
Compositional Changes in the Vaginal Bacterial Microbiome of Healthy Pregnant Women across the Three Gestational Trimesters in Ismailia, Egypt

Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Participant Enrollment
2.3. Sample Collection
2.4. The pH Measurement
2.5. DNA Extraction
2.6. PCR Amplification of 16S rRNA Gene and MiSeq Illumina Sequencing
2.7. Bioinformatic Analysis
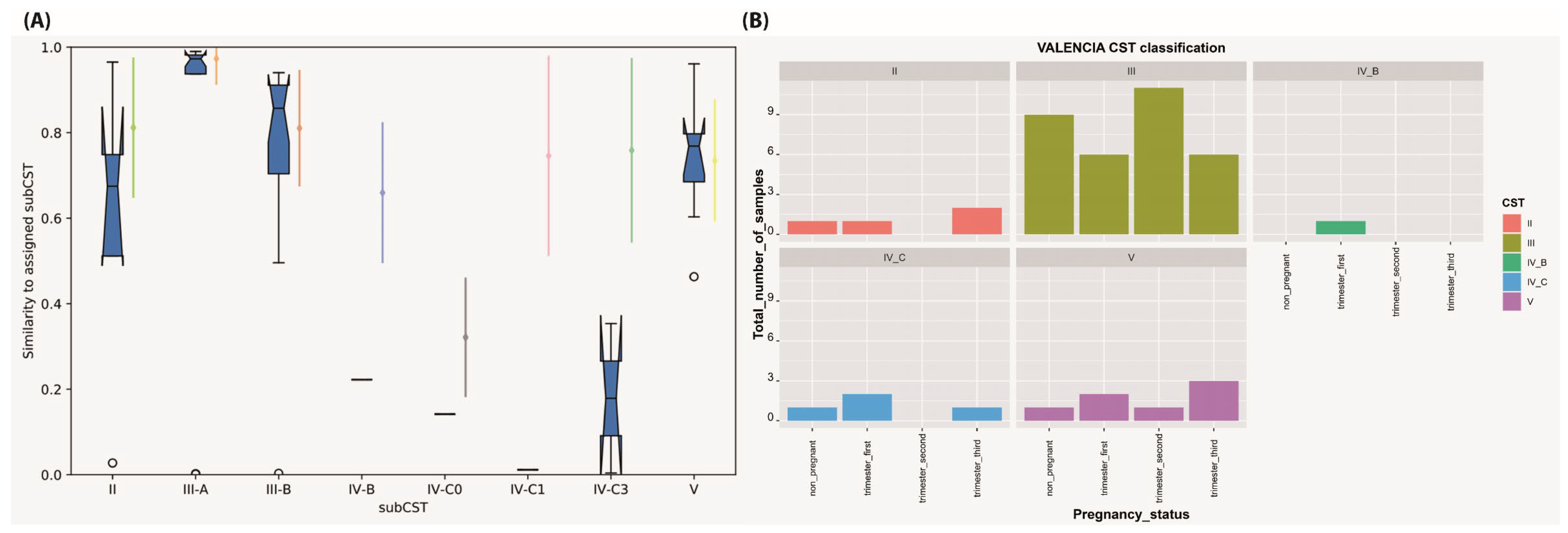
2.8. Community State Types (CST) Assignment
2.9. Data Availability
3. Results
3.1. Sequencing Data Profiles
3.2. Taxonomical Classification, Core Microbiome and Bacterial Biomarkers
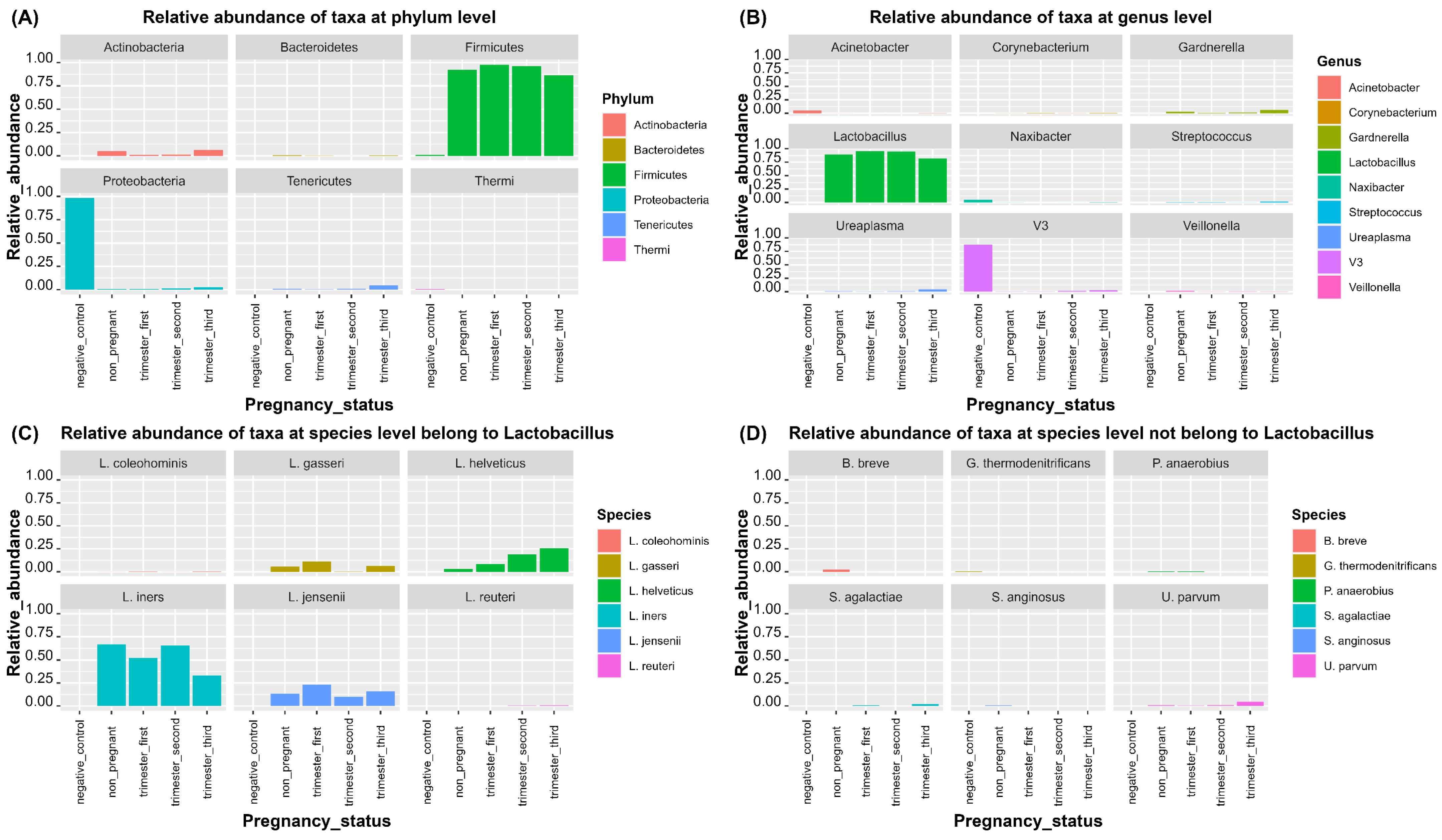
3.2.1. Taxonomical Classification
3.2.2. Bacterial Biomarkers
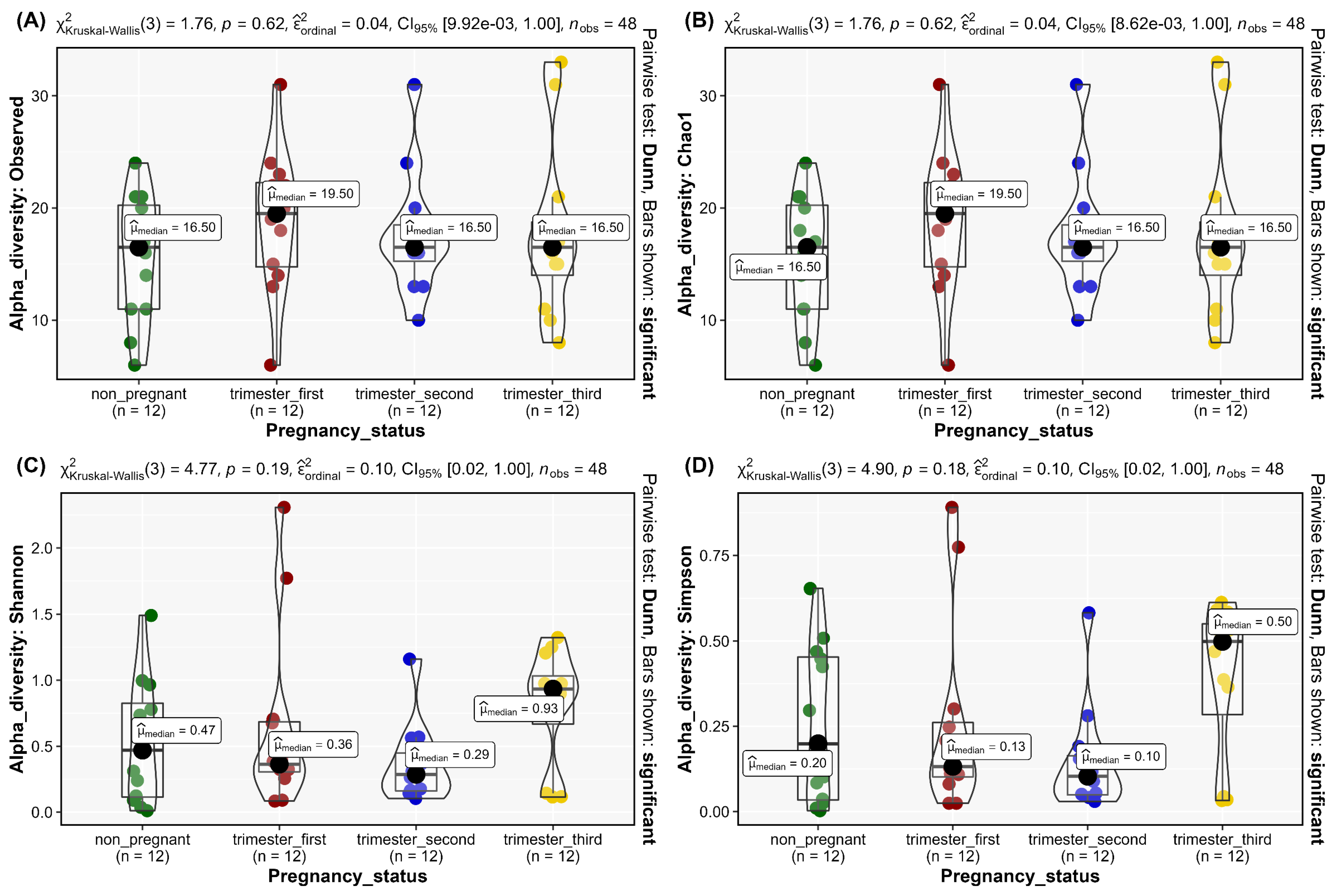
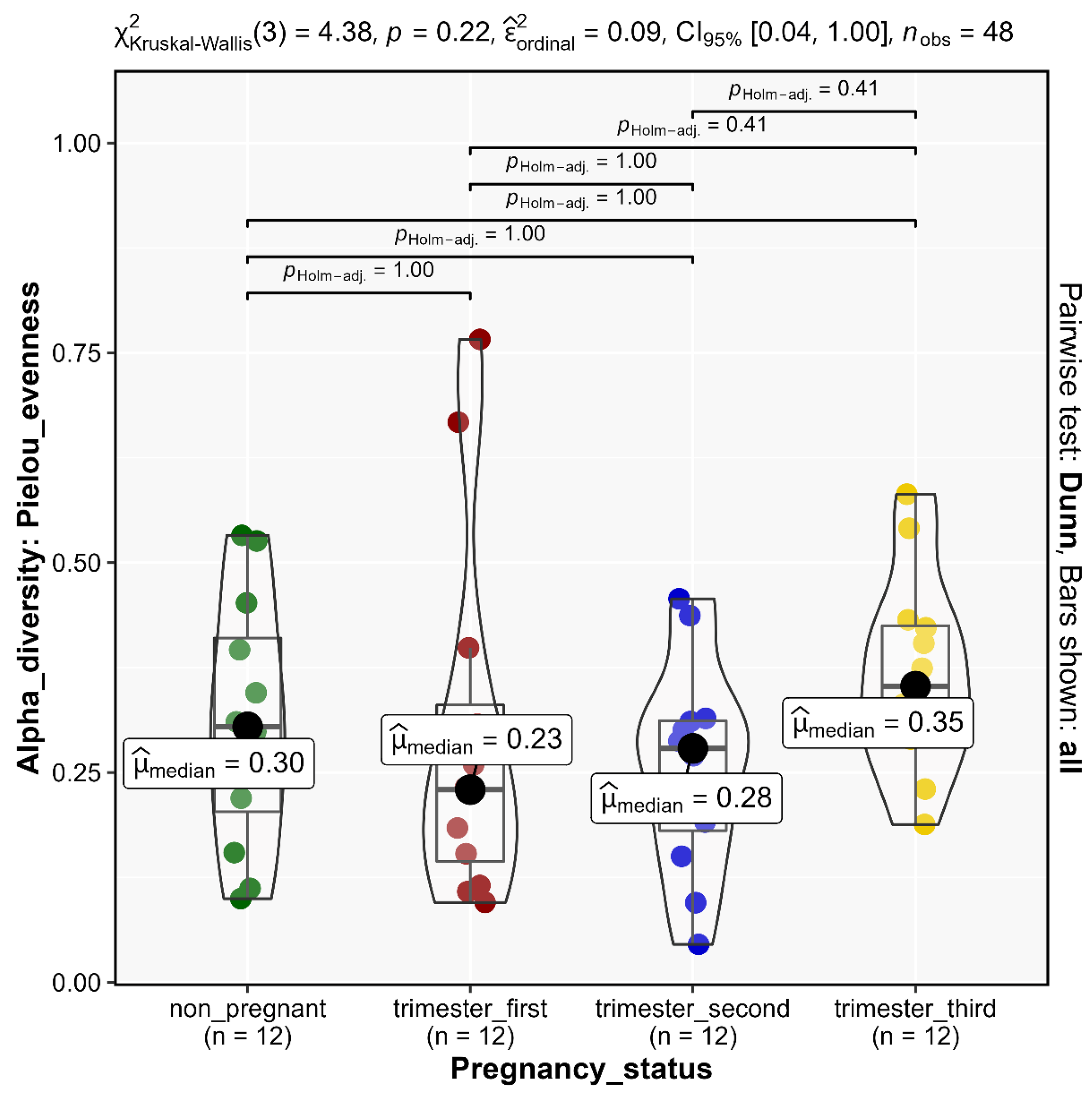
3.3. Alpha Diversity
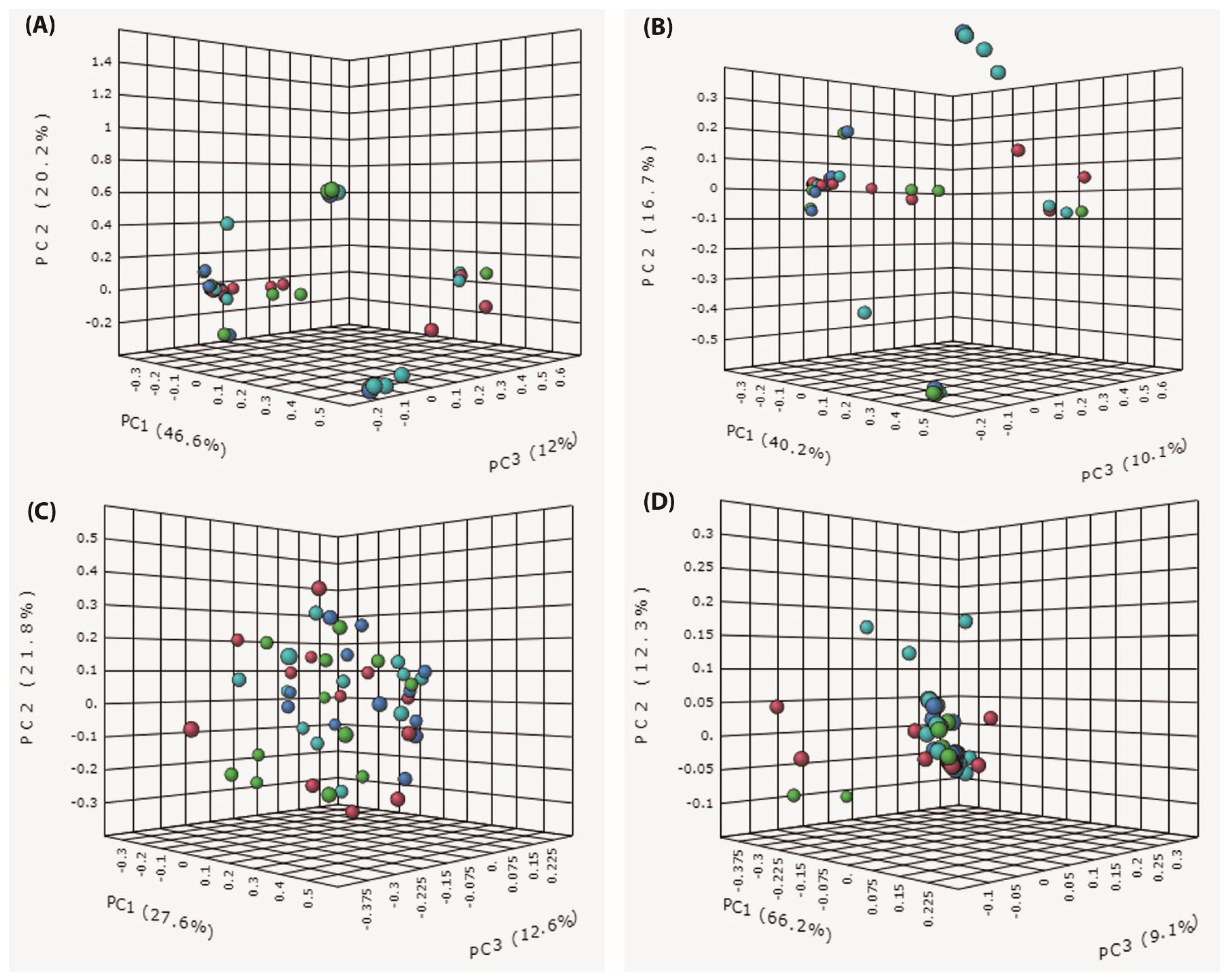
3.4. Beta Diversity
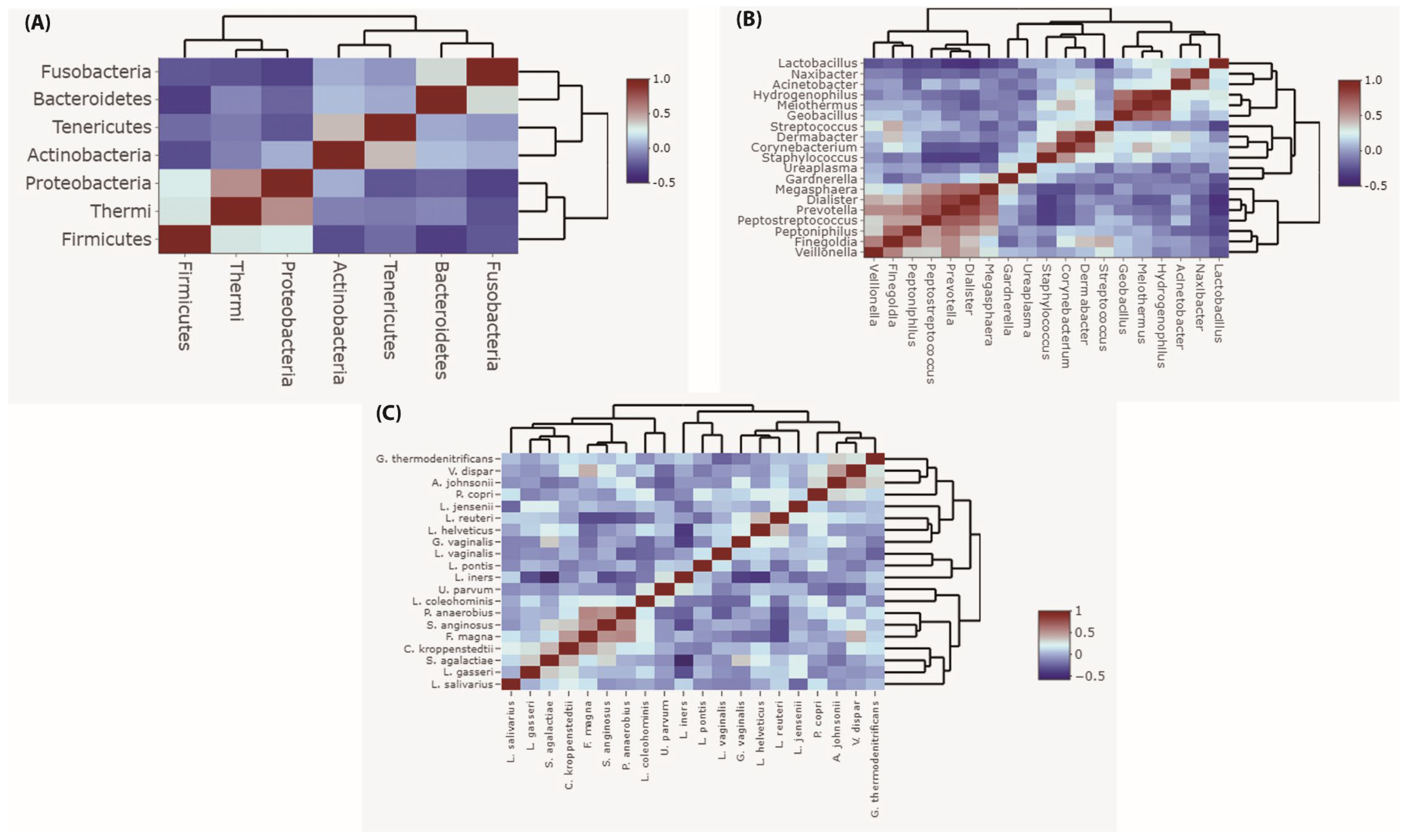
3.5. Correlation of Vaginal Microbiota during Pregnancy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethical Statement
References
- Bernabeu, A.; Lledo, B.; Díaz, M.C.; Lozano, F.M.; Ruiz, V.; Fuentes, A.; Lopez-Pineda, A.; Moliner, B.; Castillo, J.C.; Ortiz, J.A.; et al. Effect of the vaginal microbiome on the pregnancy rate in women receiving assisted reproductive treatment. J. Assist. Reprod. Genet. 2019, 36, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, L.; Verwijs, M.C.; Care, A.; Sharp, A.; Ivandic, J.; Poljak, B.; Roberts, D.; Bronowski, C.; Gill, A.C.; Darby, A.C.; et al. Vaginal bacterial load in the second trimester is associated with early preterm birth recurrence: A nested case-control study. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 2061–2072. [Google Scholar] [CrossRef] [PubMed]
- Grewal, K.; MacIntyre, D.A.; Bennett, P.R. The reproductive tract microbiota in pregnancy. Biosci. Rep. 2021, 41, BSR20203908. [Google Scholar] [CrossRef] [PubMed]
- Hočevar, K.; Maver, A.; Vidmar Šimic, M.; Hodžić, A.; Haslberger, A.; Premru Seršen, T.; Peterlin, B. Vaginal Microbiome Signature Is Associated with Spontaneous Preterm Delivery. Front. Med. 2019, 6, 201. [Google Scholar] [CrossRef]
- Juliana, N.C.A.; Peters, R.P.H.; Al-Nasiry, S.; Budding, A.E.; Morré, S.A.; Ambrosino, E. Composition of the vaginal microbiota during pregnancy in women living in sub-Saharan Africa: A PRISMA-compliant review. BMC Pregnancy Childbirth 2021, 21, 596. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chi, X.Z.; Zhang, L.; Chen, R.; Cao, J.R.; Sun, X.Y.; Yang, H.Q.; Liao, Q.P. Vaginal microbiome analysis of healthy women during different periods of gestation. Biosci. Rep. 2020, 40, BSR20201766. [Google Scholar] [CrossRef]
- Liu, L.; Chen, J.; Chen, Y.; Jiang, S.; Xu, H.; Zhan, H.; Ren, Y.; Xu, D.; Xu, Z.; Chen, D. Characterization of Vaginal Microbiota in Third Trimester Premature Rupture of Membranes Patients through 16S rDNA Sequencing. Pathogens 2022, 11, 847. [Google Scholar] [CrossRef]
- Ng, S.; Chen, M.; Kundu, S.; Wang, X.; Zhou, Z.; Zheng, Z.; Qing, W.; Sheng, H.; Wang, Y.; He, Y.; et al. Large-scale characterisation of the pregnancy vaginal microbiome and sialidase activity in a low-risk Chinese population. NPJ Biofilms Microbiomes 2021, 7, 89. [Google Scholar] [CrossRef]
- Nunn, K.L.; Witkin, S.S.; Schneider, G.M.; Boester, A.; Nasioudis, D.; Minis, E.; Gliniewicz, K.; Forney, L.J. Changes in the Vaginal Microbiome during the Pregnancy to Postpartum Transition. Reprod. Sci. 2021, 28, 1996–2005. [Google Scholar] [CrossRef]
- Serrano, M.G.; Parikh, H.I.; Brooks, J.P.; Edwards, D.J.; Arodz, T.J.; Edupuganti, L.; Huang, B.; Girerd, P.H.; Bokhari, Y.A.; Bradley, S.P.; et al. Racioethnic diversity in the dynamics of the vaginal microbiome during pregnancy. Nat. Med. 2019, 25, 1001–1011. [Google Scholar] [CrossRef]
- Severgnini, M.; Morselli, S.; Camboni, T.; Ceccarani, C.; Laghi, L.; Zagonari, S.; Patuelli, G.; Pedna, M.F.; Sambri, V.; Foschi, C.; et al. A Deep Look at the Vaginal Environment During Pregnancy and Puerperium. Front. Cell. Infect. Microbiol. 2022, 12, 838405. [Google Scholar] [CrossRef]
- Sroka-Oleksiak, A.; Gosiewski, T.; Pabian, W.; Gurgul, A.; Kapusta, P.; Ludwig-Słomczyńska, A.H.; Wołkow, P.P.; Brzychczy-Włoch, M. Next-Generation Sequencing as a Tool to Detect Vaginal Microbiota Disturbances during Pregnancy. Microorganisms 2020, 8, 1813. [Google Scholar] [CrossRef]
- Yao, T.; Wang, Z.; Liang, X.; Liu, C.; Yu, Z.; Han, X.; Liu, R.; Liu, Y.; Liu, C.; Chen, L. Signatures of vaginal microbiota by 16S rRNA gene: Potential bio-geographical application in Chinese Han from three regions of China. Int. J. Leg. Med. 2021, 135, 1213–1224. [Google Scholar] [CrossRef]
- Zhou, X.; Brown, C.J.; Abdo, Z.; Davis, C.C.; Hansmann, M.A.; Joyce, P.; Foster, J.A.; Forney, L.J. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME J. 2007, 1, 121–133. [Google Scholar] [CrossRef] [Green Version]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4680–4687. [Google Scholar] [CrossRef] [Green Version]
- DiGiulio, D.B.; Callahan, B.J.; McMurdie, P.J.; Costello, E.K.; Lyell, D.J.; Robaczewska, A.; Sun, C.L.; Goltsman, D.S.; Wong, R.J.; Shaw, G.; et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc. Natl. Acad. Sci. USA 2015, 112, 11060–11065. [Google Scholar] [CrossRef] [Green Version]
- MacIntyre, D.A.; Chandiramani, M.; Lee, Y.S.; Kindinger, L.; Smith, A.; Angelopoulos, N.; Lehne, B.; Arulkumaran, S.; Brown, R.; Teoh, T.G.; et al. The vaginal microbiome during pregnancy and the postpartum period in a European population. Sci. Rep. 2015, 5, 8988. [Google Scholar] [CrossRef] [Green Version]
- Romero, R.; Hassan, S.S.; Gajer, P.; Tarca, A.L.; Fadrosh, D.W.; Nikita, L.; Galuppi, M.; Lamont, R.F.; Chaemsaithong, P.; Miranda, J.; et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2014, 2, 4. [Google Scholar] [CrossRef] [Green Version]
- Freitas, A.C.; Chaban, B.; Bocking, A.; Rocco, M.; Yang, S.; Hill, J.E.; Money, D.M. The vaginal microbiome of pregnant women is less rich and diverse, with lower prevalence of Mollicutes, compared to non-pregnant women. Sci. Rep. 2017, 7, 9212. [Google Scholar] [CrossRef] [Green Version]
- Siiteri, P.K.; MacDonald, P.C. Placental estrogen biosynthesis during human pregnancy. J. Clin. Endocrinol. Metab. 1966, 26, 751–761. [Google Scholar] [CrossRef]
- Fettweis, J.M.; Serrano, M.G.; Girerd, P.H.; Jefferson, K.K.; Buck, G.A. A new era of the vaginal microbiome: Advances using next-generation sequencing. Chem. Biodivers. 2012, 9, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Amabebe, E.; Anumba, D.O.C. The Vaginal Microenvironment: The Physiologic Role of Lactobacilli. Front. Med. 2018, 5, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, M.; Ishiyama, A.; Tabata, M.; Sasaki, Y.; Yoneda, S.; Shiozaki, A.; Saito, S. Relationship between cervical mucus interleukin-8 concentrations and vaginal bacteria in pregnancy. Am. J. Reprod. Immunol. 2004, 52, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Witkin, S.S.; Linhares, I.M. Why do lactobacilli dominate the human vaginal microbiota? BJOG Int. J. Obstet. Gynaecol. 2017, 124, 606–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillier, S.L.; Nugent, R.P.; Eschenbach, D.A.; Krohn, M.A.; Gibbs, R.S.; Martin, D.H.; Cotch, M.F.; Edelman, R.; Pastorek, J.G., II; Rao, A.V.; et al. Association between bacterial vaginosis and preterm delivery of a low-birth-weight infant. The Vaginal Infections and Prematurity Study Group. New Engl. J. Med. 1995, 333, 1737–1742. [Google Scholar] [CrossRef] [PubMed]
- Flynn, C.A.; Helwig, A.L.; Meurer, L.N. Bacterial vaginosis in pregnancy and the risk of prematurity: A meta-analysis. J. Fam. Pract. 1999, 48, 885–892. [Google Scholar]
- Fettweis, J.M.; Brooks, J.P.; Serrano, M.G.; Sheth, N.U.; Girerd, P.H.; Edwards, D.J.; Strauss, J.F.; The Vaginal Microbiome, C.; Jefferson, K.K.; Buck, G.A. Differences in vaginal microbiome in African American women versus women of European ancestry. Microbiol. 2014, 160, 2272–2282. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, K.; Riehle, K.; Ma, J.; Segata, N.; Mistretta, T.A.; Coarfa, C.; Raza, S.; Rosenbaum, S.; Van den Veyver, I.; Milosavljevic, A.; et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS ONE 2012, 7, e36466. [Google Scholar] [CrossRef]
- Nasioudis, D.; Forney, L.J.; Schneider, G.M.; Gliniewicz, K.; France, M.; Boester, A.; Sawai, M.; Scholl, J.; Witkin, S.S. Influence of Pregnancy History on the Vaginal Microbiome of Pregnant Women in their First Trimester. Sci. Rep. 2017, 7, 10201. [Google Scholar] [CrossRef]
- Walther-António, M.R.; Jeraldo, P.; Berg Miller, M.E.; Yeoman, C.J.; Nelson, K.E.; Wilson, B.A.; White, B.A.; Chia, N.; Creedon, D.J. Pregnancy’s stronghold on the vaginal microbiome. PLoS ONE 2014, 9, e98514. [Google Scholar] [CrossRef] [Green Version]
- Forney, L.J.; Gajer, P.; Williams, C.J.; Schneider, G.M.; Koenig, S.S.; McCulle, S.L.; Karlebach, S.; Brotman, R.M.; Davis, C.C.; Ault, K.; et al. Comparison of self-collected and physician-collected vaginal swabs for microbiome analysis. J. Clin. Microbiol. 2010, 48, 1741–1748. [Google Scholar] [CrossRef]
- Schachter, J.; McCormack, W.M.; Chernesky, M.A.; Martin, D.H.; Van Der Pol, B.; Rice, P.A.; Hook, E.W., III; Stamm, W.E.; Quinn, T.C.; Chow, J.M. Vaginal swabs are appropriate specimens for diagnosis of genital tract infection with Chlamydia trachomatis. J. Clin. Microbiol. 2003, 41, 3784–3789. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Oliver, A.; LaMere, B.; Weihe, C.; Wandro, S.; Lindsay, K.L.; Wadhwa, P.D.; Mills, D.A.; Pride, D.T.; Fiehn, O.; Northen, T.; et al. Cervicovaginal Microbiome Composition Is Associated with Metabolic Profiles in Healthy Pregnancy. mBio 2020, 11. [Google Scholar] [CrossRef]
- Fettweis, J.M.; Serrano, M.G.; Brooks, J.P.; Edwards, D.J.; Girerd, P.H.; Parikh, H.I.; Huang, B.; Arodz, T.J.; Edupuganti, L.; Glascock, A.L.; et al. The vaginal microbiome and preterm birth. Nat. Med. 2019, 25, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Romero, R.; Hassan, S.S.; Gajer, P.; Tarca, A.L.; Fadrosh, D.W.; Bieda, J.; Chaemsaithong, P.; Miranda, J.; Chaiworapongsa, T.; Ravel, J. The vaginal microbiota of pregnant women who subsequently have spontaneous preterm labor and delivery and those with a normal delivery at term. Microbiome 2014, 2, 18. [Google Scholar] [CrossRef] [Green Version]
- Hemalatha, R.; Ramalaxmi, B.A.; Swetha, E.; Balakrishna, N.; Mastromarino, P. Evaluation of vaginal pH for detection of bacterial vaginosis. Indian J. Med. Res. 2013, 138, 354–359. [Google Scholar]
- Mohamed, I.; Zakeer, S.; Azab, M.; Hanora, A. Changes in Vaginal Microbiome in Pregnant and Nonpregnant Women with Bacterial Vaginosis: Toward Microbiome Diagnostics? Omics: A J. Integr. Biol. 2020, 24, 602–614. [Google Scholar] [CrossRef]
- Mattei, V.; Murugesan, S.; Al Hashmi, M.; Mathew, R.; James, N.; Singh, P.; Kumar, M.; Lakshmanan, A.P.; Terranegra, A.; Al Khodor, S.; et al. Evaluation of Methods for the Extraction of Microbial DNA From Vaginal Swabs Used for Microbiome Studies. Front. Cell. Infect. Microbiol. 2019, 9, 197. [Google Scholar] [CrossRef]
- Illumina. 16S Metagenomic Sequencing Library Preparation Protocol: Preparing 16S Ribosomal RNA Gene Amplicons for the Illumina MiSeq System. Part no. 15044223 Rev B. Illumina, San Diego, CA, USA. Available online: https://www.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 12 June 2020).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Mohsen, A.; Park, J.; Chen, Y.A.; Kawashima, H.; Mizuguchi, K. Impact of quality trimming on the efficiency of reads joining and diversity analysis of Illumina paired-end reads in the context of QIIME1 and QIIME2 microbiome analysis frameworks. BMC Bioinform. 2019, 20, 581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef] [PubMed]
- France, M.T.; Ma, B.; Gajer, P.; Brown, S.; Humphrys, M.S.; Holm, J.B.; Waetjen, L.E.; Brotman, R.M.; Ravel, J. VALENCIA: A nearest centroid classification method for vaginal microbial communities based on composition. Microbiome 2020, 8, 166. [Google Scholar] [CrossRef]
- A framework for human microbiome research. Nature 2012, 486, 215–221. [CrossRef] [Green Version]
- O’Hanlon, D.E.; Moench, T.R.; Cone, R.A. Vaginal pH and microbicidal lactic acid when lactobacilli dominate the microbiota. PLoS ONE 2013, 8, e80074. [Google Scholar] [CrossRef]
- Borges, S.; Silva, J.; Teixeira, P. The role of lactobacilli and probiotics in maintaining vaginal health. Arch. Gynecol. Obstet. 2014, 289, 479–489. [Google Scholar] [CrossRef]
- Pino, A.; Bartolo, E.; Caggia, C.; Cianci, A.; Randazzo, C.L. Detection of vaginal lactobacilli as probiotic candidates. Sci. Rep. 2019, 9, 3355. [Google Scholar] [CrossRef] [Green Version]
- Gautam, R.; Borgdorff, H.; Jespers, V.; Francis, S.C.; Verhelst, R.; Mwaura, M.; Delany-Moretlwe, S.; Ndayisaba, G.; Kyongo, J.K.; Hardy, L.; et al. Correlates of the molecular vaginal microbiota composition of African women. BMC Infect. Dis. 2015, 15, 86. [Google Scholar] [CrossRef] [Green Version]
- Jespers, V.; van de Wijgert, J.; Cools, P.; Verhelst, R.; Verstraelen, H.; Delany-Moretlwe, S.; Mwaura, M.; Ndayisaba, G.F.; Mandaliya, K.; Menten, J.; et al. The significance of Lactobacillus crispatus and L. vaginalis for vaginal health and the negative effect of recent sex: A cross-sectional descriptive study across groups of African women. BMC Infect. Dis. 2015, 15, 115. [Google Scholar] [CrossRef] [Green Version]
- Holm, J.B.; France, M.T.; Ma, B.; McComb, E.; Robinson, C.K.; Mehta, A.; Tallon, L.J.; Brotman, R.M.; Ravel, J. Comparative Metagenome-Assembled Genome Analysis of “Candidatus Lachnocurva vaginae”, Formerly Known as Bacterial Vaginosis-Associated Bacterium-1 (BVAB1). Front. Cell. Infect. Microbiol. 2020, 10, 117. [Google Scholar] [CrossRef]
- Callahan, B.J.; DiGiulio, D.B.; Goltsman, D.S.A.; Sun, C.L.; Costello, E.K.; Jeganathan, P.; Biggio, J.R.; Wong, R.J.; Druzin, M.L.; Shaw, G.M.; et al. Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women. Proc. Natl. Acad. Sci. USA 2017, 114, 9966–9971. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; You, Y.A.; Kim, Y.H.; Kwon, E.; Ansari, A.; Kim, S.M.; Lee, G.; Hur, Y.M.; Jung, Y.J.; Kim, K.; et al. Ureaplasma and Prevotella colonization with Lactobacillus abundance during pregnancy facilitates term birth. Sci. Rep. 2022, 12, 10148. [Google Scholar] [CrossRef]
- Gilbert, N.M.; Foster, L.R.; Cao, B.; Yin, Y.; Mysorekar, I.U.; Lewis, A.L. Gardnerella vaginalis promotes group B Streptococcus vaginal colonization, enabling ascending uteroplacental infection in pregnant mice. Am. J. Obstet. Gynecol. 2021, 224, 530.e531. [Google Scholar] [CrossRef]
- Ling, Z.; Liu, X.; Luo, Y.; Wu, X.; Yuan, L.; Tong, X.; Li, L.; Xiang, C. Associations between vaginal pathogenic community and bacterial vaginosis in Chinese reproductive-age women. PLoS ONE 2013, 8, e76589. [Google Scholar] [CrossRef]
- Muzny, C.A.; Łaniewski, P.; Schwebke, J.R.; Herbst-Kralovetz, M.M. Host-vaginal microbiota interactions in the pathogenesis of bacterial vaginosis. Curr. Opin. Infect. Dis. 2020, 33, 59–65. [Google Scholar] [CrossRef]
- Muzny, C.A.; Taylor, C.M.; Swords, W.E.; Tamhane, A.; Chattopadhyay, D.; Cerca, N.; Schwebke, J.R. An Updated Conceptual Model on the Pathogenesis of Bacterial Vaginosis. J. Infect. Dis. 2019, 220, 1399–1405. [Google Scholar] [CrossRef]
- Teixeira, G.S.; Soares-Brandão, K.L.K.; Branco, K.; Sampaio, J.L.M.; Nardi, R.M.D.; Mendonça, M.; Almeida, R.B.; Farias, L.M.; Carvalho, M.A.R.; Nicoli, J.R. Antagonism and synergism in Gardnerella vaginalis strains isolated from women with bacterial vaginosis. J. Med. Microbiol. 2010, 59, 891–897. [Google Scholar] [CrossRef]
- Torcia, M.G. Interplay among Vaginal Microbiome, Immune Response and Sexually Transmitted Viral Infections. Int. J. Mol. Sci. 2019, 20, 266. [Google Scholar] [CrossRef] [Green Version]
- Graspeuntner, S.; Loeper, N.; Künzel, S.; Baines, J.F.; Rupp, J. Selection of validated hypervariable regions is crucial in 16S-based microbiota studies of the female genital tract. Sci. Rep. 2018, 8, 9678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prince, A.L.; Chu, D.M.; Seferovic, M.D.; Antony, K.M.; Ma, J.; Aagaard, K.M. The perinatal microbiome and pregnancy: Moving beyond the vaginal microbiome. Cold Spring Harb. Perspect. Med. 2015, 5, a023051. [Google Scholar] [CrossRef] [PubMed]
- Estaki, M.; Jiang, L.; Bokulich, N.A.; McDonald, D.; González, A.; Kosciolek, T.; Martino, C.; Zhu, Q.; Birmingham, A.; Vázquez-Baeza, Y.; et al. QIIME 2 Enables Comprehensive End-to-End Analysis of Diverse Microbiome Data and Comparative Studies with Publicly Available Data. Curr. Protoc. Bioinform. 2020, 70, e100. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Chang, T.H.; Lin, F.M.; Liang, C.; Chiu, C.M.; Yang, T.L.; Yang, T.; Huang, C.Y.; Cheng, Y.N.; Chang, Y.A.; et al. Vaginal microbiome variances in sample groups categorized by clinical criteria of bacterial vaginosis. BMC Genom. 2018, 19, 876. [Google Scholar] [CrossRef] [PubMed]
- Ceccarani, C.; Foschi, C.; Parolin, C.; D’Antuono, A.; Gaspari, V.; Consolandi, C.; Laghi, L.; Camboni, T.; Vitali, B.; Severgnini, M.; et al. Diversity of vaginal microbiome and metabolome during genital infections. Sci. Rep. 2019, 9, 14095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Kang, G.U.; Jeon, S.Y.; Tagele, S.B.; Pham, H.Q.; Kim, M.S.; Ahmad, S.; Jung, D.R.; Park, Y.J.; Han, H.S.; et al. Vaginal Microbiome-Based Bacterial Signatures for Predicting the Severity of Cervical Intraepithelial Neoplasia. Diagnostics 2020, 10, 1013. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumari, N.; Talukdar, D.; Kothidar, A.; Sarkar, M.; Mehta, O.; Kshetrapal, P.; Wadhwa, N.; Thiruvengadam, R.; Desiraju, B.K.; et al. The Vaginal Microbial Signatures of Preterm Birth Delivery in Indian Women. Front. Cell. Infect. Microbiol. 2021, 11, 622474. [Google Scholar] [CrossRef]
- Wang, J.; Li, Z.; Ma, X.; Du, L.; Jia, Z.; Cui, X.; Yu, L.; Yang, J.; Xiao, L.; Zhang, B.; et al. Translocation of vaginal microbiota is involved in impairment and protection of uterine health. Nat. Commun. 2021, 12, 4191. [Google Scholar] [CrossRef]
- Hyman, R.W.; Fukushima, M.; Jiang, H.; Fung, E.; Rand, L.; Johnson, B.; Vo, K.C.; Caughey, A.B.; Hilton, J.F.; Davis, R.W.; et al. Diversity of the vaginal microbiome correlates with preterm birth. Reprod. Sci. 2014, 21, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Peterson, D.; Bonham, K.S.; Rowland, S.; Pattanayak, C.W.; Klepac-Ceraj, V. Comparative Analysis of 16S rRNA Gene and Metagenome Sequencing in Pediatric Gut Microbiomes. Front. Microbiol. 2021, 12, 670336. [Google Scholar] [CrossRef]
- Feehily, C.; Crosby, D.; Walsh, C.J.; Lawton, E.M.; Higgins, S.; McAuliffe, F.M.; Cotter, P.D. Shotgun sequencing of the vaginal microbiome reveals both a species and functional potential signature of preterm birth. NPJ Biofilms Microbiomes 2020, 6, 50. [Google Scholar] [CrossRef]
- O’Callaghan, J.L.; Willner, D.; Buttini, M.; Huygens, F.; Pelzer, E.S. Limitations of 16S rRNA Gene Sequencing to Characterize Lactobacillus Species in the Upper Genital Tract. Front. Cell Dev. Biol. 2021, 9, 641921. [Google Scholar] [CrossRef]
- Giampaoli, S.; DeVittori, E.; Valeriani, F.; Berti, A.; Romano Spica, V. Informativeness of NGS Analysis for Vaginal Fluid Identification. J. Forensic Sci. 2017, 62, 192–196. [Google Scholar] [CrossRef]
- Ladetto, M.; Brüggemann, M.; Monitillo, L.; Ferrero, S.; Pepin, F.; Drandi, D.; Barbero, D.; Palumbo, A.; Passera, R.; Boccadoro, M.; et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia 2014, 28, 1299–1307. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CST/Pregnancy Status | II | III | IV-B | IV-C | V | Total | |||
|---|---|---|---|---|---|---|---|---|---|
| III-A | III-B | IV-C0 | IV-C1 | IV-C3 | |||||
| Non-pregnant control group | 1 (8.33%) | 4 (33.33%) | 5 (41.67%) | - | - | - | 1 (8.33%) | 1 (8.33%) | 12 |
| Trimester—first | 1 (8.33%) | 4 (33.33%) | 2 (16.67%) | 1 (8.33%) | 1 (8.33%) | - | 1 (8.33%) | 2 (16.67%) | 12 |
| Trimester—second | - | 5 (41.67%) | 6 (50%) | - | - | - | - | 1 (8.33%) | 12 |
| Trimester—third | 2 (16.67%) | 2 (16.67%) | 4 (33.33%) | - | - | 1 (8.33%) | - | 3 (25%) | 12 |
| Total | 4 (8.33%) | 15 (31.25%) | 17 (35.42%) | 1 (2.08%) | 1 (2.08%) | 1 (2.08%) | 2 (4.17%) | 7 (14.58%) | 48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amin, M.E.; Azab, M.; Hanora, A.; Atwa, K.; Shabayek, S. Compositional Changes in the Vaginal Bacterial Microbiome of Healthy Pregnant Women across the Three Gestational Trimesters in Ismailia, Egypt. Microorganisms 2023, 11, 139. https://doi.org/10.3390/microorganisms11010139
Amin ME, Azab M, Hanora A, Atwa K, Shabayek S. Compositional Changes in the Vaginal Bacterial Microbiome of Healthy Pregnant Women across the Three Gestational Trimesters in Ismailia, Egypt. Microorganisms. 2023; 11(1):139. https://doi.org/10.3390/microorganisms11010139
Chicago/Turabian StyleAmin, Mariam E., Marwa Azab, Amro Hanora, Khaled Atwa, and Sarah Shabayek. 2023. "Compositional Changes in the Vaginal Bacterial Microbiome of Healthy Pregnant Women across the Three Gestational Trimesters in Ismailia, Egypt" Microorganisms 11, no. 1: 139. https://doi.org/10.3390/microorganisms11010139