
ApoB100 and Atherosclerosis: What’s New in the 21st Century?

 , , , , and
, , , , and
Abstract
:1. Introduction
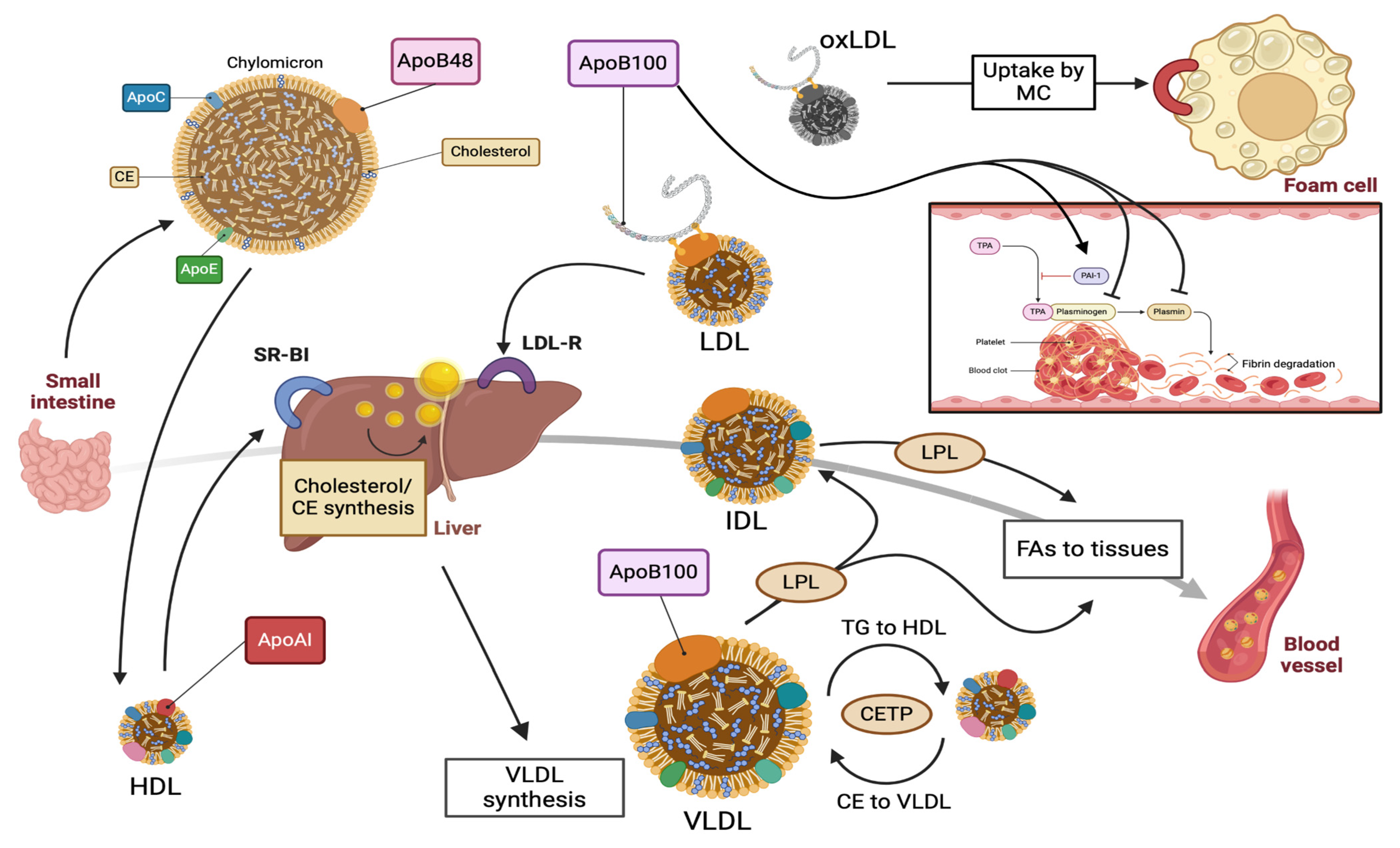
2. ApoB100: Structure, Function, and Metabolism
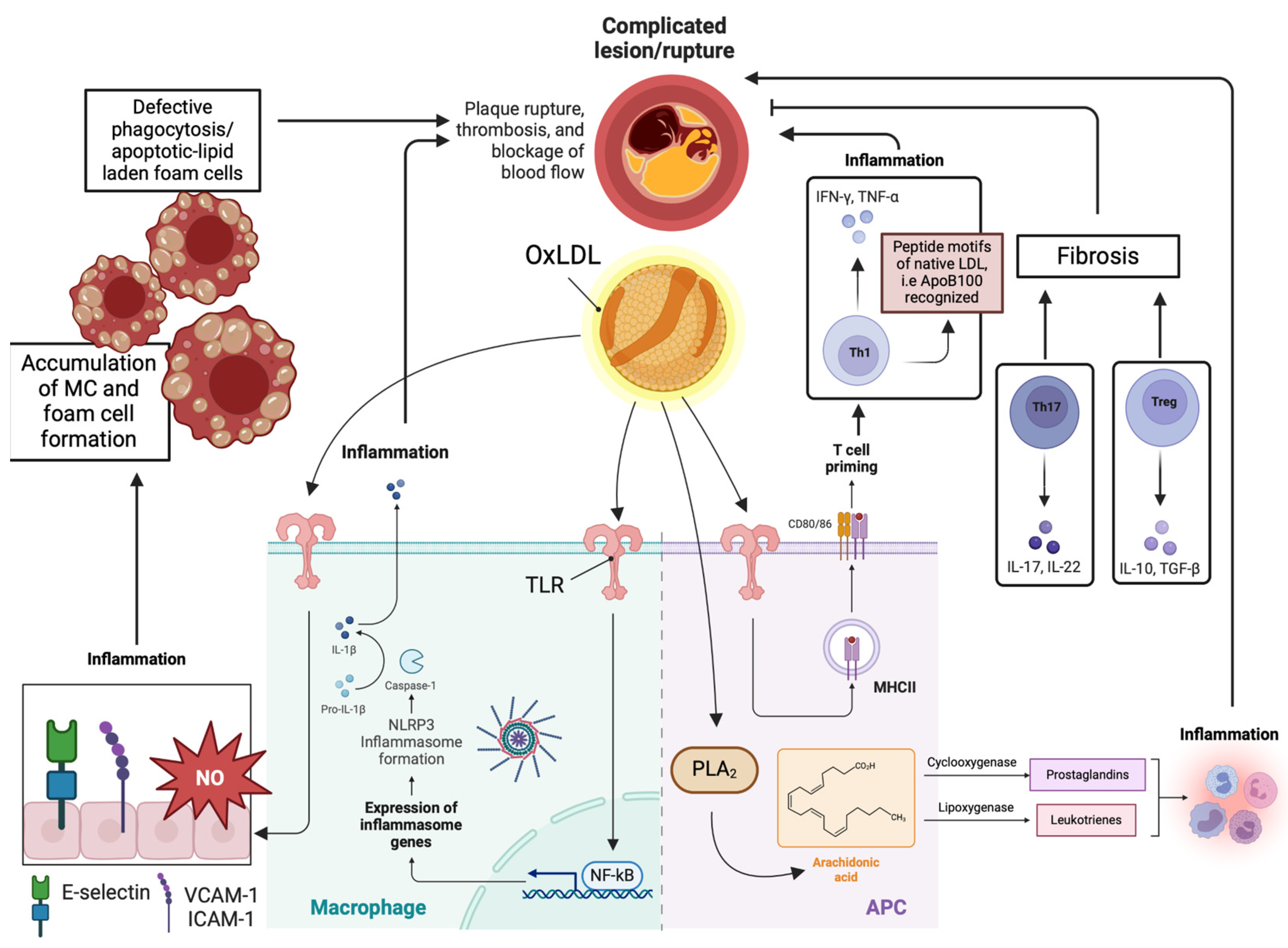
3. Molecular Mechanisms Regarding the Pathophysiology of Atherosclerosis
4. Novel Predictors of Cardiovascular Burden: The Role of ApoB100
4.1. ApoB100
4.2. The ApoB100/ApoA1 Ratio
4.3. Non-HDL-C
4.4. Autoantibodies against ApoB100: A Novel Biomarker of the Vulnerable Atheromatous Plaque
5. ApoB100 in Different Pathological Conditions
5.1. Diabetes Mellitus
5.2. Obesity
5.3. Liver Steatosis (NAFLD/MAFLD)
5.4. Polycystic Ovary Syndrome (PCOS)
5.5. Peripheral Artery Disease (PAD)
5.6. Chronic Kidney Disease (CKD)
6. ApoB Gene Mutations and Cardiovascular Risk
6.1. Familial Defective Apolipoprotein B100 (FDB)
6.2. ApoB-Related Familial Hypobetalipoproteinemia (FHBL)
7. The Effects of Lipid-Modifying Interventions on ApoB-100
7.1. Lifestyle Interventions
7.2. Traditional Hypolipidemic Agents
7.2.1. Statins
7.2.2. Ezetimibe
7.2.3. Fibrates
7.2.4. Omega-3 Fatty Acids (FAs)
7.3. Lipoprotein Apheresis (LA)
7.4. The New Era of Lipid-Modifying Therapies
7.4.1. Mipomersen and Lomitapide
7.4.2. Bempedoic Acid (BA)
7.4.3. PCSK9 Inhibition
7.4.4. Angiopoietin-Like 3 Protein (ANGPTL3) Inhibitors
7.4.5. Cholesteryl Ester Transfer Protein (CETP) Inhibitors
8. Immunomodulation of ApoB100 in the Treatment of Atherosclerosis
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Zheng, C.; Ikewaki, K.; Walsh, B.W.; Sacks, F.M. Metabolism of apoB lipoproteins of intestinal and hepatic origin during constant feeding of small amounts of fat. J. Lipid. Res. 2006, 47, 1771–1779. [Google Scholar] [CrossRef]
- Shen, H.; Damcott, C.M.; Rampersaud, E.; Pollin, T.I.; Horenstein, R.B.; McArdle, P.F.; Peyser, P.A.; Bielak, L.F.; Post, W.S.; Chang, Y.P.; et al. Familial defective apolipoprotein B-100 and increased low-density lipoprotein cholesterol and coronary artery calcification in the old order amish. Arch. Intern. Med. 2010, 170, 1850–1855. [Google Scholar] [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Fernando, S.; Schwarz, N.; Tan, J.T.; Bursill, C.A.; Psaltis, P.J. Inflammation as a Therapeutic Target in Atherosclerosis. J. Clin. Med. 2019, 8, 1109. [Google Scholar] [CrossRef]
- Behbodikhah, J.; Ahmed, S.; Elyasi, A.; Kasselman, L.J.; De Leon, J.; Glass, A.D.; Reiss, A.B. Apolipoprotein B and Cardiovascular Disease: Biomarker and Potential Therapeutic Target. Metabolites 2021, 11, 690. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Gu, Z.W.; Yang, M.; Gotto, A.M., Jr. Primary structure of apoB-100. Chem. Phys. Lipids. 1994, 67–68, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Blasiole, D.A.; Davis, R.A.; Attie, A.D. The physiological and molecular regulation of lipoprotein assembly and secretion. Mol. Biosyst. 2007, 3, 608–619. [Google Scholar] [CrossRef]
- Hussain, M.M.; Rava, P.; Walsh, M.; Rana, M.; Iqbal, J. Multiple functions of microsomal triglyceride transfer protein. Nutr. Metab. 2012, 9, 14. [Google Scholar] [CrossRef]
- Koerner, C.M.; Roberts, B.S.; Neher, S.B. Endoplasmic reticulum quality control in lipoprotein metabolism. Mol. Cell. Endocrinol. 2019, 498, 110547. [Google Scholar] [CrossRef]
- Fisher, E.A. The degradation of apolipoprotein B100: Multiple opportunities to regulate VLDL triglyceride production by different proteolytic pathways. Biochim. Biophys. Acta. 2012, 1821, 778–781. [Google Scholar] [CrossRef]
- Dashti, M.; Kulik, W.; Hoek, F.; Veerman, E.C.; Peppelenbosch, M.P.; Rezaee, F. A phospholipidomic analysis of all defined human plasma lipoproteins. Sci. Rep. 2011, 1, 139. [Google Scholar] [CrossRef] [PubMed]
- Furtado, J.D.; Ruotolo, G.; Nicholls, S.J.; Dullea, R.; Carvajal-Gonzalez, S.; Sacks, F.M. Pharmacological Inhibition of CETP (Cholesteryl Ester Transfer Protein) Increases HDL (High-Density Lipoprotein) That Contains ApoC3 and Other HDL Subspecies Associated with Higher Risk of Coronary Heart Disease. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Linton, M.F.; Tao, H.; Linton, E.F.; Yancey, P.G. SR-BI: A Multifunctional Receptor in Cholesterol Homeostasis and Atherosclerosis. Trends. Endocrinol. Metab. 2017, 28, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Dominiczak, M.H.; Caslake, M.J. Apolipoproteins: Metabolic role and clinical biochemistry applications. Ann. Clin. Biochem. 2011, 48, 498–515. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.Y. Metabolism and Modification of Apolipoprotein B-Containing Lipoproteins Involved in Dyslipidemia and Atherosclerosis. Biol. Pharm. Bull. 2016, 39, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.; Borén, J. Mechanism of lipoprotein retention by the extracellular matrix. Curr. Opin. Lipidol. 2004, 15, 505–514. [Google Scholar] [CrossRef]
- Reyes-Soffer, G.; Ginsberg, H.N.; Berglund, L.; Duell, P.B.; Heffron, S.P.; Kamstrup, P.R.; Lloyd-Jones, D.M.; Marcovina, S.M.; Yeang, C.; Koschinsky, M.L. American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Radiology and Intervention; and Council on Peripheral Vascular Disease. Lipoprotein(a): A Genetically Determined, Causal, and Prevalent Risk Factor for Atherosclerotic Cardiovascular Disease: A Scientific Statement from the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2022, 42, e48–e60. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Genetic evidence that lipoprotein(a) associates with atherosclerotic stenosis rather than venous thrombosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1732–1741. [Google Scholar] [CrossRef]
- Boffa, M.B.; Koschinsky, M.L. Lipoprotein (a): Truly a direct prothrombotic factor in cardiovascular disease? J. Lipid. Res. 2016, 57, 745–757. [Google Scholar] [CrossRef]
- Katakami, N. Mechanism of Development of Atherosclerosis and Cardiovascular Disease in Diabetes Mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39. [Google Scholar] [CrossRef]
- Hafiane, A. Vulnerable Plaque, Characteristics, Detection, and Potential Therapies. J. Cardiovasc. Dev. Dis. 2019, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, G.R.Y.; Zurek, M.; Puylaert, P.; Martinet, W. Programmed death of macrophages in atherosclerosis: Mechanisms and therapeutic targets. Nat. Rev. Cardiol. 2024. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, J.P.; Major, A.S. How Oxidized Low-Density Lipoprotein Activates Inflammatory Responses. Crit. Rev. Immunol. 2018, 38, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Grundtman, C.; Wick, G. The autoimmune concept of atherosclerosis. Curr. Opin. Lipidol. 2011, 22, 327–334. [Google Scholar] [CrossRef]
- Wolf, D.; Gerhardt, T.; Winkels, H.; Michel, N.A.; Pramod, A.B.; Ghosheh, Y.; Brunel, S.; Buscher, K.; Miller, J.; McArdle, S.; et al. Pathogenic Autoimmunity in Atherosclerosis Evolves from Initially Protective Apolipoprotein B100-Reactive CD4+ T-Regulatory Cells. Circulation 2020, 142, 1279–1293. [Google Scholar] [CrossRef]
- Roy, P.; Sidney, J.; Lindestam Arlehamn, C.S.; Phillips, E.; Mallal, S.; Armstrong Suthahar, S.S.; Billitti, M.; Rubiro, P.; Marrama, D.; Drago, F.; et al. Immunodominant MHC-II (Major Histocompatibility Complex II) Restricted Epitopes in Human Apolipoprotein B. Circ. Res. 2022, 131, 258–276. [Google Scholar] [CrossRef]
- Kotlyarov, S.; Kotlyarova, A. Involvement of Fatty Acids and Their Metabolites in the Development of Inflammation in Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 1308. [Google Scholar] [CrossRef]
- Reustle, A.; Torzewski, M. Role of p38 MAPK in Atherosclerosis and Aortic Valve Sclerosis. Int. J. Mol. Sci. 2018, 19, 3761. [Google Scholar] [CrossRef] [PubMed]
- Waksman, R.; Merdler, I.; Case, B.C.; Waksman, O.; Porto, I. Targeting inflammation in atherosclerosis: Overview, strategy and directions. EuroIntervention 2024, 20, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation and the pathogenesis of atherosclerosis. Vascul. Pharmacol. 2023, 154, 107255. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Karasawa, T.; Takahashi, M. Role of NLRP3 Inflammasomes in Atherosclerosis. J. Atheroscler. Thromb. 2017, 24, 443–451. [Google Scholar] [CrossRef]
- Galimberti, F.; Casula, M.; Olmastroni, E. Apolipoprotein B compared with low-density lipoprotein cholesterol in the atherosclerotic cardiovascular diseases risk assessment. Pharmacol. Res. 2023, 195, 106873. [Google Scholar] [CrossRef]
- Fan, J.; Liu, Y.; Yin, S.; Chen, N.; Bai, X.; Ke, Q.; Shen, J.; Xia, M. Small dense LDL cholesterol is associated with metabolic syndrome traits independently of obesity and inflammation. Nutr. Metab. 2019, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, H.; Matsumoto, T.; Yoshino, G. Influence of Metabolic Syndrome on Small, Dense LDL, and Subclinical Atherosclerosis in Older Subjects. Gerontol. Geriatr. Med. 2023, 9, 23337214231179847. [Google Scholar] [CrossRef]
- Arsenault, B.J.; Lemieux, I.; Després, J.P.; Wareham, N.J.; Luben, R.; Kastelein, J.J.; Khaw, K.T.; Boekholdt, S.M. Cholesterol levels in small LDL particles predict the risk of coronary heart disease in the EPIC-Norfolk prospective population study. Eur. Heart. J. 2007, 28, 2770–2777. [Google Scholar] [CrossRef]
- Walldius, G.; Jungner, I.; Holme, I.; Aastveit, A.H.; Kolar, W.; Steiner, E. High apolipoprotein B, low apolipoprotein A-I, and improvement in the prediction of fatal myocardial infarction (AMORIS study): A prospective study. Lancet 2001, 358, 2026–2033. [Google Scholar] [CrossRef]
- Lim, Y.; Yoo, S.; Lee, S.A.; Chin, S.O.; Heo, D.; Moon, J.C.; Moon, S.; Boo, K.; Kim, S.T.; Seo, H.M.; et al. Apolipoprotein B Is Related to Metabolic Syndrome Independently of Low Density Lipoprotein Cholesterol in Patients with Type 2 Diabetes. Endocrinol. Metab. 2015, 30, 208–215. [Google Scholar] [CrossRef]
- Marston, N.A.; Giugliano, R.P.; Melloni, G.E.M.; Park, J.G.; Morrill, V.; Blazing, M.A.; Ference, B.; Stein, E.; Stroes, E.S.; Braunwald, E.; et al. Association of Apolipoprotein B-Containing Lipoproteins and Risk of Myocardial Infarction in Individuals with and without Atherosclerosis: Distinguishing Between Particle Concentration, Type, and Content. JAMA Cardiol. 2022, 7, 250–256. [Google Scholar] [CrossRef]
- Yun, S.Y.; Rim, J.H.; Kang, H.; Lee, S.G.; Lim, J.B. Associations of LDL Cholesterol, Non-HDL Cholesterol, and Apolipoprotein B with Cardiovascular Disease Occurrence in Adults: Korean Genome and Epidemiology Study. Ann. Lab. Med. 2023, 43, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 Guideline on the Management of Blood Cholesterol: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 73, 3168–3209. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. ESC Scientific Document Group. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart. J. 2020, 41, 111–188. [Google Scholar] [CrossRef] [PubMed]
- Pearson, G.J.; Thanassoulis, G.; Anderson, T.J.; Barry, A.R.; Couture, P.; Dayan, N.; Francis, G.A.; Genest, J.; Grégoire, J.; Grover, S.A.; et al. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can. J. Cardiol. 2021, 37, 1129–1150. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Yokote, K.; Arai, H.; Iida, M.; Ishigaki, Y.; Ishibashi, S.; Umemoto, S.; Egusa, G.; Ohmura, H.; Okamura, T.; et al. Committee for Epidemiology and Clinical Management of Atherosclerosis. Japan Atherosclerosis Society (JAS) Guidelines for Prevention of Atherosclerotic Cardiovascular Diseases 2017. J. Atheroscler. Thromb. 2018, 25, 846–984. [Google Scholar] [CrossRef]
- Rousset, X.; Shamburek, R.; Vaisman, B.; Amar, M.; Remaley, A.T. Lecithin cholesterol acyltransferase: An anti- or pro-atherogenic factor? Curr. Atheroscler. Rep. 2011, 13, 249–256. [Google Scholar] [CrossRef]
- Zhu, L.; Lu, Z.; Zhu, L.; Ouyang, X.; Yang, Y.; He, W.; Feng, Y.; Yi, F.; Song, Y. Lipoprotein ratios are better than conventional lipid parameters in predicting coronary heart disease in Chinese Han people. Kardiol Pol. 2015, 73, 931–938. [Google Scholar] [CrossRef]
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J.; et al. INTERHEART Study Investigators. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 364, 937–952. [Google Scholar] [CrossRef]
- Yaseen, R.I.; El-Leboudy, M.H.; El-Deeb, H.M. The relation between ApoB/ApoA-1 ratio and the severity of coronary artery disease in patients with acute coronary syndrome. Egypt. Heart. J. 2021, 73, 24. [Google Scholar] [CrossRef]
- Walldius, G.; Jungner, I. Apolipoprotein B and apolipoprotein A-I: Risk indicators of coronary heart disease and targets for lipid-modifying therapy. J. Intern. Med. 2004, 255, 188–205. [Google Scholar] [CrossRef]
- Bodde, M.C.; Hermans, M.P.J.; Jukema, J.W.; Schalij, M.J.; Lijfering, W.M.; Rosendaal, F.R.; Romijn, F.P.H.T.M.; Ruhaak, L.R.; van der Laarse, A.; Cobbaert, C.M. Apolipoproteins A1, B, and apoB/apoA1 ratio are associated with first ST-segment elevation myocardial infarction but not with recurrent events during long-term follow-up. Clin. Res. Cardiol. 2019, 108, 520–538. [Google Scholar] [CrossRef]
- Walldius, G.; Aastveit, A.H.; Jungner, I. Stroke mortality and the apoB/apoA-I ratio: Results of the AMORIS prospective study. J. Intern. Med. 2006, 259, 259–266. [Google Scholar] [CrossRef]
- Song, Y.; Yang, Y.; Zhang, J.; Wang, Y.; He, W.; Zhang, X.; Zhu, J.; Lu, Z. The apoB100/apoAI ratio is independently associated with the severity of coronary heart disease: A cross sectional study in patients undergoing coronary angiography. Lipids. Health Dis. 2015, 14, 150. [Google Scholar] [CrossRef]
- Nayak, P.; Panda, S.; Thatoi, P.K.; Rattan, R.; Mohapatra, S.; Mishra, P.K. Evaluation of Lipid Profile and Apolipoproteins in Essential Hypertensive Patients. J. Clin. Diagn. Res. 2016, 10, BC01–BC04. [Google Scholar] [CrossRef]
- Zhan, X.; Chen, Y.; Yan, C.; Liu, S.; Deng, L.; Yang, Y.; Qiu, P.; Pan, D.; Zeng, B.; Chen, Q. Apolipoprotein B/apolipoprotein A1 ratio and mortality among incident peritoneal dialysis patients. Lipids. Health Dis. 2018, 17, 117. [Google Scholar] [CrossRef] [PubMed]
- Carr, S.S.; Hooper, A.J.; Sullivan, D.R.; Burnett, J.R. Non-HDL-cholesterol and apolipoprotein B compared with LDL-cholesterol in atherosclerotic cardiovascular disease risk assessment. Pathology 2019, 51, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Sniderman, A.D.; Williams, K.; Contois, J.H.; Monroe, H.M.; McQueen, M.J.; de Graaf, J.; Furberg, C.D. A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ. Cardiovasc. Qual. Outcomes 2011, 4, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Langlois, M.R.; Sniderman, A.D. Non-HDL Cholesterol or apoB: Which to Prefer as a Target for the Prevention of Atherosclerotic Cardiovascular Disease? Curr. Cardiol. Rep. 2020, 22, 67. [Google Scholar] [CrossRef] [PubMed]
- Marchini, T.; Malchow, S.; Caceres, L.; El Rabih, A.A.H.; Hansen, S.; Mwinyella, T.; Spiga, L.; Piepenburg, S.; Horstmann, H.; Olawale, T.; et al. Circulating Autoantibodies Recognizing Immunodominant Epitopes from Human Apolipoprotein B Associate with Cardiometabolic Risk Factors, but Not with Atherosclerotic Disease. Front. Cardiovasc. Med. 2022, 9, 826729. [Google Scholar] [CrossRef] [PubMed]
- Fagerberg, B.; Prahl Gullberg, U.; Alm, R.; Nilsson, J.; Fredrikson, G.N. Circulating autoantibodies against the apolipoprotein B-100 peptides p45 and p210 in relation to the occurrence of carotid plaques in 64-year-old women. PLoS ONE 2015, 10, e0120744. [Google Scholar] [CrossRef]
- Sjögren, P.; Fredrikson, G.N.; Samnegard, A.; Ericsson, C.G.; Ohrvik, J.; Fisher, R.M.; Nilsson, J.; Hamsten, A. High plasma concentrations of autoantibodies against native peptide 210 of apoB-100 are related to less coronary atherosclerosis and lower risk of myocardial infarction. Eur. Heart. J. 2008, 29, 2218–2226. [Google Scholar] [CrossRef]
- Sveen, K.A.; Smith, G.; Björkbacka, H.; Orho-Melander, M.; Engström, G.; Gonçalves, I.; Melander, O.; Nilsson, J.; Bengtsson, E. High levels of autoantibodies against apoB100 p210 are associated with lower incidence of atrial fibrillation in women. J. Intern. Med. 2022, 291, 207–217. [Google Scholar] [CrossRef]
- Zeng, Z.; Cao, B.; Guo, X.; Li, W.; Li, S.; Chen, J.; Zhou, W.; Zheng, C.; Wei, Y. Apolipoprotein B-100 peptide 210 antibody inhibits atherosclerosis by regulation of macrophages that phagocytize oxidized lipid. Am. J. Transl. Res. 2018, 10, 1817–1828. [Google Scholar]
- Egusa, G. Autoantibodies Against ApoB-100 as a New Marker of Coronary Vulnerable Plaque. J. Atheroscler. Thromb. 2021, 28, 1020–1022. [Google Scholar] [CrossRef]
- Aslan, I.; Kucuksayan, E.; Aslan, M. Effect of insulin analog initiation therapy on LDL/HDL subfraction profile and HDL associated enzymes in type 2 diabetic patients. Lipids. Health Dis. 2013, 12, 54. [Google Scholar] [CrossRef]
- Taskinen, M.R.; Barter, P.J.; Ehnholm, C.; Sullivan, D.R.; Mann, K.; Simes, J.; Best, J.D.; Hamwood, S.; Keech, A.C. FIELD study investigators. Ability of traditional lipid ratios and apolipoprotein ratios to predict cardiovascular risk in people with type 2 diabetes. Diabetologia 2010, 53, 1846–1855. [Google Scholar] [CrossRef]
- Zhang, P.; Gao, J.; Pu, C.; Zhang, Y. Apolipoprotein status in type 2 diabetes mellitus and its complications (Review). Mol. Med. Rep. 2017, 16, 9279–9286. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Luo, Z.; Yu, L.; Yang, Y.; Chen, Y.; Luo, X.; Lai, F.; Song, Y. Associations of the APOB rs693 and rs17240441 polymorphisms with plasma APOB and lipid levels: A meta-analysis. Lipids. Health Dis. 2017, 16, 166. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.T.; Li, Y.; Ma, Y.T.; Yang, Y.N.; Ma, X.; Li, X.M.; Liu, F.; Chen, B.D. Association between apolipoprotein B genetic polymorphism and the risk of calcific aortic stenosis in Chinese subjects, in Xinjiang, China. Lipids. Health Dis. 2018, 17, 40. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, R.A.; Al-Zahrani, M.H.; Balgoon, M.J.; Alkhattabi, N.A. Prevalence of ApoB100 rs693 gene polymorphism in metabolic syndrome among female students at King Abdulaziz University. Saudi. J. Biol. Sci. 2021, 28, 3249–3253. [Google Scholar] [CrossRef]
- Pan, J.; Gao, F.; Bao, Y.; Zhang, L.; Tu, Y.; Jia, W. Non-high-density lipoprotein cholesterol is associated more closely with albuminuria in Chinese type 2 diabetic patients with normal renal function, compared with traditional lipid parameters. J. Clin. Lipidol. 2012, 6, 382–387. [Google Scholar] [CrossRef]
- Taskinen, M.R.; Matikainen, N.; Björnson, E.; Söderlund, S.; Inkeri, J.; Hakkarainen, A.; Parviainen, H.; Sihlbom, C.; Thorsell, A.; Andersson, L.; et al. Contribution of intestinal triglyceride-rich lipoproteins to residual atherosclerotic cardiovascular disease risk in individuals with type 2 diabetes on statin therapy. Diabetologia 2023, 66, 2307–2319. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S. Mechanisms by which elevated resistin levels accelerate atherosclerotic cardiovascular disease. Rheumatol. Curr. Res. 2013, 3, 115. [Google Scholar] [CrossRef]
- Skogsberg, J.; Dicker, A.; Rydén, M.; Aström, G.; Nilsson, R.; Bhuiyan, H.; Vitols, S.; Mairal, A.; Langin, D.; Alberts, P.; et al. ApoB100-LDL acts as a metabolic signal from liver to peripheral fat causing inhibition of lipolysis in adipocytes. PLoS ONE 2008, 3, e3771. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Gan, S.K.; Wong, A.T.; Barrett, P.H.; Watts, G.F. Association between skeletal muscle fat content and very-low-density lipoprotein-apolipoprotein B-100 transport in obesity: Effect of weight loss. Diabetes. Obes. Metab. 2014, 16, 994–1000. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kim, Y.J.; Khang, A.R.; Eckel, R.H. Postprandial dyslipidemia after a standardized high-fat meal in BMI-matched healthy individuals, and in subjects with prediabetes or type 2 diabetes. Clin. Nutr. 2021, 40, 5538–5546. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.S.; Guo, B.B.; Lin, F.F.; Zeng, L.M.; Wang, T.; Dang, X.Y.; Yang, Y.; Hu, Y.H.; Liu, J.; Wang, H.Y. A novel low systemic diacylglycerol acyltransferase 1 inhibitor, Yhhu2407, improves lipid metabolism. Eur. J. Pharm. Sci. 2021, 158, 105683. [Google Scholar] [CrossRef]
- Amor, A.J.; Pinyol, M.; Sola, E.; Catalan, M.; Cofan, M.; Herreras, Z. Relationship between noninvasive scores of nonalcoholic fatty liver disease and nuclear magnetic resonance lipoprotein abnormalities: A focus on atherogenic dyslipidemia. J. Clin. Lipidol. 2017, 11, 551–561.e557. [Google Scholar] [CrossRef] [PubMed]
- Lucero, D.; Miksztowicz, V.; Gualano, G.; Longo, C.; Landeira, G.; Alvarez, E. Nonalcoholic fatty liver disease associated with metabolic syndrome: Influence of liver fibrosis stages on characteristics of very low-density lipoproteins. Clinica. Chimica. Acta 2017, 473, 1–8. [Google Scholar] [CrossRef]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.E.; Attie, A.D.; Biddinger, S.B. The regulation of ApoB metabolism by insulin. Trends. Endocrinol. Metab. 2013, 24, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Vergès, B. Pathophysiology of diabetic dyslipidaemia: Where are we? Diabetologia 2015, 58, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D. The LDL modification hypothesis of atherogenesis: An update. J. Lipid. Res. 2009, 50, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.S.; Guo, S.E.; Lin, H.S.; Hsu, J.T.; Lin, Y.S.; Lin, T.H.; Huang, T.J.; Chen, M.Y.; Chung, C.M. Impact of Apolipoprotein B on Hepatosteatosis in a Population Infected with Hepatitis C Virus: A Cross-Sectional Observational Study. Obes. Facts. 2016, 9, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Vine, D.F.; Wang, Y.; Jetha, M.M.; Ball, G.D.; Proctor, S.D. Impaired ApoB-Lipoprotein and Triglyceride Metabolism in Obese Adolescents with Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2017, 102, 970–982. [Google Scholar] [CrossRef]
- Vine, D.; Proctor, E.; Weaver, O.; Ghosh, M.; Maximova, K.; Proctor, S. A Pilot Trial: Fish Oil and Metformin Effects on ApoB-Remnants and Triglycerides in Women with Polycystic Ovary Syndrome. J. Endocr. Soc. 2021, 5, bvab114. [Google Scholar] [CrossRef]
- Wang, D.; Zhao, H.; Xing, C.; Lv, B.; Wang, X.; He, B. Androgens exacerbate hepatic triglyceride accumulation in rats with polycystic ovary syndrome by downregulating MTTP expression. Endocrine 2023. [Google Scholar] [CrossRef]
- Kamper, M.; Manns, C.C.; Plieschnig, J.A.; Schneider, W.J.; Ivessa, N.E.; Hermann, M. Estrogen enhances secretion of apolipoprotein B-100 containing lipoproteins by BeWo cells. Biochimie 2015, 112, 121–128. [Google Scholar] [CrossRef]
- Bertoia, M.L.; Pai, J.K.; Lee, J.H.; Taleb, A.; Joosten, M.M.; Mittleman, M.A.; Yang, X.; Witztum, J.L.; Rimm, E.B.; Tsimikas, S.; et al. Oxidation-specific biomarkers and risk of peripheral artery disease. J. Am. Coll. Cardiol. 2013, 61, 2169–2179. [Google Scholar] [CrossRef]
- Bertrand, C.; Saulnier, P.J.; Potier, L.; Croyal, M.; Blanchard, V.; Gand, E.; Ragot, S.; Schneider, F.; Bocock, O.; Baillet-Blanco, L.; et al. SURDIAGENE Study Group. Plasma concentrations of lipoproteins and risk of lower-limb peripheral artery disease in people with type 2 diabetes: The SURDIAGENE study. Diabetologia 2021, 64, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Tsimihodimos, V.; Mitrogianni, Z.; Elisaf, M. Dyslipidemia associated with chronic kidney disease. Open Cardiovasc. Med. J. 2011, 5, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Kim, D.K.; Oh, K.H.; Joo, K.W.; Lim, C.S.; Kim, Y.S.; Han, S.S. Apolipoprotein B is a risk factor for end-stage renal disease. Clin. Kidney J. 2020, 14, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Trevisan, R.; Dodesini, A.R.; Lepore, G. Lipids and renal disease. J. Am. Soc. Nephrol. 2006, 17, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Keane, W.F.; Tomassini, J.E.; Neff, D.R. Lipid abnormalities in patients with chronic kidney disease: Implications for the pathophysiology of atherosclerosis. J. Atheroscler. Thromb. 2013, 20, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, B.; Lin, L.; Lei, F.; Sun, T.; Zhang, X.; Song, X.; Huang, X.; Zeng, Q.; Cai, J.; et al. The association of apolipoprotein B with chronic kidney disease in the Chinese population. Front. Endocrinol. 2023, 14, 1083614. [Google Scholar] [CrossRef] [PubMed]
- Twisk, J.; Gillian-Daniel, D.L.; Tebon, A.; Wang, L.; Barrett, P.H.; Attie, A.D. The role of the LDL receptor in apolipoprotein B secretion. J. Clin. Investig. 2000, 105, 521–532. [Google Scholar] [CrossRef]
- Andersen, L.H.; Miserez, A.R.; Ahmad, Z.; Andersen, R.L. Familial defective apolipoprotein B-100: A review. J. Clin. Lipidol. 2016, 10, 1297–1302. [Google Scholar] [CrossRef]
- Whitfield, A.J.; Barrett, P.H.; van Bockxmeer, F.M.; Burnett, J.R. Lipid disorders and mutations in the APOB gene. Clin. Chem. 2004, 50, 1725–1732. [Google Scholar] [CrossRef]
- Futema, M.; Taylor-Beadling, A.; Williams, M.; Humphries, S.E. Genetic testing for familial hypercholesterolemia-past, present, and future. J. Lipid. Res. 2021, 62, 100139. [Google Scholar] [CrossRef]
- Rogozik, J.; Główczyńska, R.; Grabowski, M. Genetic backgrounds and diagnosis of familial hypercholesterolemia. Clin. Genet. 2024, 105, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Vogt, A.; Keller, C.; Heigl, C.; Weiss, N.; Zöllner, N. Two forms of familial hypercholesterolemia: Differences in cardiovascular risk factors, cardiac and extracardiac atherosclerosis. Dtsch. Med. Wochenschr. 2014, 139, 2573–2577. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, G.; Lin, X.; Yue, P. Familial hypobetalipoproteinemia: Genetics and metabolism. Cell. Mol. Life. Sci. 2005, 62, 1372–1378. [Google Scholar] [CrossRef]
- Welty, F.K. Hypobetalipoproteinemia and abetalipoproteinemia. Curr. Opin. Lipidol. 2014, 25, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hegele, R.A. Abetalipoproteinemia and homozygous hypobetalipoproteinemia: A framework for diagnosis and management. J. Inherit. Metab. Dis. 2014, 37, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, G. Familial hypobetalipoproteinemia: A review. J. Lipid. Res. 2003, 44, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Sankatsing, R.R.; Fouchier, S.W.; de Haan, S.; Hutten, B.A.; de Groot, E.; Kastelein, J.J.; Stroes, E.S. Hepatic and cardiovascular consequences of familial hypobetalipoproteinemia. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1979–1984. [Google Scholar] [CrossRef] [PubMed]
- Peloso, G.M.; Nomura, A.; Khera, A.V.; Chaffin, M.; Won, H.H.; Ardissino, D.; Danesh, J.; Schunkert, H.; Wilson, J.G.; Samani, N.; et al. Rare Protein-Truncating Variants in APOB, Lower Low-Density Lipoprotein Cholesterol, and Protection Against Coronary Heart Disease. Circ. Genom. Precis. Med. 2019, 12, e002376. [Google Scholar] [CrossRef]
- Welty, F.K. Hypobetalipoproteinemia and abetalipoproteinemia: Liver disease and cardiovascular disease. Curr. Opin. Lipidol. 2020, 31, 49–55. [Google Scholar] [CrossRef]
- Richard, C.; Couture, P.; Ooi, E.M.; Tremblay, A.J.; Desroches, S.; Charest, A.; Lichtenstein, A.H.; Lamarche, B. Effect of Mediterranean diet with and without weight loss on apolipoprotein B100 metabolism in men with metabolic syndrome. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 433–438. [Google Scholar] [CrossRef]
- Lamarche, B.; Couture, P. Dietary fatty acids, dietary patterns, and lipoprotein metabolism. Curr. Opin. Lipidol. 2015, 26, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Wood, G.; Taylor, E.; Ng, V.; Murrell, A.; Patil, A.; van der Touw, T.; Wolden, M.; Andronicos, N.; Smart, N.A. Estimating the Effect of Aerobic Exercise Training on Novel Lipid Biomarkers: A Systematic Review and Multivariate Meta-Analysis of Randomized Controlled Trials. Sports Med. 2023, 53, 871–886. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Wang, S.; Jacobson, T.A. Meta-analysis of comparison of effectiveness of lowering apolipoprotein B versus low-density lipoprotein cholesterol and nonhigh-density lipoprotein cholesterol for cardiovascular risk reduction in randomized trials. Am. J. Cardiol. 2012, 110, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Corsini, A. Clinical Pharmacology of Statins: An Update. Curr. Atheroscler. Rep. 2020, 22, 26. [Google Scholar] [CrossRef] [PubMed]
- Watts, G.F.; Chan, D.C.; Barrett, P.H.; O’Neill, F.H.; Thompson, G.R. Effect of a statin on hepatic apolipoprotein B-100 secretion and plasma campesterol levels in the metabolic syndrome. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Lamon-Fava, S.; Diffenderfer, M.R.; Barrett, P.H.; Buchsbaum, A.; Matthan, N.R.; Lichtenstein, A.H.; Dolnikowski, G.G.; Horvath, K.; Asztalos, B.F.; Zago, V.; et al. Effects of different doses of atorvastatin on human apolipoprotein B-100, B-48, and A-I metabolism. J. Lipid. Res. 2007, 48, 1746–1753. [Google Scholar] [CrossRef] [PubMed]
- Amarenco, P.; Bogousslavsky, J.; Callahan, A., 3rd; Goldstein, L.B.; Hennerici, M.; Rudolph, A.E.; Sillesen, H.; Simunovic, L.; Szarek, M.; Welch, K.M.; et al. Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) Investigators. High-dose atorvastatin after stroke or transient ischemic attack. N. Engl. J. Med. 2006, 355, 549–559. [Google Scholar] [CrossRef]
- Byun, Y.S.; Yang, X.; Bao, W.; DeMicco, D.; Laskey, R.; Witztum, J.L.; Tsimikas, S. SPARCL Trial Investigators. Oxidized Phospholipids on Apolipoprotein B-100 and Recurrent Ischemic Events Following Stroke or Transient Ischemic Attack. J. Am. Coll. Cardiol. 2017, 69, 147–158. [Google Scholar] [CrossRef]
- Chemello, A.; Gallo, A.; Lambert, G. Lipoprotein(a) is a residual cardiovascular risk factor in statin treated stroke survivors: Insights from the SPARCL trial. Atherosclerosis 2022, 355, P29. [Google Scholar] [CrossRef]
- Nagai, T.; Tomizawa, T.; Nakajima, K.; Mori, M. Effect of bezafibrate or pravastatin on serum lipid levels and albuminuria in NIDDM patients. J. Atheroscler. Thromb. 2000, 7, 91–96. [Google Scholar] [CrossRef]
- Jeong, H.S.; Hong, S.J.; Cho, J.M.; Han, K.H.; Cha, D.H.; Jo, S.H.; Kang, H.J.; Choi, S.Y.; Choi, C.U.; Cho, E.J.; et al. A Multicenter, Randomized, Double-blind, Active-controlled, Factorial Design, Phase III Clinical Trial to Evaluate the Efficacy and Safety of Combination Therapy of Pitavastatin and Ezetimibe Versus Monotherapy of Pitavastatin in Patients with Primary Hypercholesterolemia. Clin. Ther. 2022, 44, 1310–1325. [Google Scholar] [CrossRef]
- Le, N.A.; Diffenderfer, M.R.; Thongtang, N.; Ooi, E.M.; Barrett, P.H.; Horvath, K.V.; Dolnikowski, G.G.; Asztalos, B.F.; Schaefer, E.J.; Brown, W.V. Rosuvastatin Enhances the Catabolism of LDL apoB-100 in Subjects with Combined Hyperlipidemia in a Dose Dependent Manner. Lipids 2015, 50, 447–458. [Google Scholar] [CrossRef]
- Jones, P.H.; Hunninghake, D.B.; Ferdinand, K.C.; Stein, E.A.; Gold, A.; Caplan, R.J.; Blasetto, J.W. Statin Therapies for Elevated Lipid Levels Compared Across Doses to Rosuvastatin Study Group. Effects of rosuvastatin versus atorvastatin, simvastatin, and pravastatin on non-high-density lipoprotein cholesterol, apolipoproteins, and lipid ratios in patients with hypercholesterolemia: Additional results from the STELLAR trial. Clin. Ther. 2004, 26, 1388–1399. [Google Scholar] [CrossRef]
- Camilleri, E.; van Rein, N.; van Vlijmen, B.J.M.; Biedermann, J.S.; Kruip, M.J.H.A.; Leebeek, F.W.; van der Meer, F.J.; Cobbaert, C.M.; Cannegieter, S.C.; Lijfering, W.M. Influence of rosuvastatin on apolipoproteins and coagulation factor levels: Results from the STAtin Reduce Thrombophilia trial. Res. Pract. Thromb. Haemost. 2023, 7, 100063. [Google Scholar] [CrossRef]
- Vavlukis, M.; Vavlukis, A. Adding ezetimibe to statin therapy: Latest evidence and clinical implications. Drugs. Context. 2018, 7, 212534. [Google Scholar] [CrossRef]
- Genest, J. Combination of statin and ezetimibe for the treatment of dyslipidemias and the prevention of coronary artery disease. Can. J. Cardiol. 2006, 22, 863–868. [Google Scholar] [CrossRef]
- Tremblay, A.J.; Lamarche, B.; Cohn, J.S.; Hogue, J.C.; Couture, P. Effect of ezetimibe on the in vivo kinetics of apoB-48 and apoB-100 in men with primary hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1101–1106. [Google Scholar] [CrossRef]
- Tremblay, A.J.; Lamarche, B.; Hogue, J.C.; Couture, P. Effects of ezetimibe and simvastatin on apolipoprotein B metabolism in males with mixed hyperlipidemia. J. Lipid. Res. 2009, 50, 1463–1471. [Google Scholar] [CrossRef]
- Banach, M.; Surma, S.; Toth, P.P. endorsed by the International Lipid Expert Panel (ILEP). 2023: The year in cardiovascular disease—The year of new and prospective lipid lowering therapies. Can we render dyslipidemia a rare disease by 2024? Arch. Med. Sci. 2023, 19, 1602–1615. [Google Scholar] [CrossRef]
- Lin, X.; Ma, P.; Yang, C.; Wang, J.; He, K.; Chen, G.; Huang, W.; Fan, J.; Xian, X.; Wang, Y.; et al. Dietary-Induced Elevations of Triglyceride-Rich Lipoproteins Promote Atherosclerosis in the Low-Density Lipoprotein Receptor Knockout Syrian Golden Hamster. Front. Cardiovasc. Med. 2021, 8, 738060. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.; Benderly, M.; Goldbourt, U. Update on the use of fibrates: Focus on bezafibrate. Vasc. Health Risk. Manag. 2008, 4, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.H.; Kim, S.G. Fibrates Revisited: Potential Role in Cardiovascular Risk Reduction. Diabetes. Metab. J. 2020, 44, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Rader, D.J.; Millar, J.S. The effect of PPAR-alpha agonism on apolipoprotein metabolism in humans. Atherosclerosis 2010, 210, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Demant, T.; Gaw, A.; Watts, G.F.; Durrington, P.; Buckley, B.; Imrie, C.W.; Wilson, C.; Packard, C.J.; Shepherd, J. Metabolism of apoB-100-containing lipoproteins in familial hyperchylomicronemia. J. Lipid. Res. 1993, 34, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Naruszewicz, M.; Mirkiewicz, E.; Kłosiewicz-Latoszek, L. Modification of low density lipoproteins from hypertriglyceridemic patients by macrophages in vitro and the effect of bezafibrate treatment. Atherosclerosis 1989, 79, 261–265. [Google Scholar] [CrossRef]
- Croyal, M.; Kaabia, Z.; León, L.; Ramin-Mangata, S.; Baty, T.; Fall, F.; Billon-Crossouard, S.; Aguesse, A.; Hollstein, T.; Sullivan, D.R.; et al. Fenofibrate decreases plasma ceramide in type 2 diabetes patients: A novel marker of CVD? Diabetes. Metab. 2018, 44, 143–149. [Google Scholar] [CrossRef]
- Pan, M.; Cederbaum, A.I.; Zhang, Y.L.; Ginsberg, H.N.; Williams, K.J.; Fisher, E.A. Lipid peroxidation and oxidant stress regulate hepatic apolipoprotein B degradation and VLDL production. J. Clin. Investig. 2004, 113, 1277–1287. [Google Scholar] [CrossRef]
- Ouguerram, K.; Maugeais, C.; Gardette, J.; Magot, T.; Krempf, M. Effect of n-3 fatty acids on metabolism of apoB100-containing lipoprotein in type 2 diabetic subjects. Br. J. Nutr. 2006, 96, 100–106. [Google Scholar] [CrossRef]
- Bell, T.A., 3rd; Kelley, K.; Wilson, M.D.; Sawyer, J.K.; Rudel, L.L. Dietary fat-induced alterations in atherosclerosis are abolished by ACAT2-deficiency in ApoB100 only, LDLr-/- mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1396–1402. [Google Scholar] [CrossRef]
- Kayikcioglu, M. LDL Apheresis and Lp (a) Apheresis: A Clinician’s Perspective. Curr. Atheroscler. Rep. 2021, 23, 15. [Google Scholar] [CrossRef]
- Yaroustovsky, M.; Abramyan, M.; Rogalskaya, E.; Komardina, E. The use of lipoprotein apheresis for the treatment of high-risk patients with elevated lipoprotein(a) and hypercholesterolemia. Vessel Plus. 2019, 3, 9. [Google Scholar] [CrossRef]
- Franchini, M.; Capuzzo, E.; Liumbruno, G.M. Lipoprotein apheresis for the treatment of elevated circulating levels of lipoprotein(a): A critical literature review. Blood. Transfus. 2016, 14, 413–418. [Google Scholar] [CrossRef]
- Chambergo-Michilot, D.; Alur, A.; Kulkarni, S.; Agarwala, A. Mipomersen in Familial Hypercholesterolemia: An Update on Health-Related Quality of Life and Patient-Reported Outcomes. Vasc. Health Risk Manag. 2022, 18, 73–80. [Google Scholar] [CrossRef]
- McGowan, M.P.; Tardif, J.C.; Ceska, R.; Burgess, L.J.; Soran, H.; Gouni-Berthold, I.; Wagener, G.; Chasan-Taber, S. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS ONE 2012, 7, e49006. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.A.; Dufour, R.; Gagne, C.; Gaudet, D.; East, C.; Donovan, J.M.; Chin, W.; Tribble, D.L.; McGowan, M. Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: Results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation 2012, 126, 2283–2292. [Google Scholar] [CrossRef]
- Cuchel, M.; Raal, F.J.; Hegele, R.A.; Al-Rasadi, K.; Arca, M.; Averna, M.; Bruckert, E.; Freiberger, T.; Gaudet, D.; Harada-Shiba, M.; et al. 2023 Update on European Atherosclerosis Society Consensus Statement on Homozygous Familial Hypercholesterolaemia: New treatments and clinical guidance. Eur. Heart J. 2023, 44, 2277–2291. [Google Scholar] [CrossRef]
- Cuchel, M.; Bloedon, L.T.; Szapary, P.O.; Kolansky, D.M.; Wolfe, M.L.; Sarkis, A.; Millar, J.S.; Ikewaki, K.; Siegelman, E.S.; Gregg, R.E.; et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N. Engl. J. Med. 2007, 356, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Blom, D.J.; Raal, F.J.; Santos, R.D.; Marais, A.D. Lomitapide and Mipomersen-Inhibiting Microsomal Triglyceride Transfer Protein (MTP) and apoB100 Synthesis. Curr. Atheroscler. Rep. 2019, 21, 48. [Google Scholar] [CrossRef]
- Biolo, G.; Vinci, P.; Mangogna, A.; Landolfo, M.; Schincariol, P.; Fiotti, N.; Mearelli, F.; Di Girolamo, F.G. Mechanism of action and therapeutic use of bempedoic acid in atherosclerosis and metabolic syndrome. Front. Cardiovasc. Med. 2022, 9, 1028355. [Google Scholar] [CrossRef]
- Michaeli, D.T.; Michaeli, J.C.; Albers, S.; Boch, T.; Michaeli, T. Established and Emerging Lipid-Lowering Drugs for Primary and Secondary Cardiovascular Prevention. Am. J. Cardiovasc. Drugs. 2023, 23, 477–495. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Banach, M.; Mancini, G.B.J.; Lepor, N.E.; Hanselman, J.C.; Zhao, X.; Leiter, L.A. Efficacy and safety of bempedoic acid added to ezetimibe in statin-intolerant patients with hypercholesterolemia: A randomized, placebo-controlled study. Atherosclerosis 2018, 277, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, K.; Jankowski, P.; Kosior, D.A. PCSK9 inhibitors—From discovery of a single mutation to a groundbreaking therapy of lipid disorders in one decade. Arch. Med. Sci. 2017, 13, 914–929. [Google Scholar] [CrossRef] [PubMed]
- Bellino, M.; Galasso, G.; Silverio, A.; Tedeschi, M.; Formisano, C.; Romei, S.; Esposito, L.; Cancro, F.P.; Vassallo, M.G.; Accarino, G.; et al. Soluble PCSK9 Inhibition: Indications, Clinical Impact, New Molecular Insights and Practical Approach-Where Do We Stand? J. Clin. Med. 2023, 12, 2922. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Tavori, H.; Brown, P.E.; Linton, M.F.; He, J.; Giunzioni, I.; Fazio, S. Proprotein convertase subtilisin kexin type 9 promotes intestinal overproduction of triglyceride-rich apolipoprotein B lipoproteins through both low-density lipoprotein receptor-dependent and -independent mechanisms. Circulation 2014, 130, 431–441. [Google Scholar] [CrossRef]
- Vergès, B.; Duvillard, L.; Brindisi, M.C.; Gautier, E.; Krempf, M.; Costet, P.; Cariou, B. Lack of association between plasma PCSK9 and LDL-apoB100 catabolism in patients with uncontrolled type 2 diabetes. Atherosclerosis 2011, 219, 342–348. [Google Scholar] [CrossRef]
- Croyal, M.; Tran, T.T.; Blanchard, R.H.; Le Bail, J.C.; Villard, E.F.; Poirier, B.; Aguesse, A.; Billon-Crossouard, S.; Ramin-Mangata, S.; Blanchard, V.; et al. PCSK9 inhibition with alirocumab reduces lipoprotein(a) levels in nonhuman primates by lowering apolipoprotein(a) production rate. Clin. Sci. 2018, 132, 1075–1083. [Google Scholar] [CrossRef]
- Ray, K.K.; Kallend, D.; Leiter, L.A.; Raal, F.J.; Koenig, W.; Jaros, M.J.; Schwartz, G.G.; Landmesser, U.; Garcia Conde, L.; Wright, R.S. ORION-11 Investigators. Effect of inclisiran on lipids in primary prevention: The ORION-11 trial. Eur. Heart J. 2022, 43, 5047–5057. [Google Scholar] [CrossRef]
- Luo, F.; Das, A.; Khetarpal, S.A.; Fang, Z.; Zelniker, T.A.; Rosenson, R.S.; Qamar, A. ANGPTL3 inhibition, dyslipidemia, and cardiovascular diseases. Trends. Cardiovasc. Med. 2023, in press. [Google Scholar] [CrossRef]
- Raschi, E.; Casula, M.; Cicero, A.F.G.; Corsini, A.; Borghi, C.; Catapano, A. Beyond statins: New pharmacological targets to decrease LDL-cholesterol and cardiovascular events. Pharmacol. Ther. 2023, 250, 108507. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Burgess, L.J.; Ebenbichler, C.F.; Baum, S.J.; Stroes, E.S.G.; Ali, S.; Khilla, N.; McGinniss, J.; Gaudet, D.; Pordy, R. Longer-Term Efficacy and Safety of Evinacumab in Patients with Refractory Hypercholesterolemia. JAMA Cardiol. 2023, 8, 81070–81076. [Google Scholar] [CrossRef]
- Nelson, A.J.; Sniderman, A.D.; Ditmarsch, M.; Dicklin, M.R.; Nicholls, S.J.; Davidson, M.H.; Kastelein, J.J.P. Cholesteryl Ester Transfer Protein Inhibition Reduces Major Adverse Cardiovascular Events by Lowering Apolipoprotein B Levels. Int. J. Mol. Sci. 2022, 23, 9417. [Google Scholar] [CrossRef]
- Hovingh, G.K.; Kastelein, J.J.; van Deventer, S.J.; Round, P.; Ford, J.; Saleheen, D.; Rader, D.J.; Brewer, H.B.; Barter, P.J. Cholesterol ester transfer protein inhibition by TA-8995 in patients with mild dyslipidaemia (TULIP): A randomised, double-blind, placebo-controlled phase 2 trial. Lancet 2015, 386, 452–460. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ditmarsch, M.; Kastelein, J.J.; Rigby, S.P.; Kling, D.; Curcio, D.L.; Alp, N.J.; Davidson, M.H. Lipid lowering effects of the CETP inhibitor obicetrapib in combination with high-intensity statins: A randomized phase 2 trial. Nat. Med. 2022, 28, 1672–1678. [Google Scholar] [CrossRef]
- Tardif, J.C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A.; Eikelboom, J.W.; Schut, A.; Opstal, T.S.J.; The, S.H.K.; Xu, X.F.; Ireland, M.A.; Lenderink, T.; et al. LoDoCo2 Trial Investigators. Colchicine in Patients with Chronic Coronary Disease. N. Engl. J. Med. 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Abbate, A.; Trankle, C.R.; Buckley, L.F.; Lipinski, M.J.; Appleton, D.; Kadariya, D.; Canada, J.M.; Carbone, S.; Roberts, C.S.; Abouzaki, N.; et al. Interleukin-1 Blockade Inhibits the Acute Inflammatory Response in Patients with ST-Segment–Elevation Myocardial Infarction. J. Am. Heart Assoc. 2020, 9, e014941. [Google Scholar] [CrossRef] [PubMed]
- Lecis, D.; Massaro, G.; Benedetto, D.; Di Luozzo, M.; Russo, G.; Mauriello, A.; Federici, M.; Sangiorgi, G.M. Immunomodulation Therapies for Atherosclerosis: The Past, the Present, and the Future. Int. J. Mol. Sci. 2023, 24, 10979. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Wigren, M.; Shah, P.K. Vaccines against atherosclerosis. Expert Rev. Vaccines 2013, 12, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Chyu, K.Y.; Zhao, X.; Zhou, J.; Dimayuga, P.C.; Lio, N.W.; Cercek, B.; Trac, N.T.; Chung, E.J.; Shah, P.K. Immunization using ApoB-100 peptide-linked nanoparticles reduces atherosclerosis. JCI Insight. 2022, 7, e149741. [Google Scholar] [CrossRef] [PubMed]
- Gisterå, A.; Hermansson, A.; Strodthoff, D.; Klement, M.L.; Hedin, U.; Fredrikson, G.N.; Nilsson, J.; Hansson, G.K.; Ketelhuth, D.F. Vaccination against T-cell epitopes of native ApoB100 reduces vascular inflammation and disease in a humanized mouse model of atherosclerosis. J. Intern. Med. 2017, 281, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, R.; Lebens, M.; Hermansson, A.; Fredrikson, G.N.; Strodthoff, D.; Rudling, M.; Ketelhuth, D.F.; Gerdes, N.; Holmgren, J.; Nilsson, J.; et al. Intranasal immunization with an apolipoprotein B-100 fusion protein induces antigen-specific regulatory T cells and reduces atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, H.J.; Choi, J.S.; Han, J.; Kim, J.Y.; Na, H.K.; Joung, H.J.; Kim, Y.S.; Binas, B. An apolipoprotein B100 mimotope prevents obesity in mice. Clin. Sci. 2016, 130, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.K.; Choe, M.K.; Kim, H.J.; Kim, Y.S.; Binas, B.; Kim, H.J. An ApoB100-mimetic vaccine prevents obesity and liver steatosis in ApoE-/-mice. Pharmacol. Rep. 2017, 69, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Plochg, B.F.J.; Englert, H.; Rangaswamy, C.; Konrath, S.; Malle, M.; Lampalzer, S.; Beisel, C.; Wollin, S.; Frye, M.; Aberle, J.; et al. Liver damage promotes pro-inflammatory T-cell responses against apolipoprotein B-100. J. Intern. Med. 2022, 291, 648–664. [Google Scholar] [CrossRef]
- Wang, L.; Fang, X.; Yang, Z.; Li, X.; Cheng, M.; Cheng, L.; Wang, G.; Li, W.; Liu, L. LncRP11-675F6.3 responds to rapamycin treatment and reduces triglyceride accumulation via interacting with HK1 in hepatocytes by regulating autophagy and VLDL-related proteins. Acta Biochim. Biophys. Sin. 2023, 55, 1606–1617. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Lipid-Lowering Agent | Mechanism of Action | Proposed Mechanisms Regarding the Effects on ApoB |
|---|---|---|
| Statins [115,116,117,122] | HMG-CoA reductase inhibitor | 1.↓ the entry of ApoB-containing lipoproteins in plasma 2.↑ FCR of ApoB-containing lipoproteins 3.(-) PR of ApoB-containing lipoproteins |
| Ezetimibe [127,128,130] | Selective blockade of NPC1L1 | 1.↑ FCR of ApoB-containing lipoproteins 2.↓ PR of ApoB-containing lipoproteins 3.↓ expression of vascular adhesion factors in endothelial cells 4.↓ lipid uptake by macrophages |
| Fibrates [133,134,135] | PPAR-α agonism | 1.↑ FCR of VLDL-ApoB100 2.↓ PR of VLDL-ApoB100 3.↑ FCR of LDL-ApoB100 in TG < 400 mg/dL 4.↓ FCR of LDL-ApoB100 in TG > 400 mg/dL due to activation of RES: - ↑ uptake LDL-ApoB100 by macrophages due to ↑ uptake by scavenger receptors or oxidative damage |
| Omega-3 FA [137,138,139] | ↓ TGs probably due to: - ↓ lipogenic gene expression - ↑ β-oxidation of FA - ↑ LPL expression | 1. Intrahepatic degradation of ApoB100 2.↓ ACAT2 3. Alterations in sphingolipids: - ↑ CR: VLDL1- > VLDL2- > LDL - ↓ VLDL1-ApoB100 production |
| Mipomersen [143] | Antisense oligonucleotide inhibitor of ApoB100 | Selective degradation of ApoB100-mRNA |
| Lomitapide [148] | Direct inhibition of MTP in hepatocytes and enterocytes | ↓ of ApoB-containing lipoproteins |
| Bempedoic acid [149,150,151] | ATP-citrate lyase inhibitor | 1.↓ cholesterol synthesis in the liver 2. Up-regulation of LDL-R expression 3.↑ LDL-C clearance |
| Evolocumab/ Alirocumab [154,155,156] | PCSK9 inhibitors | 1.↓ PCSK9 ability to bind to the LDL-R: - Intrahepatic accumulation of LDL-R - ↑ LDL-C clearance - ↓ LDL-C levels 2.↓ stability and mRNA expression of ApoB |
| Inclisiran [157] | small interfering ribonucleic acid (siRNA) | ↓ PCSK9-mediated degradation of LDL-R: - ↑ LDL-R expression - ↑ LDL-C uptake by the liver |
| Evinacumab/ Vupanorsen [158,159,160] | ANGPTL3 inhibitors | - ↑ activity of LPL and EL - ↑ clearance of ApoB-containing lipoproteins |
| Obicetrapib [161,162,163] | CETP inhibitor | 1.↓ the rate of transfer of CE from HDL into TLRs 2.↑ cholesterol content in HDL 3.↑ formation of larger HDL particles that are more slowly catabolized 4. Depletion of cholesterol content of ApoB |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kounatidis, D.; Vallianou, N.G.; Poulaki, A.; Evangelopoulos, A.; Panagopoulos, F.; Stratigou, T.; Geladari, E.; Karampela, I.; Dalamaga, M. ApoB100 and Atherosclerosis: What’s New in the 21st Century? Metabolites 2024, 14, 123. https://doi.org/10.3390/metabo14020123
Kounatidis D, Vallianou NG, Poulaki A, Evangelopoulos A, Panagopoulos F, Stratigou T, Geladari E, Karampela I, Dalamaga M. ApoB100 and Atherosclerosis: What’s New in the 21st Century? Metabolites. 2024; 14(2):123. https://doi.org/10.3390/metabo14020123
Chicago/Turabian StyleKounatidis, Dimitris, Natalia G. Vallianou, Aikaterini Poulaki, Angelos Evangelopoulos, Fotis Panagopoulos, Theodora Stratigou, Eleni Geladari, Irene Karampela, and Maria Dalamaga. 2024. "ApoB100 and Atherosclerosis: What’s New in the 21st Century?" Metabolites 14, no. 2: 123. https://doi.org/10.3390/metabo14020123