

Krüppel-like Factor-4-Mediated Macrophage Polarization and Phenotypic Transitions Drive Intestinal Fibrosis in THP-1 Monocyte Models In Vitro
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Methods
2.2. Human Induced Pluripotent Stem (iPS) Cell-Derived Small Intestinal Organoids (HIOs)
2.3. Scratch-Wound Healing Assay
2.4. Transwell Migration Assay
2.5. RNA Extraction, Reverse Transcription, and Real-Time PCR
2.6. Western Blot Analysis
2.7. Transfection
2.8. Immunofluorescence Staining
2.9. Statistical Analysis
3. Results
3.1. Phenotypes of Macrophage Subtypes and Differences in KLF4 Expression
3.2. Effect of KLF4 Knockdown on Macrophage Polarization and Inflammatory Phenotype Induction
3.3. Macrophage Subtypes Modulate Intestinal Fibrosis
3.4. Macrophage Polarization and Fibrosis Modulation in the Intestinal Stromal Myofibroblast Environment
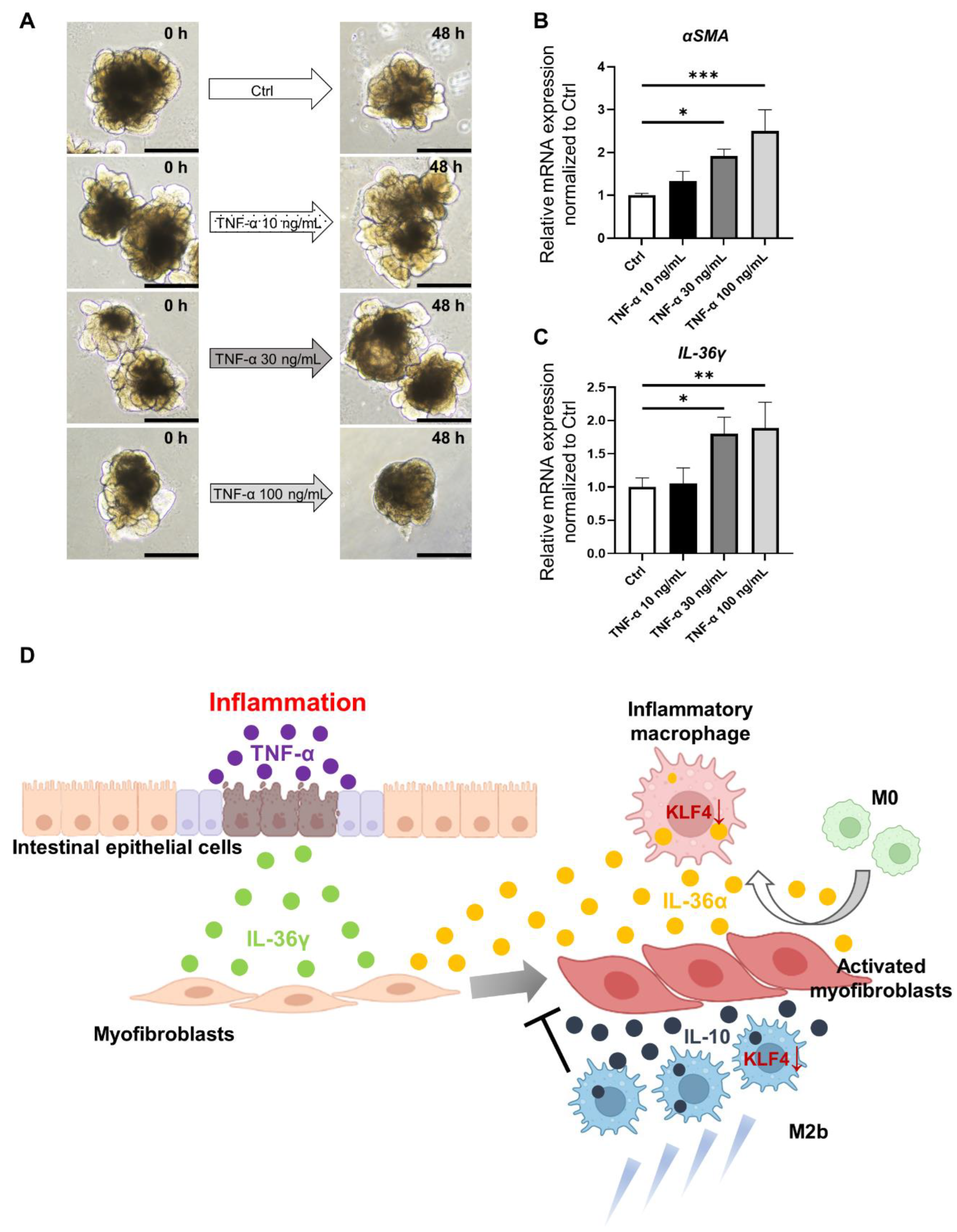
3.5. Characterization of Inflammatory and Fibrotic Responses in an IBD Model Using HIOs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Lane, E.R.; Zisman, T.L.; Suskind, D.L. The microbiota in inflammatory bowel disease: Current and therapeutic insights. J. Inflamm. Res. 2017, 10, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Baraty, B.; Lee Robertson, H.; Filyk, A.; Shen, H.; Fung, T.; Novak, K.; Ma, C.; Panaccione, R.; Achkar, J.P.; et al. Systematic review: Medical therapy for fibrostenosing Crohn’s disease. Aliment. Pharmacol. Ther. 2020, 51, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Folsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004, 53, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Latella, G.; Di Gregorio, J.; Flati, V.; Rieder, F.; Lawrance, I.C. Mechanisms of initiation and progression of intestinal fibrosis in IBD. Scand. J. Gastroenterol. 2015, 50, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mao, R.; Kurada, S.; Wang, J.; Lin, S.; Chandra, J.; Rieder, F. Pathogenesis of fibrostenosing Crohn’s disease. Transl. Res. 2019, 209, 39–54. [Google Scholar] [CrossRef]
- Satsangi, J.; Silverberg, M.S.; Vermeire, S.; Colombel, J.F. The Montreal classification of inflammatory bowel disease: Controversies, consensus, and implications. Gut 2006, 55, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Valatas, V.; Filidou, E.; Drygiannakis, I.; Kolios, G. Stromal and immune cells in gut fibrosis: The myofibroblast and the scarface. Ann. Gastroenterol. 2017, 30, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef]
- Imai, J.; Kitamoto, S.; Sugihara, K.; Nagao-Kitamoto, H.; Hayashi, A.; Morhardt, T.L.; Kuffa, P.; Higgins, P.D.R.; Barnich, N.; Kamada, N. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol. 2019, 12, 632–643. [Google Scholar] [CrossRef]
- Scheibe, K.; Kersten, C.; Schmied, A.; Vieth, M.; Primbs, T.; Carle, B.; Knieling, F.; Claussen, J.; Klimowicz, A.C.; Zheng, J.; et al. Inhibiting Interleukin 36 Receptor Signaling Reduces Fibrosis in Mice With Chronic Intestinal Inflammation. Gastroenterology 2019, 156, 1082–1097.e1011. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Sica, A.; Mantovani, A.; Locati, M. Macrophage activation and polarization. Front. Biosci. 2008, 13, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Xiong, J.; Lu, Y.; Wang, L.; Tian, T. SENP1-KLF4 signalling regulates LPS-induced macrophage M1 polarization. FEBS J. 2023, 290, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, A.M.; Yang, V.W. Kruppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Orzechowska, E.J.; Katano, T.; Bialkowska, A.B.; Yang, V.W. Interplay among p21(Waf1/Cip1), MUSASHI-1 and Kruppel-like factor 4 in activation of Bmi1-Cre(ER) reserve intestinal stem cells after gamma radiation-induced injury. Sci. Rep. 2020, 10, 18300. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Sharma, N.; Kapadia, F.; Zhou, G.; Lu, Y.; Hong, H.; Paruchuri, K.; Mahabeleshwar, G.H.; Dalmas, E.; Venteclef, N.; et al. Kruppel-like factor 4 regulates macrophage polarization. J. Clin. Investig. 2011, 121, 2736–2749. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Lu, X.; Ren, J.; Privratsky, J.R.; Yang, B.; Rudemiller, N.P.; Zhang, J.; Griffiths, R.; Jain, M.K.; Nedospasov, S.A.; et al. KLF4 in Macrophages Attenuates TNFalpha-Mediated Kidney Injury and Fibrosis. J. Am. Soc. Nephrol. 2019, 30, 1925–1938. [Google Scholar] [CrossRef] [PubMed]
- Onozato, D.; Ogawa, I.; Kida, Y.; Mizuno, S.; Hashita, T.; Iwao, T.; Matsunaga, T. Generation of Budding-Like Intestinal Organoids from Human Induced Pluripotent Stem Cells. J. Pharm. Sci. 2021, 110, 2637–2650. [Google Scholar] [CrossRef]
- Onozato, D.; Yamashita, M.; Nakanishi, A.; Akagawa, T.; Kida, Y.; Ogawa, I.; Hashita, T.; Iwao, T.; Matsunaga, T. Generation of Intestinal Organoids Suitable for Pharmacokinetic Studies from Human Induced Pluripotent Stem Cells. Drug Metab. Dispos. 2018, 46, 1572–1580. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Tugal, D.; Liao, X.; Jain, M.K. Transcriptional control of macrophage polarization. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.; Lohler, J.; Rennick, D.; Rajewsky, K.; Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993, 75, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.H.; Wang, X.; Balaji, S.; Butte, M.J.; Bollyky, P.L.; Keswani, S.G. The Role of the Anti-Inflammatory Cytokine Interleukin-10 in Tissue Fibrosis. Adv. Wound Care 2020, 9, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, P.; Rajasingh, J.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ. Res. 2009, 104, e9–e18. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Huang, S.; Wang, L.; Wu, Z.; Liang, M.; Li, H.; Lv, L.; Li, W.; Wu, Z. M2b Macrophages Regulate Cardiac Fibroblast Activation and Alleviate Cardiac Fibrosis After Reperfusion Injury. Circ. J. 2020, 84, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Boutet, M.A.; Bart, G.; Penhoat, M.; Amiaud, J.; Brulin, B.; Charrier, C.; Morel, F.; Lecron, J.C.; Rolli-Derkinderen, M.; Bourreille, A.; et al. Distinct expression of interleukin (IL)-36alpha, beta and gamma, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin. Exp. Immunol. 2016, 184, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The interleukin-1 family: Back to the future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [PubMed]
- Medina-Contreras, O.; Harusato, A.; Nishio, H.; Flannigan, K.L.; Ngo, V.; Leoni, G.; Neumann, P.A.; Geem, D.; Lili, L.N.; Ramadas, R.A.; et al. Cutting Edge: IL-36 Receptor Promotes Resolution of Intestinal Damage. J. Immunol. 2016, 196, 34–38. [Google Scholar] [CrossRef]
- Mora-Buch, R.; Dotti, I.; Planell, N.; Calderon-Gomez, E.; Jung, P.; Masamunt, M.C.; Llach, J.; Ricart, E.; Batlle, E.; Panes, J.; et al. Epithelial IL-1R2 acts as a homeostatic regulator during remission of ulcerative colitis. Mucosal Immunol. 2016, 9, 950–959. [Google Scholar] [CrossRef]
- Nishida, A.; Hidaka, K.; Kanda, T.; Imaeda, H.; Shioya, M.; Inatomi, O.; Bamba, S.; Kitoh, K.; Sugimoto, M.; Andoh, A. Increased Expression of Interleukin-36, a Member of the Interleukin-1 Cytokine Family, in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2016, 22, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Scheibe, K.; Backert, I.; Wirtz, S.; Hueber, A.; Schett, G.; Vieth, M.; Probst, H.C.; Bopp, T.; Neurath, M.F.; Neufert, C. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut 2017, 66, 823–838. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Chang, E.B. Inflammatory Bowel Diseases (IBD) and the Microbiome-Searching the Crime Scene for Clues. Gastroenterology 2021, 160, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Narula, N.; Wong, E.C.L.; Dehghan, M.; Mente, A.; Rangarajan, S.; Lanas, F.; Lopez-Jaramillo, P.; Rohatgi, P.; Lakshmi, P.V.M.; Varma, R.P.; et al. Association of ultra-processed food intake with risk of inflammatory bowel disease: Prospective cohort study. BMJ 2021, 374, n1554. [Google Scholar] [CrossRef] [PubMed]
- de Souza, H.S.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Rieder, F.; Fiocchi, C. Intestinal fibrosis in IBD--a dynamic, multifactorial process. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ji, Y.; Jiang, R.; Fang, C.; Shi, G.; Cheng, L.; Zuo, Y.; Ye, Y.; Su, X.; Li, J.; et al. Reduced smooth muscle-fibroblasts transformation potentially decreases intestinal wound healing and colitis-associated cancer in ageing mice. Signal Transduct. Target. Ther. 2023, 8, 294. [Google Scholar] [CrossRef]
- Warzecha, Z.; Dembinski, A.; Ceranowicz, P.; Konturek, P.C.; Niemiec, J.; Stachura, J.; Tomaszewska, R.; Konturek, S.J. The influence of sensory nerves and CGRP on the pancreatic regeneration after repeated episodes of acute pancreatitis in rats. J. Physiol. Pharmacol. 2000, 51, 449–461. [Google Scholar]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanno, T.; Katano, T.; Shimura, T.; Tanaka, M.; Nishie, H.; Fukusada, S.; Ozeki, K.; Ogawa, I.; Iwao, T.; Matsunaga, T.; et al. Krüppel-like Factor-4-Mediated Macrophage Polarization and Phenotypic Transitions Drive Intestinal Fibrosis in THP-1 Monocyte Models In Vitro. Medicina 2024, 60, 713. https://doi.org/10.3390/medicina60050713
Kanno T, Katano T, Shimura T, Tanaka M, Nishie H, Fukusada S, Ozeki K, Ogawa I, Iwao T, Matsunaga T, et al. Krüppel-like Factor-4-Mediated Macrophage Polarization and Phenotypic Transitions Drive Intestinal Fibrosis in THP-1 Monocyte Models In Vitro. Medicina. 2024; 60(5):713. https://doi.org/10.3390/medicina60050713
Chicago/Turabian StyleKanno, Takuya, Takahito Katano, Takaya Shimura, Mamoru Tanaka, Hirotada Nishie, Shigeki Fukusada, Keiji Ozeki, Isamu Ogawa, Takahiro Iwao, Tamihide Matsunaga, and et al. 2024. "Krüppel-like Factor-4-Mediated Macrophage Polarization and Phenotypic Transitions Drive Intestinal Fibrosis in THP-1 Monocyte Models In Vitro" Medicina 60, no. 5: 713. https://doi.org/10.3390/medicina60050713