Effect of Alloying Elements on the Mechanical Properties of Mo3Si


1
School of Mechanical Engineering, Jiangsu University of Technology, Changzhou 213001, China
2
School of Materials Engineering, Jiangsu University of Technology, Changzhou 213001, China
3
School of Materials Science and Engineering, Central South University, Changsha 410083, China
*
Author to whom correspondence should be addressed.
Metals 2021, 11(1), 129; https://doi.org/10.3390/met11010129
Submission received: 8 December 2020
/
Revised: 29 December 2020
/
Accepted: 8 January 2021
/
Published: 11 January 2021
Abstract
:Molybdenum silicides are attractive high-temperature structural materials because of their excellent thermal stability and outstanding oxidation resistance at high temperatures. First-principles calculations were employed to investigate the effect of alloying elements (Cr, Nb, V, W, Al, Ga, and Ge) on the mechanical properties of Mo3Si. The structural stabilities of doped Mo3Si were calculated, showing that the Pm-3n structure was stable at the investigated low-doping concentration. The calculated elastic constants have also evaluated some essential mechanical properties of doped Mo3Si. Cr- and V-doping decreased the elastic modulus, while Al- and Nb-doping slightly increased the shear and Young’s modulus of Mo3Si. Furthermore, V-, Al- and Nb-doping decreased the B/G and Poisson ratio, suggesting that these elements could form strong covalent bonds, and decrease shear deformation and alloy ductility. Based on the three-dimensional contours and two-dimensional projection of the elastic modulus, Cr- and V-doping exhibited a significant influence on the anisotropy of the shear and Young’s modulus. According to charge density and density of states, the electronic structures of alloyed Mo3Si were further analyzed to reveal the doping effects.
1. Introduction
Molybdenum silicides are attractive high-temperature structural materials because of their excellent thermal stability and outstanding oxidation resistance at high temperatures [1,2,3,4,5,6]. However, their brittleness at room temperature has restricted their engineering applications incredibly. For the Mo-Si system, there are three stable compounds: MoSi2, Mo5Si3, and Mo3Si. Notably, some studies [1,2,3,4] have focused on Mo5Si3 and MoSi2 over the past few decades, while the research about Mo3Si (another molybdenum silicide) is still limited, attributed to its relatively low fracture toughness and room temperature strength. The low fracture toughness of Mo3Si is ascribed to its few active slip systems [7]. New doped Mo3Si with alloying elements should be developed to acquire adequate fracture toughness and ductility.
Alloying elements close to molybdenum in the periodic table, including Nb and W, has attracted great interest in the last few decades. Ray [6] investigated the effects of Nb and W on the stability of Mo3Si by combining theoretical calculations and experiments. Rosales [7,8] studied the effect of niobium on some mechanical behaviors of Mo3Si by the arc-melting technique. Their results suggested that Nb can strengthen Mo3Si due to lattice distortion [7], which is favorable to wear resistance [8]. Xu [9] proposed that Mo addition can improve the anti-damage properties and the hardness of Mo3Si-based coatings by the double glow-discharge plasma approach. Furthermore, the influences of some alloying elements (including B, Al, and Cr) on the oxidation resistance of Mo3Si are being investigated [10,11,12].
First-principles calculations [13,14,15,16,17] can provide a more in-depth explanation and prediction of the relationship between mechanical properties and alloying effect for molybdenum silicides. In our early work [18], the effects of Cr, Nb, and W on the elastic properties of MoSi2 were studied by the first-principles. Hu [19] proposed that MoSi2 doped with Al enhances the ductility by the first-principles calculations, whereas Ge affects the ductility adversely. However, the alloying influences on the mechanical behaviors of Mo3Si are still limited, especially these neighboring elements of silicon in the periodic table. Only these lattice constants [20] and some thermodynamic properties [20,21] of Mo3Si have been investigated using first-principles calculations. This work analyzed the elastic properties of Mo3Si alloyed with different concentrations of Cr, Nb, V, W, Al, Ga, and Ge from first-principles calculations. Remarkably, Cr, Nb, V, and W substitute preferentially on Mo sites, while Al, Ga, and Ge substitute preferentially on Si sites.
2. Computational Methods
These calculations were carried out with the use of Perdew-Burke-Ernzerh of ex-change–correlation (XC) functional [22] based on density functional theory (DFT) in the VASP (Vienna Ab-initio Simulation Package) [23,24].. The electron-core interaction was described by the projector augmented wave method (PAW). The electronic configurations of elements were: Mo 4p64d55s1, Si 3s23p2, Cr 3d54s1, Nb 4s24p64d45s1, V 3p63d44s1, W 5p65d46s2, Al 3s23p1, Ga 3d104s24p1, and Ge 3d104s24p2. The energy cut-off was 420 eV, which was considered enough in our early research [18,20].
Mo3Si belonged to an A15 cubic structure (space group: Pm-3n). Mo and Si occupied 6c (0.25, 0, 0.5) and 2a (0, 0, 0) of the Wyckoff positions. The crystal structure of Mo3Si is shown in Figure 1. These 2 × 2 × 1, 2 × 1 × 1, and 1 × 1 × 1 supercells of Mo3Si were established to study the alloying effects at the concentrations of 3.125, 6.25, and 12.5 at.%, respectively. Cr, Nb, V, and W are doped on the Mo site, while Al, Ga, and Ge are doped on the Si site. Stress-strain method was used to evaluate the elastic constants. Several different strain modes were imposed on the crystal structure and the Cauchy stress tensor for each strain mode was evaluated, and then the related elastic constants were identified as the coefficients in strain-stress relations. The 16 × 16 × 16 Monkhorst-Pack k-mesh [25] generated for the conventional cell of Mo3Si and the scaled-down k-meshes were used for the corresponding supercells. The force convergence criterion per atom was 2 × 10−2 eV/Å.
3. Results and Discussion
3.1. Structural Stability
It is of great significance to assess the structural stability for a doped system [18,19]. The formation enthalpies of the doped Mo3Si were calculated to evaluate their phase stability as follows:
where p, q, r are the molar fractions of Mo, Si, and alloying elements X in the alloy, and , , , are the energies of these pure elements and the corresponding alloy, respectively.
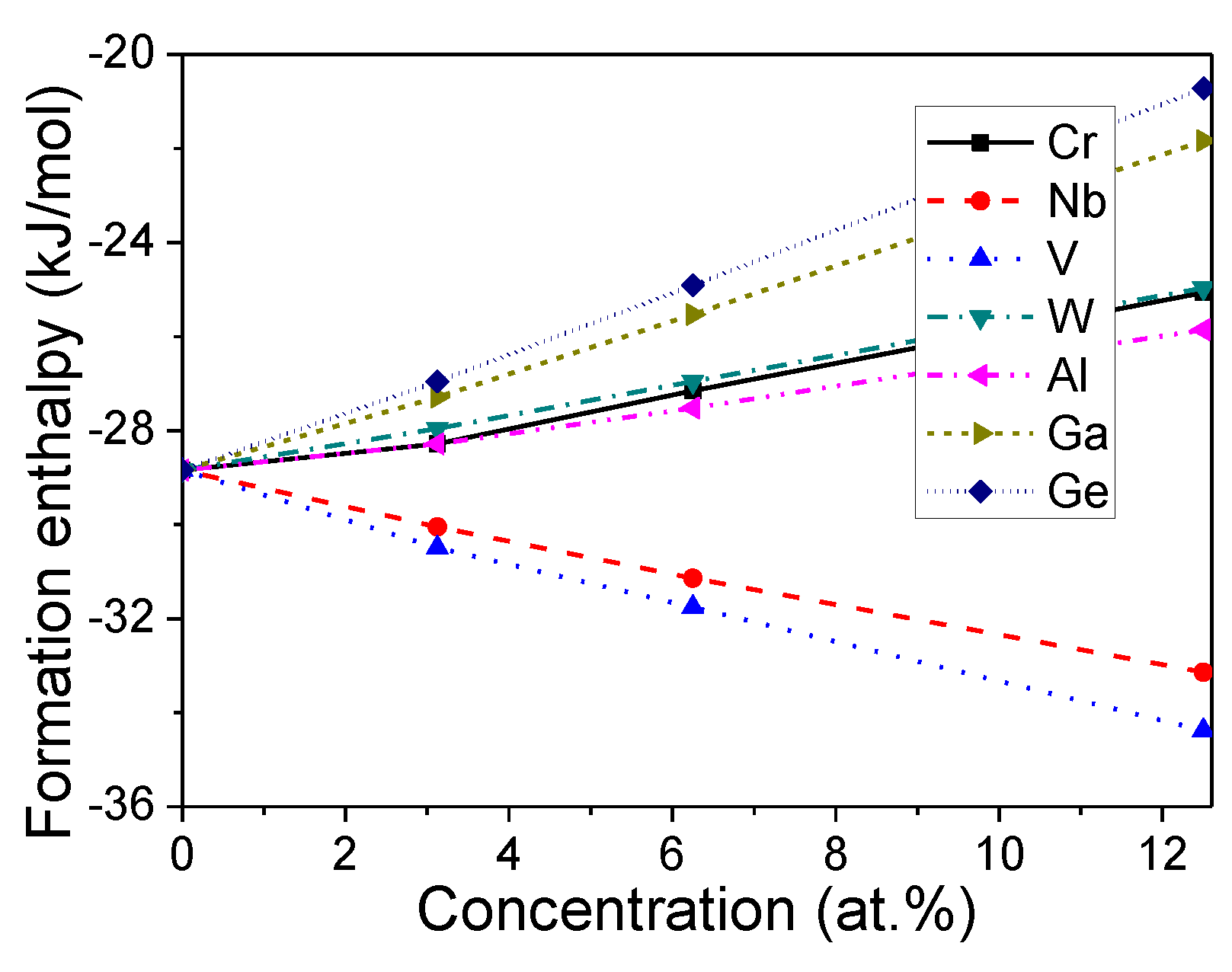
The formation enthalpy of undoped Mo3Si with the Pm-3n structure was 28.82 kJ/mol-atom, which agreed with the experimental value (−30.5 kJ/mol-atom [26]) and previous DFT calculation (−27.99 kJ/mol-atom [27]).
Figure 2 shows that the formation enthalpies of Mo3Si doped with Cr, W, Al, Ga, and Ge increase with the increasing doping concentration, while those of Mo3Si doped with Nb and V decrease with the increasing doping concentration. It is suggested that Nb and V help structural stability, while the other doping elements (Cr, W, Al, Ga, and Ge) decrease structural stability. Ray [6] proposed that Nb substitution (≤27.5 at.%) stabilizes Mo3Si, while W-doping destabilizes Mo3Si. Our calculations about the formation enthalpy are in accord with Ray’s experiments [6].
These formation enthalpies of doped Mo3Si are negative, suggesting that doped Mo3Si with a Pm-3n structure is stable at the investigated low-doping concentration (≤12.5 at.%). This work investigated the alloying effects of different elements under low-doping concentrations (≤12.5 at.%) on Mo3Si.
Table 1 shows the elastic constants of Mo3Si. The calculated elastic constants of Mo3Si in the present work agree with the previous DFT calculations [9,21,28,29]. For a cubic structure, the mechanical criteria of stability are , , and [30]. The calculated elastic constants of doped Mo3Si conform to these stability conditions above, suggesting that doped Mo3Si is mechanically stable.
3.2. Mechanical Properties
The bulk and shear modulus of a cubic structure can be evaluated based on the Voigt-Reuss-Hill approximation as follows [31,32]:
where are the calculated elastic constants of Mo3Si; and are the isotropised bulk and shear modulus. Here, these subscripts represent Voigt, Reuss, and Hill approximation, respectively.
The isotropised Poisson ratio [33] and isotropised Young’s modulus are calculated as:
The Debye temperature [34] and wave velocities are assessed by these calculated mechanical properties as:
where , and are longitudinal, transverse, and average wave velocities; is the atom number; is the density; is the weight; , , and are the Avagadro’s, Planck’s, and Boltzmann’s constants, respectively.
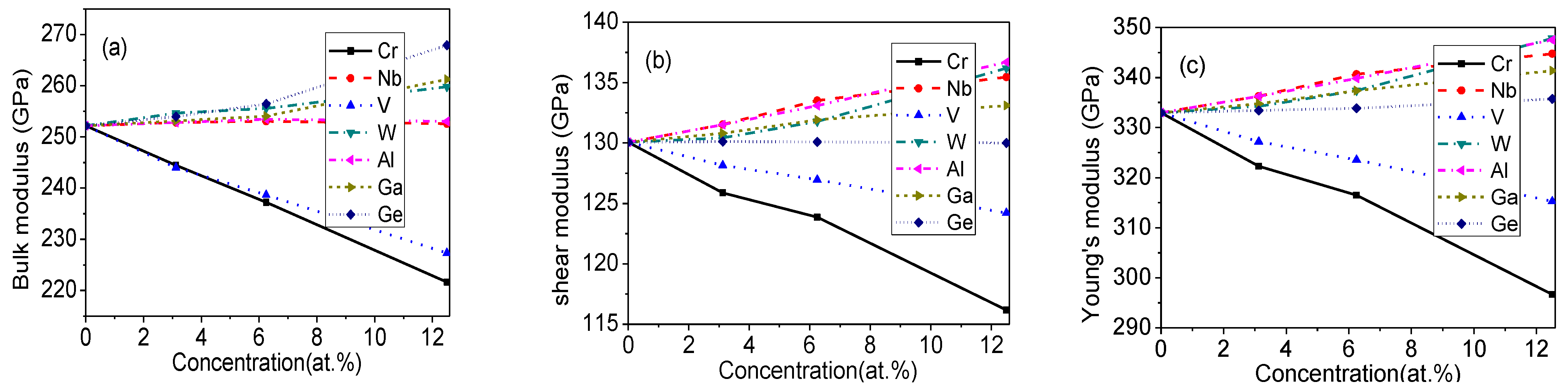
According to Equations (2) and (3), the elastic moduli of Mo3Si doped with elements are calculated and given in Figure 3. The and of undoped Mo3Si are 252.1 and 130.1 GPa, respectively, which agrees with Xu’s calculation [9] (248.9 and 133.5 GPa) and Zhong’s calculation [21] (245.5 and 133.2 GPa).
In Figure 3, the elastic moduli of Cr- and V-doped Mo3Si decrease distinctly, while those of W- and Ga-doped Mo3Si increase moderately. The bulk modulus of Ge-doped Mo3Si increases obviously, but its and change little. The and of Al- and Nb-doped Mo3Si increase slightly, but their bulk moduli are almost unchanged.
There is an empirical equation () showing that the shear modulus is approximately proportional to Vickers hardness . It is suggested that Al-, Nb-, W- and Ga-doping increase the hardness of Mo3Si. Unfortunately, there is still a lack of experimental data from the literature about the mechanical properties of doped Mo3Si. Rosales [7] stated that Nb should enhance the hardness and decrease the toughness according to an arc-melting technique. Our findings are in accord with Rosales’ viewpoints [7].
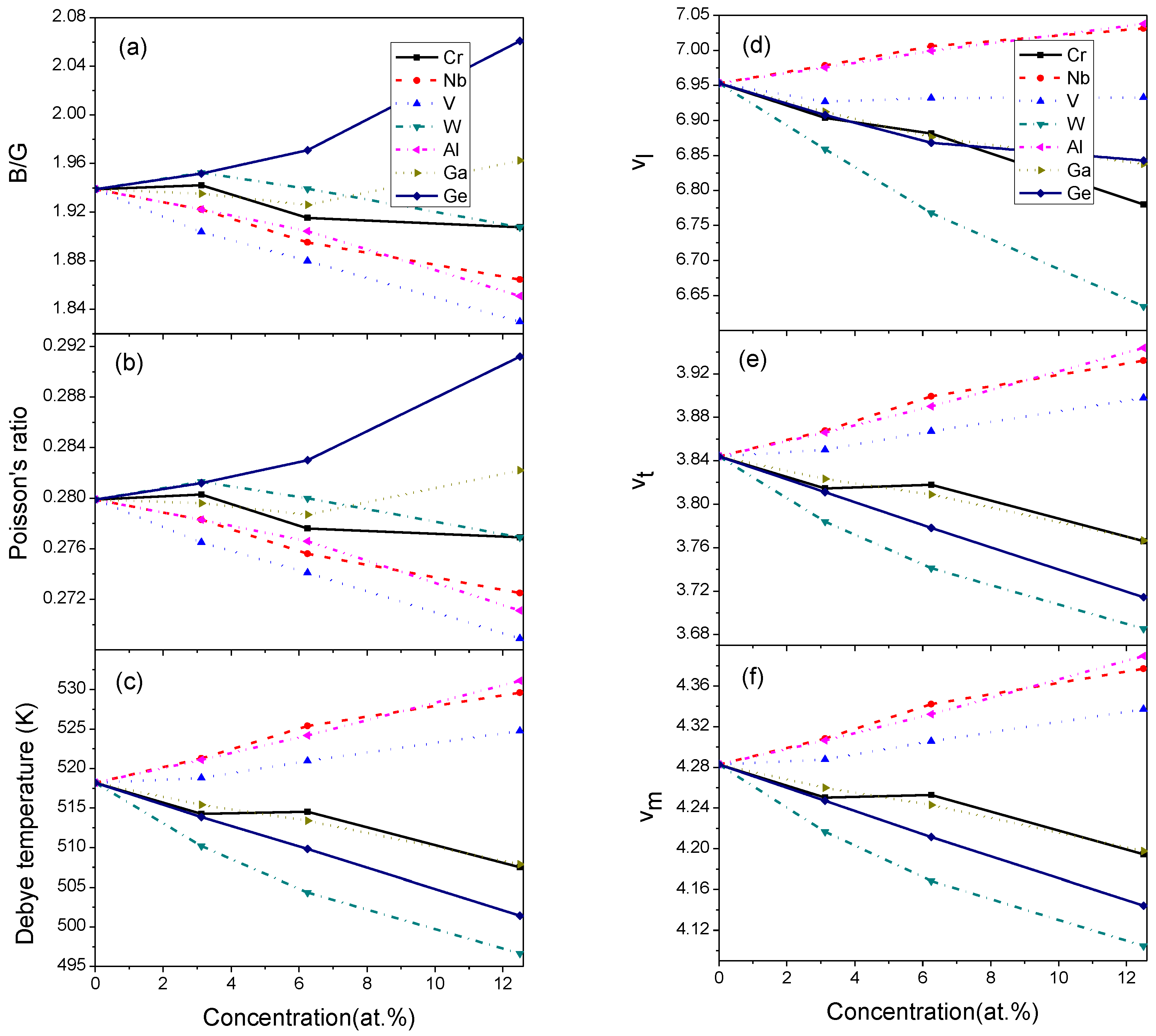
The alloy’s ductility or brittleness can be evaluated by B/G and [35]. Figure 4 shows the B/G and of alloyed Mo3Si. The B/G and of undoped Mo3Si are higher than 1.75 and 0.26, respectively, suggesting that the ductility of Mo3Si is not low. The B/G and of Mo3Si decrease with increasing V, Al, and Nb, and increase with the increasing Ge. The effect of Cr, W, and Ga on the B/G and of Mo3Si is relatively small. It is suggested that V-, Al-, and Nb-doping can form strong covalent bonds, and decrease shear deformation and alloy ductility. Rosales [7] proposed that Nb should strengthen the solid solution and reduce the toughness of Mo3Si, which is in line with our calculations.
Figure 4 shows the wave velocities and Debye temperature of doped Mo3Si. The Debye temperature of doped Mo3Si is 518.2 K, consistent with Zhong’s calculation (522.2 K) [21]. These alloying elements exhibit different effects on the wave velocities and the Debye temperature of Mo3Si. Compared to undoped Mo3Si, the wave velocities and Debye temperatures of Nb-, Al- and V-doped Mo3Si increase, while those of Cr-, W-, Ga- and Ge-doped Mo3Si decrease. Among these alloying elements, Mo3Si doped with Al and Nb exhibit high wave velocities and Debye temperatures, and those of W-doped Mo3Si reduce strikingly.
3.3. Anisotropic Elasticity
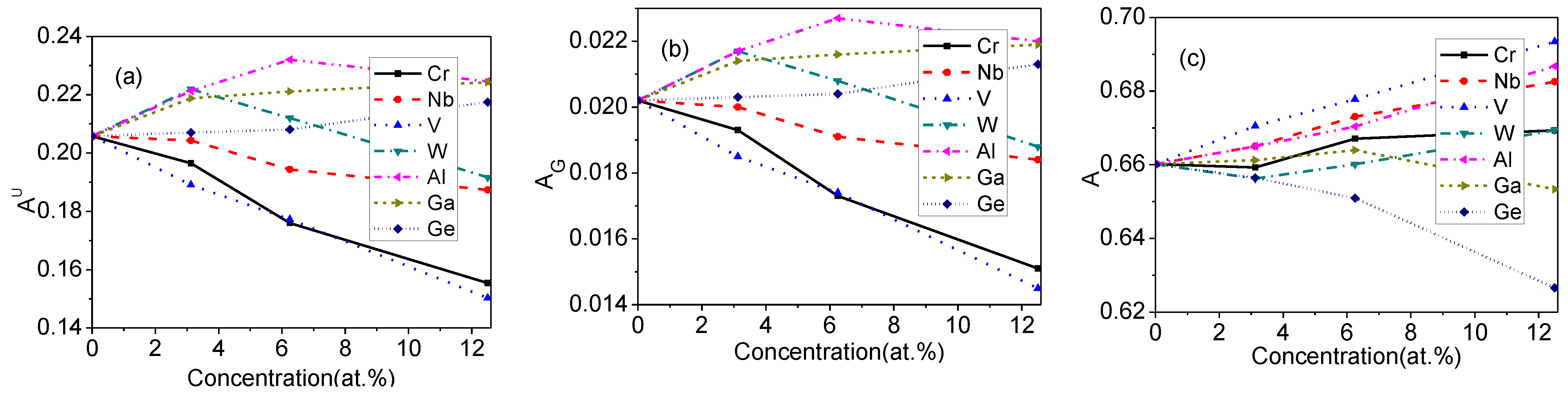
Some mechanical anisotropic indexes of doped Mo3Si, such as the universal anisotropic index (), the percent anisotropy of the bulk and shear modulus ( and ), and the Zener’s shear anisotropy index (), are denoted as:
According to Equation (3), the of doped Mo3Si is zero, suggesting that the bulk modulus is elastically isotropic. Figure 5 shows the mechanical anisotropic indexes, , , and of doped Mo3Si. It is seen that the shear modulus exhibits strong directional dependence. The percent anisotropy of the shear modulus decreases by Mo-site doping (Cr, Nb, and V), but increases by Si-site doping (Al, Ga, and Ge). The of W-doped Mo3Si increases slightly at relatively low concentrations (≤6.25 at.%), but decreases at high concentrations (12.5 at.%). The Zener’s shear anisotropy index increases by Mo-site doping (Cr, Nb, and V) and Al-doping, but decreases by Ge-doping obviously. The of W-doped Mo3Si shows a fluctuation, which is similar to the situation of .
In Figure 5, the of Mo3Si doped with Cr and V decrease, that of Al-doped Mo3Si increases obviously, and that of W-doped Mo3Si shows a fluctuation. It is also seen from Figure 5 that the of Mo3Si doped with V increases, and that of Mo3Si doped with Ge clearly decreases. The directionality of the mechanical properties for Cr-, V-, Al and Ge-doped Mo3Si should be different from that of undoped Mo3Si, attributed to differential physical properties and electronic structures between the alloying elements (Cr, V, Al and Ge) and these substituted atoms (Mo or Si).
Anisotropic elasticity as a function of crystallographic orientation is studied according to the three-dimensional surfaces of an elastic modulus. The and [36,37,38,39] of a cubic structure in spherical coordinates can be calculated by:
where are the matrix compliance constants which can be obtained by solving the inverse matrix of elastic matrix; , , and are the direction cosines for three coordinates, respectively.
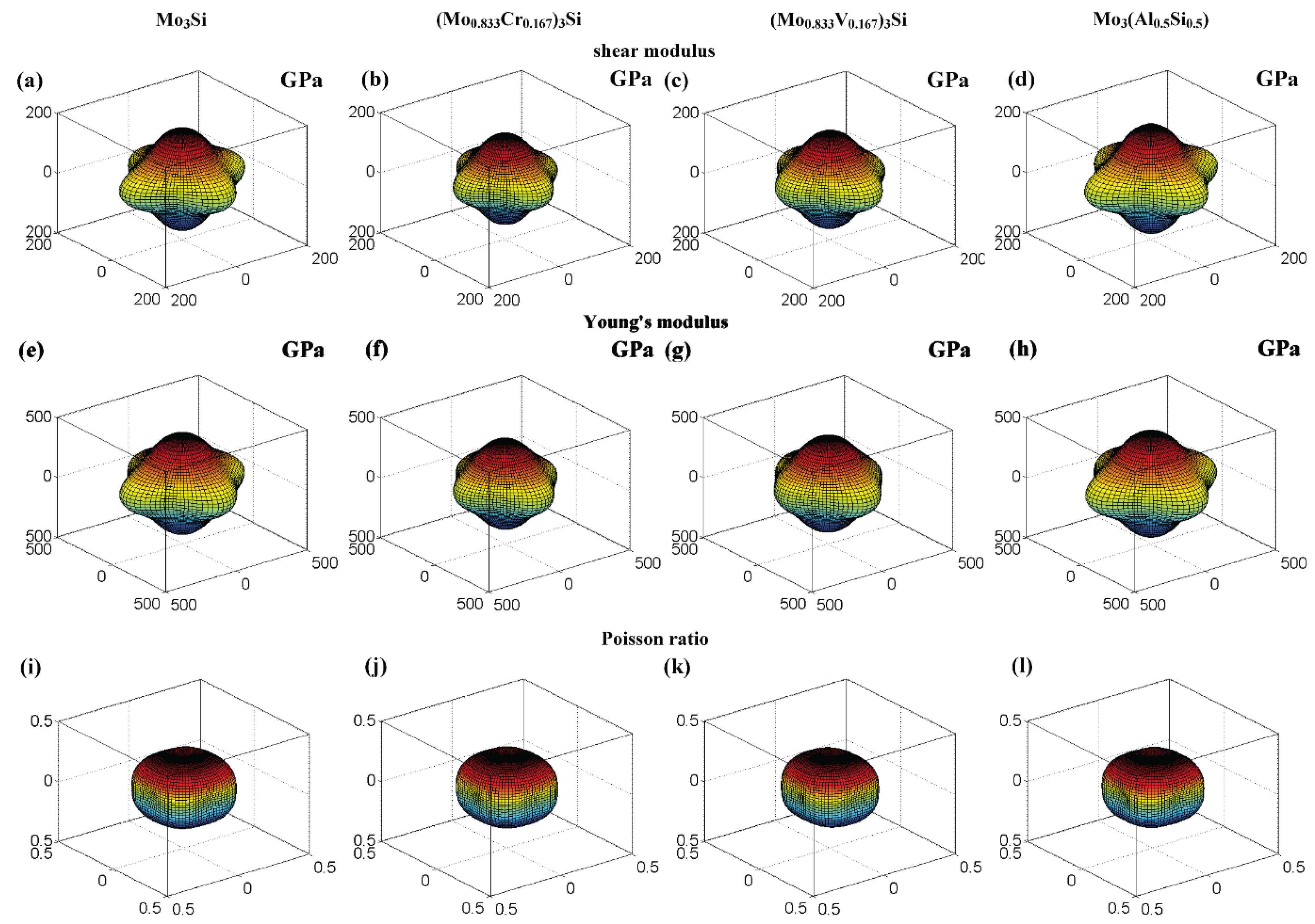
According to Equation (6), the bulk modulus is elastically isotropic. Combined with Equations (3) and (6), the shear modulus and Poisson ratio at different directions can also be calculated using spherical coordinates. Moreover, the calculation formula of the shear modulus and Poisson ratio for a cubic structure can also be referred in Refs. [33,39]. Figure 6 shows the 3D surfaces of the shear modulus, Young’s modulus and Poisson ratio for Mo3Si alloyed with 12.5 at.% Cr, V, and Al. These surfaces of Cr- and V-doped Mo3Si are different from those of undoped Mo3Si, showing the significant effect of Cr and V on elastic anisotropy. The anisotropy of shear and Young’s modulus for Mo3Si doped with Al strengthens slightly. The calculations also show that the 3D contours of the Poisson ratio for doped Mo3Si are very close to those of undoped Mo3Si, suggesting that the effect of doping with the alloying elements on the anisotropy of Poisson ratio is small.
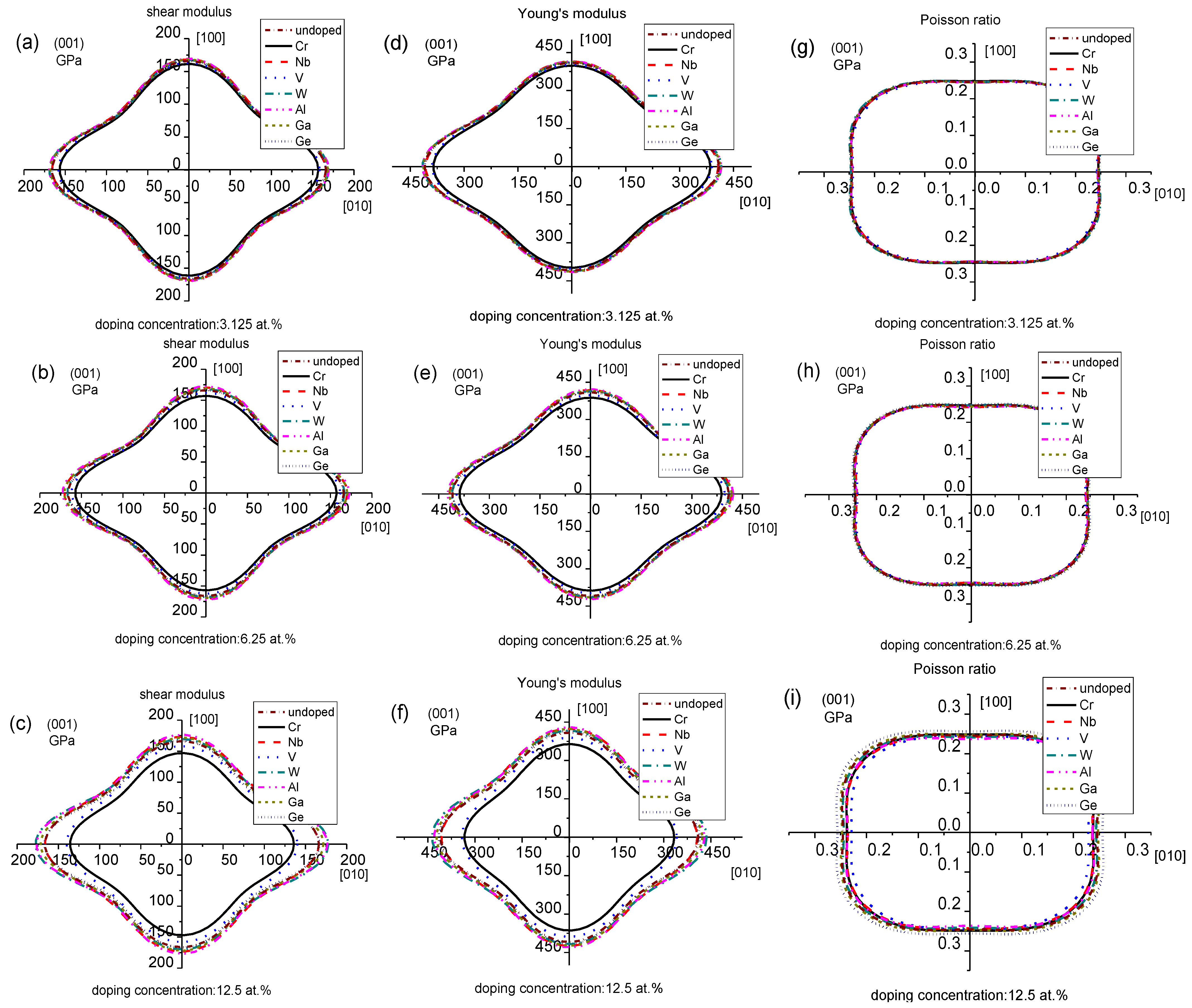
In order to have a better understanding of the origin of the changes in the mechanical properties along different directions, the 2D projections of the shear modulus, Young’s modulus and Poisson ratio at the (001) plane are given in Figure 7. Cr- and V-doping can decrease the shear and Young’s modulus for Mo3Si and affect the anisotropy of the shear and Young’s modulus. This trend becomes evident with the increased Cr- and V-doping concentrations, consistent with the above mechanical properties and elastic anisotropy. The anisotropy of Mo3Si doped with other elements changes little, and that of the shear and Young’s modulus for Mo3Si doped with Al increases mildly. These findings are in accord with the calculated anisotropic elasticity. It is also seen that the of Mo3Si doped with 12.5 at.% V, Al, and Nb decrease evidently, and that of Mo3Si doped with 12.5 at.% Ge increases, which agrees well with the above calculated Poisson ratio in Figure 4.
3.4. Electronic Structures
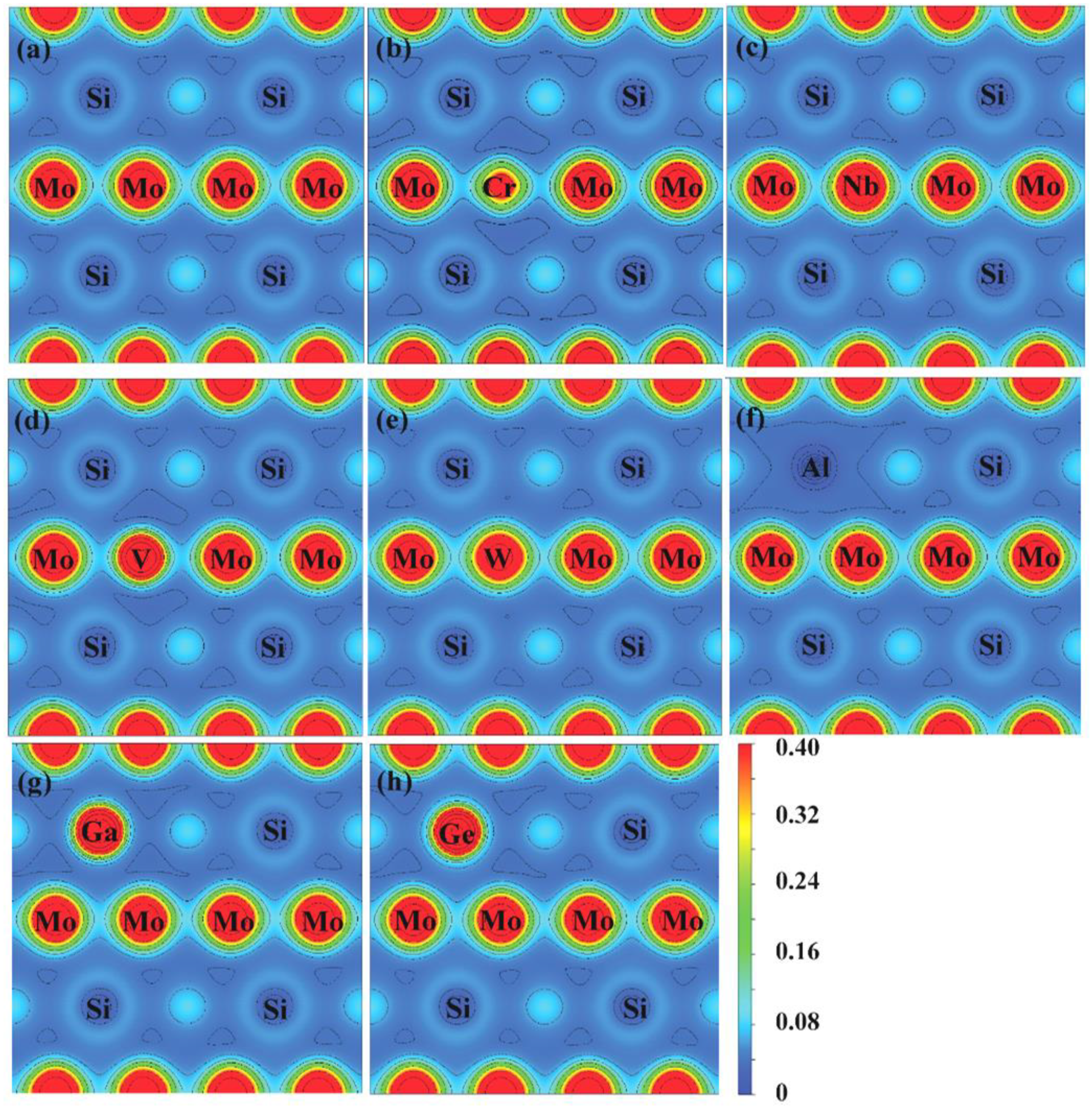
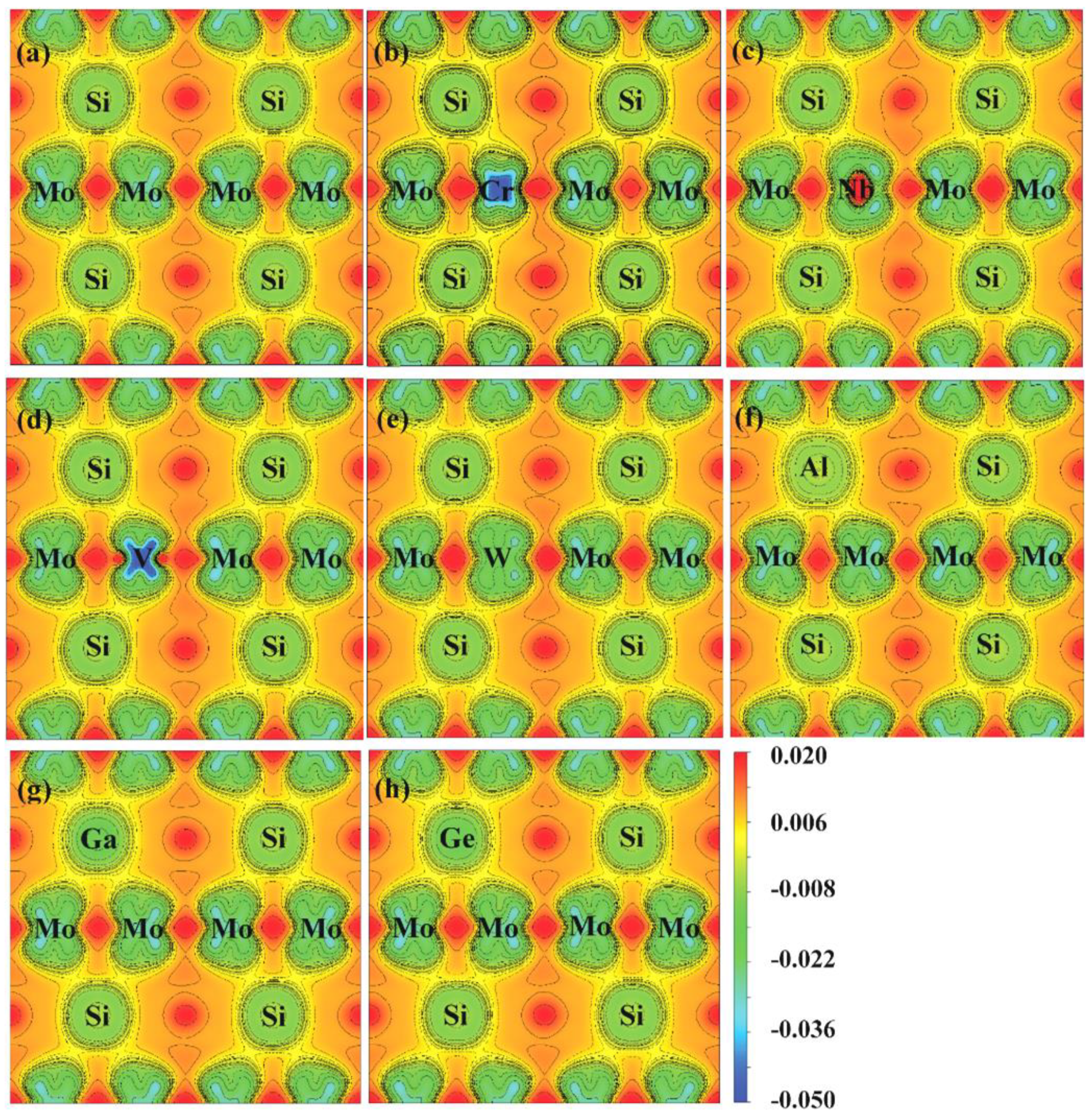
Figure 8 and Figure 9 show the charge densities and deformation charge densities of undoped and doped Mo3Si, respectively. The charge densities show the elongated contours between Si and Mo, suggesting a robust covalent bonding generated by hybridizing Si-3p and Mo-4d. Figure 9 shows different charge densities with these doped elements, which affects the mechanical properties of Mo3Si.
In Figure 9, Cr- and V-doping reduce the charge accumulation between Si and Mo compared to those of undoped Mo3Si, suggesting their low elastic modulus. Nb-doping increases the charge accumulation slightly, indicating that Nb can strengthen the elastic modulus of Mo3Si. These findings agree with the above mechanical properties in Section 3.2.
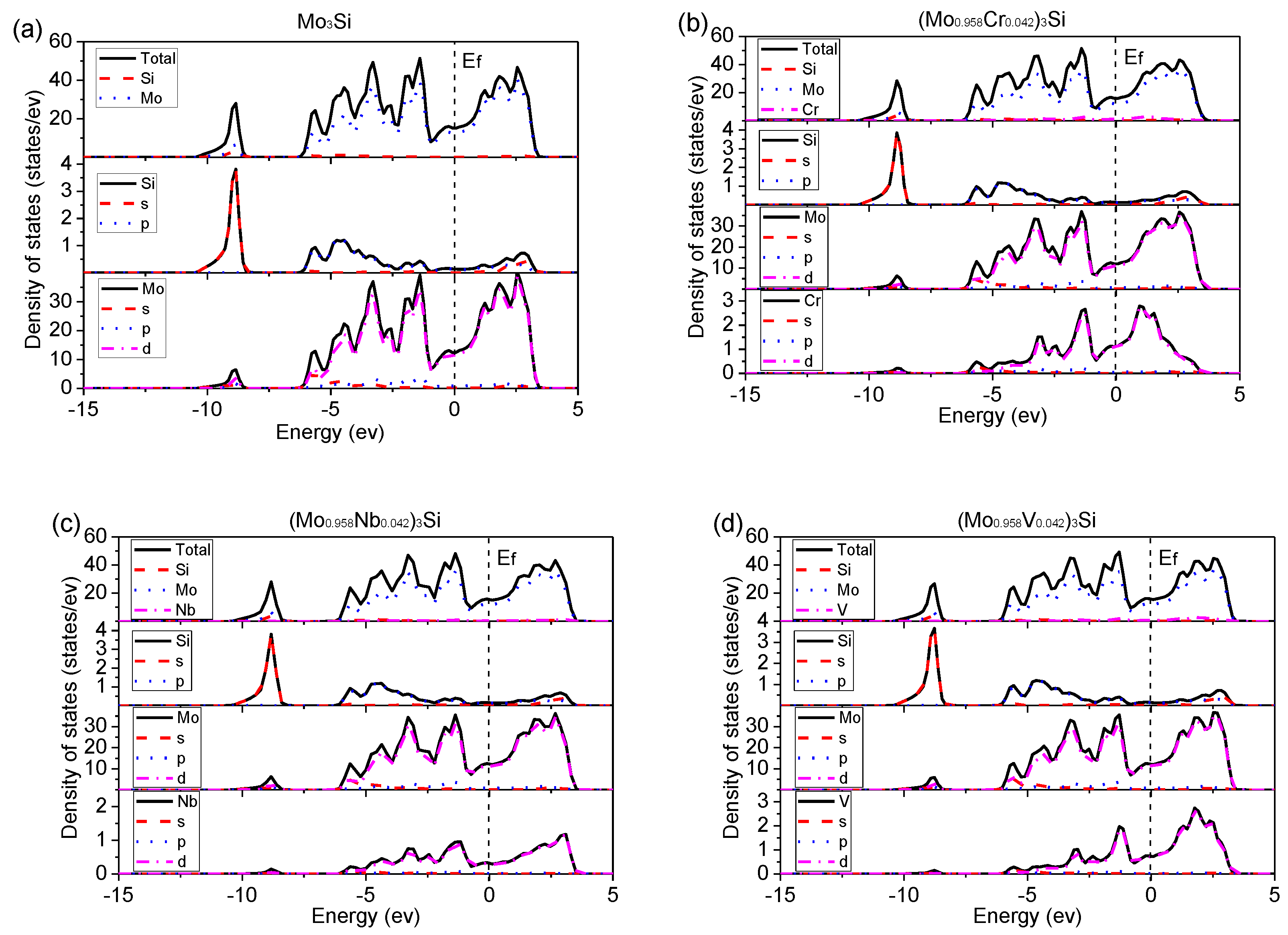
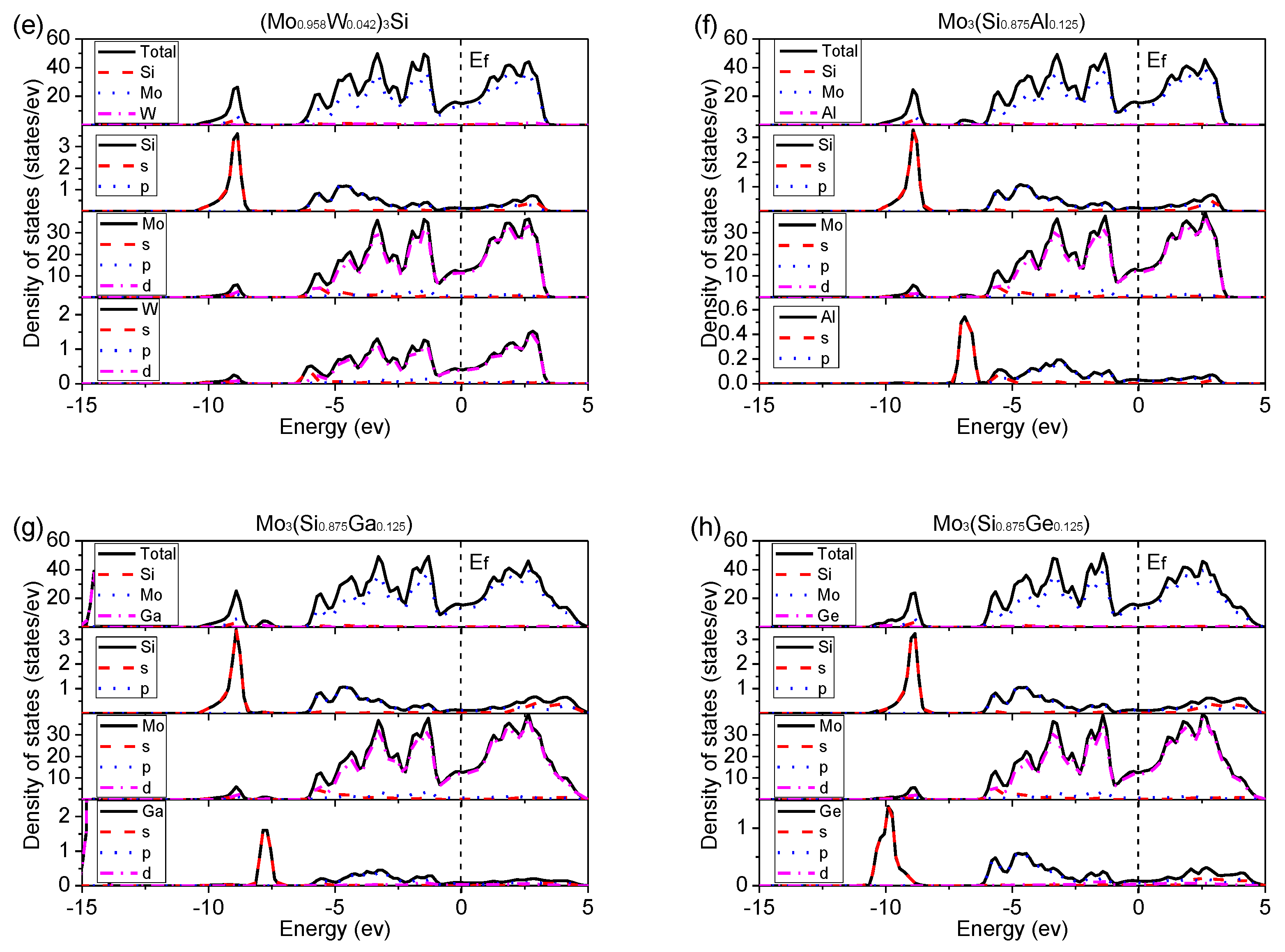
Figure 10 shows the density of states (DOS) of undoped and doped Mo3Si with alloying elements. In Figure 10a, there is no gap and non-zero DOS at the Fermi level for undoped Mo3Si, which shows its metallic behaviors. Furthermore, there is an overlap between Mo-4d and Si-3p for undoped Mo3Si. Two peaks are divided by the Femi level called the pseudogap. These phenomena suggest a strong covalent bonding between Si and Mo, consistent with the above charge results. The same situations also occur in doped Mo3Si, suggesting strong covalent bonding caused by the d-p hybridization between Mo-site atoms (Mo, Cr, Nb, V, and W) and Si-site atoms (Si, Al, Ga, and Ge).
In Figure 10, the DOS of doped Mo3Si is close to that of undoped Mo3Si. However, there are some small changes in DOS with different doped elements. A slight shift of the peaks to low energy for V- and Nb-doped Mo3Si suggests that V- and Nb-doping increase the structural stability of Mo3Si, following the above formation enthalpies in Section 3.1.
In Figure 9c,d, some 4d anti-bonding of V and Nb moves away from the Fermi level and increases the gap between their anti-bonding and bonding states. It is suggested that V and Nb induce dislocation-slip energy, unfavorable to the ductility. As mentioned above, Nb’s adverse effect on toughness has been confirmed in Rosales’ experiments [7]. Moreover, a high DOS at the Fermi level for Ge- and Ga-doped Mo3Si indicates that Ge- and Ga-doping strengthen metallic bonding and the ductility, which conforms to the calculated B/G and (See Section 3.2).
4. Conclusions
First-principles calculations were employed to investigate the effect of alloying elements (Cr, Nb, V, W, Al, Ga, and Ge) on the mechanical properties of Mo3Si. The structural stabilities of doped Mo3Si were calculated, which suggested that the Pm-3n structure was stable at these investigated low-doping concentrations (≤12.5 at.%). The calculated elastic constants have been used to evaluate some critical mechanical properties of Mo3Si doped with alloying elements.
Compared to undoped Mo3Si, Cr- and V-doping drastically decreased the elastic modulus, while Al- and Nb-doping slightly increased the shear and Young’s modulus. Moreover, V-, Al- and Nb-doping decreased the B/G and Poisson ratio, suggesting that these elements could form strong covalent bonds, and decrease shear deformation and the alloy’s ductility. The wave velocities and Debye temperatures of Nb-, Al- and V-doped Mo3Si increased, while those of Cr-, W-, Ga- and Ge-doped Mo3Si decreased.
Based on the three-dimensional contours and two-dimensional projection of the elastic modulus, Cr- and V-doping exhibited a significant influence on the anisotropy of the shear and Young’s modulus. The anisotropy of Mo3Si doped with the other elements changed little, and that of the shear modulus for Mo3Si doped with Al increased mildly. The electronic structures of doped Mo3Si were also investigated according to the charge density and DOS, which helped us gain a deeper understanding of doping behaviors.
Author Contributions
Conceptualization, Y.J.; methodology, S.S.; investigation, W.B.; supervision, S.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Higher Education Science Foundation of Jiangsu Province of China (17KJA430006).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gu, S.; Sun, S.; Li, X.; Lei, W.; Rashad, M.; Yin, L.; Wang, Y.; Chen, L.; Jiang, Y. The structure and stability of the low-index surfaces of D8m-Mo5Si3 by first-principles calculations. Ceram. Int. 2020, 46, 877–887. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, P.; Zhang, C.-M. Structure, mechanical, electronic and thermodynamic properties of Mo5Si3 from first-principles calculations. Ceram. Int. 2018, 44, 12357–12362. [Google Scholar] [CrossRef]
- Sun, S.; Li, X.; Wang, H.; Jiang, Y.; Yi, D. Adsorption of oxygen atom on MoSi2 (110) surface. Appl. Surf. Sci. 2016, 382, 239–248. [Google Scholar] [CrossRef]
- Dharmawardhana, C.C.; Zhou, J.; Taylor, M.; Miao, J.; Perepezko, J.H.; Heinz, H. Reactive modeling of Mo3Si oxidation and resulting silica morphology. Acta Mater. 2020, 187, 93–102. [Google Scholar] [CrossRef]
- Gnesin, I.; Gnesin, B. Composition of the Mo-Mo3Si alloys obtained via various methods. Int. J. Refract. Met. Hard Mater. 2020, 88, 105188. [Google Scholar] [CrossRef]
- Ray, P.K.; Ye, Y.; Akinc, M.; Kramer, M. Effect of Nb and W substitutions on the stability of the A15 Mo3Si phase. J. Alloy. Compd. 2012, 537, 65–70. [Google Scholar] [CrossRef]
- Rosales, I.; Garcia-Ramirez, M.J.; Diaz-Reyes, C.; Martínez, H. Defects analysis on the strengthening of solid solution Nb ad-ditions in Mo3Si alloys. J. Mater. Res. Technol. 2019, 8, 99–104. [Google Scholar] [CrossRef]
- Rosales, I.; Martinez-Valencia, H.; Ponce, D.; Ruiz, J. Wear performance of Nb-alloyed, pulsed plasma nitrided Mo3Si intermetallic. Int. J. Refract. Met. Hard Mater. 2007, 25, 250–255. [Google Scholar] [CrossRef]
- Xu, J.; Hu, W.; Yan, Y.; Lu, X.; Munroe, P.; Xie, Z.-H. Microstructure and mechanical properties of a Mo-toughened Mo3Si-based in situ nanocomposite. Vacuum 2014, 109, 112–119. [Google Scholar] [CrossRef]
- Deng, X.; Zhang, G.; Wang, T.; Ren, S.; Li, Z.; Song, P.; Shi, Y. Characterization and oxidation resistance of B-modified Mo3Si coating on Mo substrate. J. Alloy. Compd. 2019, 807, 151693. [Google Scholar] [CrossRef]
- Rosales, I.; Martinez-Valencia, H.; Bahena, D.; Ruiz, J.; Guardián, R.; Colin, J. Oxidation performance of Mo3Si with Al additions. Corros. Sci. 2009, 51, 534–538. [Google Scholar] [CrossRef]
- Ochiai, S. Improvement of the oxidation-proof property and the scale structure of Mo3Si intermetallic alloy through the addi-tion of chromium and aluminum elements. Intermetallics 2006, 14, 1351–1357. [Google Scholar] [CrossRef]
- Dong, N.; Jia, R.; Yang, J.; Wang, J.; Liu, J.; Fang, X.; Han, P.-D. The Effects of Co and W on Structural Stability and Mechanical Properties of Austenitic Heat-Resistant Steel Sanicro 25: A First-Principle Study. Metals 2020, 10, 1051. [Google Scholar] [CrossRef]
- Cheng, J.; Yun, Y.; Wang, J.; Rui, J.; Shun, W.; Du, Y.-L. Effect of Nb on the Microstructure and Mechanical Properties of Ti2Cu Intermetallic through the First-Principle Calculations and Experimental Investigation. Metals 2020, 10, 547. [Google Scholar] [CrossRef]
- Bi, W.; Sun, S.; Bei, S.; Jiang, Y. Segregation of S at Mo(001)/MoSi2(001) interface. Ceram. Int. 2020, 46, 5050–5057. [Google Scholar] [CrossRef]
- Li, X.-P.; Sun, S.-P.; Yu, Y.; Wang, H.-J.; Jiang, Y.; Yi, D.-Q. Composition and temperature dependences of site occupation for Al, Cr, W, and Nb in MoSi2. Chin. Phys. B 2015, 24, 120502. [Google Scholar] [CrossRef]
- Sun, S.; Li, X.; Wang, H.; Jiang, Y.; Yi, D. Prediction on anisotropic elasticity, sound velocity, and thermodynamic properties of MoSi2 under pressure. J. Alloy. Compd. 2015, 652, 106–115. [Google Scholar] [CrossRef]
- Sun, S.; Li, X.; Zhang, Y.; Wang, H.; Yu, Y.; Yong, J.; Yi, D. Prediction of the mechanical properties of MoSi2 doped with Cr, Nb and W from first-principles calculations. J. Alloy. Compd. 2017, 714, 459–466. [Google Scholar] [CrossRef]
- Hu, H.; Wu, X.; Wang, R.; Li, W.; Liu, Q. First principles study on the phase stability and mechanical properties of MoSi2 al-loyed with Al, Mg and Ge. Intermetallics 2015, 67, 26–34. [Google Scholar]
- Sun, S.P.; Zhu, J.L.; Gu, S.; Wang, H.J.; Jiang, Y.; Yi, D.Q. First-principles investigation of activity and solubility of Si in Mo solid solution. Int. J. Mod. Phys. B 2018, 32, 1850305. [Google Scholar] [CrossRef]
- Zhong, S.-Y.; Chen, Z.; Wang, M.; Chen, D. Structural, elastic and thermodynamic properties of Mo3Si and Mo3Ge. Eur. Phys. J. B 2016, 89, 6. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Fujiwara, H.; Ueda, Y. Thermodynamic properties of molybdenum silicides by molten electrolyte EMF measurements. J. Alloy. Compd. 2007, 441, 168–173. [Google Scholar] [CrossRef]
- Chen, Y.; Hammerschmidt, T.; Pettifor, D.; Shang, J.-X.; Zhang, Y. Influence of vibrational entropy on structural stability of Nb–Si and Mo–Si systems at elevated temperatures. Acta Mater. 2009, 57, 2657–2664. [Google Scholar] [CrossRef]
- Tütüncü, H.; Bağcı, S.; Srivastava, G.P.; Bagci, S. Electronic structure, phonons, and electron-phonon interaction in Mo3Si. Phys. Rev. B 2010, 82, 214510. [Google Scholar] [CrossRef]
- Ma, N.; Cooper, B.R.; Kang, B.S. Tight-binding study of thermal expansions for Mo3Si. J. Appl. Phys. 2006, 99, 053514. [Google Scholar] [CrossRef] [Green Version]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Oxford University Press: London, UK, 1985. [Google Scholar]
- Wu, Z.; Zhao, E.; Xiang, H.; Hao, X.; Liu, X.; Meng, J. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles. Phys. Rev. B 2007, 76, 054115. [Google Scholar] [CrossRef]
- Hill, R. The Elastic Behaviour of a Crystalline Aggregate. Proc. Phys. Soc. Sect. A 1952, 65, 349–354. [Google Scholar] [CrossRef]
- Gorodtsov, V.A.; Lisovenko, D.S. Auxetics among Materials with Cubic Anisotropy. Mech. Solids 2020, 55, 461–474. [Google Scholar] [CrossRef]
- Anderson, O.L. A simplified method for calculating the debye temperature from elastic constants. J. Phys. Chem. Solids 1963, 24, 909–917. [Google Scholar] [CrossRef]
- Pugh, S. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B. Density functional theory for calculation of elastic properties of or-thorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891. [Google Scholar] [CrossRef]
- Li, S.; Quan, H.; Yu, S. Structural, electronic, and elastic properties of orthorhombic NaB3H8: A first-principles investigation. Solid State Commun. 2019, 299, 113653. [Google Scholar] [CrossRef]
- Gaillac, R.; Pullumbi, P.; Coudert, F.-X. ELATE: An open-source online application for analysis and visualization of elastic tensors. J. Phys. Condens. Matter 2016, 28, 275201. [Google Scholar] [CrossRef]
- Goldstein, R.V.; Gorodtsov, V.A.; Lisovenko, D.S. Shear modulus of cubic crystals. Lett. Mater. 2012, 2, 21–24. [Google Scholar] [CrossRef]
Figure 1.
Crystal structure of Mo3Si.

Figure 2.
Formation enthalpies of doped Mo3Si.

Figure 3.
Mechanical properties of doped Mo3Si. (a) bulk modulus; (b) shear modulus; (c) Young’s modulus.
Figure 3.
Mechanical properties of doped Mo3Si. (a) bulk modulus; (b) shear modulus; (c) Young’s modulus.

Figure 4.
B/G, Poisson ratio, wave velocities and Debye temperatures of doped Mo3Si. (a) B/G (b) Poisson ratio (c) Debye temperatures (d) (e) (f) .
Figure 4.
B/G, Poisson ratio, wave velocities and Debye temperatures of doped Mo3Si. (a) B/G (b) Poisson ratio (c) Debye temperatures (d) (e) (f) .

Figure 5.
Mechanical anisotropic indexes of doped Mo3Si. (a) ; (b) ; (c) .

Figure 6.
Three-dimensional contours of the shear modulus, Young’s modulus and Poisson ratio for undoped and doped Mo3Si. The distance between zero and any point on the surfaces is equal to the elastic modulus or Poisson ratio in that direction. (a–d) Shear modulus; (e–h) Young’s modulus; (i–l) Poisson ratio.
Figure 6.
Three-dimensional contours of the shear modulus, Young’s modulus and Poisson ratio for undoped and doped Mo3Si. The distance between zero and any point on the surfaces is equal to the elastic modulus or Poisson ratio in that direction. (a–d) Shear modulus; (e–h) Young’s modulus; (i–l) Poisson ratio.

Figure 7.
Two-dimensional projections of the shear modulus, Young’s modulus and Poisson ratio for undoped and doped Mo3Si. (a–c) Shear modulus; (d–f) Young’s modulus; (g–i) Poisson ratio.
Figure 7.
Two-dimensional projections of the shear modulus, Young’s modulus and Poisson ratio for undoped and doped Mo3Si. (a–c) Shear modulus; (d–f) Young’s modulus; (g–i) Poisson ratio.

Figure 8.
Charge densities of undoped and doped Mo3Si. (a) undoped; (b) Cr; (c) Nb; (d) V; (e) W; (f) Al; (g) Ga; (h) Ge.
Figure 8.
Charge densities of undoped and doped Mo3Si. (a) undoped; (b) Cr; (c) Nb; (d) V; (e) W; (f) Al; (g) Ga; (h) Ge.

Figure 9.
Deformation charge densities of undoped and doped Mo3Si. (a) Undoped; (b) Cr; (c) Nb; (d) V; (e) W; (f) Al; (g) Ga; (h) Ge.
Figure 9.
Deformation charge densities of undoped and doped Mo3Si. (a) Undoped; (b) Cr; (c) Nb; (d) V; (e) W; (f) Al; (g) Ga; (h) Ge.

Figure 10.
Density of states (DOS) of undoped and doped Mo3Si. (a) Mo3Si; (b) (Mo0.958Cr0.042)3Si; (c) (Mo0.958Nb0.042)3Si; (d) (Mo0.958V0.042)3Si; (e) (Mo0.958W0.042)3Si; (f) Mo3(Si0.875Al0.125); (g) Mo3(Si0.875Ga0.125); (h) Mo3(Si0.875Ge0.125).
Figure 10.
Density of states (DOS) of undoped and doped Mo3Si. (a) Mo3Si; (b) (Mo0.958Cr0.042)3Si; (c) (Mo0.958Nb0.042)3Si; (d) (Mo0.958V0.042)3Si; (e) (Mo0.958W0.042)3Si; (f) Mo3(Si0.875Al0.125); (g) Mo3(Si0.875Ga0.125); (h) Mo3(Si0.875Ge0.125).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bi, W.; Sun, S.; Bei, S.; Jiang, Y. Effect of Alloying Elements on the Mechanical Properties of Mo3Si. Metals 2021, 11, 129. https://doi.org/10.3390/met11010129
AMA Style
Bi W, Sun S, Bei S, Jiang Y. Effect of Alloying Elements on the Mechanical Properties of Mo3Si. Metals. 2021; 11(1):129. https://doi.org/10.3390/met11010129
Chicago/Turabian StyleBi, Wei, Shunping Sun, Shaoyi Bei, and Yong Jiang. 2021. "Effect of Alloying Elements on the Mechanical Properties of Mo3Si" Metals 11, no. 1: 129. https://doi.org/10.3390/met11010129
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.