
Characterizing the Alteration in Rumen Microbiome and Carbohydrate-Active Enzymes Profile with Forage of Muskoxen Rumen through Comparative Metatranscriptomics

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design and Sample Collection
2.2. RNA Extraction
2.3. Total RNA Library Construction and Sequencing
2.4. Sequences Analysis of Microbiome
2.5. Mining of Carbohydrate-Active Enzymes for Muskoxen Fed Triticale Straw
2.6. Statistical Analysis
3. Results
3.1. Metatranscriptomics Sequence Data Statistics
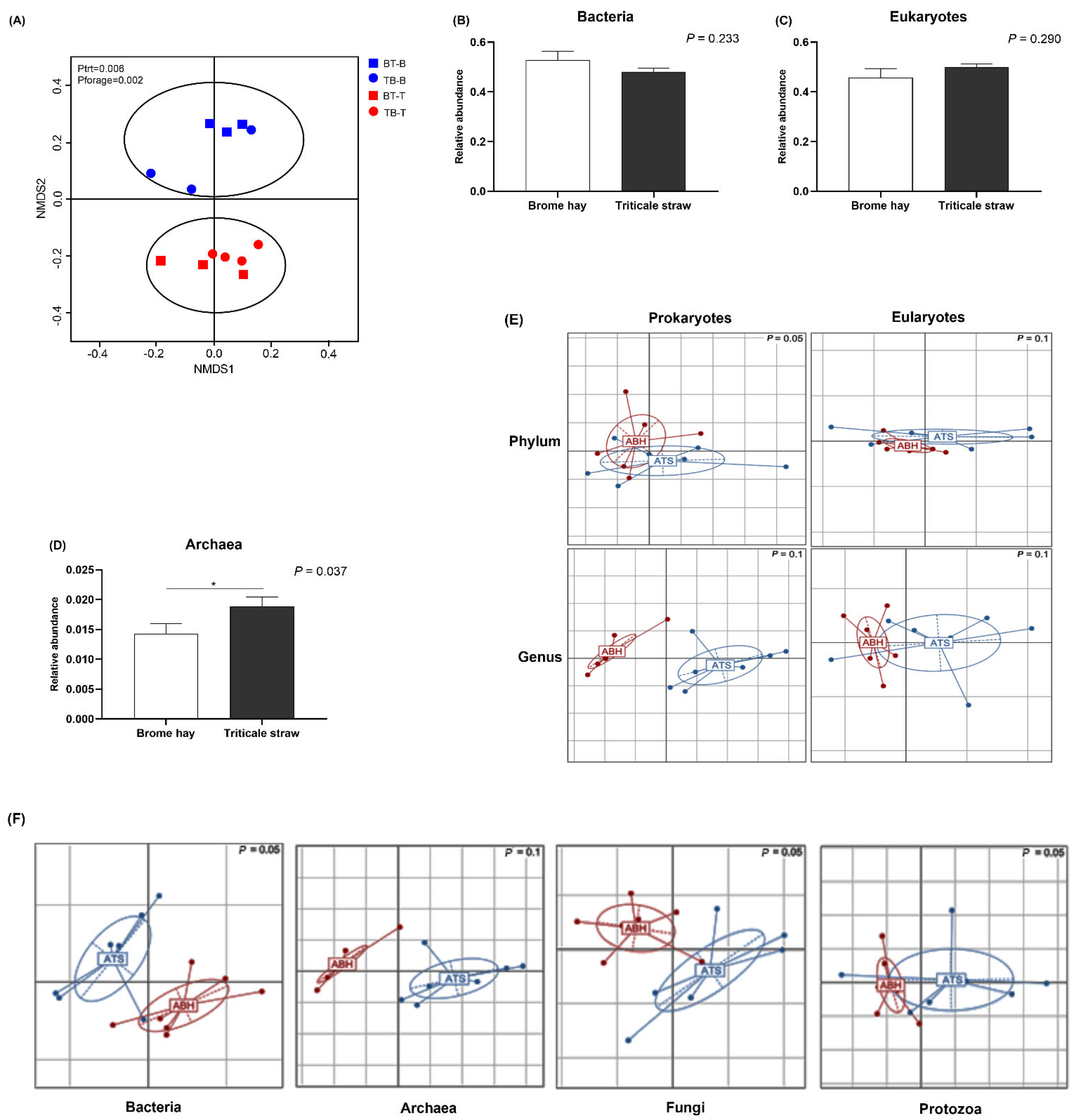
3.2. Total Community Structure and Diversity
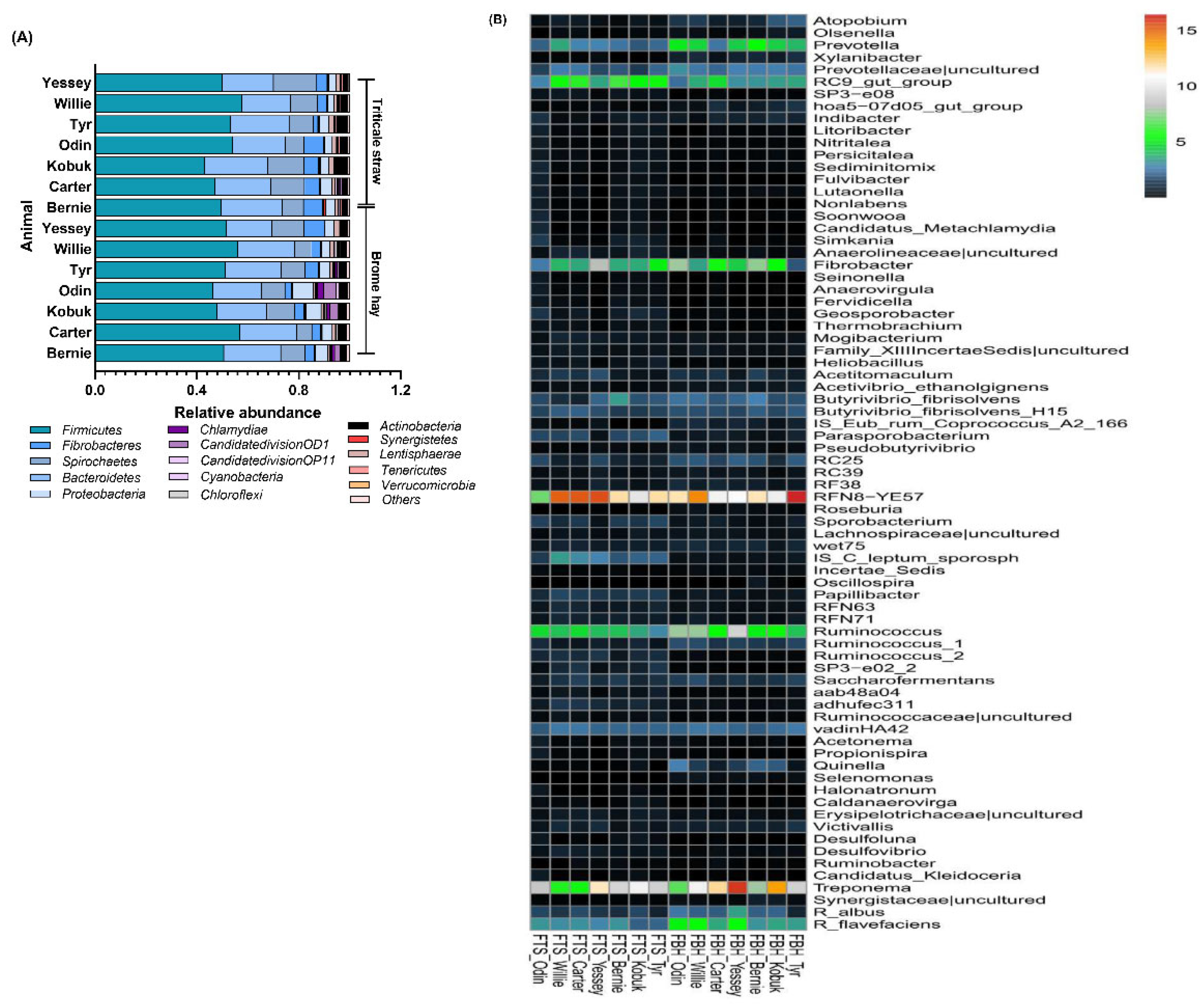
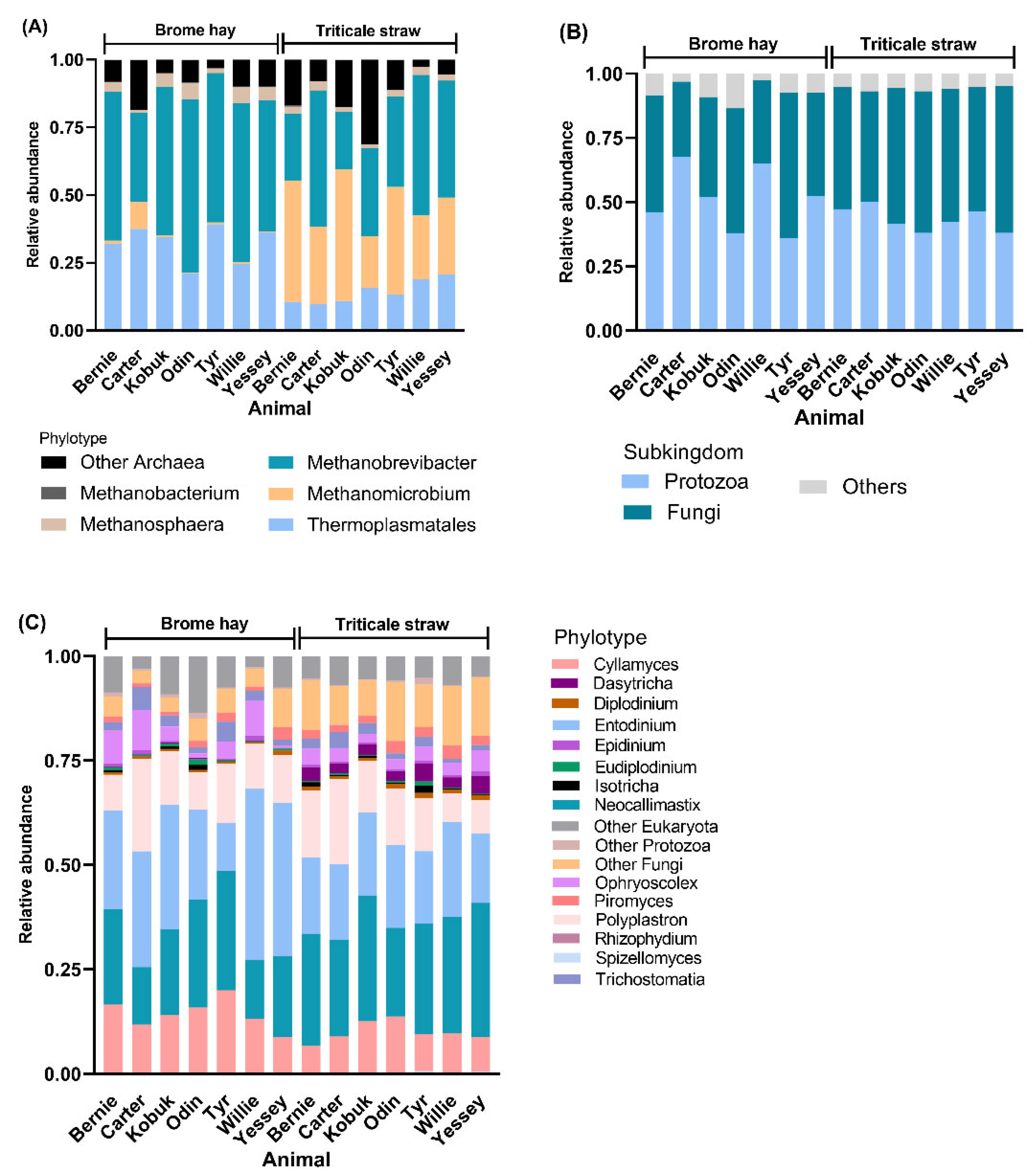
3.3. Highly Abundant Microbes in Muskoxen Rumen
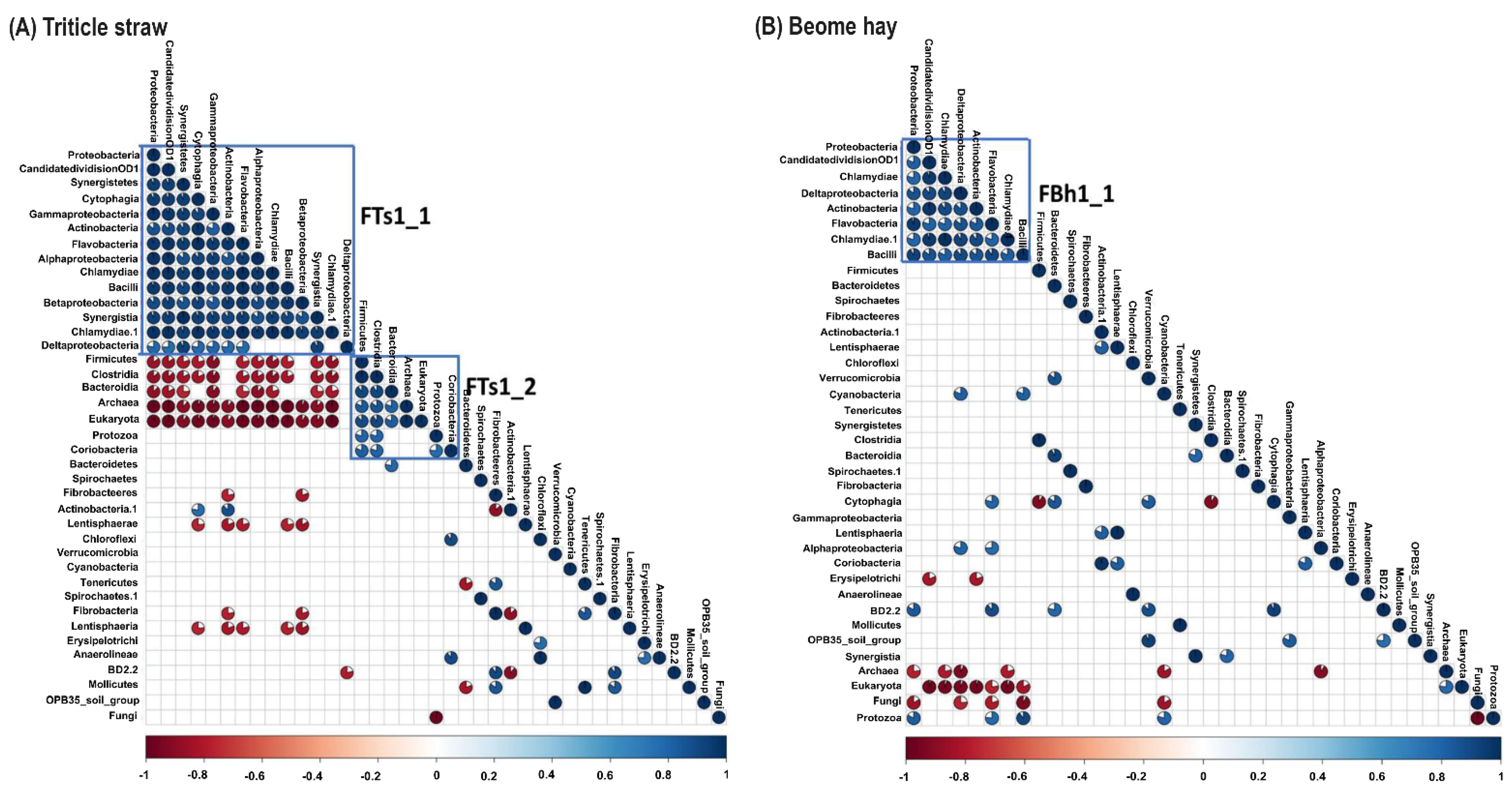
3.4. Unfolding Microbial Diversity in Response to Dietary Changes and Ruminant Animal
3.5. CAZymes Profile of Muskoxen Rumen Microbiome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dijkstra, J.; Tamminga, S. Simulation of the effects of diet on the contribution of rumen protozoa to degradation of fibre in the rumen. Br. J. Nutr. 1995, 74, 617–634. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Indugu, N.; Vecchiarelli, B.; Pitta, D.W. Associative patterns among anaerobic fungi, methanogenic archaea, and bacterial communities in response to changes in diet and age in the rumen of dairy cows. Front. Microbiol. 2015, 6, 781. [Google Scholar] [CrossRef] [Green Version]
- Kittelmann, S.; Seedorf, H.; Walters, W.A.; Clemente, J.C.; Knight, R.; Gordon, J.I.; Janssen, P.H. Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities. PLoS ONE 2013, 8, e47879. [Google Scholar] [CrossRef]
- Brulc, J.M.; Antonopoulos, D.A.; Miller, M.E.; Wilson, M.K.; Yannarell, A.C.; Dinsdale, E.A.; Edwards, R.E.; Frank, E.D.; Emerson, J.B.; Wacklin, P.; et al. Gene-centric metagenomics of the fiber-adherent bovine rumen microbiome reveals forage specific glycoside hydrolases. Proc. Natl. Acad. Sci. USA 2009, 106, 1948–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, K.J.; Mcallister, T.A. Compartmentation in the Rumen; Springer: Berlin/Heidelberg, Germany, 1997; pp. 492–522. [Google Scholar]
- Kong, Y.; Teather, R.; Forster, R. Composition, spatial distribution, and diversity of the bacterial communities in the rumen of cows fed different forages. FEMS Microbiol. Ecol. 2010, 74, 612–622. [Google Scholar] [CrossRef]
- Koike, S.; Yoshitani, S.; Kobayashi, Y.; Tanaka, K. Phylogenetic analysis of fiber-associated rumen bacterial community and PCR detection of uncultured bacteria. FEMS Microbiol. Lett. 2003, 229, 23–30. [Google Scholar] [CrossRef]
- Aurilia, V.; Martin, J.C.; McCrae, S.I.; Scott, K.P.; Rincon, M.T.; Flint, H.J. Three multidomain esterases from the cellulolytic rumen anaerobe Ruminococcus flavefaciens 17 that carry divergent dockerin sequences. Microbiology 2000, 146, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.K. Draft Genome Sequence of Fervidicella metallireducens Strain AeBT, an Iron-Reducing Thermoanaerobe from the Great Artesian Basin. Genome Announc. 2014, 2, e00345-14. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Kim, S.J.; Min, U.G.; Lee, Y.J.; Kim, S.G.; Jung, M.Y.; Seo, Y.S.; Rhee, S.K. Geosporobacter ferrireducens sp. nov., an anaerobic iron-reducing bacterium isolated from an oil-contaminated site. Antonie Van Leeuwenhoek 2015, 107, 971–977. [Google Scholar] [CrossRef]
- Paillard, D.; McKain, N.; Chaudhary, L.C.; Walker, N.D.; Pizette, F.; Koppova, I.; McEwan, N.R.; Kopecny, J.; Vercoe, P.E.; Louis, P.; et al. Relation between phylogenetic position, lipid metabolism and butyrate production by different Butyrivibrio-like bacteria from the rumen. Antonie Van Leeuwenhoek 2007, 91, 417–422. [Google Scholar] [CrossRef]
- Greening, R.C.; Leedle, J.A. Enrichment and isolation of Acetitomaculum ruminis, gen. nov., sp. nov.: Acetogenic bacteria from the bovine rumen. Arch. Microbiol. 1989, 151, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Blix, A.S.; Ness, J.; Lian, H.; Callaghan, T.V. Experimental reduction of food quality is not compensated with increased food intake in high-Arctic muskoxen (Ovibos moschatus). Br. J. Nutr. 2012, 108, 1337–1340. [Google Scholar] [CrossRef] [Green Version]
- Gunn, A. A Comparison of digestive Tract Morphology in muskoxen and caribou from Victoria Island, Northwest Territories, Canada. Rangifer 1992, 17, 17–19. [Google Scholar] [CrossRef] [Green Version]
- Ishaq, S.L.; Sundset, M.A.; Crouse, J.; Wright, A.G. High-throughput DNA sequencing of the moose rumen from different geographical locations reveals a core ruminal methanogenic archaeal diversity and a differential ciliate protozoal diversity. Microb. Genom. 2015, 1, e000034. [Google Scholar] [CrossRef]
- Pope, P.B.; Mackenzie, A.K.; Gregor, I.; Smith, W.; Sundset, M.A.; McHardy, A.C.; Morrison, M.; Eijsink, V.G. Metagenomics of the Svalbard reindeer rumen microbiome reveals abundance of polysaccharide utilization loci. PLoS ONE 2012, 7, e38571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruninger, R.J.; Sensen, C.W.; McAllister, T.A.; Forster, R.J. Diversity of rumen bacteria in canadian cervids. PLoS ONE 2014, 9, e89682. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Flores, A.; Sundset, M.A.; Pope, P.B.; Hagen, L.H.; Bockwoldt, M. First insight into the faecal microbiota of the high Arctic muskoxen (Ovibos moschatus). Microb. Genom. 2016, 2, e000066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, M.; Wang, P.; O’Toole, N.; Barboza, P.S.; Ungerfeld, E.; Leigh, M.B.; Selinger, L.B.; Butler, G.; Tsang, A.; McAllister, T.A.; et al. Snapshot of the eukaryotic gene expression in muskoxen rumen—A metatranscriptomic approach. PLoS ONE 2011, 6, e20521. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Xia, Y.; Seviour, R.; Forster, R.; McAllister, T.A. Biodiversity and composition of methanogenic populations in the rumen of cows fed alfalfa hay or triticale straw. FEMS Microbiol. Ecol. 2013, 84, 302–315. [Google Scholar] [CrossRef] [Green Version]
- Petri, R.M.; Schwaiger, T.; Penner, G.B.; Beauchemin, K.A.; Forster, R.J.; McKinnon, J.J.; McAllister, T.A. Characterization of the core rumen microbiome in cattle during transition from forage to concentrate as well as during and after an acidotic challenge. PLoS ONE 2013, 8, e83424. [Google Scholar] [CrossRef] [Green Version]
- Fouts, D.E.; Szpakowski, S.; Purushe, J.; Torralba, M.; Waterman, R.C.; MacNeil, M.D.; Alexander, L.J.; Nelson, K.E. Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen. PLoS ONE 2012, 7, e48289. [Google Scholar] [CrossRef]
- Kittelmann, S.; Janssen, P.H. Characterization of rumen ciliate community composition in domestic sheep, deer, and cattle, feeding on varying diets, by means of PCR-DGGE and clone libraries. FEMS Microbiol. Ecol. 2011, 75, 468–481. [Google Scholar] [CrossRef] [Green Version]
- Ishaq, S.L.; Wright, A.D. Design and validation of four new primers for next-generation sequencing to target the 18S rRNA genes of gastrointestinal ciliate protozoa. Appl. Environ. Microbiol. 2014, 80, 5515–5521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Liu, X.; Groenewald, J.Z. Phylogeny of anaerobic fungi (phylum Neocallimastigomycota), with contributions from yak in China. Antonie Van Leeuwenhoek 2017, 110, 87–103. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Elekwachi, C.; Jiao, J.; Wang, M.; Tang, S.; Zhou, C.; Tan, Z.; Forster, R.J. Changes in Metabolically Active Bacterial Community during Rumen Development, and Their Alteration by Rhubarb Root Powder Revealed by 16S rRNA Amplicon Sequencing. Front. Microbiol. 2017, 8, 159. [Google Scholar] [CrossRef]
- Wang, Z.; Elekwachi, C.O.; Jiao, J.; Wang, M.; Tang, S.; Zhou, C.; Tan, Z.; Forster, R.J. Investigation and manipulation of metabolically active methanogen community composition during rumen development in black goats. Sci. Rep. 2017, 7, 422. [Google Scholar] [CrossRef] [PubMed]
- Tymensen, L.D.; McAllister, T.A. Community structure analysis of methanogens associated with rumen protozoa reveals bias in universal archaeal primers. Appl. Environ. Microbiol. 2012, 78, 4051–4056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitta, D.W.; Kumar, S.; Veiccharelli, B.; Parmar, N.; Reddy, B.; Joshi, C.G. Bacterial diversity associated with feeding dry forage at different dietary concentrations in the rumen contents of Mehshana buffalo (Bubalus bubalis) using 16S pyrotags. Anaerobe 2014, 25, 31–41. [Google Scholar] [CrossRef]
- Turner, T.R.; Ramakrishnan, K.; Walshaw, J.; Heavens, D.; Alston, M.; Swarbreck, D.; Osbourn, A.; Grant, A.; Poole, P.S. Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. ISME J. 2013, 7, 2248–2258. [Google Scholar] [CrossRef] [Green Version]
- Comtet-Marre, S.; Parisot, N.; Lepercq, P.; Chaucheyras-Durand, F.; Mosoni, P.; Peyretaillade, E.; Bayat, A.R.; Shingfield, K.J.; Peyret, P.; Forano, E. Metatranscriptomics Reveals the Active Bacterial and Eukaryotic Fibrolytic Communities in the Rumen of Dairy Cow Fed a Mixed Diet. Front. Microbiol. 2017, 8, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Tian, Y.; Li, J.; Luo, Y.; Liu, D.; Zheng, H.; Wang, J.; Dong, Z.; Hu, S.; Huang, L. Metatranscriptomic analyses of plant cell wall polysaccharide degradation by microorganisms in the cow rumen. Appl. Environ. Microbiol. 2015, 81, 1375–1386. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.D.; Patel, A.K.; Parmar, N.R.; Shah, T.M.; Patel, J.B.; Pandya, P.R.; Joshi, C.G. Microbial and Carbohydrate Active Enzyme profile of buffalo rumen metagenome and their alteration in response to variation in the diet. Gene 2014, 545, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Qi, M.; Barboza, P.; Leigh, M.B.; Ungerfeld, E.; Selinger, L.B.; McAllister, T.A.; Forster, R.J. Isolation of high-quality total RNA from rumen anaerobic bacteria and fungi, and subsequent detection of glycoside hydrolases. Can. J. Microbiol. 2011, 57, 590–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Gilna, P.; Li, W. Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 2009, 25, 1338–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W. Analysis and comparison of very large metagenomes with fast clustering and functional annotation. BMC Bioinform. 2009, 10, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Warnecke, F.; Luginbühl, P.; Ivanova, N.; Ghassemian, M.; Richardson, T.H.; Stege, J.T.; Cayouette, M.; McHardy, A.C.; Djordjevic, G.; Aboushadi, N.; et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature 2007, 450, 560–565. [Google Scholar] [CrossRef]
- Bohra, V.; Tikariha, H.; Dafale, N.A. Genomically Defined Paenibacillus polymyxa ND24 for Efficient Cellulase Production Utilizing Sugarcane Bagasse as a Substrate. Appl. Biochem. Biotechnol. 2019, 187, 266–281. [Google Scholar] [CrossRef]
- Dray, S.; Dufour, A. The ade4 Package: Implementing the Duality Diagram for Ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Craig, W.M.; Broderick, G.A.; Ricker, D.B. Quantitation of microorganisms associated with the particulate phase of ruminal ingesta. J. Nutr. 1987, 117, 56. [Google Scholar] [CrossRef]
- Olubobokun, J.A.; Craig, W.M. Quantity and characteristics of microorganisms associated with ruminal fluid or particles. J. Anim. Sci. 1990, 68, 3360–3370. [Google Scholar] [CrossRef]
- Mcallister, T.A.; Newbold, C.J. Redirecting rumen fermentation to reduce methanogenesis. Aust. J. Exp. Agric. 2008, 48, 7–13. [Google Scholar] [CrossRef]
- Carberry, C.A.; Waters, S.M.; Kenny, D.A.; Creevey, C.J. Rumen methanogenic genotypes differ in abundance according to host residual feed intake phenotype and diet type. Appl. Environ. Microbiol. 2014, 80, 586–594. [Google Scholar] [CrossRef] [Green Version]
- Grilli, D.J.; Fliegerova, K.; Kopecny, J.; Lama, S.P.; Egea, V.; Sohaefer, N.; Pereyra, C.; Ruiz, M.S.; Sosa, M.A.; Arenas, G.N.; et al. Analysis of the rumen bacterial diversity of goats during shift from forage to concentrate diet. Anaerobe 2016, 42, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Ben David, Y.; Dassa, B.; Borovok, I.; Lamed, R.; Koropatkin, N.M.; Martens, E.C.; White, B.A.; Bernalier-Donadille, A.; Duncan, S.H.; Flint, H.J.; et al. Ruminococcal cellulosome systems from rumen to human. Environ. Microbiol. 2015, 17, 3407–3426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarakanov, B.V.; Lavlinskii, D. [Cellulosolytic bacteria of the genus Ruminococcus from cattle rumen]. Mikrobiologiia 1998, 67, 518–521. [Google Scholar] [PubMed]
- Xie, G.; Bruce, D.C.; Challacombe, J.F.; Chertkov, O.; Detter, J.C.; Gilna, P.; Han, C.S.; Lucas, S.; Misra, M.; Myers, G.L.; et al. Genome sequence of the cellulolytic gliding bacterium Cytophaga hutchinsonii. Appl. Environ. Microbiol. 2007, 73, 3536–3546. [Google Scholar] [CrossRef] [Green Version]
- Ransom-Jones, E.; Jones, D.L.; McCarthy, A.J.; McDonald, J.E. The Fibrobacteres: An important phylum of cellulose-degrading bacteria. Microb. Ecol. 2012, 63, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Sijpesteijn, A.K. On Ruminococcus flavefaciens, a cellulose-decomposing bacterium from the rumen of sheep and cattle. J. Gen. Microbiol. 1951, 5, 869–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klieve, A.V.; O’Leary, M.N.; McMillen, L.; Ouwerkerk, D. Ruminococcus bromii, identification and isolation as a dominant community member in the rumen of cattle fed a barley diet. J. Appl. Microbiol. 2007, 103, 2065–2073. [Google Scholar] [CrossRef] [PubMed]
- Christopherson, M.R.; Dawson, J.A.; Stevenson, D.M.; Cunningham, A.C.; Bramhacharya, S.; Weimer, P.J.; Kendziorski, C.; Suen, G. Unique aspects of fiber degradation by the ruminal ethanologen Ruminococcus albus 7 revealed by physiological and transcriptomic analysis. BMC Genom. 2014, 15, 1066. [Google Scholar] [CrossRef] [Green Version]
- Mrazek, J.; Tepsic, K.; Avgustin, G.; Kopecny, J. Diet-dependent shifts in ruminal butyrate-producing bacteria. Folia Microbiol. 2006, 51, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Kasperowicz, A.; Stan-Glasek, K.; Guczynska, W.; Pristas, P.; Javorsky, P.; Vandzurova, A.; Michalowski, T. beta-Fructofuranosidase and sucrose phosphorylase of rumen bacterium Pseudobutyrivibrio ruminis strain 3. World J. Microbiol. Biotechnol. 2012, 28, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Hang, S.; Mao, S.; Zhu, W. Diversity of butyrivibrio group bacteria in the rumen of goats and its response to the supplementation of garlic oil. Asian-Australas. J. Anim. Sci. 2014, 27, 179–186. [Google Scholar] [CrossRef]
- Kane, M.D.; Breznak, J.A. Acetonema longum gen. nov. sp. nov., an H2/CO2 acetogenic bacterium from the termite, Pterotermes occidentis. Arch. Microbiol. 1991, 156, 91–98. [Google Scholar] [CrossRef]
- Ueki, A.; Watanabe, M.; Ohtaki, Y.; Kaku, N.; Ueki, K. Description of Propionispira arcuata sp. nov., isolated from a methanogenic reactor of cattle waste, reclassification of Zymophilus raffinosivorans and Zymophilus paucivorans as Propionispira raffinosivorans comb. nov. and Propionispira paucivorans comb. nov. and emended description of the genus Propionispira. Int. J. Syst. Evol. Microbiol. 2014, 64, 3571–3577. [Google Scholar] [CrossRef] [Green Version]
- Woyke, T.; Teeling, H.; Ivanova, N.N.; Huntemann, M.; Richter, M.; Gloeckner, F.O.; Boffelli, D.; Anderson, I.J.; Barry, K.W.; Shapiro, H.J.; et al. Symbiosis insights through metagenomic analysis of a microbial consortium. Nature 2006, 443, 950–955. [Google Scholar] [CrossRef]
- Peltier, T.C.; Barboza, P.S.; Blake, J.E. Seasonal hyperphagia does not reduce digestive efficiency in an Arctic grazer. Physiol. Biochem. Zool. 2003, 76, 471–483. [Google Scholar] [CrossRef]
- Mechichi, T.; Labat, M.; Garcia, J.L.; Thomas, P.; Patel, B.K. Sporobacterium olearium gen. nov., sp. nov., a new methanethiol-producing bacterium that degrades aromatic compounds, isolated from an olive mill wastewater treatment digester. Int. J. Syst. Bacteriol. 1999, 49 Pt 4, 1741–1748. [Google Scholar] [CrossRef] [Green Version]
- Thompson, T.E.; Conrad, R.; Zeikus, J.G. Regulation of carbon and electron flow in Propionispira arboris: Physiological function of hydrogenase and its role in homopropionate formation. FEMS Microbiol. Lett. 1984, 22, 265–271. [Google Scholar] [CrossRef]
- Krzmarzick, M.J.; Crary, B.B.; Harding, J.J.; Oyerinde, O.O.; Leri, A.C.; Myneni, S.C.; Novak, P.J. Natural niche for organohalide-respiring Chloroflexi. Appl. Environ. Microbiol. 2012, 78, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Bachar, A.; Omoregie, E.; Wit, R.D.; Jonkers, H.M. Diversity and Function of Chloroflexus-Like Bacteria in a Hypersaline Microbial Mat: Phylogenetic Characterization and Impact on Aerobic Respiration. Appl. Environ. Microbiol. 2007, 73, 3975–3983. [Google Scholar] [CrossRef] [Green Version]
- Klouche, N.; Fardeau, M.L.; Lascourreges, J.F.; Cayol, J.L.; Hacene, H.; Thomas, P.; Magot, M. Geosporobacter subterraneus gen. nov., sp. nov., a spore-forming bacterium isolated from a deep subsurface aquifer. Int. J. Syst. Evol. Microbiol. 2007, 57, 1757–1761. [Google Scholar] [CrossRef] [PubMed]
- Ciranna, A.; Santala, V.; Karp, M. Biohydrogen production in alkalithermophilic conditions: Thermobrachium celere as a case study. Bioresour. Technol. 2011, 102, 8714–8722. [Google Scholar] [CrossRef] [PubMed]
- Fonty, G.; Williams, A.G.; Bonnemoy, F.; Morvan, B.; Withers, S.E.; Gouet, P. Effect of Methanobrevibacter sp MF1 inoculation on glycoside hydrolase and polysaccharide depolymerase activities, wheat straw degradation and volatile fatty acid concentrations in the rumen of gnotobiotically-reared lambs. Anaerobe 1997, 3, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.L.; Wolin, M.J.; Zhao, H.X.; Bryant, M.P. Characteristics of methanogens isolated from bovine rumen. Appl. Environ. Microbiol. 1986, 51, 201–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rea, S.; Bowman, J.P.; Popovski, S.; Pimm, C.; Wright, A.D. Methanobrevibacter millerae sp. nov. and Methanobrevibacter olleyae sp. nov., methanogens from the ovine and bovine rumen that can utilize formate for growth. Int. J. Syst. Evol. Microbiol. 2007, 57, 450–456. [Google Scholar] [CrossRef]
- Lee, J.H.; Kumar, S.; Lee, G.H.; Chang, D.H.; Rhee, M.S.; Yoon, M.H.; Kim, B.C. Methanobrevibacter boviskoreani sp. nov., isolated from the rumen of Korean native cattle. Int. J. Syst. Evol. Microbiol. 2013, 63, 4196–4201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanner, R.S.; Wolfe, R.S. Nutritional requirements of Methanomicrobium mobile. Appl. Environ. Microbiol. 1988, 54, 625–628. [Google Scholar] [CrossRef] [Green Version]
- McAllister, T.A.; Bae, H.D.; Jones, G.A.; Cheng, K.J. Microbial attachment and feed digestion in the rumen. J. Anim. Sci. 1994, 72, 3004–3018. [Google Scholar] [CrossRef] [Green Version]
- Hernández, C.; Alamilla-Ortiz, Z.L.; Escalante, A.E.; Navarro-Díaz, M.; Carrillo-Reyes, J.; Moreno-Andrade, I.; Valdez-Vazquez, I. Heat-shock treatment applied to inocula for H2 production decreases microbial diversities, interspecific interactions and performance using cellulose as substrate. Int. J. Hydrogen Energy 2019, 44, 13126–13134. [Google Scholar] [CrossRef]
- Dollhofer, V.; Podmirseg, S.M.; Callaghan, T.M.; Griffith, G.W.; Fliegerová, K. Anaerobic Fungi and Their Potential for Biogas Production. Adv. Biochem. Eng. Biotechnol. 2015, 151, 41–61. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zheng, X.; Pilgaard, B.; Holck, J.; Muschiol, J.; Li, S.; Lange, L. Identification and characterization of GH11 xylanase and GH43 xylosidase from the chytridiomycetous fungus, Rhizophlyctis rosea. Appl. Microbiol. Biotechnol. 2019, 103, 777–791. [Google Scholar] [CrossRef] [Green Version]
- Akin, D.E.; Amos, H.E. Mode of attack on orchardgrass leaf blades by rumen protozoa. Appl. Environ. Microbiol. 1979, 37, 332–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, M.W.; Zhang, Q.; Yang, Y.; Zou, C.; Li, L.; Liang, X.; Wei, S.; Lin, B. Ruminal fermentation and microbial community differently influenced by four typical subtropical forages in vitro. Anim. Nutr. 2018, 4, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef]
- Jose, V.L.; More, R.P.; Appoothy, T.; Arun, A.S. In depth analysis of rumen microbial and carbohydrate-active enzymes profile in Indian crossbred cattle. Syst. Appl. Microbiol. 2017, 40, 160–170. [Google Scholar] [CrossRef]
- Artzi, L.; Dadosh, T.; Milrot, E.; Moraïs, S.; Levin-Zaidman, S.; Morag, E.; Bayer, E.A. Colocalization and Disposition of Cellulosomes in Clostridium clariflavum as Revealed by Correlative Superresolution Imaging. mBio 2018, 9, e00012-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Zhu, Y.; Luo, Y.; Song, L.; Liu, D.; Liu, L.; Chen, F.; Wang, M.; Li, J.; Zeng, X.; et al. Metagenomic insights into the fibrolytic microbiome in yak rumen. PLoS ONE 2012, 7, e40430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kala, A.; Kamra, D.N.; Agarwal, N.; Chaudhary, L.C.; Joshi, C.G. Insights into Metatranscriptome, and CAZymes of Buffalo Rumen Supplemented with Blend of Essential Oils. Indian J. Microbiol. 2020, 60, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Anju, K.; Kamra, D.; Chaudhary, L.; Neeta, A. Metagenomics and CAZymes in rumen: A review. Indian J. Anim. Nutr. 2019, 36, 1–10. [Google Scholar]
- Bernardes, A.; Pellegrini, V.O.A.; Curtolo, F.; Camilo, C.M.; Mello, B.L.; Johns, M.A.; Scott, J.L.; Guimaraes, F.E.C.; Polikarpov, I. Carbohydrate binding modules enhance cellulose enzymatic hydrolysis by increasing access of cellulases to the substrate. Carbohydr. Polym. 2019, 211, 57–68. [Google Scholar] [CrossRef]
- Mota, T.R.; Oliveira, D.; Marchiosi, R.; Ferrarese-Filho, O.; Santos, W. Plant cell wall composition and enzymatic deconstruction. AIMS Bioeng. 2018, 5, 63–77. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef]
- Biddle, A.; Stewart, L.; Blanchard, J.; Leschine, S. Untangling the genetic basis of fibrolytic specialization by Lachnospiraceae and Ruminococcaceae in diverse gut communities. Diversity 2013, 5, 627–640. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
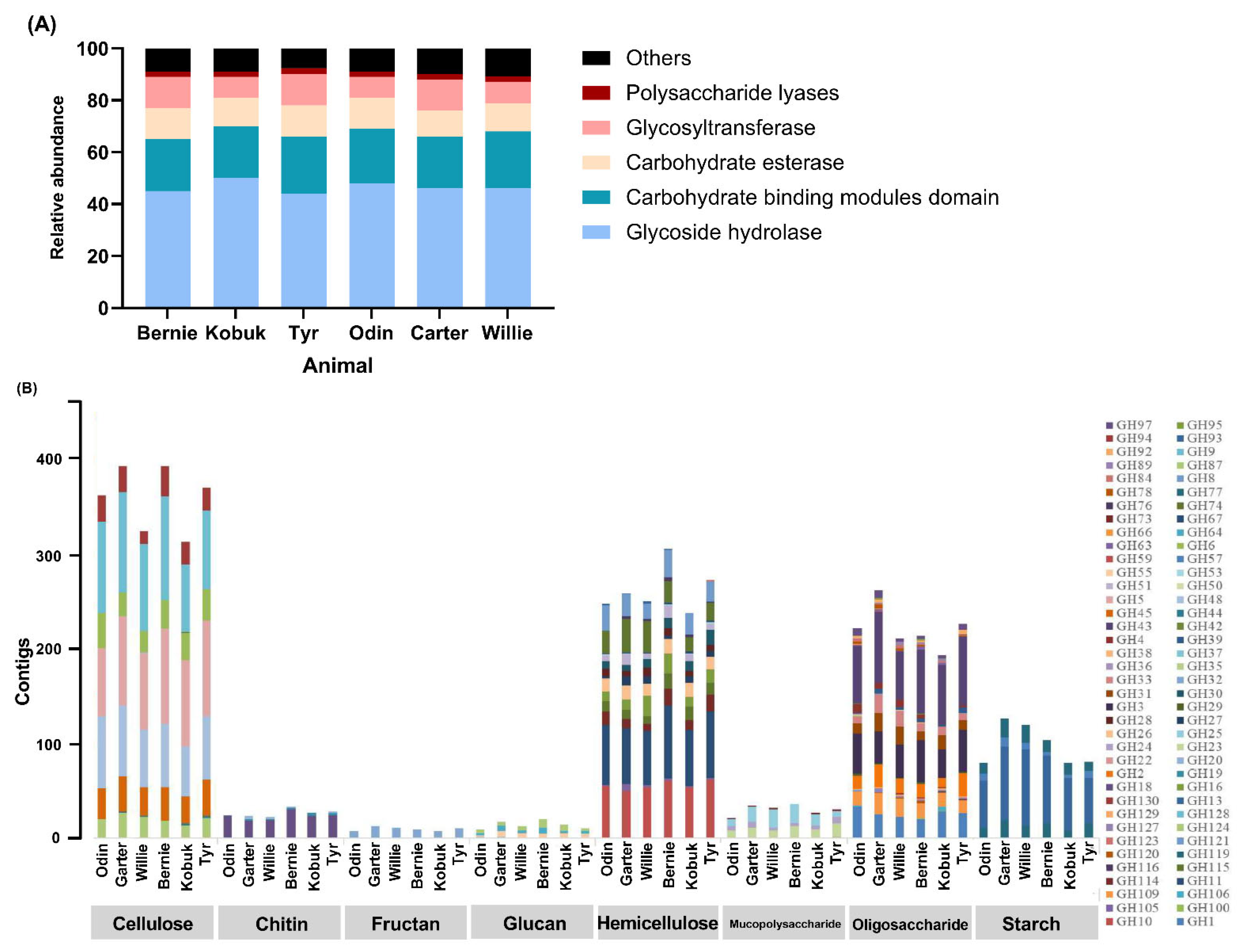{kind=link}
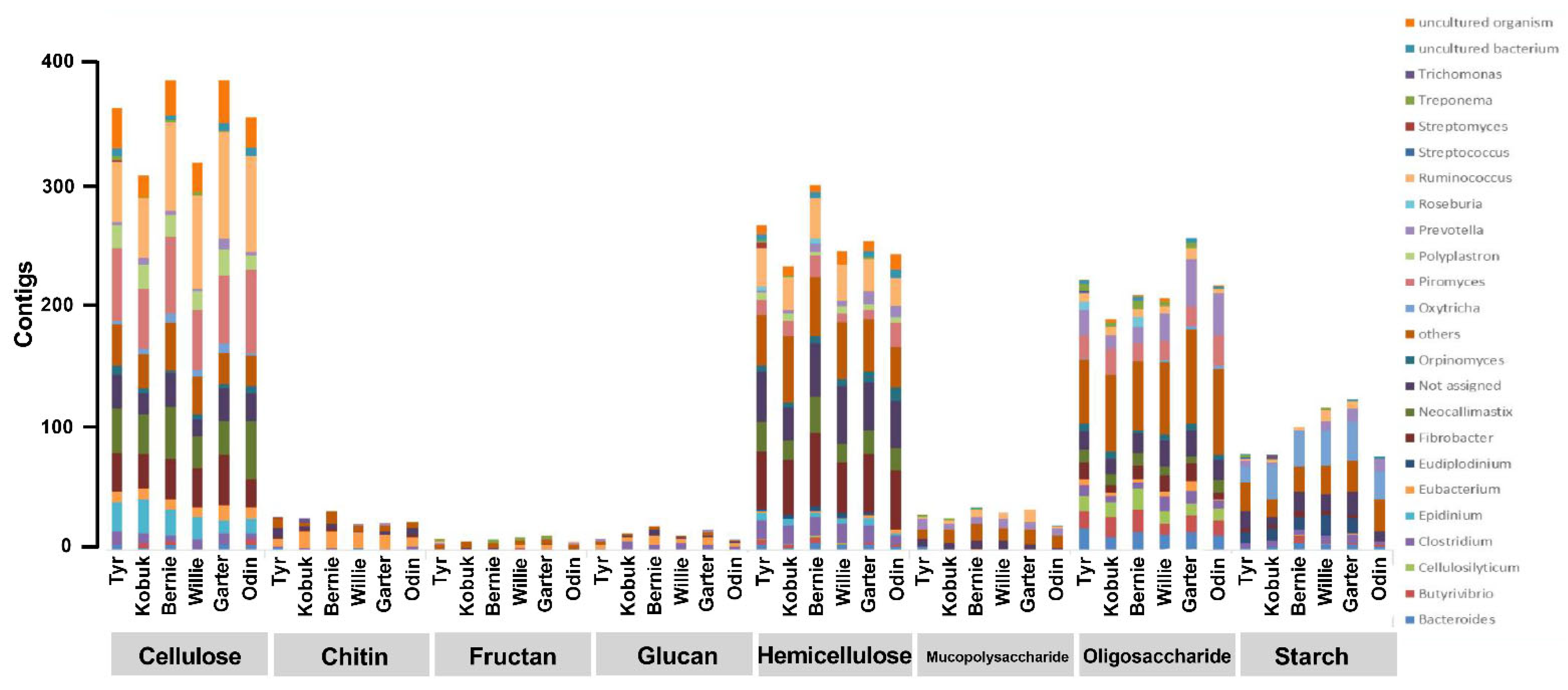{kind=link}
| Item | Triticale Straw | Brome Hay | |
|---|---|---|---|
| Bacteria | Phylum | Proteobacteria, CandidatedivisionOD1, Chloroflexi, and Chlamydiae | Synergistetes, Tenericutes, and Cyanobacteria |
| Class | Chlamydiae, Deltaproteobacteria, Anaerolineae, Actinobacteria, and Betaproteobacteria | Coriobacteria, BD2-2, Synergistia, and Mollicutes | |
| Order | Chlamydiales, Micrococcales, Rickettsiales, Anaerolineales, Desulfovibrionales, Bacillales, Burkholderiales, Rhodobacterales, and Thermoanaerobacterales | Coriobacteriales, RF9, Synergistales, Rhodospirillales, and Aeromonadales | |
| Family | Ruminococcaceae, Rikenellaceae, BS11_gut_group, Clostridiaceae, Simkaniaceae, Anaerolineaceae, Desulfovibrionaceae, Heliobacteriaceae, Peptococcaceae, Thermoactinomycetaceae, Cytophagaceae, Family_IIIIncertaeSedis, Desulfobacteraceae, Flammeovirgaceae, Cystobacterineae, Flavobacteriaceae, and Parachlamydiaceae | Prevotellaceae, Veillonellaceae, Coriobacteriaceae, S24-7, Synergistaceae, Rhodospirillaceae, and Succinivibrionaceae | |
| Genus | IS_C_leptum_sporosph, RC9_gut_group, Parasporobacterium, Papillibacter, Ruminococcus_2, SP3-e02_2, Sporobacterium, adhufec311, Geosporobacter, Simkania, SP3_e08, Thermobrachium, RFN63, Desulfovibrio, Anaerolineaceae|uncultured, Fervidicella, Heliobacillus, RFN71, aab48a04, Anaerovirgula, Erysipelotrichaceae|uncultured, Ruminococcaceae|uncultured, Seinonella, Nonlabens, Mogibacterium, Desulfoluna, Persicitalea, Caldanaerovirga, Fulvibacter, Candidatus_Kleidoceria, Sediminitomix, Candidatus_Metachlamydia, and Propionispira | Ruminococcus, Prevotella, Quinella, Atopobium, RC25, Ruminococcus_1, Xylanibacter, IS_Eub_rum_Coprococcus_A2_166, hoa5-07d05_gut_group, Selenomonas, Roseburia, Acetivibrio_ethanolgignens, RF38, Olsenella, Incertae_Sedis, Synergistaceae|uncultured, Pseudobutyrivibrio, and wet75 | |
| Species | R. flavefaciens, and R.albus | ||
| Fungi | Cyllamyces | Spizellomyces | |
| Protozoa | Entodinium | Dasytricha, and Diplodinium | |
| Archaea | Methanomicrobium | Thermoplasmatales and Methanobrevibacter |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Elekwachi, C.O.; Bai, S.; Luo, Y.; Zhang, K.; Forster, R.J. Characterizing the Alteration in Rumen Microbiome and Carbohydrate-Active Enzymes Profile with Forage of Muskoxen Rumen through Comparative Metatranscriptomics. Microorganisms 2022, 10, 71. https://doi.org/10.3390/microorganisms10010071
Wu X, Elekwachi CO, Bai S, Luo Y, Zhang K, Forster RJ. Characterizing the Alteration in Rumen Microbiome and Carbohydrate-Active Enzymes Profile with Forage of Muskoxen Rumen through Comparative Metatranscriptomics. Microorganisms. 2022; 10(1):71. https://doi.org/10.3390/microorganisms10010071
Chicago/Turabian StyleWu, Xiaofeng, Chijioke O. Elekwachi, Shiping Bai, Yuheng Luo, Keying Zhang, and Robert J. Forster. 2022. "Characterizing the Alteration in Rumen Microbiome and Carbohydrate-Active Enzymes Profile with Forage of Muskoxen Rumen through Comparative Metatranscriptomics" Microorganisms 10, no. 1: 71. https://doi.org/10.3390/microorganisms10010071