
The Role of Soil Microbial Diversity in the Conservation of Native Seed Bacterial Microbiomes

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
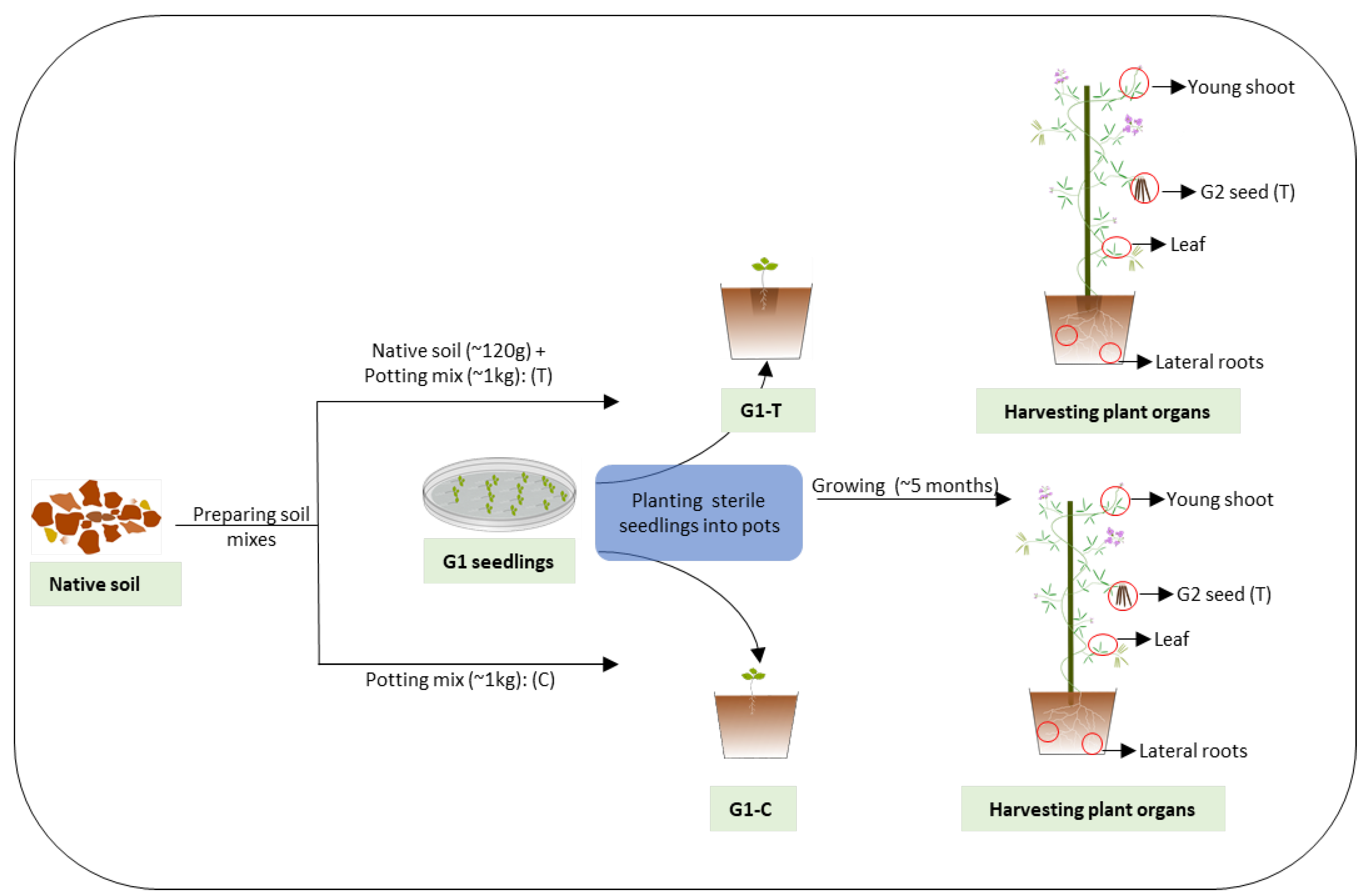
2.1. Glycine Seed Collection
2.2. Plant Growth and Sample Collection
2.3. Microbial DNA Extraction and Amplicon Library Construction
2.4. Bioinformatic Analysis of 16S rRNA Gene Amplicon Library Sequences
3. Results
3.1. 16S rRNA Gene Sequencing
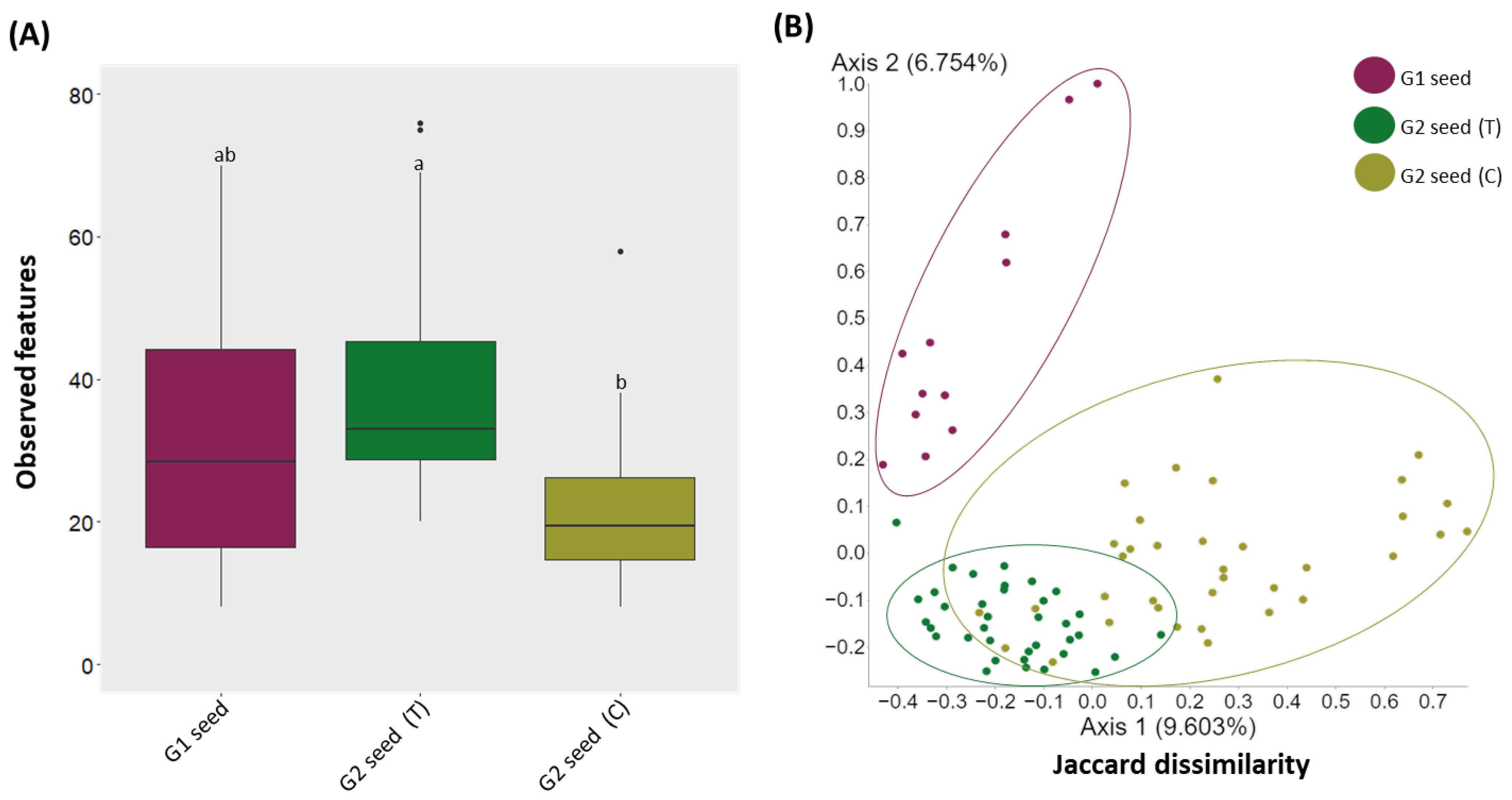
3.2. Microbiome Profiling. Temporal Variation in the G. clandestina Seed Microbiome
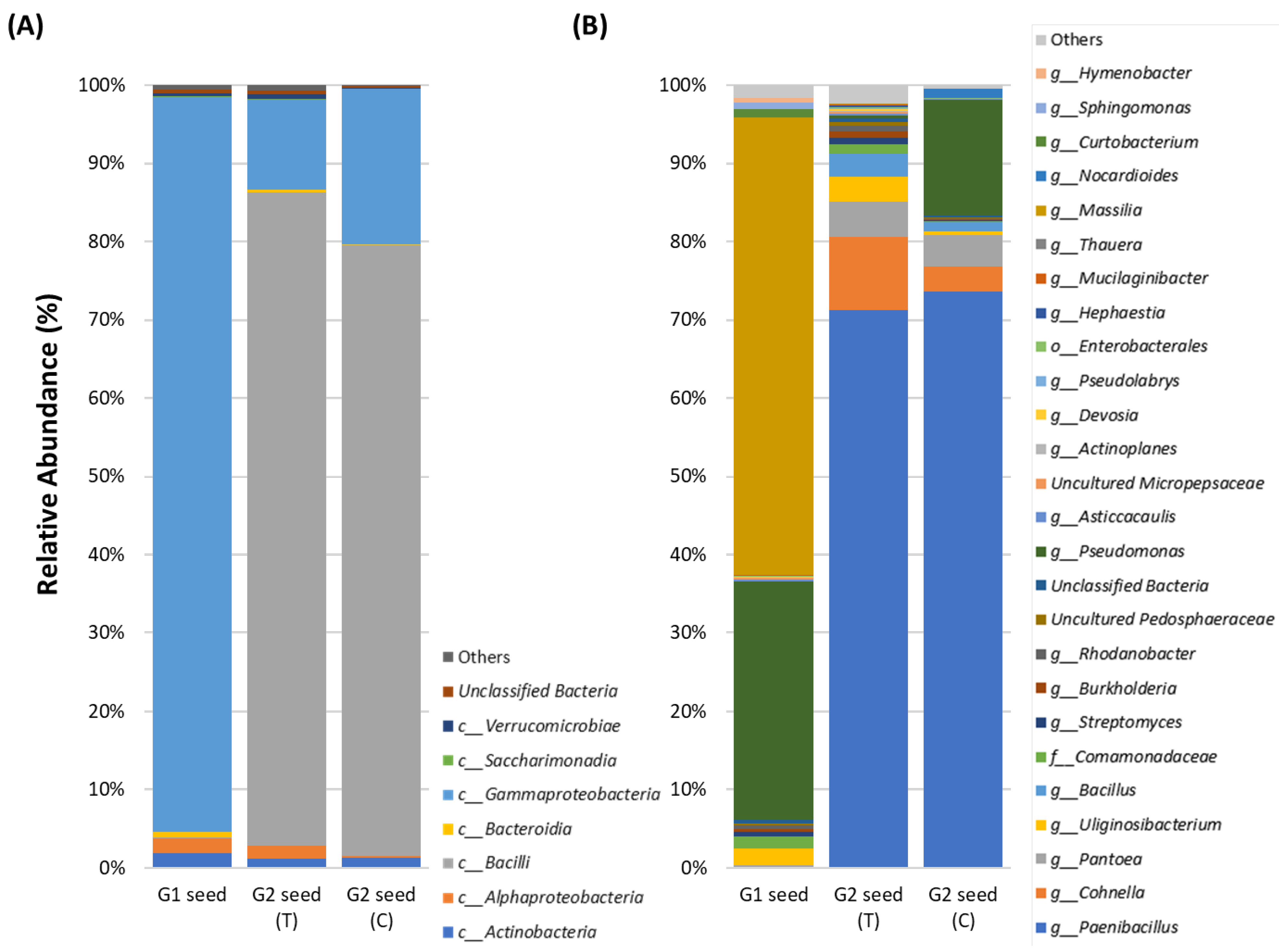
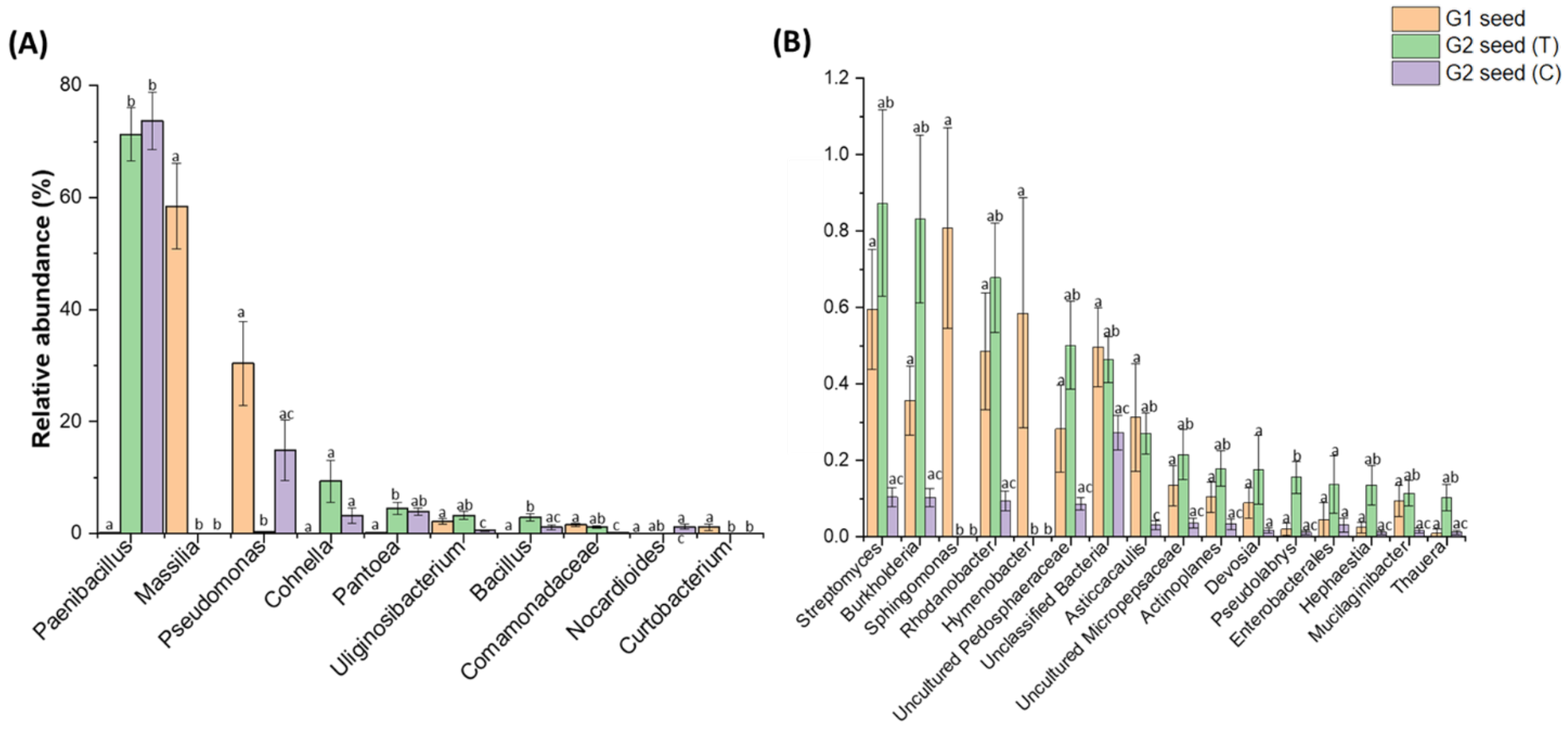
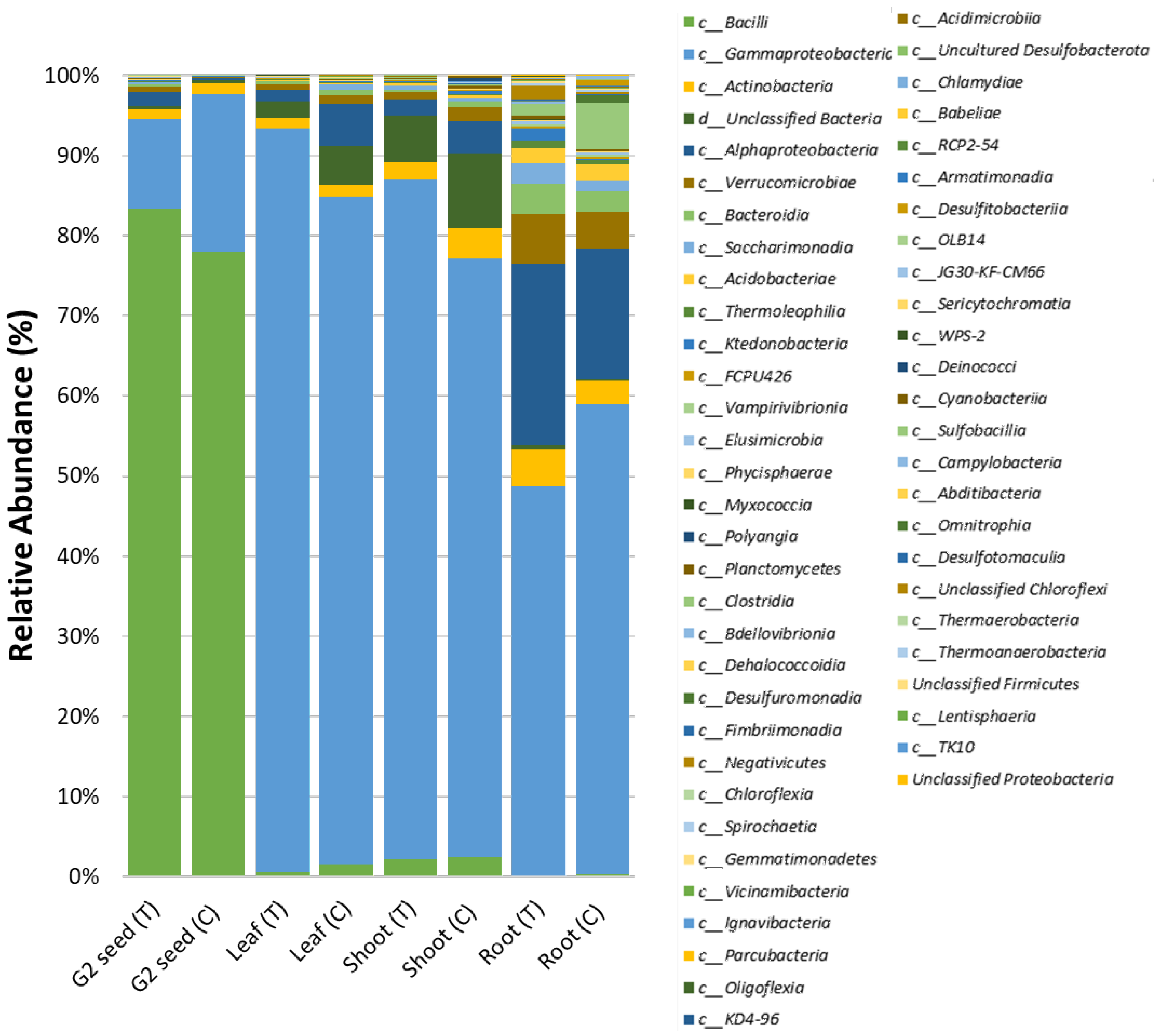
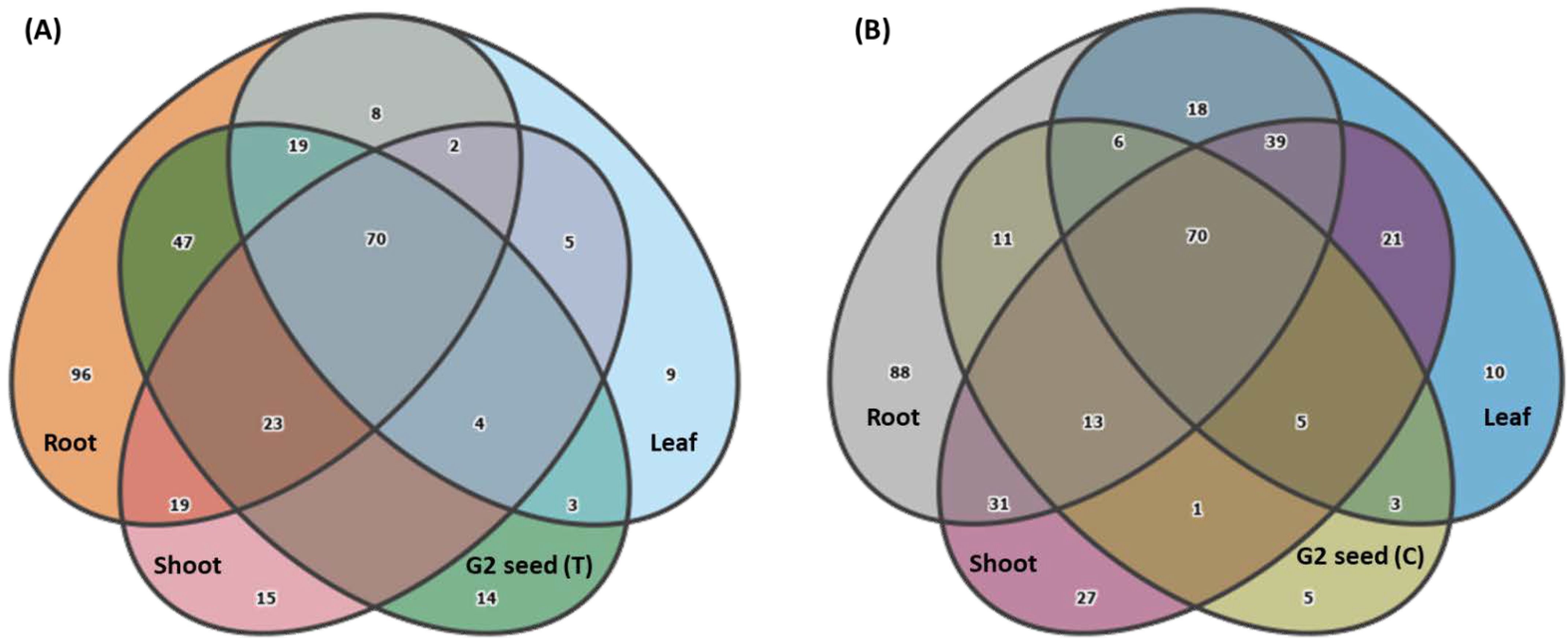
3.3. G. clandestina Seed Microbiome Composition G1 and G2 (T and C)
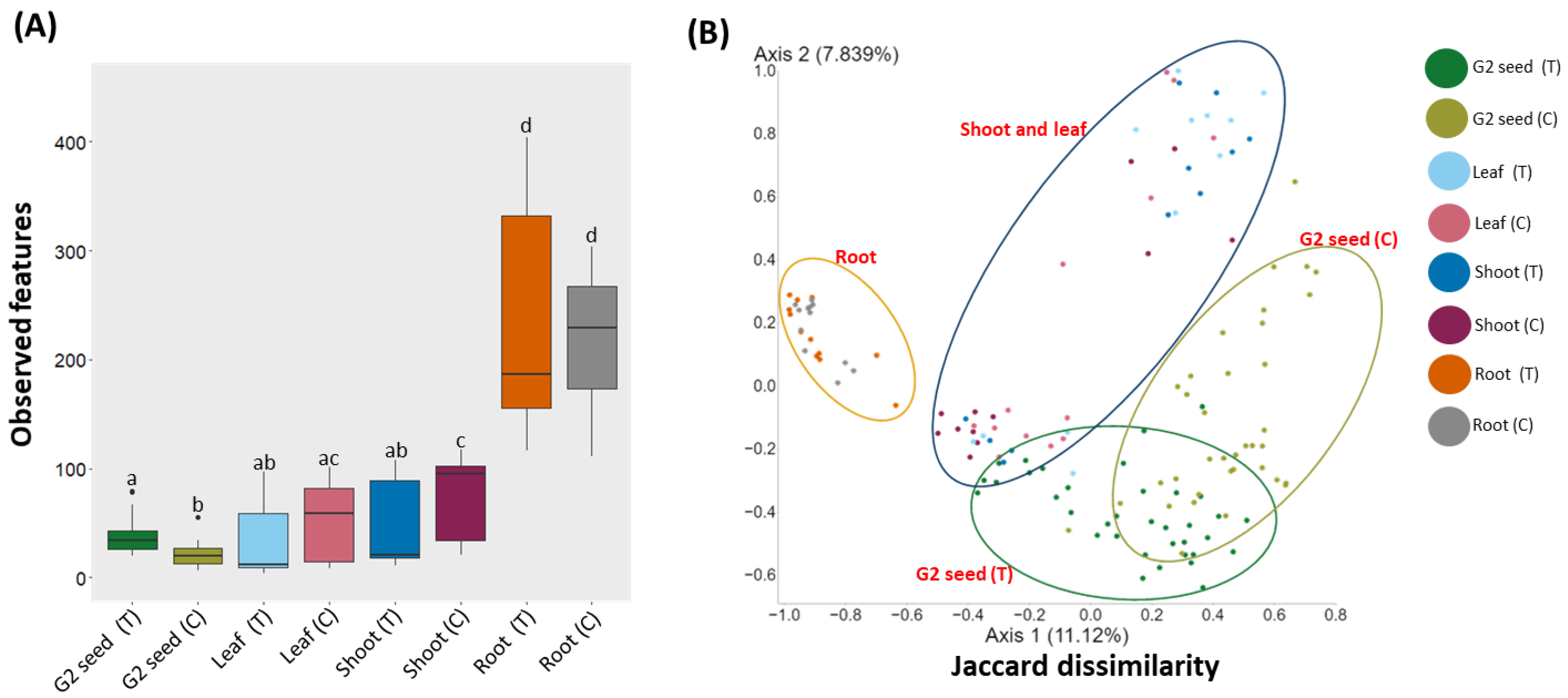
3.4. Microbiome Profiling, Spatial Variation in the G. clandestina Microbiome
3.5. G. clandestina Microbiome Composition across G1 Plant Organs and G2 (T and C) Seed
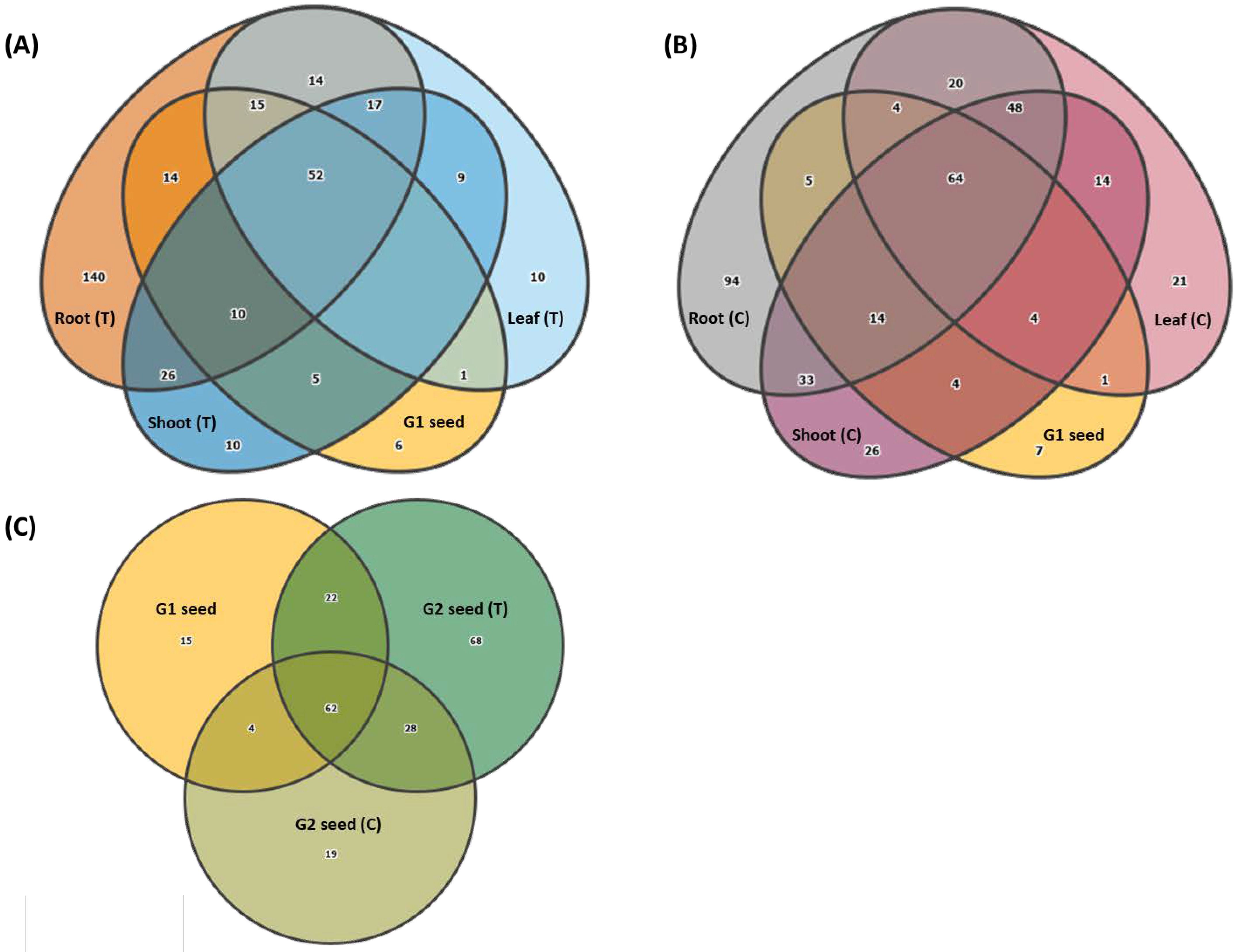
3.6. Vertical Transmission of G1 Seed Microbiota across G1 Plant Organs and G2 Seed
4. Discussion
4.1. G. clandestina Seed Microbiota Composition
4.2. Effect of Soil Type on Composition and Vertical Transmission of Seed Microbiota
4.3. Effect of Soil Type on Redistribution of Bacterial Communities among Plant Organs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Hardoim, P.R.; Van Overbeek, L.S.; Berg, G.; Pirttilä, A.M.; Compant, S.; Campisano, A.; Döring, M.; Sessitsch, A. The hidden world within plants: Ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mol. Biol. Rev. 2015, 79, 293–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, T.R.; James, E.K.; Poole, P.S. The plant microbiome. Genome Biol. 2013, 14, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochefort, A.; Simonin, M.; Marais, C.; Guillerm-Erckelboudt, A.-Y.; Barret, M.; Sarniguet, A. Transmission of Seed and Soil Microbiota to Seedling. Msystems 2021, 6, e00446-21. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lee, K.K.; Jeon, J.; Harris, W.A.; Lee, Y.-H. Domestication of Oryza species eco-evolutionarily shapes bacterial and fungal communities in rice seed. Microbiome 2020, 8, 20. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, R.; Khan, A.L.; Bilal, S.; Asaf, S.; Lee, I.-J. What Is There in Seeds? Vertically Transmitted Endophytic Resources for Sustainable Improvement in Plant Growth. Front. Plant Sci. 2018, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Walitang, D.I.; Kim, C.G.; Jeon, S.; Kang, Y.; Sa, T. Conservation and transmission of seed bacterial endophytes across generations following crossbreeding and repeated inbreeding of rice at different geographic locations. Microbiol. Open 2019, 8, e00662. [Google Scholar] [CrossRef]
- Yandigeri, M.S.; Meena, K.K.; Singh, D.; Malviya, N.; Singh, D.P.; Solanki, M.K.; Yadav, A.K.; Arora, D.K. Drought-tolerant endophytic actinobacteria promote growth of wheat (Triticum aestivum) under water stress conditions. Plant Growth Regul. 2012, 68, 411–420. [Google Scholar] [CrossRef]
- Murphy, B.R.; Jadwiszczak, M.J.; Soldi, E.; Hodkinson, T.R. Endophytes from the crop wild relative Hordeum secalinum L. improve agronomic traits in unstressed and salt-stressed barley. Cogent Food Agric. 2018, 4, 1549195. [Google Scholar] [CrossRef]
- Díaz Herrera, S.; Grossi, C.; Zawoznik, M.; Groppa, M.D. Wheat seeds harbour bacterial endophytes with potential as plant growth promoters and biocontrol agents of Fusarium graminearum. Microbiol. Res. 2016, 186–187, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Mann, R.; Sawbridge, T.; Kaur, J.; Auer, D.; Spangenberg, G. Novel Xanthomonas Species From the Perennial Ryegrass Seed Microbiome–Assessing the Bioprotection Activity of Non-pathogenic Relatives of Pathogens. Front. Microbiol. 2020, 11, 1991. [Google Scholar] [CrossRef] [PubMed]
- Ridout, M.E.; Schroeder, K.L.; Hunter, S.S.; Styer, J.; Newcombe, G. Priority effects of wheat seed endophytes on a rhizosphere symbiosis. Symbiosis 2019, 78, 19–31. [Google Scholar] [CrossRef]
- Johnston-Monje, D.; Lundberg, D.S.; Lazarovits, G.; Reis, V.M.; Raizada, M.N. Bacterial populations in juvenile maize rhizospheres originate from both seed and soil. Plant Soil 2016, 405, 337–355. [Google Scholar] [CrossRef] [Green Version]
- Moroenyane, I.; Tremblay, J.; Yergeau, E. Soybean microbiome recovery after disruption is modulated by the seed and not the soil microbiome. Phytobiomes J. 2021, 5, 418–431. [Google Scholar] [CrossRef]
- Ke, J.; Wang, B.; Yoshikuni, Y. Microbiome engineering: Synthetic biology of plant-associated microbiomes in sustainable agriculture. Trends Biotechnol. 2021, 39, 244–261. [Google Scholar] [CrossRef]
- Pérez-Jaramillo, J.E.; Mendes, R.; Raaijmakers, J.M. Impact of plant domestication on rhizosphere microbiome assembly and functions. Plant Mol. Biol. 2016, 90, 635–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez Jaramillo, J.E.; Carrion, V.J.; Bosse, M.; Ferrão, L.F.V.; De Hollander, M.; Garcia, A.A.F.; Ramirez, C.A.; Mendez, R.; Raaijmakers, J.M. Linking rhizosphere microbiome composition of wild and domesticated Phaseolus vulgaris to genotypic and root phenotypic traits. ISME J. 2017, 11, 2244–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullaeva, Y.; Ambika Manirajan, B.; Honermeier, B.; Schnell, S.; Cardinale, M. Domestication affects the composition, diversity, and co-occurrence of the cereal seed microbiota. J. Adv. Res. 2021, 31, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Özkurt, E.; Hassani, M.A.; Sesiz, U.; Künzel, S.; Dagan, T.; Özkan, H.; Stukenbrock, E.H. Seed-derived microbial colonization of wild Emmer and domesticated bread Wheat (Triticum dicoccoides and T. aestivum) seedlings shows pronounced differences in overall diversity and composition. mBio 2020, 11, e02637-20. [Google Scholar] [CrossRef] [PubMed]
- Mammadov, J.; Buyyarapu, R.; Guttikonda, S.K.; Parliament, K.; Abdurakhmonov, I.Y.; Kumpatla, S.P. Wild Relatives of Maize, Rice, Cotton, and Soybean: Treasure Troves for Tolerance to Biotic and Abiotic Stresses. Front. Plant Sci. 2018, 9, 886. [Google Scholar] [CrossRef] [PubMed]
- Wassermann, B.; Cernava, T.; Müller, H.; Berg, C.; Berg, G. Seeds of native alpine plants host unique microbial communities embedded in cross-kingdom networks. Microbiome 2019, 7, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, G.; Raaijmakers, J.M. Saving seed microbiomes. ISME J. 12 2018, 12, 1167–1170. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, R.K.; Agrawal, A. Revised genebank standards for management of plant genetic resources. Indian J. Agric. Sci. 2015, 85, 157–165. [Google Scholar]
- Köberl, M.; Wagner, P.; Müller, H.; Matzer, R.; Unterfrauner, H.; Cernava, T.; Berg, G. Unraveling the Complexity of Soil Microbiomes in a Large-Scale Study Subjected to Different Agricultural Management in Styria. Front. Microbiol. 2020, 11, 1052. [Google Scholar] [CrossRef] [PubMed]
- Naylor, D.; Fansler, S.; Brislawn, C.; Nelson, W.C.; Hofmockel, K.S.; Jansson, J.K.; McClure, R.; Harwood, C.S. Deconstructing the Soil Microbiome into Reduced-Complexity Functional Modules. mBio 2020, 11, e01349-20. [Google Scholar] [CrossRef] [PubMed]
- Chandel, A.; Mann, R.; Kaur, J.; Norton, S.; Edwards, J.; Spangenberg, G.; Sawbridge, T. Implications of Seed Vault Storage Strategies for Conservation of Seed Bacterial Microbiomes. Front. Microbiol. 2021, 12, 784796. [Google Scholar] [CrossRef]
- Morales Moreira, Z.P.; Helgason, B.L.; Germida, J.J. Environment has a Stronger Effect than Host Plant Genotype in Shaping Spring Brassica napus Seed Microbiomes. Phytobiomes J. 2021, 5, 220–230. [Google Scholar] [CrossRef]
- Johnston-Monje, D.; Gutiérrez, J.P.; Lopez-Lavalle, L.A.B. Seed-Transmitted Bacteria and Fungi Dominate Juvenile Plant Microbiomes. Front. Microbiol. 2021, 12, 737616. [Google Scholar] [CrossRef] [PubMed]
- Rascovan, N.; Carbonetto, B.; Perrig, D.; Diaz, M.; Canciani, W.; Abalo, M.; Alloati, J.; Gonzalez-Anta, G.; Vazquez, M.P. Integrated analysis of root microbiomes of soybean and wheat from agricultural fields. Sci. Rep. 2016, 6, 28084. [Google Scholar] [CrossRef] [PubMed]
- Kämpfer, P.; Glaeser, S.P.; McInroy, J.A.; Busse, H.J. Cohnella rhizosphaerae sp. nov., isolated from the rhizosphere environment of Zea mays. Int. J. Adv. Sci. Technol. 2014, 64, 1811–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Hewezi, T.; Lebeis, S.L.; Pantalone, V.; Grewal, P.S.; Staton, M.E. Soil indigenous microbiome and plant genotypes cooperatively modify soybean rhizosphere microbiome assembly. BMC Microbiol. 2019, 19, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Kumari, S.; Vaishnav, A.; Choudhary, D.K.; Sharma, K.P. Isolation and characterization of plant growth promoting bacteria from soybean rhizosphere and their effect on soybean plant growth promotion. Int. J. Adv. Sci. Tech. Res. 2016, 6, 397–410. [Google Scholar] [CrossRef] [Green Version]
- Hone, H.; Mann, R.; Yang, G.; Kaur, J.; Tannenbaum, I.; Li, T.; Spangenberg, G.; Sawbridge, T. Profiling, isolation and characterisation of beneficial microbes from the seed microbiomes of drought tolerant wheat. Sci. Rep. 2021, 11, 11916. [Google Scholar] [CrossRef]
- Ofek, M.; Hadar, Y.; Minz, D. Ecology of Root Colonizing Massilia (Oxalobacteraceae). PLoS ONE 2012, 7, e40117. [Google Scholar] [CrossRef] [PubMed]
- Tannenbaum, I.; Kaur, J.; Mann, R.; Sawbridge, T.; Rodoni, B.; Spangenberg, G. Profiling the Lolium perenne microbiome: From seed to seed. Phytobiomes J. 2020, 4, 281–289. [Google Scholar] [CrossRef]
- Hardoim, P.R.; Andreote, F.D.; Reinhold-Hurek, B.; Sessitsch, A.; van Overbeek, L.S.; van Elsas, J.D. Rice root-associated bacteria: Insights into community structures across 10 cultivars. FEMS Microbiol. Ecol. 2011, 77, 154–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shade, A.; Jones, S.E.; Caporaso, J.G.; Handelsman, J.; Knight, R.; Fierer, N.; Gilbert, J.A. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio 2014, 5, e01371-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jousset, A.; Bienhold, C.; Chatzinotas, A.; Gallien, L.; Gobet, A.; Kurm, V.; Küsel, K.; Rillig, M.C.; Rivett, D.W.; Salles, J.F. Where less may be more: How the rare biosphere pulls ecosystems strings. ISME J. 2017, 11, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, A.; Zachow, C.; Müller, H.; Grand, A.; Temme, N.; Tilcher, R.; Berg, G. Understanding the Impact of Cultivar, Seed Origin, and Substrate on Bacterial Diversity of the Sugar Beet Rhizosphere and Suppression of Soil-Borne Pathogens. Front. Plant Sci. 2020, 11, 1450. [Google Scholar] [CrossRef] [PubMed]
- Barret, M.; Briand, M.; Bonneau, S.; Préveaux, A.; Valière, S.; Bouchez, O.; Hunault, G.; Simoneau, P.; Jacques, M.-A. Emergence Shapes the Structure of the Seed Microbiota. Appl. Environ. Microbiol. 2015, 81, 1257–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Danzberger, J.; Schöler, A.; Schröder, P.; Schloter, M.; Radl, V. Dominant Groups of Potentially Active Bacteria Shared by Barley Seeds become Less Abundant in Root Associated Microbiome. Front. Plant Sci. 2017, 8, 1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, T.; Chen, S. Colonization of wheat, maize and cucumber by Paenibacillus polymyxa WLY78. PLoS ONE 2017, 12, e0169980. [Google Scholar] [CrossRef] [PubMed]
- Timmusk, S.; Wagner, E.G.H. The plant-growth-promoting rhizobacterium Paenibacillus polymyxa induces changes in Arabidopsis thaliana gene expression: A possible connection between biotic and abiotic stress responses. Mol. Plant-Microbe Interact. 1999, 12, 951–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kefela, T.; Gachomo, E.W.; Kotchoni, S.O. Paenibacillus polymyxa, Bacillus licheniformis and Bradyrhizobium japonicum IRAT FA3 promote faster seed germination rate, growth and disease resistance under pathogenic pressure. J. Plant Biochem. Physiol. 2015, 3, 145. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Agnihotri, R.K.; Vamil, R.; Sharma, R. Effect of phytohormones on seed germination and seedling growth of Coriandrum sativum L. Pak. J. Biol. Sci. (PJBS) 2014, 17, 594–596. [Google Scholar] [CrossRef] [PubMed]
- Goggin, D.E.; Emery, R.N.; Kurepin, L.V.; Powles, S.B. A potential role for endogenous microflora in dormancy release, cytokinin metabolism and the response to fluridone in Lolium rigidum seeds. Ann. Bot. 2015, 115, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Li, J.; Ji, M.; Wu, Q.; Wu, X.; Ma, Y.; Sui, W.; Zhao, L.; Zhang, X. Non-synchronous Structural and Functional Dynamics During the Coalescence of Two Distinct Soil Bacterial Communities. Front. Microbiol. 2019, 10, 1125. [Google Scholar] [CrossRef]
- Ramoneda, J.; Le Roux, J.; Stadelmann, S.; Frossard, E.; Frey, B.; Gamper, H.A. Coalescence of rhizobial communities in soil interacts with fertilization and determines the assembly of rhizobia in root nodules. bioRxiv 2020. [Google Scholar] [CrossRef]
- Rocca, J.D.; Simonin, M.; Bernhardt, E.S.; Washburne, A.D.; Wright, J.P. Rare microbial taxa emerge when communities collide: Freshwater and marine microbiome responses to experimental mixing. Ecology 2020, 101, e02956. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.M.; Becker-Uncapher, I.; Carlson, M.; Fierer, N. Variable influences of soil and seed-associated bacterial communities on the assembly of seedling microbiomes. ISME J. 2021, 15, 2748–2762. [Google Scholar] [CrossRef] [PubMed]
- Wippel, K.; Tao, K.; Niu, Y.; Zgadzaj, R.; Kiel, N.; Guan, R.; Dahms, E.; Zhang, P.; Jensen, D.B.; Logemann, E.; et al. Host preference and invasiveness of commensal bacteria in the Lotus and Arabidopsis root microbiota. Nat. Microbiol. 2021, 6, 1150–1162. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.B. The seed microbiome: Origins, interactions, and impacts. Plant Soil 2018, 422, 7–34. [Google Scholar] [CrossRef]
- Moran, N.A. Symbiosis. Curr. Biol. 2006, 16, 866–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston-Monje, D.; Mousa, W.K.; Lazarovits, G.; Raizada, M.N. Impact of swapping soils on the endophytic bacterial communities of pre-domesticated, ancient and modern maize. BMC Plant Biol. 2014, 14, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, E.B. Microbial dynamics and interactions in the spermosphere. Annu. Rev. Phytopathol. 2004, 42, 271–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, R.; Noel, Z.A.; Benucci, G.M.N.; Chilvers, M.I.; Trail, F.; Bonito, G. Crop Management Impacts the Soybean (Glycine max) Microbiome. Front. Microbiol. 2020, 11, 1116. [Google Scholar] [CrossRef] [PubMed]
- Gdanetz, K.; Trail, F. The wheat microbiome under four management strategies, and potential for endophytes in disease protection. Phytobiomes 2017, 1, 158–168. [Google Scholar] [CrossRef] [Green Version]
- Bergelson, J.; Mittelstrass, J.; Horton, M.W. Characterizing both bacteria and fungi improves understanding of the Arabidopsis root microbiome. Sci. Rep. 2019, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Cregger, M.A.; Veach, A.M.; Yang, Z.K.; Crouch, M.J.; Vilgalys, R.; Tuskan, G.A.; Schadt, C.W. The Populus holobiont: Dissecting the effects of plant niches and genotype on the microbiome. Microbiome 2018, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Samuel, S.O.; Suzuki, K.; Asiloglu, R.; Harada, N. Soil-root interface influences the assembly of the endophytic bacterial community in rice plants. Biol. Fertil. Soils 2021, 58, 35–48. [Google Scholar] [CrossRef]
- Lindemann, J.; Upper, C. Aerial dispersal of epiphytic bacteria over bean plants. Appl. Environ. Microbiol. 1985, 50, 1229–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, E.; Bernhart, M.; Müller, H.; Winkler, J.; Berg, G. The Cucurbita pepo seed microbiome: Genotype-specific composition and implications for breeding. Plant Soil 2018, 422, 35–49. [Google Scholar] [CrossRef]
- Kong, H.G.; Song, G.C.; Ryu, C.-M. Inheritance of seed and rhizosphere microbial communities through plant–soil feedback and soil memory. Environ. Microbiol. Rep. 2019, 11, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Mohd Din, A.R.J.; Rosli, M.A.; Mohamad Azam, Z.; Othman, N.Z.; Sarmidi, M.R. Paenibacillus polymyxa Role Involved in Phosphate Solubilization and Growth Promotion of Zea mays under Abiotic Stress Condition. Proc. Natl. Acad. Sci. India Sect. B Biol. Sci. 2020, 90, 63–71. [Google Scholar] [CrossRef]
- Widdig, M.; Schleuss, P.-M.; Weig, A.R.; Guhr, A.; Biederman, L.A.; Borer, E.T.; Crawley, M.J.; Kirkman, K.P.; Seabloom, E.W.; Wragg, P.D.; et al. Nitrogen and Phosphorus Additions Alter the Abundance of Phosphorus-Solubilizing Bacteria and Phosphatase Activity in Grassland Soils. Front. Environ. Sci. 2019, 7, 185. [Google Scholar] [CrossRef] [Green Version]
- Kang, A.; Zhang, N.; Xun, W.; Dong, X.; Xiao, M.; Liu, Z.; Xu, Z.; Feng, H.; Zou, J.; Shen, Q.; et al. Nitrogen fertilization modulates beneficial rhizosphere interactions through signaling effect of nitric oxide. Plant Physiol. 2021, 188, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Mitter, B.; Pfaffenbichler, N.; Flavell, R.; Compant, S.; Antonielli, L.; Petric, A.; Berninger, T.; Naveed, M.; Sheibani-Tezerji, R.; von Maltzahn, G.; et al. A New Approach to Modify Plant Microbiomes and Traits by Introducing Beneficial Bacteria at Flowering into Progeny Seeds. Front. Microbiol. 2017, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prado, A.; Marolleau, B.; Vaissière, B.E.; Barret, M.; Torres-Cortes, G. Insect pollination: An ecological process involved in the assembly of the seed microbiota. Sci. Rep. 2020, 10, 3575. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandel, A.; Mann, R.; Kaur, J.; Norton, S.; Auer, D.; Edwards, J.; Spangenberg, G.; Sawbridge, T. The Role of Soil Microbial Diversity in the Conservation of Native Seed Bacterial Microbiomes. Microorganisms 2022, 10, 750. https://doi.org/10.3390/microorganisms10040750
Chandel A, Mann R, Kaur J, Norton S, Auer D, Edwards J, Spangenberg G, Sawbridge T. The Role of Soil Microbial Diversity in the Conservation of Native Seed Bacterial Microbiomes. Microorganisms. 2022; 10(4):750. https://doi.org/10.3390/microorganisms10040750
Chicago/Turabian StyleChandel, Ankush, Ross Mann, Jatinder Kaur, Sally Norton, Desmond Auer, Jacqueline Edwards, German Spangenberg, and Timothy Sawbridge. 2022. "The Role of Soil Microbial Diversity in the Conservation of Native Seed Bacterial Microbiomes" Microorganisms 10, no. 4: 750. https://doi.org/10.3390/microorganisms10040750