
The Crowded Uterine Horn Mouse Model for Examining Postnatal Metabolic Consequences of Intrauterine Growth Restriction vs. Macrosomia in Siblings


Abstract
:
1. Introduction
2. Results
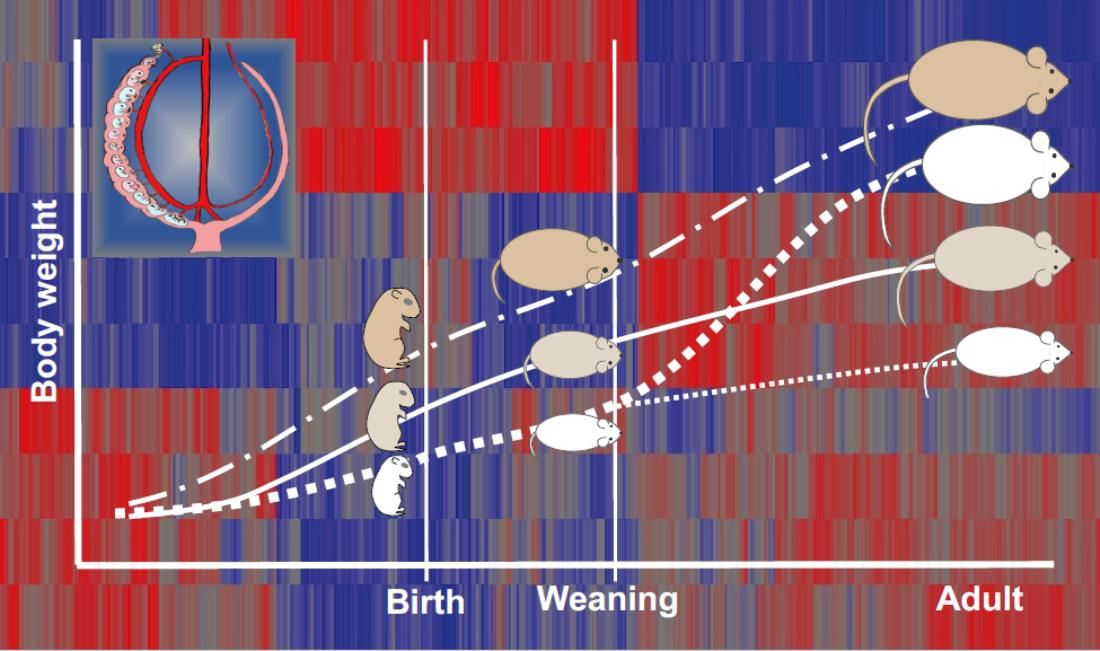
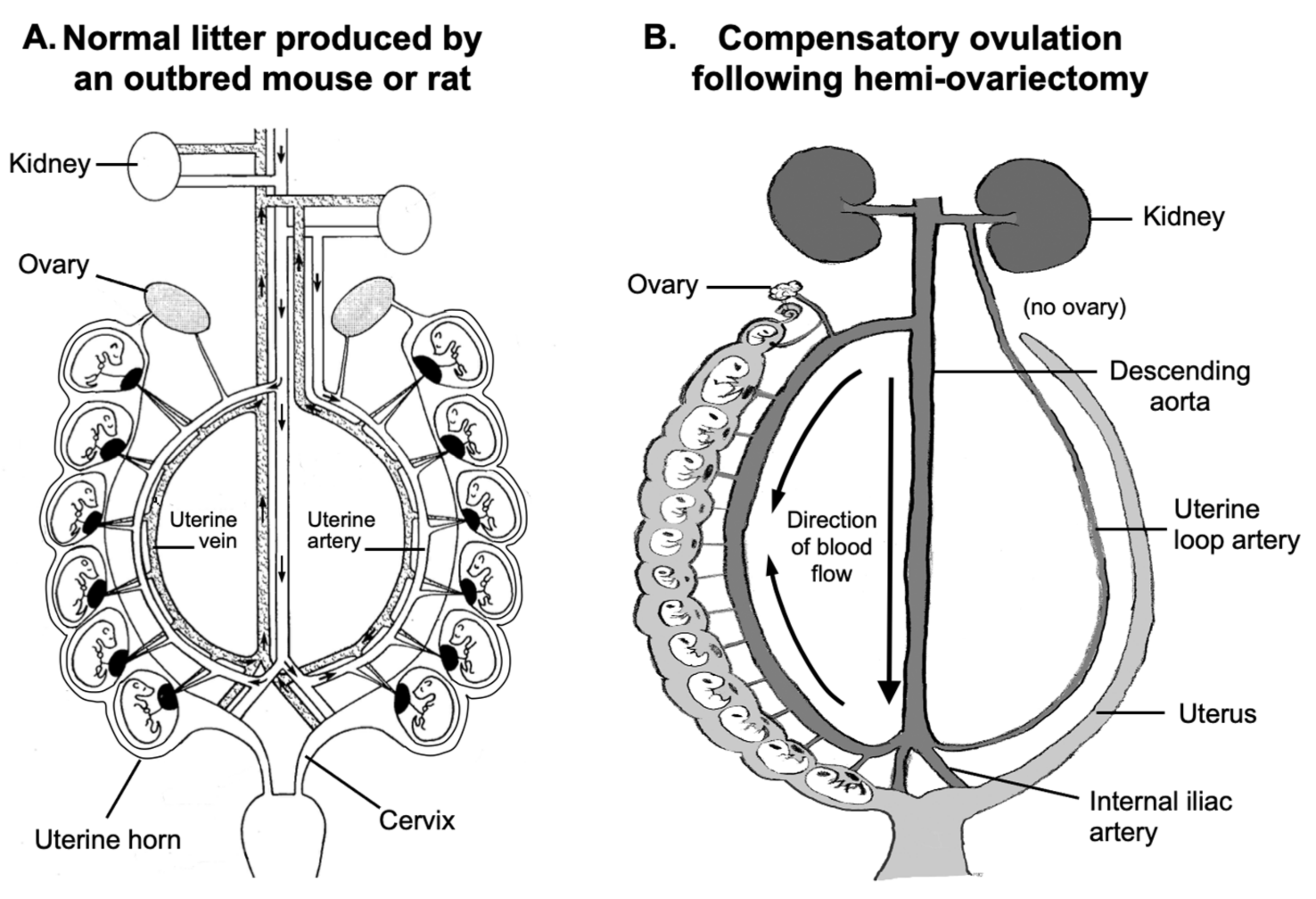
2.1. Placental Blood Flow and Fetal Growth
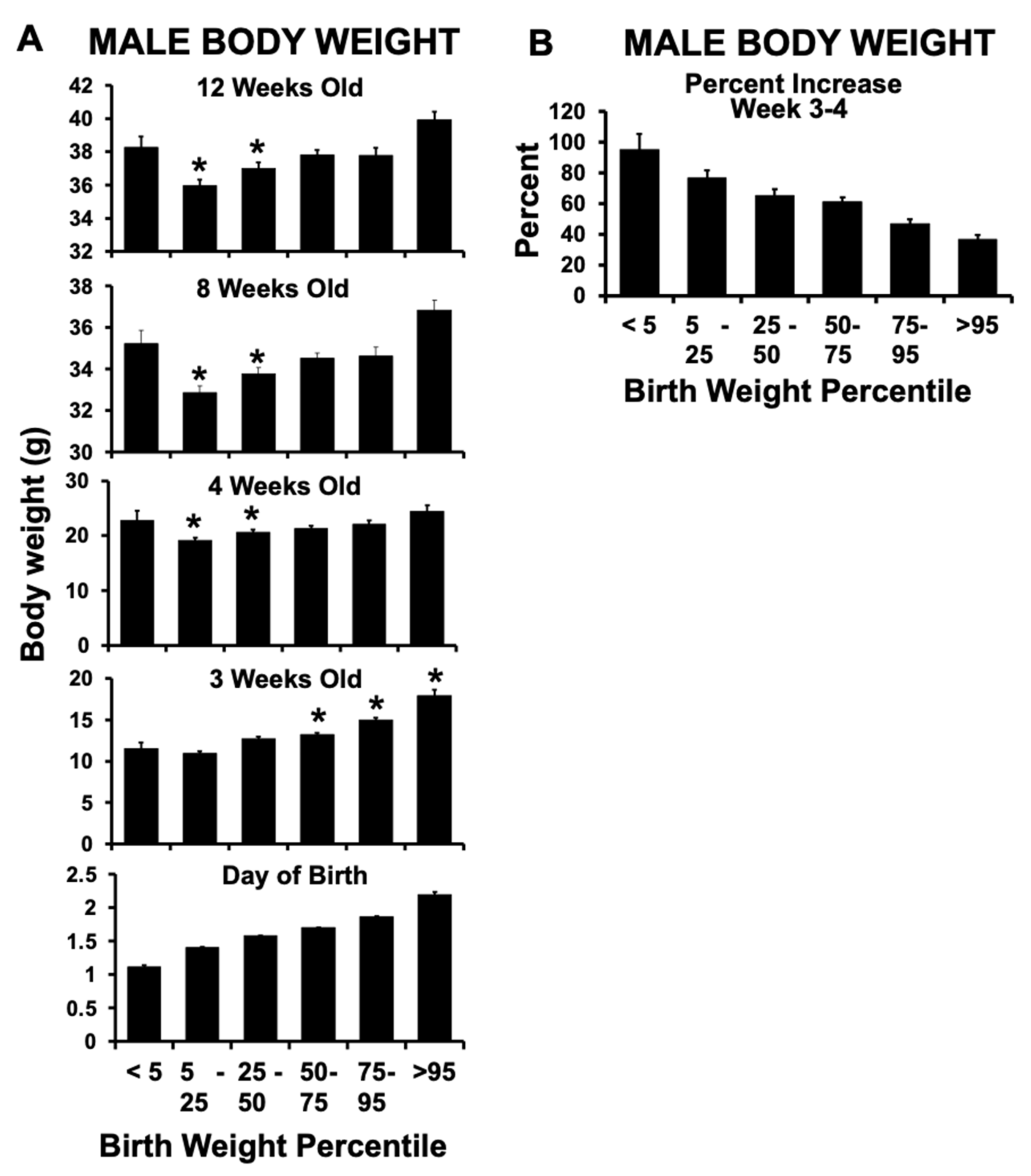
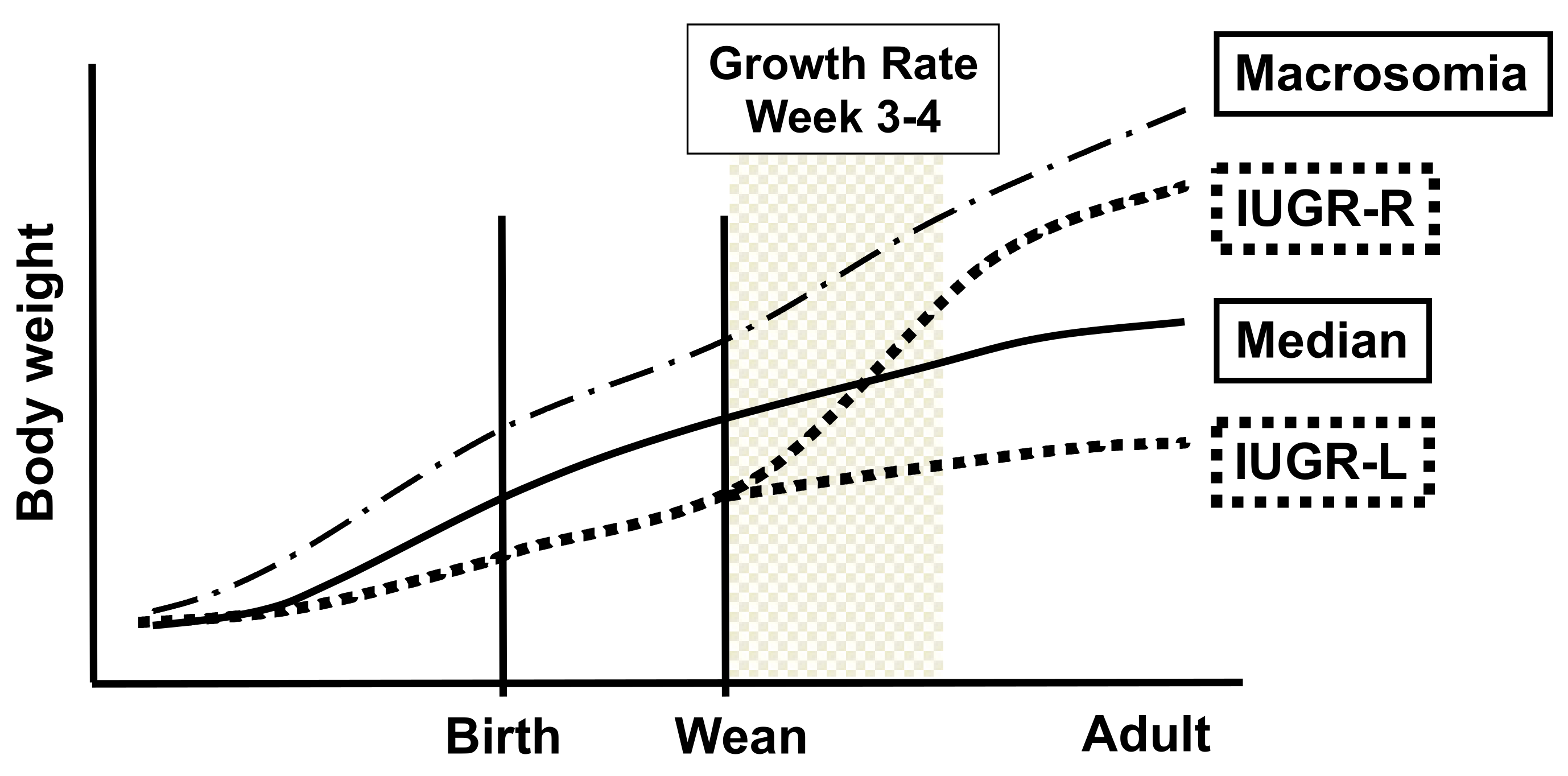
2.2. Birth Weight Criteria and Postnatal Growth Rate
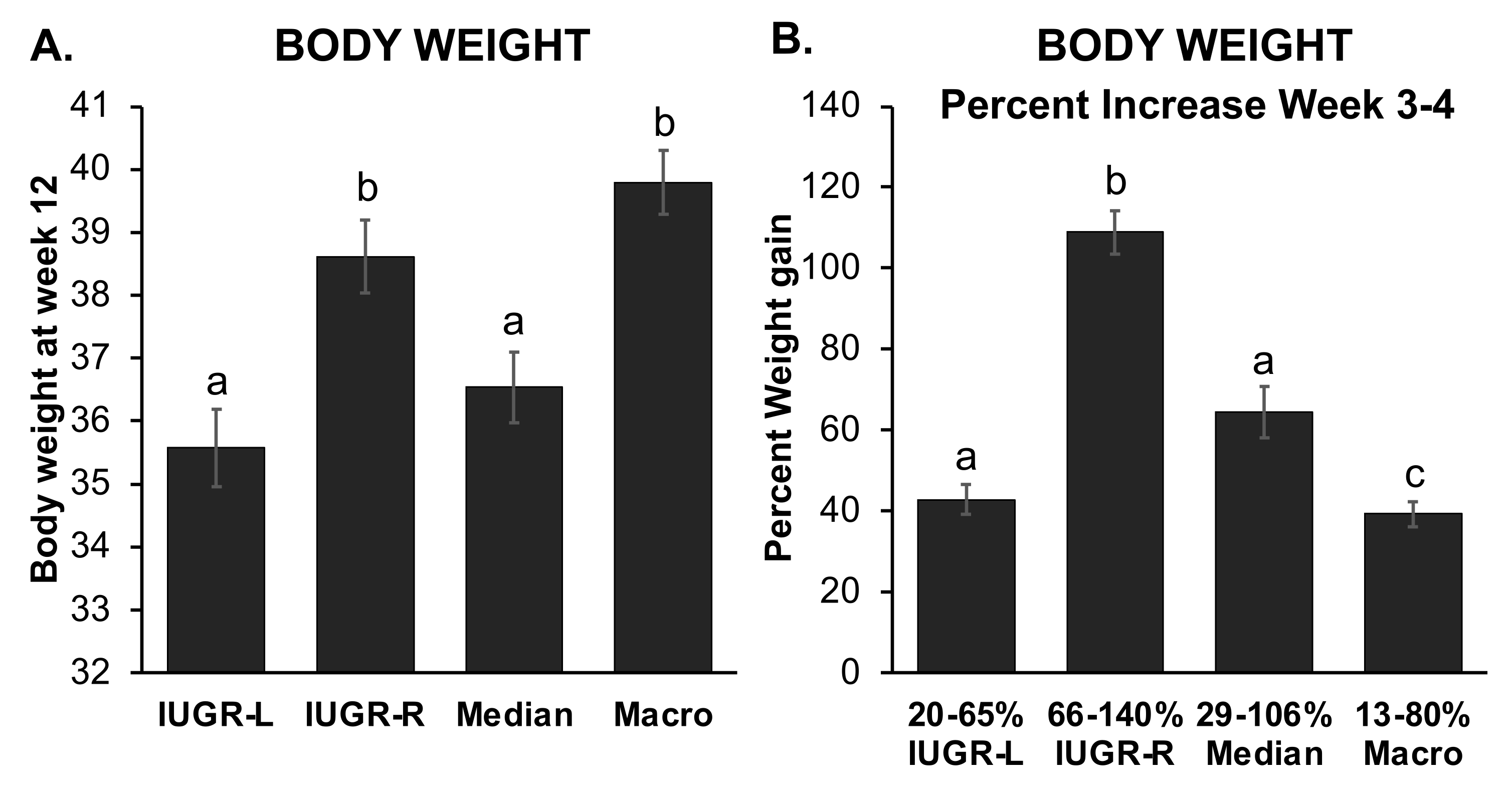
2.3. Different Sub-Groups of IUGR Males
2.4. Experiments on Block 1 and Block 2 Animals When 6 Months Old
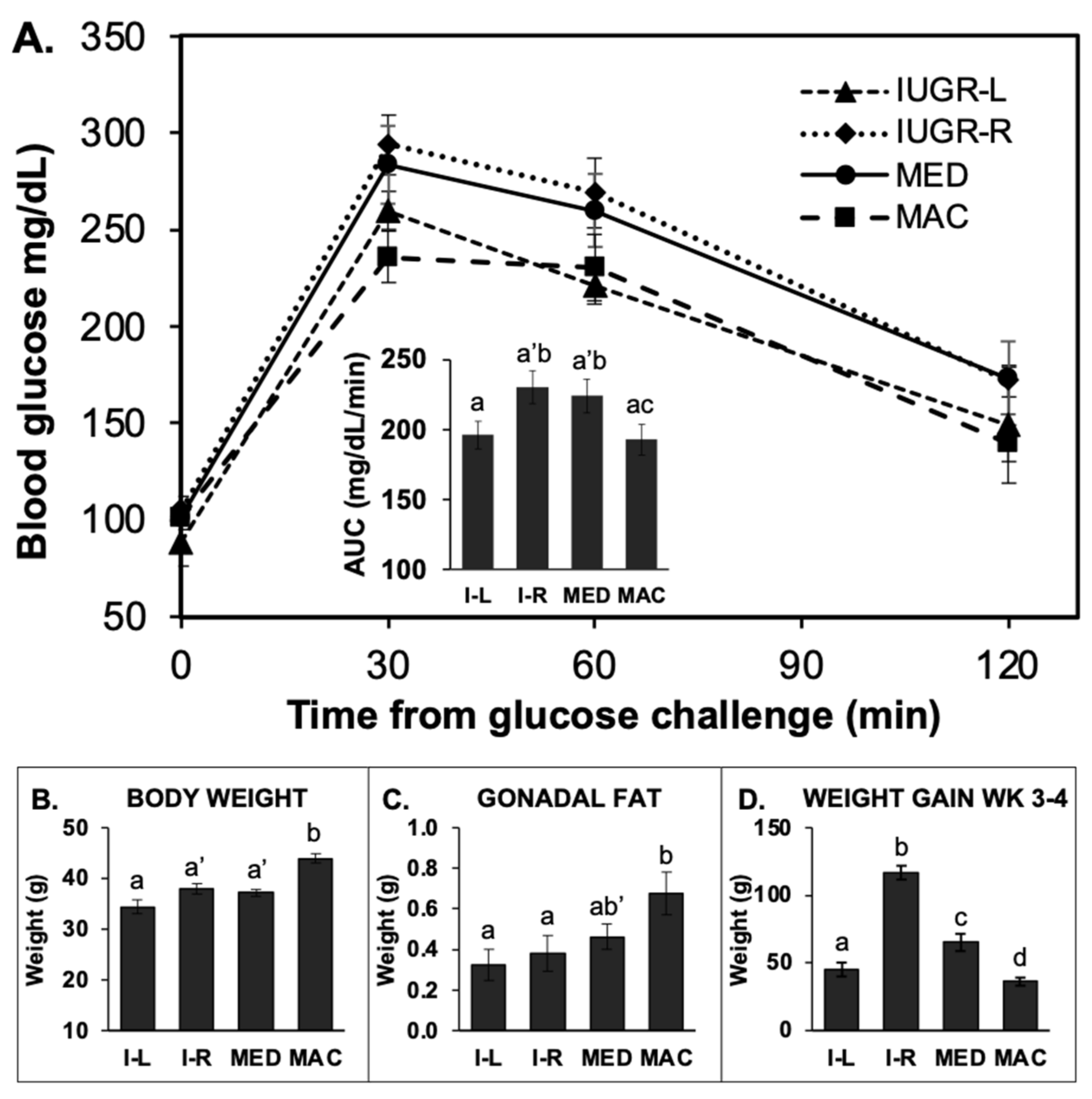
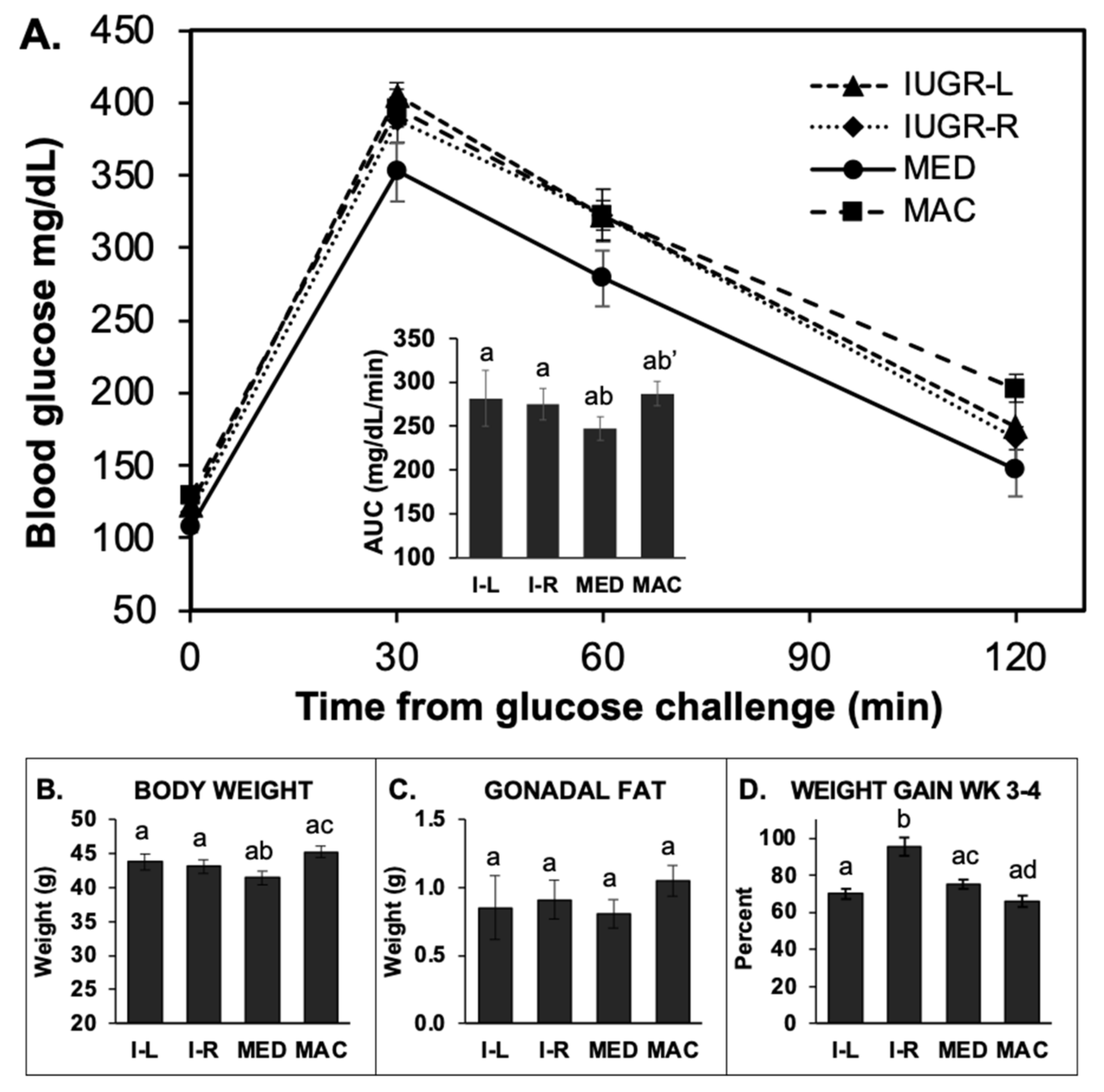
2.5. Glucose Tolerance Test (GTT)
2.5.1. GTT in 14-h Fasted Males
2.5.2. GTT in 4-h Fasted (Block 2) Males
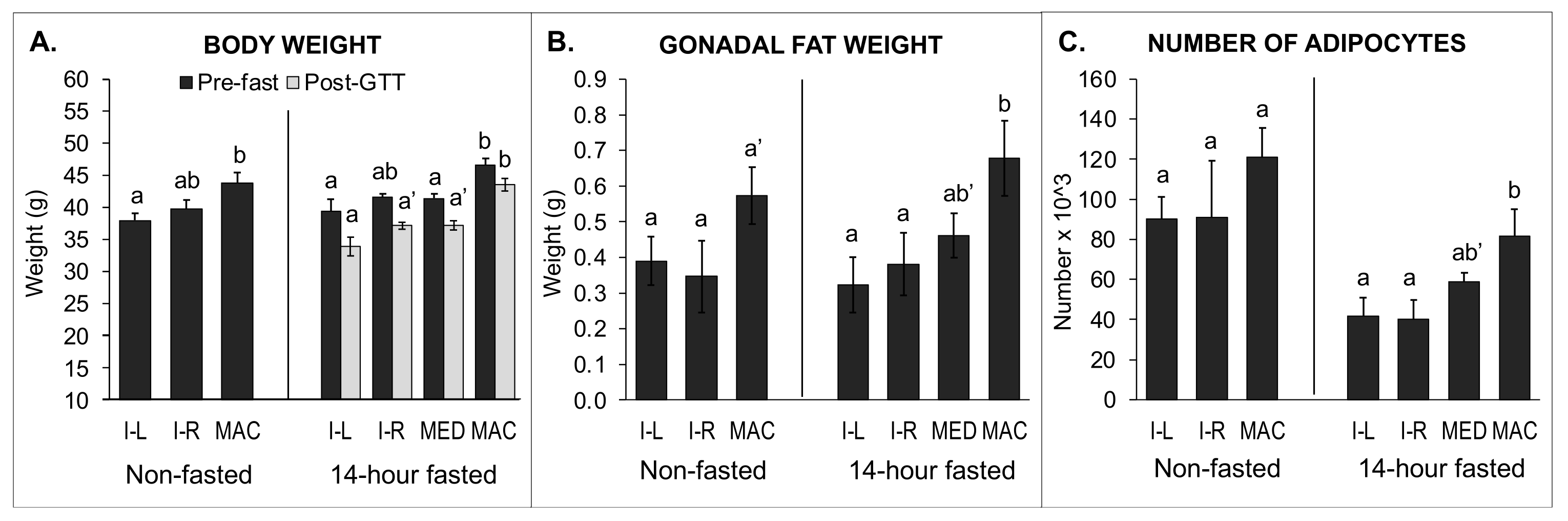
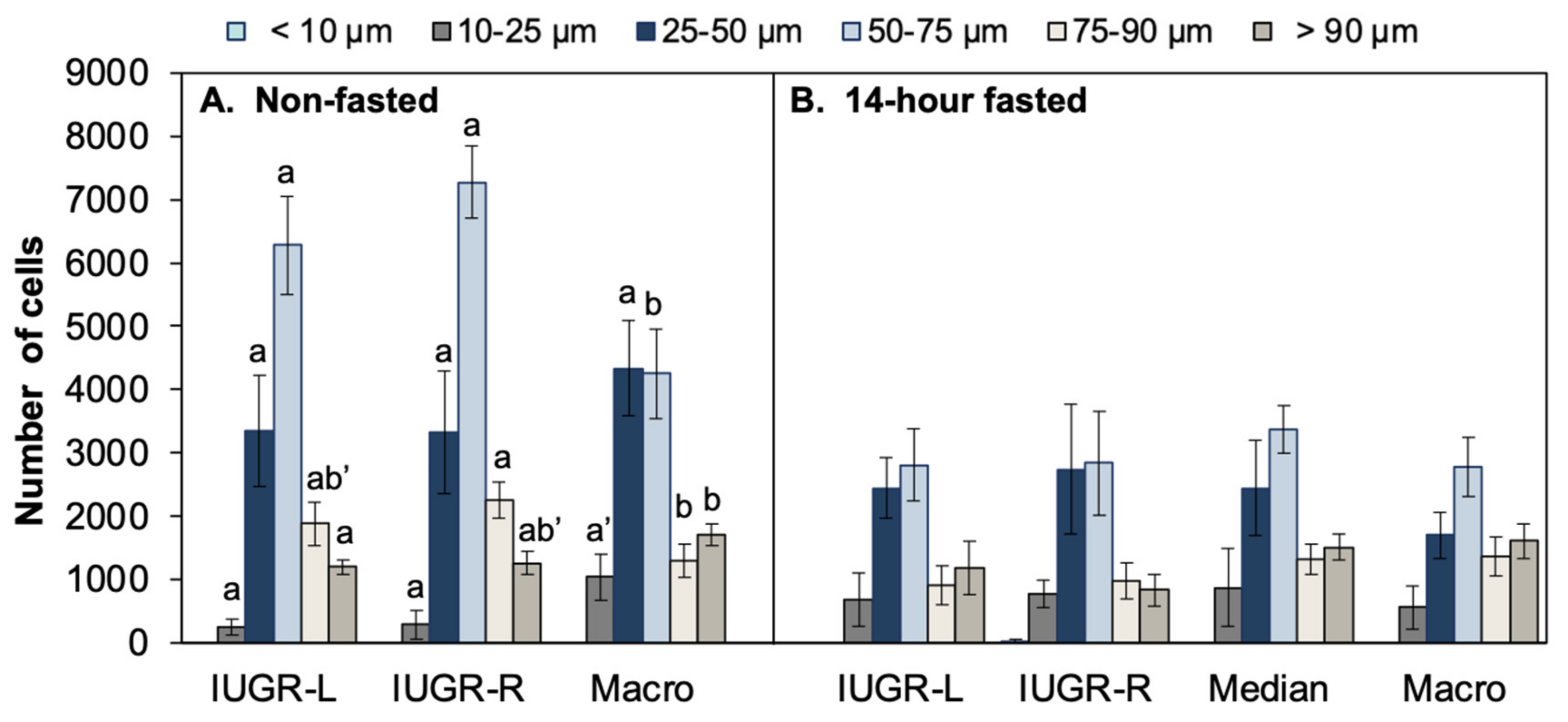
2.6. Body Weight, Organ Weights, Fat Pad Weights, and Gonadal Fat Pad Adipocyte Number and Size: Comparison of 14-h Fasted and Non-Fasted Block 1 Males
2.6.1. Data from 14-h Fasted Males
2.6.2. Data from Non-Fasted Males
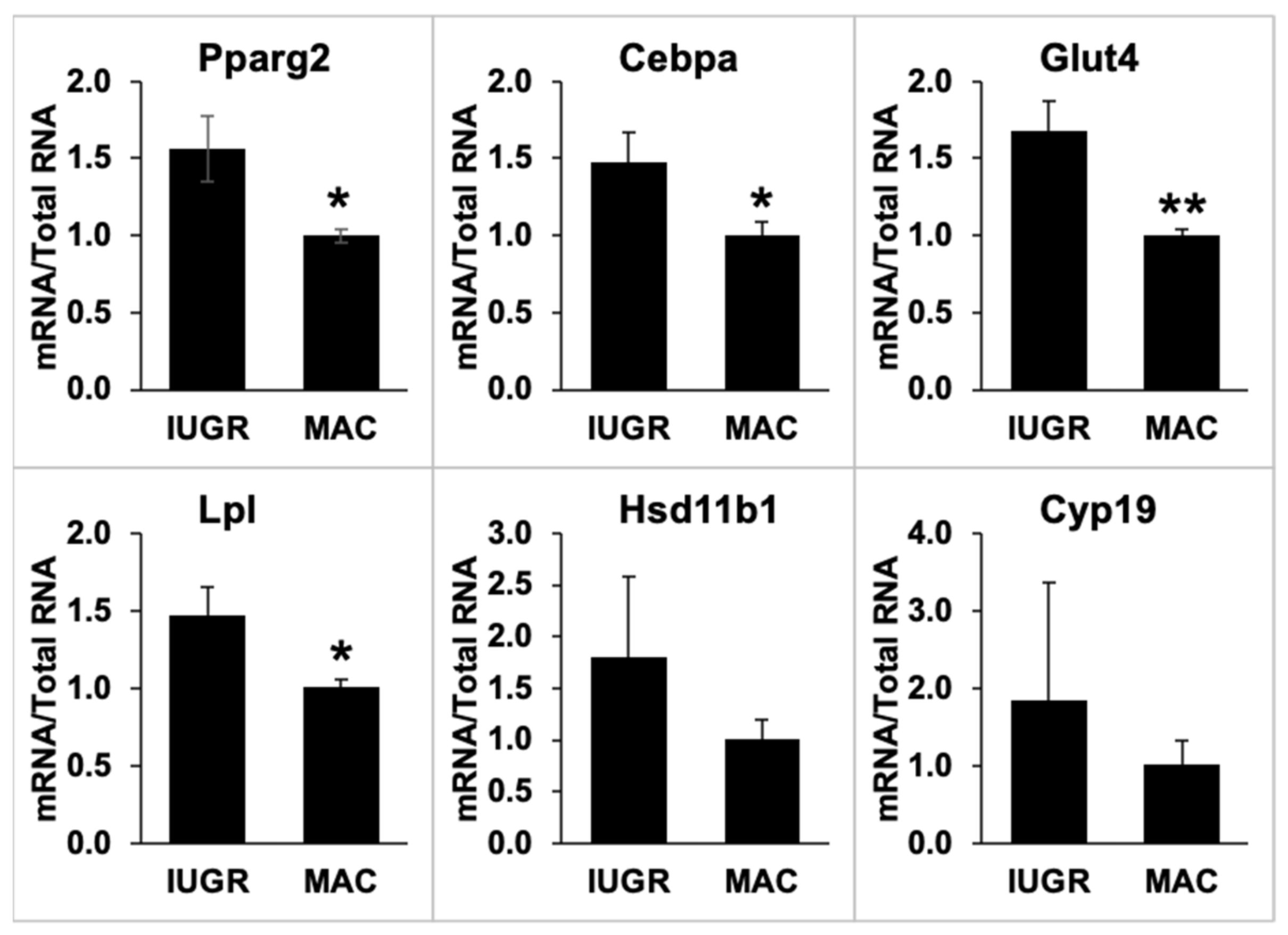
2.7. Gonadal Adipose Tissue Gene Expression by qPCR
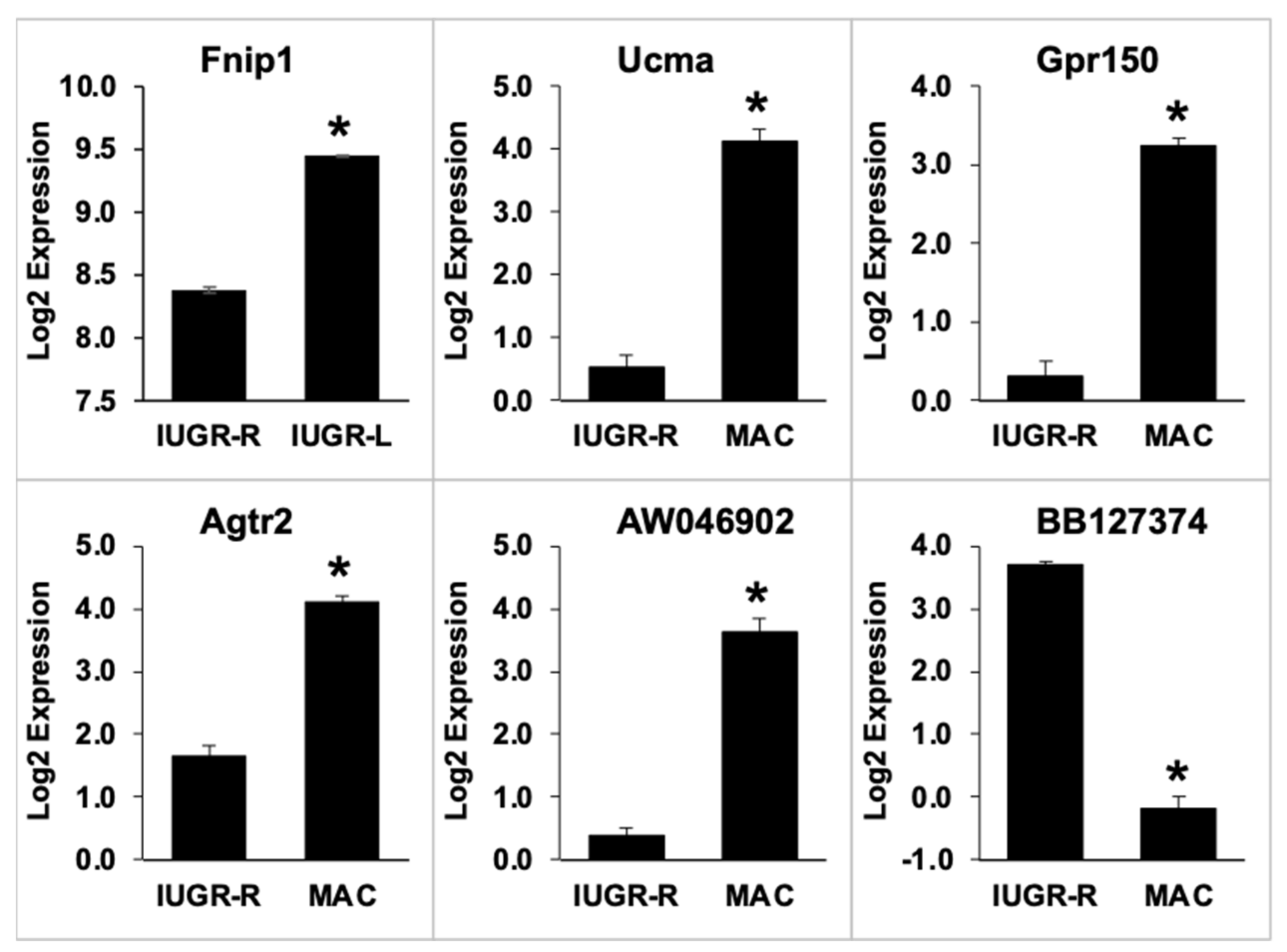
2.8. Gonadal Adipose Tissue Gene Expression by Microarray Analysis
2.8.1. Direct Comparison of Gene Expression in IUGR-L, IUGR-R and Macrosomic Males
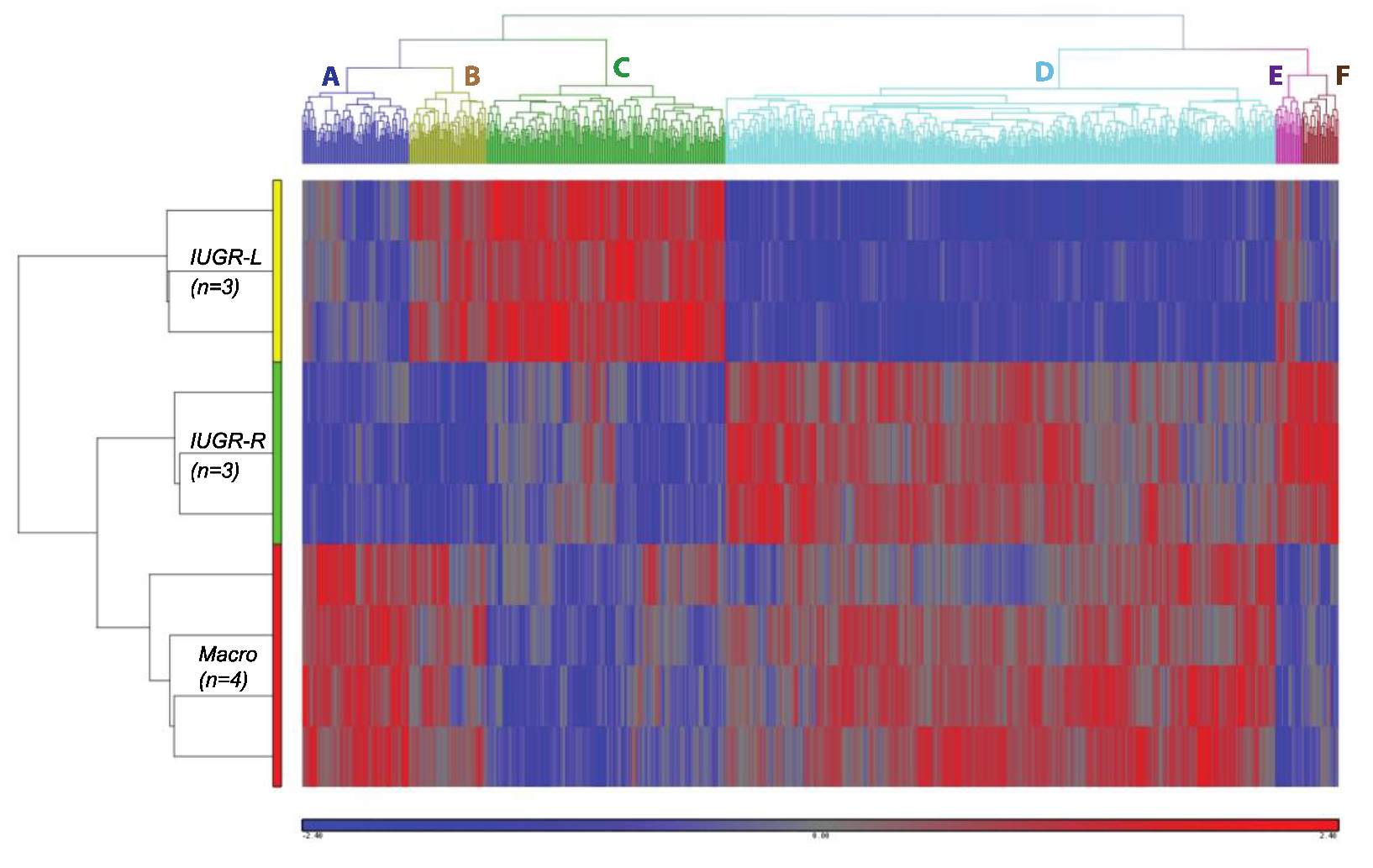
2.8.2. Clustering Analysis of Differentially Expressed Genes
3. Discussion
4. Materials and Methods
4.1. Animal Husbandry
4.2. Hemi-Ovariectomy Procedure
4.3. Mating of Females and Determination of Offspring Body Weights
4.4. Experimental Animals
4.4.1. Block 1 Animals
4.4.2. Block 2 Animals
4.5. Glucose Tolerance Test (GTT)
4.6. Collection of Gonadal Fat
4.7. Analysis of Fat Cell Number and Volume
4.8. Analysis of Gonadal Fat Gene Expression
4.8.1. qPCR Assay
4.8.2. Microarray Analysis
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 2017, 68, 3–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, S.; Bollani, L.; Decembrino, L.; Di Comite, A.; Angelini, M.; Stronati, M. Short-term and long-term sequelae in intrauterine growth retardation (IUGR). J. Matern. Fetal. Neonatal. Med. 2013, 26, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J.; vom Saal, F.S. Role of nutrition and environmental endocrine disrupting chemicals during the perinatal period on the aetiology of obesity. Mol. Cell. Endocrinol. 2009, 304, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. The malnourished baby and infant. Br. Med. Bull. 2001, 60, 69–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fall, C.H. The Genesis of ‘Fetal Origins of Adult Disease’. Int. J. Diab. Dev. Ctries. 2001, 21, 5. [Google Scholar]
- Baird, J.; Fisher, D.; Lucas, P.; Kleijnen, J.; Roberts, H.; Law, C. Being big or growing fast: Systematic review of size and growth in infancy and later obesity. Bmj 2005, 331, 929. [Google Scholar] [CrossRef] [Green Version]
- Styrud, J.; Eriksson, U.J.; Grill, V.; Swenne, I. Experimental intrauterine growth retardation in the rat causes a reduction of pancreatic B-cell mass, which persists into adulthood. Biol. Neonate 2005, 88, 122–128. [Google Scholar] [CrossRef]
- Jones, A.P.; Friedman, M.I. Obesity and adipocyte abnormalities in offspring of rats undernourished during pregnancy. Science 1982, 215, 1518–1519. [Google Scholar] [CrossRef]
- Vuguin, P.M. Animal Models for Small for Gestational Age and Fetal Programing of Adult Disease. Horm. Res. 2007, 68, 113–123. [Google Scholar] [CrossRef] [Green Version]
- Woodward-Lopez, G.; Davis, R.L.; Gerstein, D.E.; Crawford, P.B. Obesity Dietary and Developmental Influences; CRC Press: Boca Raton, FL, USA, 2006; p. 336. [Google Scholar]
- Ravelli, G.P.; Stein, Z.A.; Susser, M.W. Obesity in young men after famine exposure in utero and early infancy. N. Engl. J. Med. 1976, 295, 349–353. [Google Scholar] [CrossRef]
- Jansson, T.; Ylven, K.; Wennergren, M.; Powell, T.L. Glucose transport and system A activity in syncytiotrophoblast microvillous and basal plasma membranes in intrauterine growth restriction. Placenta 2002, 23, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Ogata, E.S.; Finley, S.L. Selective ligation of uterine artery branches accelerates fetal growth in the rat. Pediatr. Res. 1988, 24, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, H.; Gauldie, J. The acute phase response. Immunol. Today 1994, 15, 74–80. [Google Scholar] [CrossRef]
- Priante, E.; Verlato, G.; Giordano, G.; Stocchero, M.; Visentin, S.; Mardegan, V.; Baraldi, E. Intrauterine Growth Restriction: New Insight from the Metabolomic Approach. Metabolites 2019, 9, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oken, E.; Gillman, M.W. Fetal origins of obesity. Obes. Res. 2003, 11, 496–506. [Google Scholar] [CrossRef]
- Taveras, E.M.; Rifas-Shiman, S.L.; Belfort, M.B.; Kleinman, K.P.; Oken, E.; Gillman, M.W. Weight status in the first 6 months of life and obesity at 3 years of age. Pediatrics 2009, 123, 1177–1183. [Google Scholar] [CrossRef] [Green Version]
- Overview. Elsevier Point of Care Editorial Board. Intrauterine Growth Restriction. Available online: https://www-clinicalkey-com.proxy.mul.missouri.edu/#!/content/clinical_overview/67-s2.0-e1056319-3828-4963-b385-e379fb2f33a9 (accessed on 16 November 2021).
- McLaren, A.; Michie, D. Control of pre-natal growth in mammals. Nature 1960, 187, 363–365. [Google Scholar] [CrossRef]
- vom Saal, F.S.; Dhar, M.D. Blood flow in the uterine loop artery and loop vein is bi-directional in the mouse: Implications for intrauterine transport of steroids. Physiol. Behav. 1992, 52, 163–171. [Google Scholar] [CrossRef]
- Even, M.D.; Laughlin, M.H.; Krause, G.F.; vom Saal, F.S. Differences in blood flow to uterine segments and placentae in relation to sex, intrauterine location and side in pregnant rats. J. Reprod. Fertil. 1994, 102, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Coe, B.L.; Kirkpatrick, J.R.; Taylor, J.A.; vom Saal, F.S. A new ‘crowded uterine horn’ mouse model for examining the relationship between foetal growth and adult obesity. Basic Clin. Pharm. Toxicol. 2008, 102, 162–167. [Google Scholar] [CrossRef]
- vom Saal, F.S.; Finch, C.E.; Nelson, J.F. Natural history and mechanisms of reproductive aging in humans, laboratory rodents and other selected vertebrates. In The Physiology of Reproduction, 2nd ed.; Knobil, E., Neil, J.D., Eds.; Raven Press: New York, NY, USA, 1994; Volume 2, pp. 1213–1314. [Google Scholar]
- vom Saal, F.S. Variation in phenotype due to random intrauterine positioning of male and female fetuses in rodents. J. Reprod. Fertil. 1981, 62, 633–650. [Google Scholar] [CrossRef] [Green Version]
- Jensen, T.L.; Kiersgaard, M.K.; Sorensen, D.B.; Mikkelsen, L.F. Fasting of mice: A review. Lab. Anim. 2013, 47, 225–240. [Google Scholar] [CrossRef]
- Pan, D.Z.; Miao, Z.; Comenho, C.; Rajkumar, S.; Koka, A.; Lee, S.H.T.; Alvarez, M.; Kaminska, D.; Ko, A.; Sinsheimer, J.S.; et al. Identification of TBX15 as an adipose master trans regulator of abdominal obesity genes. Genome Med. 2021, 13, 123. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulos, S.; Blair, A.R.; Deluca, N.; Fam, B.C.; Proietto, J. Evaluating the glucose tolerance test in mice. Am. J. Endocrinol. Metab. 2008, 295, E1323–E1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.A.; Sommerfeld-Sager, J.M.; Meng, C.X.; Nagel, S.C.; Shioda, T.; Vom Saal, F.S. Reduced body weight at weaning followed by increased post-weaning growth rate interacts with part-per-trillion fetal serum concentrations of bisphenol A (BPA) to impair glucose tolerance in male mice. PLoS ONE 2018, 13, e0208846. [Google Scholar] [CrossRef] [Green Version]
- vom Saal, F.S.; Vandenberg, L.N. Update on the Health Effects of Bisphenol A: Overwhelming Evidence of Harm. Endocrinology 2021, 162, bqaa171. [Google Scholar] [CrossRef]
- Xue, B.; Sukumaran, S.; Nie, J.; Jusko, W.J.; DuBois, D.C.; Almon, R.R. Adipose Tissue Deficiency and Chronic Inflammation in Diabetic Goto-Kakizaki Rats. PLoS ONE 2011, 6, e17386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ailhaud, G.; Grimaldi, P.; Negrel, R. Cellular and molecular aspects of adipose tissue development. Annu. Rev. Nutr. 1992, 12, 207–233. [Google Scholar] [CrossRef]
- Baba, M.; Hong, S.-B.; Sharma, N.; Warren, M.B.; Nickerson, M.L.; Iwamatsu, A.; Zhen, W.; Burke, T.R.J.; Linehan, W.M.; Schmidt, L.S.; et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 15552–15557. [Google Scholar] [CrossRef] [Green Version]
- Linehan, W.M.; Srinivasan, R.; Schmidt, L.S. The genetic basis of kidney cancer: A metabolic disease. Nat. Rev. Urol. 2010, 7, 277–285. [Google Scholar] [CrossRef]
- Blanchard, P.-G.; Festuccia, W.T.; Houde, V.P.; St-Pierre, P.; Brule, S.; Turcotte, V.; Cote, M.; Bellmann, K.; Marette, A.; Deshaies, Y. Major involvement of mTOR in the PPARγ-induced stimulation of adipose tissue lipid uptake and fat accretion. J. Lipid Res. 2012, 53, 1117–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-H.; Li, F.; Takahashi, N. The renin angiotensin system and the metabolic syndrome. Open Hypertens. J. 2010, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Masseira, F.; Lamande, N.; Ailhaud, G.; Teboul, M.; Moustaid-Moussa, N.; Gasc, J.M.; Quignard-Boulange, A. Deficiency of angiotensisn type 2 receptor rescues obesity but not hypertension induced by overexpression of angiotensinogen in adipose tissue. Endocrinology 2009, 150, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Hafizi, S.; Wang, X.; Chester, A.; Yacoub, M.H.; Proud, C.G. ANG II activates effectors of mTOR via PI3-K signaling in human coronary smooth muscle cells. Am. J. Physiol.—Heart Circ. Physiol. 2004, 287, H1232–H1238. [Google Scholar] [CrossRef]
- Pulakat, L.; DeMarco, V.G.; Whaley-Connell, A.; Sowers, J.R. The Impact of Overnutrition on Insulin Metabolic Signaling in the Heart and the Kidney. Cardiorenal. Med. 2011, 1, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Brey, C.W.; Nelder, M.P.; Hailemariam, T.; Gaugler, R.; Hashmi, S. Krüppel-like family of transcription factors: An emerging new frontier in fat biology. Int. J. Biol. Sci. 2009, 5, 622–636. [Google Scholar] [CrossRef]
- Pei, H.; Yao, Y.; Yang, Y.; Liao, K.; Wu, J.-R. Kruppel-like factor KLF9 regulates PPARg transactivation at the middle stage of adipogenesis. Cell Death Differ. 2011, 18, 315–327. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, S. Role of kruppel-like transcription factors in adipogenesis. Dev. Biol. 2013, 474, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Wei, H.; Song, T.; Yang, Y.; Zhang, F.; Zhou, Y.; Peng, J.; Jiang, S. KLF13 promotes porcine adipocyte differentiation through PPARγ activation. Cell Biosci. 2015, 5, 28. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Yea, S.; Li, S.; Chen, Z.; Narla, G.; Banck, M.; Laborda, J.; Tan, S.; Friedman, J.M.; Friedman, S.L.; et al. Kruppel-like Factor-6 Promotes Preadipocyte Differentiation through Histone Deacetylase 3-dependent Repression of DLK1. J. Biol. Chem. 2005, 280, 26941–26952. [Google Scholar] [CrossRef] [Green Version]
- Verboven, K.; Wouters, K.; Gaens, K.; Hansen, D.; Bijnen, M.; Wetzels, S.; Stehouwer, C.D.; Goossens, G.H.; Schalkwijk, C.G.; Blaak, E.E.; et al. Abdominal subcutaneous and visceral adipocyte size, lipolysis and inflammation relate to insulin resistance in male obese humans. Sci. Rep. 2018, 8, 4677. [Google Scholar] [CrossRef]
- Liu, F.; Wang, H.; Zhu, D.; Bi, Y. Adipose Morphology: A Critical Factor in Regulation of Human Metabolic Diseases and Adipose Tissue Dysfunction. Obes. Surg. 2020, 30, 5086–5100. [Google Scholar] [CrossRef]
- Ruschke, K.; Illes, M.; Kern, M.; Kloting, I.; Fasshauer, M.; Schon, M.R.; Kosacka, H.; Fitzl, G.; Kovacs, P.; Stumvoll, M.; et al. Repin1 maybe involved in the regulation of cell size and glucose transport in adipocytes. Biochem. Biophys. Res. Commun. 2010, 400, 246–251. [Google Scholar] [CrossRef]
- Hales, C.N.; Barker, D.J. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef] [Green Version]
- Gluckman, P.D.; Hanson, M.A.; Beedle, A.S. Early life events and their consequences for later disease: A life history and evolutionary perspective. Am. J. Hum. Biol. 2007, 19, 1–19. [Google Scholar] [CrossRef]
- Martinez, M.A.; Castro, I.; Rovira, J.; Ares, S.; Rodriguez, J.M.; Cunha, S.C.; Casal, S.; Fernandes, J.O.; Schuhmacher, M.; Nadal, M. Early-life intake of major trace elements, bisphenol A, tetrabromobisphenol A and fatty acids: Comparing human milk and commercial infant formulas. Environ. Res. 2019, 169, 246–255. [Google Scholar] [CrossRef]
- Ruiz, D.; Padmanabhan, V.; Sargis, R.M. Stress, Sex, and Sugar: Glucocorticoids and Sex-Steroid Crosstalk in the Sex-Specific Misprogramming of Metabolism. J. Endocr. Soc. 2020, 4, bvaa087. [Google Scholar] [CrossRef]
- Palanza, P.; Paterlini, S.; Brambilla, M.M.; Ramundo, G.; Caviola, G.; Gioiosa, L.; Parmigiani, S.; Vom Saal, F.S.; Ponzi, D. Sex-biased impact of endocrine disrupting chemicals on behavioral development and vulnerability to disease: Of mice and children. Neurosci. Biobehav. Rev. 2021, 121, 29–46. [Google Scholar] [CrossRef]
- Sieli, P.T.; Jasarevic, E.; Warzak, D.A.; Mao, J.; Ellersieck, M.R.; Liao, C.; Kannan, K.; Collet, S.H.; Toutain, P.L.; vom Saal, F.S.; et al. Comparison of serum bisphenol A concentrations in mice exposed to bisphenol A through the diet versus oral bolus exposure. Env. Health Perspect. 2011, 119, 1260–1265. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, J.; Gallian, E. Methods for the determination of adipose cell size in man and animals. J. Lipid Res. 1968, 9, 110–119. [Google Scholar] [CrossRef]
- Kump, D.S.; Booth, F.W. Sustained rise in triacylglycerol synthesis and increased epididymal fat mass when rats cease voluntary wheel running. J. Physiol. 2005, 565, 911–925. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.D. The Coulter Principle: Foundation for an Industry. JALA 2003, 8, 10. [Google Scholar] [CrossRef]
- Bustin, S.A. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 2000, 25, 169–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell. 1999, 4, 611–617. [Google Scholar] [CrossRef]
- Jones, J.R.; Barrick, C.; Kim, K.-A.; Lindner, J.; Blondeau, B.; Fuimoto, Y.; Shiota, M.; Kesterson, R.A.; Kahn, B.B.; Magnuson, M.A. Deletion of PPAR in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2005, 102, 6207–6212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaestner, K.H.; Christy, R.J.; Lane, M.D. Mouse insulin-responsive glucose transporter gene: Characterization of the gene and trans-activation by the CCAAT/enhancer binding protein. Proc. Natl. Acad. Sci. USA 1990, 87, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Moseti, D.; Regassa, A.; Kim, W.-K. Molecular Regulation of Adipogenesis and Potential Anti-Adipogenic Bioactive Molecules. Int. J. Mol. Sci. 2016, 17, 124–147. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, A.M.; Orlando, R.A. Role of adipocyte-derived lipoprotein lipase in adipocyte hypertrophy. Nutr. Metabolism 2007, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Rosmond, R. Association studies of genetic polymorphisms in central obesity: A critical review. Int. J. Obes. 2003, 27, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Latil, A.; Bieche, I.; Vidaud, D.; Lidereau, R.; Berthon, P.; Cussenot, O.; Vidaud, M. Evaluation of androgen, estrogen (ER alpha and ER beta), and progesterone receptor expression in human prostate cancer by real-time quantitative reverse transcription-polymerase chain reaction assays. Cancer Res. 2001, 61, 1919–1926. [Google Scholar]
- Richter, C.A.; Taylor, J.A.; Ruhlen, R.R.; Welshons, W.V.; vom Saal, F.S. Estradiol and bisphenol A stimulate androgen receptor and estrogen receptor gene expression in fetal mouse prostate cells. Environ. Health Perspect. 2007, 115, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups Compared | KEGG Pathway | Ratio | Direction | p-Value | Gene Identifier | Gene ID |
|---|---|---|---|---|---|---|
| IUGR-L | Type 2 Diabetes Mellitus | 9.5 | Up | 0.002 | NM_008840 | Pik3cd |
| Vs. IUGR-R | down-regulated in IUGR-R | 4.31 | Down | 0.047 | BE943756 | Cacna1a |
| z score = 3.86 | 3.45 | Up | 0.019 | NM_010438 | Hk1 | |
| 2.92 | Down | 0.045 | BB184171 | Mapk8 | ||
| 2.11 | Down | 0.004 | BB205102 | Pik3cg | ||
| 2.11 | Down | 0.002 | BB048682 | Cacna1d | ||
| 2.03 | Down | 0.038 | BE947490 | Mapk10 | ||
| Fat digestion and absorption | 3.04 | Up | 0.025 | BG070618 | - | |
| down-regulated in IUGR-R | 3.01 | Down | 0.047 | NM_011128 | Pnliprp2 | |
| z-score = 2.12 | 2.56 | Down | 0.023 | NM_025469 | Clps | |
| 2.05 | Down | 0.006 | AI326372 | Pnlip | ||
| Macrosomic vs. | Carbohydrate digestion and absorption | 5.67 | Up | 0.019 | NM_008840 | Pik3cd |
| IUGR-R | 2.89 | Down | 0.011 | BI696040 | Atp1a1 | |
| up-regulated in IUGR-R | 2.42 | Up | 0.028 | NM_019741 | Slc2a5 | |
| z score = 4.67 | 2.22 | Up | 0.037 | BC027319 | Atp1b1 | |
| 2.2 | Up | 0.043 | BC027319 | Atp1b1 | ||
| Fat digestion and absorption | 6.49 | Up | 0.048 | AI194999 | Apoa1 | |
| up-regulated in IUGR-R | 4.96 | Up | 0.015 | NM_007980 | Fabp2 | |
| z-score = 4.09 | 4.29 | Up | 0.006 | AI527359 | Apoa1 | |
| 3.41 | Up | 0.019 | NM_025469 | Clps |
| Gene Name | Sequence (5′–3′) | |
|---|---|---|
| Hsd11b1 | Forward | GCAGCATTGCCGTCATCTC |
| Reverse | GAACCCATCCAGAGCAAACTTG | |
| Probe | TGGCTGGGAAAATGACCCAGCCTATG | |
| Cyp19 | Forward | CCGAGCCTTTGGAGAACAATT |
| Reverse | TCCACACAAACTTCCACCATTC | |
| Probe | TTTCTTTATGAAAGCTCTGACGGGCCCT |
| Gene Name | Assay ID | Context Sequence |
|---|---|---|
| Pparg | Mm00440945_m1 | TCAGTGGAGACCGCCCAGGCTTGCT |
| Cebpa | Mm00514283_s1 | ACCAGCCACCGCCGCCACCGCCACC |
| Lpl | Mm00434764_m1 | ATGGATGGACGGTAACGGGAATGTA |
| Glut4 (Slc2a4) | Mm00436615_m1 | CTGCTGCTGCTGGAACGGGTTCCAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taylor, J.A.; Coe, B.L.; Shioda, T.; vom Saal, F.S. The Crowded Uterine Horn Mouse Model for Examining Postnatal Metabolic Consequences of Intrauterine Growth Restriction vs. Macrosomia in Siblings. Metabolites 2022, 12, 102. https://doi.org/10.3390/metabo12020102
Taylor JA, Coe BL, Shioda T, vom Saal FS. The Crowded Uterine Horn Mouse Model for Examining Postnatal Metabolic Consequences of Intrauterine Growth Restriction vs. Macrosomia in Siblings. Metabolites. 2022; 12(2):102. https://doi.org/10.3390/metabo12020102
Chicago/Turabian StyleTaylor, Julia A., Benjamin L. Coe, Toshi Shioda, and Frederick S. vom Saal. 2022. "The Crowded Uterine Horn Mouse Model for Examining Postnatal Metabolic Consequences of Intrauterine Growth Restriction vs. Macrosomia in Siblings" Metabolites 12, no. 2: 102. https://doi.org/10.3390/metabo12020102