, 13457. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Parasite | Glucose Metabolism Genes | Hypoxia-Related Genes | References |
|---|---|---|---|
| Enteric Parasites | |||
| Entamoeba histolytica | Pgm2, Gpt2, Pdk3, LDHA, ENO1, ENO2, PFKFB3, GAPDH, ALDOA | Vegfa, Cgref1, Prelid2, Grk3, Celsr3, Atg9b, Icam1, Gpr160, Jdp2, Ciart, Wdr45b, Slc25a36, Il6ra, Cpne8, Tpd52, Bnip3, Gpt2, Slc8a1, Celsr3, Ackr4, Ppan, Hspbap1, Lancl3, Ccng2, Matr3, Taf9b, Piga, Kcnj2, Cntnap1, Gpnmb, HIF1a, MT1G, MT1H, MT1P, MT2A, MT3, IER3, JUN, FOS, JUNB, JUND, HSP70, DDIT3, CRYAB | [55,56] |
| Giardia duodenalis | ENO1, ENO1, ALDOA, ALDOC | JUN, FOS, IER3, ANKRD37, GADD45A, IATF, MIR210HG, SLC2A3, NOS2, HILPDA | [58,59] |
| Cryptosporidium spp. | - | HIF1a, HSP70 | [60,61] |
| Tissue and Blood Parasites | |||
| Leishmania spp. | Hk2, Pfkl, Ldha, Kh3, Gapdh, Eno2, Pdha1 | Hif1a, Hif2a, Vefga, Pik3ca, Plog1, Trf, Vhl, Plog2, Cul2, Serpine1, Bcl2, Mtor, Erbb2, Insr, Nos2, Tlr4, Cdkn1α, Hmox1, Akt3, Eif4e2, Il6, Angtp1, Rps6kb2, Egln3, Ifngr1, Mknk1, Eloc, Rps6, Rela, Egln1, Cdkn1b, Rbx1, Prkca, Camk2b, miRNA210 | [62,63,64,65,66] |
| Toxoplasma gondii | HK2, PFK1, PGK1, ENO1 | Hif1a/HIF1a, EPO, IER3, JUNB, ITF, ICAM1, BNIP3. TFR, GADD45 | [67,68,69,70] |
| Plasmodium sp. | - | Vegf, Hif1a | [71] |
| Trypanosoma spp. | PFKFB3, PFKP, ENO1, ENO2, GAPDH, HK2, PGM1, HK1, LDHA, ALDOA, PKM, PGK1 | HIF1a, VEGFB, NOCT, BHLHE40, GPI, SLC2A3, FOS, SLC2A1, GLRX, PGF, TPI1, MAFF | [72] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeMichele, E.; Sosnowski, O.; Buret, A.G.; Allain, T. Regulatory Functions of Hypoxia in Host–Parasite Interactions: A Focus on Enteric, Tissue, and Blood Protozoa. Microorganisms 2023, 11, 1598. https://doi.org/10.3390/microorganisms11061598
DeMichele E, Sosnowski O, Buret AG, Allain T. Regulatory Functions of Hypoxia in Host–Parasite Interactions: A Focus on Enteric, Tissue, and Blood Protozoa. Microorganisms. 2023; 11(6):1598. https://doi.org/10.3390/microorganisms11061598
Chicago/Turabian StyleDeMichele, Emily, Olivia Sosnowski, Andre G. Buret, and Thibault Allain. 2023. "Regulatory Functions of Hypoxia in Host–Parasite Interactions: A Focus on Enteric, Tissue, and Blood Protozoa" Microorganisms 11, no. 6: 1598. https://doi.org/10.3390/microorganisms11061598