
Protein Phosphatase 2A as a Therapeutic Target in Pulmonary Diseases
 ,
,
Abstract
:1. Introduction
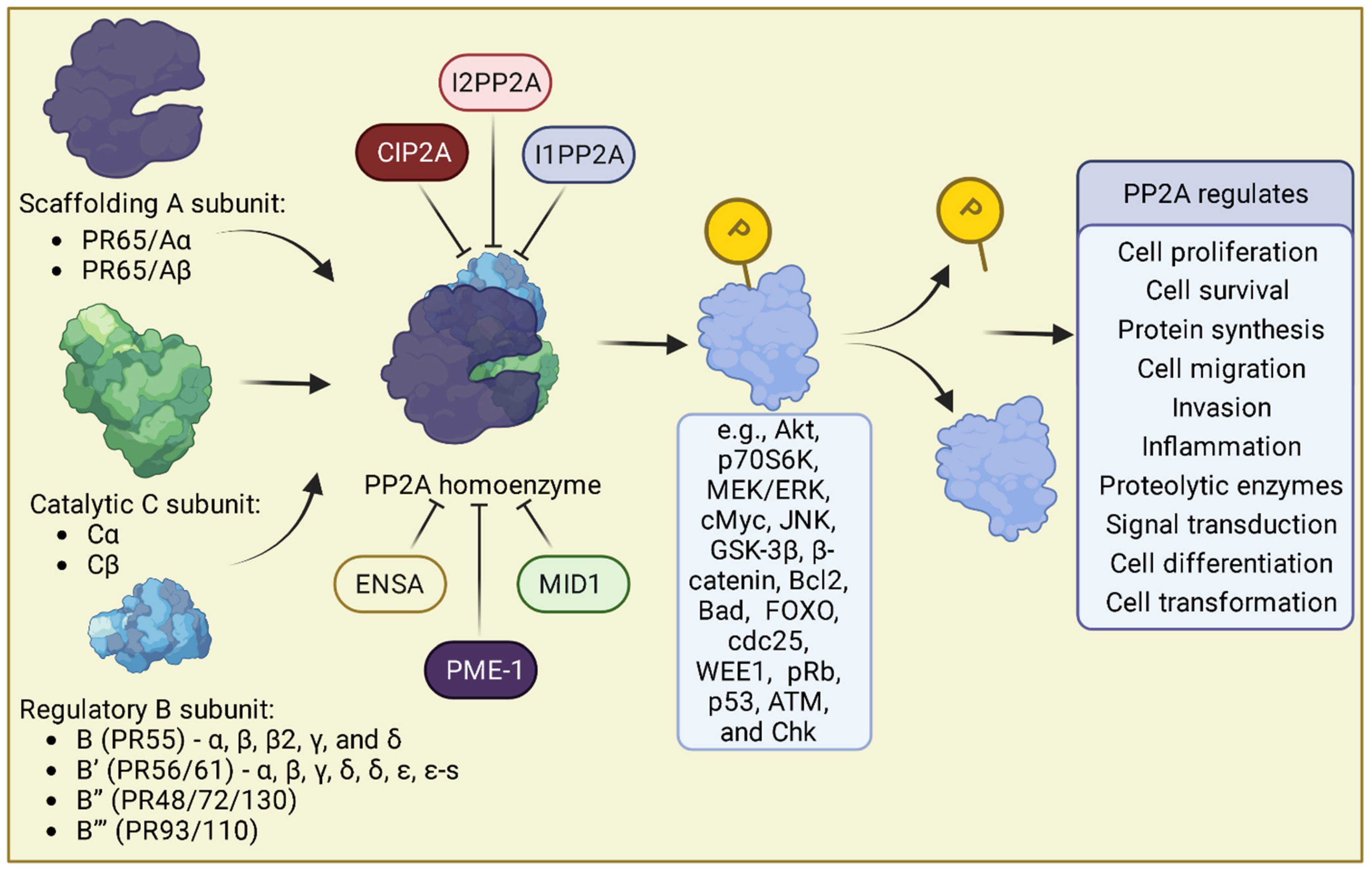
2. Protein Phosphatase 2A
PP2A Subunits
3. The Activation Status of PP2A in Pulmonary Diseases
3.1. PP2A in Cigarette Smoke-Induced COPD
3.2. PP2A Responses in Alpha-1 Antitrypsin Deficiency
3.3. PP2A in Asthma
3.4. PP2A in Pulmonary Fibrosis
3.5. PP2A in Lung Cancer
4. Endogenous Inhibitors of PP2A in Pulmonary Diseases
4.1. Cancerous Inhibitor of PP2A (CIP2A)
4.2. Inhibitor 2 of PP2A (I2PP2A/SET)
4.3. Ubiquitin E3 Ligase Midline 1 (MID1)
4.4. Protein Phosphatase Methylesterase 1 (PME-1)
4.5. Newly Discovered PP2A Regulators
5. Potential Approaches to Activate PP2A
5.1. Indirect Activation of PP2A via Targeting Endogenous Inhibitors
5.2. Inhibiting SET with FTY720
5.3. Next-Generation SET Inhibitors
5.4. Inhibiting CIP2A
5.4.1. Erlotinib Derivatives
5.4.2. Metformin
5.4.3. Other Newer CIP2A Inhibitors
5.5. Inhibiting PME-1
5.6. Direct Activation of PP2A
5.7. Unknown Mechanism of Targeting PP2A
6. Potential Therapeutic Benefits of PP2A Inhibition
7. Potential Negative Impact of Systemic Targeting PP2A
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD Chronic Respiratory Disease Collaborators. Prevalence and attributable health burden of chronic respiratory diseases, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2020, 8, 585–596. [Google Scholar] [CrossRef]
- Durham, A.L.; Adcock, I.M. The relationship between COPD and lung cancer. Lung Cancer 2015, 90, 121–127. [Google Scholar] [CrossRef]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Nader, C.P.; Cidem, A.; Verrills, N.M.; Ammit, A.J. Protein phosphatase 2A (PP2A): A key phosphatase in the progression of chronic obstructive pulmonary disease (COPD) to lung cancer. Respir. Res. 2019, 20, 222. [Google Scholar] [CrossRef] [PubMed]
- Didkowska, J.; Wojciechowska, U.; Mańczuk, M.; Łobaszewski, J. Lung cancer epidemiology: Contemporary and future challenges worldwide. Ann. Transl. Med. 2016, 4, 150. [Google Scholar] [CrossRef] [PubMed]
- Quaderi, S.A.; Hurst, J.R. The unmet global burden of COPD. Glob. Health Epidemiol. Genom. 2018, 3, e4. [Google Scholar] [CrossRef]
- Turner, M.C.; Chen, Y.; Krewski, D.; Calle, E.E.; Thun, M.J. Chronic obstructive pulmonary disease is associated with lung cancer mortality in a prospective study of never smokers. Am. J. Respir. Crit. Care Med. 2007, 176, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Chen, S.; Chen, X.; Zou, W.; Liu, Z.; Wu, Y.; Hu, S. Global trends in the incidence and mortality of asthma from 1990 to 2019: An age-period-cohort analysis using the global burden of disease study 2019. Front. Public Health 2022, 10, 1036674. [Google Scholar] [CrossRef]
- Mattiuzzi, C.; Lippi, G. Worldwide asthma epidemiology: Insights from the Global Health Data Exchange database. Int. Forum. Allergy Rhinol. 2020, 10, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef]
- Zheng, Q.; Cox, I.A.; Campbell, J.A.; Xia, Q.; Otahal, P.; de Graaff, B.; Corte, T.J.; Teoh, A.K.Y.; Walters, E.H.; Palmer, A.J. Mortality and survival in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. ERJ Open Res. 2022, 8, 00591–2021. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Cho, U.S.; Xu, W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature 2007, 445, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, P.J.; Creyghton, M.P.; Bernards, R. Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta 2009, 1795, 1–15. [Google Scholar] [CrossRef]
- Khew-Goodall, Y.; Hemmings, B.A. Tissue-specific expression of mRNAs encoding alpha- and beta-catalytic subunits of protein phosphatase 2A. FEBS Lett. 1988, 238, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.R.; Hofsteenge, J.; Hemmings, B.A. Molecular cloning of cDNAs encoding two isoforms of the catalytic subunit of protein phosphatase 2A. Biochemistry 1987, 26, 7215–7220. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Bird, R.C. Selective induction of cell cycle regulatory genes cdk1 (p34cdc2), cyclins A/B, and the tumor suppressor gene Rb in transformed cells by okadaic acid. J. Cell Physiol. 1995, 164, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Everett, A.D.; Kamibayashi, C.; Brautigan, D.L. Transgenic expression of protein phosphatase 2A regulatory subunit B56gamma disrupts distal lung differentiation. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 282, L1266–L1271. [Google Scholar] [CrossRef]
- Trotman, L.C.; Alimonti, A.; Scaglioni, P.P.; Koutcher, J.A.; Cordon-Cardo, C.; Pandolfi, P.P. Identification of a tumour suppressor network opposing nuclear Akt function. Nature 2006, 441, 523–527. [Google Scholar] [CrossRef]
- Papasozomenos, S.C.; Su, Y. Rapid dephosphorylation of tau in heat-shocked fetal rat cerebral explants: Prevention and hyperphosphorylation by inhibitors of protein phosphatases PP1 and PP2A. J. Neurochem. 1995, 65, 396–406. [Google Scholar] [CrossRef]
- Lucena, R.; Alcaide-Gavilan, M.; Anastasia, S.D.; Kellogg, D.R. Wee1 and Cdc25 are controlled by conserved PP2A-dependent mechanisms in fission yeast. Cell Cycle 2017, 16, 428–435. [Google Scholar] [CrossRef]
- Scheidtmann, K.H.; Mumby, M.C.; Rundell, K.; Walter, G. Dephosphorylation of simian virus 40 large-T antigen and p53 protein by protein phosphatase 2A: Inhibition by small-t antigen. Mol. Cell Biol. 1991, 11, 1996–2003. [Google Scholar] [CrossRef]
- Huang, K.L.; Jee, D.; Stein, C.B.; Elrod, N.D.; Henriques, T.; Mascibroda, L.G.; Baillat, D.; Russell, W.K.; Adelman, K.; Wagner, E.J. Integrator Recruits Protein Phosphatase 2A to Prevent Pause Release and Facilitate Transcription Termination. Mol. Cell 2020, 80, 345–358.e349. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Qi, Y.; Hu, S.; Cao, X.; Xu, C.; Yin, Z.; Chen, X.; Li, Y.; Liu, W.; Li, J.; et al. Identification of Integrator-PP2A complex (INTAC), an RNA polymerase II phosphatase. Science 2020, 370, eabb5872. [Google Scholar] [CrossRef]
- Fianu, I.; Chen, Y.; Dienemann, C.; Dybkov, O.; Linden, A.; Urlaub, H.; Cramer, P. Structural basis of Integrator-mediated transcription regulation. Science 2021, 374, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Vervoort, S.J.; Welsh, S.A.; Devlin, J.R.; Barbieri, E.; Knight, D.A.; Offley, S.; Bjelosevic, S.; Costacurta, M.; Todorovski, I.; Kearney, C.J.; et al. The PP2A-Integrator-CDK9 axis fine-tunes transcription and can be targeted therapeutically in cancer. Cell 2021, 184, 3143–3162.e3132. [Google Scholar] [CrossRef] [PubMed]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef]
- Andrade, M.A.; Bork, P. HEAT repeats in the Huntington’s disease protein. Nat. Genet. 1995, 11, 115–116. [Google Scholar] [CrossRef]
- Fellner, T.; Lackner, D.H.; Hombauer, H.; Piribauer, P.; Mudrak, I.; Zaragoza, K.; Juno, C.; Ogris, E. A novel and essential mechanism determining specificity and activity of protein phosphatase 2A (PP2A) in vivo. Genes Dev. 2003, 17, 2138–2150. [Google Scholar] [CrossRef] [PubMed]
- Hombauer, H.; Weismann, D.; Mudrak, I.; Stanzel, C.; Fellner, T.; Lackner, D.H.; Ogris, E. Generation of active protein phosphatase 2A is coupled to holoenzyme assembly. PLoS Biol. 2007, 5, e155. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, E.; Ołdziej, S.; Ciarkowski, J. Molecular modeling of the catalytic domain of serine/threonine phosphatase-1 with the Zn2+ and Mn2+ di-nuclear ion centers in the active site. Comput. Chem. 2000, 24, 381–390. [Google Scholar] [CrossRef]
- Xing, Y.; Xu, Y.; Chen, Y.; Jeffrey, P.D.; Chao, Y.; Lin, Z.; Li, Z.; Strack, S.; Stock, J.B.; Shi, Y. Structure of protein phosphatase 2A core enzyme bound to tumor-inducing toxins. Cell 2006, 127, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Cundell, M.J.; Hutter, L.H.; Nunes Bastos, R.; Poser, E.; Holder, J.; Mohammed, S.; Novak, B.; Barr, F.A. A PP2A-B55 recognition signal controls substrate dephosphorylation kinetics during mitotic exit. J. Cell Biol. 2016, 214, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Hertz, E.P.T.; Kruse, T.; Davey, N.E.; López-Méndez, B.; Sigurðsson, J.O.; Montoya, G.; Olsen, J.V.; Nilsson, J. A Conserved Motif Provides Binding Specificity to the PP2A-B56 Phosphatase. Mol. Cell 2016, 63, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Sangodkar, J.; Farrington, C.C.; McClinch, K.; Galsky, M.D.; Kastrinsky, D.B.; Narla, G. All roads lead to PP2A: Exploiting the therapeutic potential of this phosphatase. Febs J. 2016, 283, 1004–1024. [Google Scholar] [CrossRef] [PubMed]
- Mayer, R.E.; Hendrix, P.; Cron, P.; Matthies, R.; Stone, S.R.; Goris, J.; Merlevede, W.; Hofsteenge, J.; Hemmings, B.A. Structure of the 55-kDa regulatory subunit of protein phosphatase 2A: Evidence for a neuronal-specific isoform. Biochemistry 1991, 30, 3589–3597. [Google Scholar] [CrossRef]
- Strack, S.; Chang, D.; Zaucha, J.A.; Colbran, R.J.; Wadzinski, B.E. Cloning and characterization of B delta, a novel regulatory subunit of protein phosphatase 2A. FEBS Lett. 1999, 460, 462–466. [Google Scholar] [CrossRef]
- Dagda, R.K.; Zaucha, J.A.; Wadzinski, B.E.; Strack, S. A developmentally regulated, neuron-specific splice variant of the variable subunit Bbeta targets protein phosphatase 2A to mitochondria and modulates apoptosis. J. Biol. Chem. 2003, 278, 24976–24985. [Google Scholar] [CrossRef]
- Jin, Z.; Shi, J.; Saraf, A.; Mei, W.; Zhu, G.Z.; Strack, S.; Yang, J. The 48-kDa alternative translation isoform of PP2A:B56epsilon is required for Wnt signaling during midbrain-hindbrain boundary formation. J. Biol. Chem. 2009, 284, 7190–7200. [Google Scholar] [CrossRef] [PubMed]
- Martens, E.; Stevens, I.; Janssens, V.; Vermeesch, J.; Götz, J.; Goris, J.; Van Hoof, C. Genomic organisation, chromosomal localisation tissue distribution and developmental regulation of the PR61/B’ regulatory subunits of protein phosphatase 2A in mice. J. Mol. Biol. 2004, 336, 971–986. [Google Scholar] [CrossRef]
- Zwaenepoel, K.; Louis, J.V.; Goris, J.; Janssens, V. Diversity in genomic organisation, developmental regulation and distribution of the murine PR72/B” subunits of protein phosphatase 2A. BMC Genom. 2008, 9, 393. [Google Scholar] [CrossRef] [PubMed]
- Castets, F.; Rakitina, T.; Gaillard, S.; Moqrich, A.; Mattei, M.G.; Monneron, A. Zinedin, SG2NA, and striatin are calmodulin-binding, WD repeat proteins principally expressed in the brain. J. Biol. Chem. 2000, 275, 19970–19977. [Google Scholar] [CrossRef]
- Doherty, D.F.; Nath, S.; Poon, J.; Foronjy, R.F.; Ohlmeyer, M.; Dabo, A.J.; Salathe, M.; Birrell, M.; Belvisi, M.; Baumlin, N.; et al. Protein Phosphatase 2A Reduces Cigarette Smoke-induced Cathepsin S and Loss of Lung Function. Am. J. Respir. Crit. Care Med. 2019, 200, 51–62. [Google Scholar] [CrossRef]
- Nath, S.; Ohlmeyer, M.; Salathe, M.A.; Poon, J.; Baumlin, N.; Foronjy, R.F.; Geraghty, P. Chronic Cigarette Smoke Exposure Subdues PP2A Activity by Enhancing Expression of the Oncogene CIP2A. Am. J. Respir. Cell Mol. Biol. 2018, 59, 695–705. [Google Scholar] [CrossRef]
- Geraghty, P.; Eden, E.; Pillai, M.; Campos, M.; McElvaney, N.G.; Foronjy, R.F. α1-Antitrypsin activates protein phosphatase 2A to counter lung inflammatory responses. Am. J. Respir. Crit. Care Med. 2014, 190, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Sangodkar, J.; Perl, A.; Tohme, R.; Kiselar, J.; Kastrinsky, D.B.; Zaware, N.; Izadmehr, S.; Mazhar, S.; Wiredja, D.D.; O’Connor, C.M.; et al. Activation of tumor suppressor protein PP2A inhibits KRAS-driven tumor growth. J. Clin. Investig. 2017, 127, 2081–2090. [Google Scholar] [CrossRef]
- Xu, L.; Deng, X. Suppression of cancer cell migration and invasion by protein phosphatase 2A through dephosphorylation of mu- and m-calpains. J. Biol. Chem. 2006, 281, 35567–35575. [Google Scholar] [CrossRef]
- Gupta, M.K.; Asosingh, K.; Aronica, M.; Comhair, S.; Cao, G.; Erzurum, S.; Panettieri, R.A., Jr.; Naga Prasad, S.V. Defective Resensitization in Human Airway Smooth Muscle Cells Evokes β-Adrenergic Receptor Dysfunction in Severe Asthma. PLoS ONE 2015, 10, e0125803. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Ito, K.; Kanda, A.; Tomoda, K.; Miller-Larsson, A.; Barnes, P.J.; Mercado, N. Protein tyrosine phosphatase PTP-RR regulates corticosteroid sensitivity. Respir. Res. 2016, 17, 30. [Google Scholar] [CrossRef]
- Xia, H.; Seeman, J.; Hong, J.; Hergert, P.; Bodem, V.; Jessurun, J.; Smith, K.; Nho, R.; Kahm, J.; Gaillard, P.; et al. Low α2β1 integrin function enhances the proliferation of fibroblasts from patients with idiopathic pulmonary fibrosis by activation of the β-catenin pathway. Am. J. Pathol. 2012, 181, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Collison, A.M.; Li, J.; de Siqueira, A.P.; Lv, X.; Toop, H.D.; Morris, J.C.; Starkey, M.R.; Hansbro, P.M.; Zhang, J.; Mattes, J. TRAIL signals through the ubiquitin ligase MID1 to promote pulmonary fibrosis. BMC Pulm Med. 2019, 19, 31. [Google Scholar] [CrossRef]
- Geraghty, P.; Hardigan, A.A.; Wallace, A.M.; Mirochnitchenko, O.; Thankachen, J.; Arellanos, L.; Thompson, V.; D’Armiento, J.M.; Foronjy, R.F. The glutathione peroxidase 1-protein tyrosine phosphatase 1B-protein phosphatase 2A axis. A key determinant of airway inflammation and alveolar destruction. Am. J. Respir. Cell Mol. Biol. 2013, 49, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Foronjy, R.F.; Dabo, A.J.; Taggart, C.C.; Weldon, S.; Geraghty, P. Respiratory syncytial virus infections enhance cigarette smoke induced COPD in mice. PLoS ONE 2014, 9, e90567. [Google Scholar] [CrossRef]
- Wallace, A.M.; Hardigan, A.; Geraghty, P.; Salim, S.; Gaffney, A.; Thankachen, J.; Arellanos, L.; D’Armiento, J.M.; Foronjy, R.F. Protein phosphatase 2A regulates innate immune and proteolytic responses to cigarette smoke exposure in the lung. Toxicol. Sci. 2012, 126, 589–599. [Google Scholar] [CrossRef]
- Nair, P.M.; Starkey, M.R.; Haw, T.J.; Liu, G.; Collison, A.M.; Mattes, J.; Wark, P.A.; Morris, J.C.; Verrills, N.M.; Clark, A.R.; et al. Enhancing tristetraprolin activity reduces the severity of cigarette smoke-induced experimental chronic obstructive pulmonary disease. Clin. Transl. Immunol. 2019, 8, e01084. [Google Scholar] [CrossRef]
- Kauko, O.; Westermarck, J. Non-genomic mechanisms of protein phosphatase 2A (PP2A) regulation in cancer. Int. J. Biochem. Cell Biol. 2018, 96, 157–164. [Google Scholar] [CrossRef]
- Wang, S.S.; Esplin, E.D.; Li, J.L.; Huang, L.; Gazdar, A.; Minna, J.; Evans, G.A. Alterations of the PPP2R1B gene in human lung and colon cancer. Science 1998, 282, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Li, H.H.; Cai, X.; Shouse, G.P.; Piluso, L.G.; Liu, X. A specific PP2A regulatory subunit, B56gamma, mediates DNA damage-induced dephosphorylation of p53 at Thr55. Embo J. 2007, 26, 402–411. [Google Scholar] [CrossRef]
- Kawahara, E.; Maenaka, S.; Shimada, E.; Nishimura, Y.; Sakurai, H. Dynamic regulation of extracellular signal-regulated kinase (ERK) by protein phosphatase 2A regulatory subunit B56γ1 in nuclei induces cell migration. PLoS ONE 2013, 8, e63729. [Google Scholar] [CrossRef]
- Arnold, H.K.; Sears, R.C. Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-myc accumulation. Mol. Cell Biol. 2006, 26, 2832–2844. [Google Scholar] [CrossRef]
- Ma, L.; Wen, Z.S.; Liu, Z.; Hu, Z.; Ma, J.; Chen, X.Q.; Liu, Y.Q.; Pu, J.X.; Xiao, W.L.; Sun, H.D.; et al. Overexpression and small molecule-triggered downregulation of CIP2A in lung cancer. PLoS ONE 2011, 6, e20159. [Google Scholar] [CrossRef]
- Brognard, J.; Clark, A.S.; Ni, Y.; Dennis, P.A. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001, 61, 3986–3997. [Google Scholar]
- David, O.; Jett, J.; LeBeau, H.; Dy, G.; Hughes, J.; Friedman, M.; Brody, A.R. Phospho-Akt overexpression in non-small cell lung cancer confers significant stage-independent survival disadvantage. Clin. Cancer Res. 2004, 10, 6865–6871. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Chai, Y.; Li, Y.; Liu, X.; Zhang, J. CIP2A overexpression induces autoimmune response and enhances JNK signaling pathway in human lung cancer. BMC Cancer 2015, 15, 895. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.M.; Starkey, M.R.; Haw, T.J.; Liu, G.; Horvat, J.C.; Morris, J.C.; Verrills, N.M.; Clark, A.R.; Ammit, A.J.; Hansbro, P.M. Targeting PP2A and proteasome activity ameliorates features of allergic airway disease in mice. Allergy 2017, 72, 1891–1903. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Mercado, N.; Barnes, P.J.; Ito, K. Defects of protein phosphatase 2A causes corticosteroid insensitivity in severe asthma. PLoS ONE 2011, 6, e27627. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kanda, A.; Yun, Y.; Dan Van, B.; Suzuki, K.; Sawada, S.; Asako, M.; Iwai, H. Reduced Local Response to Corticosteroids in Eosinophilic Chronic Rhinosinusitis with Asthma. Biomolecules 2020, 10, 326. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kanda, A.; Bui, D.V.; Yun, Y.; Nguyen, L.M.; Chu, H.H.; Mitani, A.; Suzuki, K.; Asako, M.; Iwai, H. Omalizumab Restores Response to Corticosteroids in Patients with Eosinophilic Chronic Rhinosinusitis and Severe Asthma. Biomedicines 2021, 9, 787. [Google Scholar] [CrossRef]
- Penberthy, K.K.; Buckley, M.W.; Arandjelovic, S.; Ravichandran, K. Ex vivo modulation of the Foxo1 phosphorylation state does not lead to dysfunction of T regulatory cells. PLoS ONE 2017, 12, e0173386. [Google Scholar] [CrossRef]
- Han, B.; Liu, Q.; Su, X.; Zhou, L.; Zhang, B.; Kang, H.; Ning, J.; Li, C.; Zhao, B.; Niu, Y.; et al. The role of PP2A /NLRP3 signaling pathway in ambient particulate matter 2.5 induced lung injury. Chemosphere 2022, 307, 135794. [Google Scholar] [CrossRef]
- Sun, L.; Hult, E.M.; Cornell, T.T.; Kim, K.K.; Shanley, T.P.; Wilke, C.A.; Agarwal, M.; Gurczynski, S.J.; Moore, B.B.; Dahmer, M.K. Loss of myeloid-specific protein phosphatase 2A enhances lung injury and fibrosis and results in IL-10-dependent sensitization of epithelial cell apoptosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L1035–L1048. [Google Scholar] [CrossRef]
- Clark, A.R.; Ohlmeyer, M. Protein phosphatase 2A as a therapeutic target in inflammation and neurodegeneration. Pharmacol. Ther. 2019, 201, 181–201. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Osaki, K.; Kanamoto, M.; Nakao, Y.; Takahashi, E.; Higuchi, T.; Kamata, H. Distinct B subunits of PP2A regulate the NF-κB signalling pathway through dephosphorylation of IKKβ, IκBα and RelA. FEBS Lett. 2017, 591, 4083–4094. [Google Scholar] [CrossRef]
- Foronjy, R.F.; Ochieng, P.O.; Salathe, M.A.; Dabo, A.J.; Eden, E.; Baumlin, N.; Cummins, N.; Barik, S.; Campos, M.; Thorp, E.B.; et al. Protein tyrosine phosphatase 1B negatively regulates S100A9-mediated lung damage during respiratory syncytial virus exacerbations. Mucosal. Immunol. 2016, 9, 1317–1329. [Google Scholar] [CrossRef]
- Foronjy, R.F.; Salathe, M.A.; Dabo, A.J.; Baumlin, N.; Cummins, N.; Eden, E.; Geraghty, P. TLR9 expression is required for the development of cigarette smoke-induced emphysema in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L154–L166. [Google Scholar] [CrossRef]
- Janga, S.R.; Hamm-Alvarez, S.F. PP2A: A Novel Target to Prevent Cathepsin S-mediated Damage in Smoking-induced Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2019, 200, 6–8. [Google Scholar] [CrossRef]
- Wilkinson, R.D.; Williams, R.; Scott, C.J.; Burden, R.E. Cathepsin S: Therapeutic, diagnostic, and prognostic potential. Biol. Chem. 2015, 396, 867–882. [Google Scholar] [CrossRef]
- Rahman, M.M.; Rumzhum, N.N.; Hansbro, P.M.; Morris, J.C.; Clark, A.R.; Verrills, N.M.; Ammit, A.J. Activating protein phosphatase 2A (PP2A) enhances tristetraprolin (TTP) anti-inflammatory function in A549 lung epithelial cells. Cell Signal 2016, 28, 325–334. [Google Scholar] [CrossRef]
- Ahn, J.H.; McAvoy, T.; Rakhilin, S.V.; Nishi, A.; Greengard, P.; Nairn, A.C. Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56delta subunit. Proc. Natl. Acad. Sci. USA 2007, 104, 2979–2984. [Google Scholar] [CrossRef]
- Li, J.J.; Tay, H.L.; Maltby, S.; Xiang, Y.; Eyers, F.; Hatchwell, L.; Zhou, H.; Toop, H.D.; Morris, J.C.; Nair, P.; et al. MicroRNA-9 regulates steroid-resistant airway hyperresponsiveness by reducing protein phosphatase 2A activity. J. Allergy Clin. Immunol. 2015, 136, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.S.; Rahman, M.M.; Rumzhum, N.N.; Oliver, B.G.; Verrills, N.M.; Ammit, A.J. Theophylline Represses IL-8 Secretion from Airway Smooth Muscle Cells Independently of Phosphodiesterase Inhibition. Novel Role as a Protein Phosphatase 2A Activator. Am. J. Respir. Cell Mol. Biol. 2016, 54, 792–801. [Google Scholar] [CrossRef]
- Barnes, P.J. Theophylline. Pharmaceuticals 2010, 3, 725–747. [Google Scholar] [CrossRef]
- Hatchwell, L.; Girkin, J.; Dun, M.D.; Morten, M.; Verrills, N.; Toop, H.D.; Morris, J.C.; Johnston, S.L.; Foster, P.S.; Collison, A.; et al. Salmeterol attenuates chemotactic responses in rhinovirus-induced exacerbation of allergic airways disease by modulating protein phosphatase 2A. J. Allergy Clin. Immunol. 2014, 133, 1720–1727. [Google Scholar] [CrossRef] [PubMed]
- Staser, K.; Shew, M.A.; Michels, E.G.; Mwanthi, M.M.; Yang, F.C.; Clapp, D.W.; Park, S.J. A Pak1-PP2A-ERM signaling axis mediates F-actin rearrangement and degranulation in mast cells. Exp. Hematol. 2013, 41, 56–66.52. [Google Scholar] [CrossRef] [PubMed]
- Kranias, G.; Watt, L.F.; Carpenter, H.; Holst, J.; Ludowyke, R.; Strack, S.; Sim, A.T.; Verrills, N.M. Protein phosphatase 2A carboxymethylation and regulatory B subunits differentially regulate mast cell degranulation. Cell Signal 2010, 22, 1882–1890. [Google Scholar] [CrossRef] [PubMed]
- Leikauf, G.D.; Kim, S.H.; Jang, A.S. Mechanisms of ultrafine particle-induced respiratory health effects. Exp. Mol. Med. 2020, 52, 329–337. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Dag, S.; Hlubek, F.; Kirchner, T. beta-catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am. J. Pathol. 1999, 155, 1033–1038. [Google Scholar] [CrossRef]
- Crawford, H.C.; Fingleton, B.M.; Rudolph-Owen, L.A.; Goss, K.J.; Rubinfeld, B.; Polakis, P.; Matrisian, L.M. The metalloproteinase matrilysin is a target of beta-catenin transactivation in intestinal tumors. Oncogene 1999, 18, 2883–2891. [Google Scholar] [CrossRef]
- Zuo, F.; Kaminski, N.; Eugui, E.; Allard, J.; Yakhini, Z.; Ben-Dor, A.; Lollini, L.; Morris, D.; Kim, Y.; DeLustro, B.; et al. Gene expression analysis reveals matrilysin as a key regulator of pulmonary fibrosis in mice and humans. Proc. Natl. Acad. Sci. USA 2002, 99, 6292–6297. [Google Scholar] [CrossRef]
- Morali, O.G.; Delmas, V.; Moore, R.; Jeanney, C.; Thiery, J.P.; Larue, L. IGF-II induces rapid beta-catenin relocation to the nucleus during epithelium to mesenchyme transition. Oncogene 2001, 20, 4942–4950. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am. J. Pathol. 2001, 159, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lu, Z.; Hay, E.D. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol. Int. 2002, 26, 463–476. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef]
- Ivaska, J.; Nissinen, L.; Immonen, N.; Eriksson, J.E.; Kähäri, V.M.; Heino, J. Integrin alpha 2 beta 1 promotes activation of protein phosphatase 2A and dephosphorylation of Akt and glycogen synthase kinase 3 beta. Mol. Cell Biol. 2002, 22, 1352–1359. [Google Scholar] [CrossRef]
- Yano, Y.; Yoshida, M.; Hoshino, S.; Inoue, K.; Kida, H.; Yanagita, M.; Takimoto, T.; Hirata, H.; Kijima, T.; Kumagai, T.; et al. Anti-fibrotic effects of theophylline on lung fibroblasts. Biochem. Biophys. Res. Commun. 2006, 341, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Hahn, H.J.; Oh, S.R.; Lee, H.J. Theophylline Attenuates BLM-Induced Pulmonary Fibrosis by Inhibiting Th17 Differentiation. Int. J. Mol. Sci. 2023, 24, 1019. [Google Scholar] [CrossRef] [PubMed]
- Ruediger, R.; Ruiz, J.; Walter, G. Human cancer-associated mutations in the Aα subunit of protein phosphatase 2A increase lung cancer incidence in Aα knock-in and knockout mice. Mol. Cell Biol. 2011, 31, 3832–3844. [Google Scholar] [CrossRef]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 2004, 6, 308–318. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, L.; Wen, Z.S.; Cheng, Y.X.; Zhou, G.B. Ethoxysanguinarine Induces Inhibitory Effects and Downregulates CIP2A in Lung Cancer Cells. ACS Med. Chem. Lett. 2014, 5, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, L.; Wen, Z.S.; Hu, Z.; Wu, F.Q.; Li, W.; Liu, J.; Zhou, G.B. Cancerous inhibitor of PP2A is targeted by natural compound celastrol for degradation in non-small-cell lung cancer. Carcinogenesis 2014, 35, 905–914. [Google Scholar] [CrossRef]
- Pavic, K.; Gupta, N.; Omella, J.D.; Derua, R.; Aakula, A.; Huhtaniemi, R.; Maatta, J.A.; Hofflin, N.; Okkeri, J.; Wang, Z.; et al. Structural mechanism for inhibition of PP2A-B56alpha and oncogenicity by CIP2A. Nat. Commun. 2023, 14, 1143. [Google Scholar] [CrossRef]
- Soofiyani, S.R.; Hejazi, M.S.; Baradaran, B. The role of CIP2A in cancer: A review and update. Biomed. Pharmacother. 2017, 96, 626–633. [Google Scholar] [CrossRef]
- Khanna, A.; Okkeri, J.; Bilgen, T.; Tiirikka, T.; Vihinen, M.; Visakorpi, T.; Westermarck, J. ETS1 mediates MEK1/2-dependent overexpression of cancerous inhibitor of protein phosphatase 2A (CIP2A) in human cancer cells. PLoS ONE 2011, 6, e17979. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.M.; Phillips, B.L.; Chan, E.K. miR-375 activates p21 and suppresses telomerase activity by coordinately regulating HPV E6/E7, E6AP, CIP2A, and 14-3-3ζ. Mol. Cancer 2014, 13, 80. [Google Scholar] [CrossRef]
- Zhao, S.; Gao, X.; Zang, S.; Li, Y.; Feng, X.; Yuan, X. MicroRNA-383-5p acts as a prognostic marker and inhibitor of cell proliferation in lung adenocarcinoma by cancerous inhibitor of protein phosphatase 2A. Oncol. Lett. 2017, 14, 3573–3579. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.M.; Patel, R.S.; Phillips, B.L.; Wang, H.; Cohen, D.M.; Reinhold, W.C.; Chang, L.J.; Yang, L.J.; Chan, E.K. Tumor suppressor miR-375 regulates MYC expression via repression of CIP2A coding sequence through multiple miRNA-mRNA interactions. Mol. Biol. Cell 2013, 24, 1638–1648, s1631–s1637. [Google Scholar] [CrossRef]
- Li, M.; Guo, H.; Damuni, Z. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry 1995, 34, 1988–1996. [Google Scholar] [CrossRef]
- Zhou, X.; Updegraff, B.L.; Guo, Y.; Peyton, M.; Girard, L.; Larsen, J.E.; Xie, X.J.; Zhou, Y.; Hwang, T.H.; Xie, Y.; et al. PROTOCADHERIN 7 Acts through SET and PP2A to Potentiate MAPK Signaling by EGFR and KRAS during Lung Tumorigenesis. Cancer Res. 2017, 77, 187–197. [Google Scholar] [CrossRef]
- Pippa, R.; Dominguez, A.; Christensen, D.J.; Moreno-Miralles, I.; Blanco-Prieto, M.J.; Vitek, M.P.; Odero, M.D. Effect of FTY720 on the SET-PP2A complex in acute myeloid leukemia; SET binding drugs have antagonistic activity. Leukemia 2014, 28, 1915–1918. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gu, Y.; Wang, H.; Yin, J.; Zheng, G.; Zhang, Z.; Lu, M.; Wang, C.; He, Z. Overexpression of PP2A inhibitor SET oncoprotein is associated with tumor progression and poor prognosis in human non-small cell lung cancer. Oncotarget 2015, 6, 14913–14925. [Google Scholar] [CrossRef]
- Liu, H.; Gu, Y.; Yin, J.; Zheng, G.; Wang, C.; Zhang, Z.; Deng, M.; Liu, J.; Jia, X.; He, Z. SET-mediated NDRG1 inhibition is involved in acquisition of epithelial-to-mesenchymal transition phenotype and cisplatin resistance in human lung cancer cell. Cell Signal 2014, 26, 2710–2720. [Google Scholar] [CrossRef]
- McConnell, J.L.; Watkins, G.R.; Soss, S.E.; Franz, H.S.; McCorvey, L.R.; Spiller, B.W.; Chazin, W.J.; Wadzinski, B.E. Alpha4 is a ubiquitin-binding protein that regulates protein serine/threonine phosphatase 2A ubiquitination. Biochemistry 2010, 49, 1713–1718. [Google Scholar] [CrossRef]
- Collison, A.; Hatchwell, L.; Verrills, N.; Wark, P.A.; de Siqueira, A.P.; Tooze, M.; Carpenter, H.; Don, A.S.; Morris, J.C.; Zimmermann, N.; et al. The E3 ubiquitin ligase midline 1 promotes allergen and rhinovirus-induced asthma by inhibiting protein phosphatase 2A activity. Nat. Med. 2013, 19, 232–237. [Google Scholar] [CrossRef]
- Zhang, L.; Li, J.; Lv, X.; Guo, T.; Li, W.; Zhang, J. MID1-PP2A complex functions as new insights in human lung adenocarcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 855–864. [Google Scholar] [CrossRef]
- Girkin, J.L.; Hatchwell, L.M.; Collison, A.M.; Starkey, M.R.; Hansbro, P.M.; Yagita, H.; Foster, P.S.; Mattes, J. TRAIL signaling is proinflammatory and proviral in a murine model of rhinovirus 1B infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L89–L99. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, S.; Qian, Z.; Su, X.; Fan, S.; Fu, J.; Liu, Y.; Yin, X.; Gao, Z.; Zhang, J.; et al. Genetic amplification of PPME1 in gastric and lung cancer and its potential as a novel therapeutic target. Cancer Biol. Ther. 2014, 15, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Yabe, R.; Miura, A.; Usui, T.; Mudrak, I.; Ogris, E.; Ohama, T.; Sato, K. Protein Phosphatase Methyl-Esterase PME-1 Protects Protein Phosphatase 2A from Ubiquitin/Proteasome Degradation. PLoS ONE 2015, 10, e0145226. [Google Scholar] [CrossRef]
- Mochida, S.; Maslen, S.L.; Skehel, M.; Hunt, T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 2010, 330, 1670–1673. [Google Scholar] [CrossRef]
- Thapa, C.; Roivas, P.; Haataja, T.; Permi, P.; Pentikainen, U. Interaction mechanism of endogenous PP2A inhibitor protein ENSA with PP2A. FEBS J. 2022, 289, 519–534. [Google Scholar] [CrossRef]
- Sakashita, S.; Li, D.; Nashima, N.; Minami, Y.; Furuya, S.; Morishita, Y.; Tachibana, K.; Sato, Y.; Noguchi, M. Overexpression of immunoglobulin (CD79a) binding protein1 (IGBP-1) in small lung adenocarcinomas and its clinicopathological significance. Pathol. Int. 2011, 61, 130–137. [Google Scholar] [CrossRef]
- Grech, G.; Blazquez-Domingo, M.; Kolbus, A.; Bakker, W.J.; Mullner, E.W.; Beug, H.; von Lindern, M. Igbp1 is part of a positive feedback loop in stem cell factor-dependent, selective mRNA translation initiation inhibiting erythroid differentiation. Blood 2008, 112, 2750–2760. [Google Scholar] [CrossRef]
- Kong, M.; Ditsworth, D.; Lindsten, T.; Thompson, C.B. Alpha4 is an essential regulator of PP2A phosphatase activity. Mol. Cell 2009, 36, 51–60. [Google Scholar] [CrossRef]
- Cornell, T.T.; Hinkovska-Galcheva, V.; Sun, L.; Cai, Q.; Hershenson, M.B.; Vanway, S.; Shanley, T.P. Ceramide-dependent PP2A regulation of TNFalpha-induced IL-8 production in respiratory epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 296, L849–L856. [Google Scholar] [CrossRef] [PubMed]
- Chalfant, C.E.; Szulc, Z.; Roddy, P.; Bielawska, A.; Hannun, Y.A. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J. Lipid Res. 2004, 45, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Chalfant, C.E.; Kishikawa, K.; Mumby, M.C.; Kamibayashi, C.; Bielawska, A.; Hannun, Y.A. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J. Biol. Chem. 1999, 274, 20313–20317. [Google Scholar] [CrossRef]
- Dobrowsky, R.T.; Kamibayashi, C.; Mumby, M.C.; Hannun, Y.A. Ceramide activates heterotrimeric protein phosphatase 2A. J. Biol. Chem. 1993, 268, 15523–15530. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Saddoughi, S.A.; Song, P.; Sultan, I.; Ponnusamy, S.; Senkal, C.E.; Snook, C.F.; Arnold, H.K.; Sears, R.C.; Hannun, Y.A.; et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J. 2009, 23, 751–763. [Google Scholar] [CrossRef]
- Ross, E.A.; Naylor, A.J.; O’Neil, J.D.; Crowley, T.; Ridley, M.L.; Crowe, J.; Smallie, T.; Tang, T.J.; Turner, J.D.; Norling, L.V.; et al. Treatment of inflammatory arthritis via targeting of tristetraprolin, a master regulator of pro-inflammatory gene expression. Ann. Rheum. Dis. 2017, 76, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Zonta, F.; Pagano, M.A.; Trentin, L.; Tibaldi, E.; Frezzato, F.; Trimarco, V.; Facco, M.; Zagotto, G.; Pavan, V.; Ribaudo, G.; et al. Lyn sustains oncogenic signaling in chronic lymphocytic leukemia by strengthening SET-mediated inhibition of PP2A. Blood 2015, 125, 3747–3755. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; MacKenzie, R.J.; Pippa, R.; Eide, C.A.; Oddo, J.; Tyner, J.W.; Sears, R.; Vitek, M.P.; Odero, M.D.; Christensen, D.J.; et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin. Cancer Res. 2014, 20, 2092–2103. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.J.; Ohkubo, N.; Oddo, J.; Van Kanegan, M.J.; Neil, J.; Li, F.; Colton, C.A.; Vitek, M.P. Apolipoprotein E and peptide mimetics modulate inflammation by binding the SET protein and activating protein phosphatase 2A. J. Immunol. 2011, 186, 2535–2542. [Google Scholar] [CrossRef]
- Switzer, C.H.; Cheng, R.Y.; Vitek, T.M.; Christensen, D.J.; Wink, D.A.; Vitek, M.P. Targeting SET/I(2)PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene 2011, 30, 2504–2513. [Google Scholar] [CrossRef]
- Wang, S.; Xie, W.; Wang, D.; Peng, Z.; Zheng, Y.; Liu, N.; Dai, W.; Wang, Y.; Wang, Z.; Yang, Y.; et al. Discovery of a small molecule targeting SET-PP2A interaction to overcome BCR-ABLT315I mutation of chronic myeloid leukemia. Oncotarget 2015, 6, 12128–12140. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J.; Baselga, J. Epidermal growth factor receptor targeting in cancer. Semin. Oncol. 2006, 33, 369–385. [Google Scholar] [CrossRef]
- Wu, Y.L.; Zhou, C.; Liam, C.K.; Wu, G.; Liu, X.; Zhong, Z.; Lu, S.; Cheng, Y.; Han, B.; Chen, L.; et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: Analyses from the phase III, randomized, open-label, ENSURE study. Ann. Oncol. 2015, 26, 1883–1889. [Google Scholar] [CrossRef]
- Yu, H.C.; Hung, M.H.; Chen, Y.L.; Chu, P.Y.; Wang, C.Y.; Chao, T.T.; Liu, C.Y.; Shiau, C.W.; Chen, K.F. Erlotinib derivative inhibits hepatocellular carcinoma by targeting CIP2A to reactivate protein phosphatase 2A. Cell Death Dis. 2014, 5, e1359. [Google Scholar] [CrossRef]
- Chao, T.T.; Wang, C.Y.; Lai, C.C.; Chen, Y.L.; Tsai, Y.T.; Chen, P.T.; Lin, H.I.; Huang, Y.C.; Shiau, C.W.; Yu, C.J.; et al. TD-19, an erlotinib derivative, induces epidermal growth factor receptor wild-type nonsmall-cell lung cancer apoptosis through CIP2A-mediated pathway. J. Pharmacol. Exp. Ther. 2014, 351, 352–358. [Google Scholar] [CrossRef]
- Chen, K.F.; Pao, K.C.; Su, J.C.; Chou, Y.C.; Liu, C.Y.; Chen, H.J.; Huang, J.W.; Kim, I.; Shiau, C.W. Development of erlotinib derivatives as CIP2A-ablating agents independent of EGFR activity. Bioorg. Med. Chem. 2012, 20, 6144–6153. [Google Scholar] [CrossRef]
- Niesvizky, R.; Flinn, I.W.; Rifkin, R.; Gabrail, N.; Charu, V.; Clowney, B.; Essell, J.; Gaffar, Y.; Warr, T.; Neuwirth, R.; et al. Community-Based Phase IIIB Trial of Three UPFRONT Bortezomib-Based Myeloma Regimens. J. Clin. Oncol. 2015, 33, 3921–3929. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.F.; Liu, C.Y.; Lin, Y.C.; Yu, H.C.; Liu, T.H.; Hou, D.R.; Chen, P.J.; Cheng, A.L. CIP2A mediates effects of bortezomib on phospho-Akt and apoptosis in hepatocellular carcinoma cells. Oncogene 2010, 29, 6257–6266. [Google Scholar] [CrossRef]
- Tseng, L.M.; Liu, C.Y.; Chang, K.C.; Chu, P.Y.; Shiau, C.W.; Chen, K.F. CIP2A is a target of bortezomib in human triple negative breast cancer cells. Breast Cancer Res. 2012, 14, R68. [Google Scholar] [CrossRef]
- Elgendy, M.; Cirò, M.; Hosseini, A.; Weiszmann, J.; Mazzarella, L.; Ferrari, E.; Cazzoli, R.; Curigliano, G.; DeCensi, A.; Bonanni, B.; et al. Combination of Hypoglycemia and Metformin Impairs Tumor Metabolic Plasticity and Growth by Modulating the PP2A-GSK3β-MCL-1 Axis. Cancer Cell 2019, 35, 798–815.e795. [Google Scholar] [CrossRef]
- Kickstein, E.; Krauss, S.; Thornhill, P.; Rutschow, D.; Zeller, R.; Sharkey, J.; Williamson, R.; Fuchs, M.; Kohler, A.; Glossmann, H.; et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 21830–21835. [Google Scholar] [CrossRef]
- Bachovchin, D.A.; Speers, A.E.; Brown, S.J.; Spicer, T.P.; Fernandez-Vega, V.; Ferguson, J.; Mohr, J.T.; Murphy, J.; Fu, G.C.; Cravatt, B.F.; et al. Probe Development Efforts to Identify Novel Inhibitors of Protein Phosphatase Methylesterase-1 (PME-1). In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Kastrinsky, D.B.; Sangodkar, J.; Zaware, N.; Izadmehr, S.; Dhawan, N.S.; Narla, G.; Ohlmeyer, M. Reengineered tricyclic anti-cancer agents. Bioorg. Med. Chem. 2015, 23, 6528–6534. [Google Scholar] [CrossRef]
- Bownes, L.V.; Marayati, R.; Quinn, C.H.; Beierle, A.M.; Hutchins, S.C.; Julson, J.R.; Erwin, M.H.; Stewart, J.E.; Mroczek-Musulman, E.; Ohlmeyer, M.; et al. Pre-Clinical Study Evaluating Novel Protein Phosphatase 2A Activators as Therapeutics for Neuroblastoma. Cancers 2022, 14, 1952. [Google Scholar] [CrossRef] [PubMed]
- Kabashima, T.; Kawaguchi, T.; Wadzinski, B.E.; Uyeda, K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proc. Natl. Acad. Sci. USA 2003, 100, 5107–5112. [Google Scholar] [CrossRef]
- Neuzil, J.; Weber, T.; Schröder, A.; Lu, M.; Ostermann, G.; Gellert, N.; Mayne, G.C.; Olejnicka, B.; Nègre-Salvayre, A.; Stícha, M.; et al. Induction of cancer cell apoptosis by alpha-tocopheryl succinate: Molecular pathways and structural requirements. Faseb. J. 2001, 15, 403–415. [Google Scholar] [CrossRef]
- Huang, P.H.; Wang, D.; Chuang, H.C.; Wei, S.; Kulp, S.K.; Chen, C.S. alpha-Tocopheryl succinate and derivatives mediate the transcriptional repression of androgen receptor in prostate cancer cells by targeting the PP2A-JNK-Sp1-signaling axis. Carcinogenesis 2009, 30, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Feschenko, M.S.; Stevenson, E.; Nairn, A.C.; Sweadner, K.J. A novel cAMP-stimulated pathway in protein phosphatase 2A activation. J. Pharmacol. Exp. Ther. 2002, 302, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Pan, L.; Groen, R.W.; Baleydier, F.; Kentsis, A.; Marineau, J.; Grebliunaite, R.; Kozakewich, E.; Reed, C.; Pflumio, F.; et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J. Clin. Investig. 2014, 124, 644–655. [Google Scholar] [CrossRef]
- Allen, S.C.; Tiwari, D. Theophylline as a systemic anti-inflammatory agent: The need for its revival as a possible adjunctive treatment for “inflammaging”. Biol. Eng. Med. 2019, 4, 1–3. [Google Scholar] [CrossRef]
- Allen, S.; Khattab, A.; Vassallo, M.; Kwan, J. Inflammation and Muscle Weakness in COPD: Considering a Renewed Role for Theophylline? Curr. Respir. Med. Rev. 2018, 14, 35–41. [Google Scholar] [CrossRef]
- Kanehara, M.; Yokoyama, A.; Tomoda, Y.; Shiota, N.; Iwamoto, H.; Ishikawa, N.; Taooka, Y.; Haruta, Y.; Hattori, N.; Kohno, N. Anti-inflammatory effects and clinical efficacy of theophylline and tulobuterol in mild-to-moderate chronic obstructive pulmonary disease. Pulm Pharmacol. Ther. 2008, 21, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Davis, M.D.; Heise, C.E.; Albert, R.; Cottens, S.; Hof, R.; Bruns, C.; Prieschl, E.; Baumruker, T.; Hiestand, P.; et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 2002, 277, 21453–21457. [Google Scholar] [CrossRef]
- Saddoughi, S.A.; Gencer, S.; Peterson, Y.K.; Ward, K.E.; Mukhopadhyay, A.; Oaks, J.; Bielawski, J.; Szulc, Z.M.; Thomas, R.J.; Selvam, S.P.; et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol. Med. 2013, 5, 105–121. [Google Scholar] [CrossRef]
- Brinkmann, V.; Billich, A.; Baumruker, T.; Heining, P.; Schmouder, R.; Francis, G.; Aradhye, S.; Burtin, P. Fingolimod (FTY720): Discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 2010, 9, 883–897. [Google Scholar] [CrossRef]
- Liu, W.; Liang, Q.; Balzar, S.; Wenzel, S.; Gorska, M.; Alam, R. Cell-specific activation profile of extracellular signal-regulated kinase 1/2, Jun N-terminal kinase, and p38 mitogen-activated protein kinases in asthmatic airways. J. Allergy Clin. Immunol. 2008, 121, 893–902.e892. [Google Scholar] [CrossRef]
- Griego, S.D.; Weston, C.B.; Adams, J.L.; Tal-Singer, R.; Dillon, S.B. Role of p38 mitogen-activated protein kinase in rhinovirus-induced cytokine production by bronchial epithelial cells. J. Immunol. 2000, 165, 5211–5220. [Google Scholar] [CrossRef]
- Das, J.; Chen, C.H.; Yang, L.; Cohn, L.; Ray, P.; Ray, A. A critical role for NF-kappa B in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat. Immunol. 2001, 2, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Beisiegel, U.; Weber, W.; Ihrke, G.; Herz, J.; Stanley, K.K. The LDL-receptor-related protein, LRP, is an apolipoprotein E-binding protein. Nature 1989, 341, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef]
- Curtiss, L.K.; Edgington, T.S. The biologic activity of the immunoregulatory lipoprotein, LDL-In is independent of its free fatty acid content. J. Immunol. 1981, 126, 1382–1386. [Google Scholar] [CrossRef] [PubMed]
- Pepe, M.G.; Curtiss, L.K. Apolipoprotein E is a biologically active constituent of the normal immunoregulatory lipoprotein, LDL-In. J. Immunol. 1986, 136, 3716–3723. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.F.; Yu, H.C.; Liu, C.Y.; Chen, H.J.; Chen, Y.C.; Hou, D.R.; Chen, P.J.; Cheng, A.L. Bortezomib sensitizes HCC cells to CS-1008, an antihuman death receptor 5 antibody, through the inhibition of CIP2A. Mol. Cancer Ther. 2011, 10, 892–901. [Google Scholar] [CrossRef]
- Huang, C.Y.; Wei, C.C.; Chen, K.C.; Chen, H.J.; Cheng, A.L.; Chen, K.F. Bortezomib enhances radiation-induced apoptosis in solid tumors by inhibiting CIP2A. Cancer Lett. 2012, 317, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Menezes, M.E.; Pannell, L.K.; Mulekar, M.S.; Honkanen, R.E.; Shevde, L.A.; Samant, R.S. DNAJB6 chaperones PP2A mediated dephosphorylation of GSK3β to downregulate β-catenin transcription target, osteopontin. Oncogene 2012, 31, 4472–4483. [Google Scholar] [CrossRef]
- Bennecib, M.; Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K. Role of protein phosphatase-2A and -1 in the regulation of GSK-3, cdk5 and cdc2 and the phosphorylation of tau in rat forebrain. FEBS Lett. 2000, 485, 87–93. [Google Scholar] [CrossRef]
- Phoenix, K.N.; Vumbaca, F.; Fox, M.M.; Evans, R.; Claffey, K.P. Dietary energy availability affects primary and metastatic breast cancer and metformin efficacy. Breast Cancer Res. Treat. 2010, 123, 333–344. [Google Scholar] [CrossRef]
- Simone, B.A.; Dan, T.; Palagani, A.; Jin, L.; Han, S.Y.; Wright, C.; Savage, J.E.; Gitman, R.; Lim, M.K.; Palazzo, J.; et al. Caloric restriction coupled with radiation decreases metastatic burden in triple negative breast cancer. Cell Cycle 2016, 15, 2265–2274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fan, X.; Su, Z.; Yuan, T.; Yin, H.; Gu, H.; Zuo, Y.; Chen, S.; Zhou, H.; Su, G. Pretreatment with metformin prevents microcystin-LR-induced tau hyperphosphorylation via mTOR-dependent PP2A and GSK-3β activation. Environ. Toxicol. 2021, 36, 2414–2425. [Google Scholar] [CrossRef]
- Katila, N.; Bhurtel, S.; Shadfar, S.; Srivastav, S.; Neupane, S.; Ojha, U.; Jeong, G.S.; Choi, D.Y. Metformin lowers α-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 2017, 125, 396–407. [Google Scholar] [CrossRef]
- Pampalakis, G.; Angelis, G.; Zingkou, E.; Vekrellis, K.; Sotiropoulou, G. A chemogenomic approach is required for effective treatment of amyotrophic lateral sclerosis. Clin. Transl. Med. 2022, 12, e657. [Google Scholar] [CrossRef] [PubMed]
- Hervás, D.; Fornés-Ferrer, V.; Gómez-Escribano, A.P.; Sequedo, M.D.; Peiró, C.; Millán, J.M.; Vázquez-Manrique, R.P. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS ONE 2017, 12, e0179283. [Google Scholar] [CrossRef]
- Liu, Z.; Ma, L.; Zhou, G.B. The main anticancer bullets of the Chinese medicinal herb, thunder god vine. Molecules 2011, 16, 5283–5297. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhou, X.; Fu, C.; Wang, Q.; Nie, T.; Zou, F.; Guo, R.; Liu, H.; Zhang, B.; Dai, M. Celastrol induces apoptosis of human osteosarcoma cells via the mitochondrial apoptotic pathway. Oncol. Rep. 2015, 34, 1129–1136. [Google Scholar] [CrossRef]
- Lee, J.H.; Won, Y.S.; Park, K.H.; Lee, M.K.; Tachibana, H.; Yamada, K.; Seo, K.I. Celastrol inhibits growth and induces apoptotic cell death in melanoma cells via the activation ROS-dependent mitochondrial pathway and the suppression of PI3K/AKT signaling. Apoptosis 2012, 17, 1275–1286. [Google Scholar] [CrossRef]
- Longin, S.; Zwaenepoel, K.; Louis, J.V.; Dilworth, S.; Goris, J.; Janssens, V. Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic Subunit. J. Biol. Chem. 2007, 282, 26971–26980. [Google Scholar] [CrossRef]
- Yu, X.X.; Du, X.; Moreno, C.S.; Green, R.E.; Ogris, E.; Feng, Q.; Chou, L.; McQuoid, M.J.; Pallas, D.C. Methylation of the protein phosphatase 2A catalytic subunit is essential for association of Balpha regulatory subunit but not SG2NA, striatin, or polyomavirus middle tumor antigen. Mol. Biol. Cell 2001, 12, 185–199. [Google Scholar] [CrossRef]
- Puustinen, P.; Junttila, M.R.; Vanhatupa, S.; Sablina, A.A.; Hector, M.E.; Teittinen, K.; Raheem, O.; Ketola, K.; Lin, S.; Kast, J.; et al. PME-1 protects extracellular signal-regulated kinase pathway activity from protein phosphatase 2A-mediated inactivation in human malignant glioma. Cancer Res. 2009, 69, 2870–2877. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Li, Z.; Chen, Y.; Stock, J.B.; Jeffrey, P.D.; Shi, Y. Structural mechanism of demethylation and inactivation of protein phosphatase 2A. Cell 2008, 133, 154–163. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Snyder, S.H. Phenothiazine drugs: Structure-activity relationships explained by a conformation that mimics dopamine. Proc. Natl. Acad. Sci. USA 1975, 72, 1899–1903. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Yang, Y.R.; Lee, S.K.; Kim, S.H.; Kim, Y.H.; Cha, J.Y.; Oh, S.W.; Ha, J.R.; Ryu, S.H.; Suh, P.G. Potential inhibition of PDK1/Akt signaling by phenothiazines suppresses cancer cell proliferation and survival. Ann. N. Y. Acad. Sci. 2008, 1138, 393–403. [Google Scholar] [CrossRef]
- Kang, U.G.; Kim, M.J.; Suh, P.G.; Ryu, S.H.; Park, J.B.; Kim, J.H.; Kim, Y.S.; Lee, Y.H. Inhibition of trifluoperazine-induced DNA fragmentation by cyclic AMP mediated signaling. Mol. Cells 1999, 9, 596–602. [Google Scholar]
- Leonard, D.; Huang, W.; Izadmehr, S.; O’Connor, C.M.; Wiredja, D.D.; Wang, Z.; Zaware, N.; Chen, Y.; Schlatzer, D.M.; Kiselar, J.; et al. Selective PP2A Enhancement through Biased Heterotrimer Stabilization. Cell 2020, 181, 688–701.e616. [Google Scholar] [CrossRef]
- Cunningham, M.P.; Essapen, S.; Thomas, H.; Green, M.; Lovell, D.P.; Topham, C.; Marks, C.; Modjtahedi, H. Coexpression, prognostic significance and predictive value of EGFR, EGFRvIII and phosphorylated EGFR in colorectal cancer. Int. J. Oncol. 2005, 27, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Atreya, C.E.; Corcoran, R.B.; Kopetz, S. Expanded RAS: Refining the patient population. J. Clin. Oncol. 2015, 33, 682–685. [Google Scholar] [CrossRef]
- Holck, S.; Nielsen, H.J.; Pedersen, N.; Larsson, L.I. Phospho-ERK1/2 levels in cancer cell nuclei predict responsiveness to radiochemotherapy of rectal adenocarcinoma. Oncotarget 2015, 6, 34321–34328. [Google Scholar] [CrossRef]
- Bownes, L.V.; Julson, J.R.; Quinn, C.H.; Hutchins, S.C.; Erwin, M.H.; Markert, H.R.; Stewart, J.E.; Mroczek-Musulman, E.; Aye, J.; Yoon, K.J.; et al. The Effects of Protein Phosphatase 2A Activation with Novel Tricyclic Sulfonamides on Hepatoblastoma. J. Pediatr. Surg. 2023, 58, 1145. [Google Scholar] [CrossRef]
- Kar, S.; Palit, S.; Ball, W.B.; Das, P.K. Carnosic acid modulates Akt/IKK/NF-κB signaling by PP2A and induces intrinsic and extrinsic pathway mediated apoptosis in human prostate carcinoma PC-3 cells. Apoptosis 2012, 17, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Lipina, C.; Hundal, H.S. Carnosic acid stimulates glucose uptake in skeletal muscle cells via a PME-1/PP2A/PKB signalling axis. Cell Signal 2014, 26, 2343–2349. [Google Scholar] [CrossRef]
- Nakamura, S.; Takamura, T.; Matsuzawa-Nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem. 2009, 284, 14809–14818. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Liu, Y.; Tang, A.; Chen, L.; Miao, D.; Yuan, X. Hepatocyte-specific ablation of PP2A catalytic subunit α attenuates liver fibrosis progression via TGF-β1/Smad signaling. Biomed. Res. Int. 2015, 2015, 794862. [Google Scholar] [CrossRef]
- Chattopadhyay, D.; Swingle, M.R.; Salter, E.A.; Wood, E.; D’Arcy, B.; Zivanov, C.; Abney, K.; Musiyenko, A.; Rusin, S.F.; Kettenbach, A.; et al. Crystal structures and mutagenesis of PPP-family ser/thr protein phosphatases elucidate the selectivity of cantharidin and novel norcantharidin-based inhibitors of PP5C. Biochem. Pharmacol. 2016, 109, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S. Medical uses of mylabris in ancient China and recent studies. J. Ethnopharmacol. 1989, 26, 147–162. [Google Scholar] [CrossRef]
- D’Arcy, B.M.; Swingle, M.R.; Papke, C.M.; Abney, K.A.; Bouska, E.S.; Prakash, A.; Honkanen, R.E. The Antitumor Drug LB-100 Is a Catalytic Inhibitor of Protein Phosphatase 2A (PPP2CA) and 5 (PPP5C) Coordinating with the Active-Site Catalytic Metals in PPP5C. Mol. Cancer Ther. 2019, 18, 556–566. [Google Scholar] [CrossRef]
- Chen, X.Y.; Cai, C.Z.; Yu, M.L.; Feng, Z.M.; Zhang, Y.W.; Liu, P.H.; Zeng, H.; Yu, C.H. LB100 ameliorates nonalcoholic fatty liver disease via the AMPK/Sirt1 pathway. World J. Gastroenterol. 2019, 25, 6607–6618. [Google Scholar] [CrossRef]
- Ho, W.S.; Sizdahkhani, S.; Hao, S.; Song, H.; Seldomridge, A.; Tandle, A.; Maric, D.; Kramp, T.; Lu, R.; Heiss, J.D.; et al. LB-100, a novel Protein Phosphatase 2A (PP2A) inhibitor, sensitizes malignant meningioma cells to the therapeutic effects of radiation. Cancer Lett. 2018, 415, 217–226. [Google Scholar] [CrossRef]
- Maggio, D.; Ho, W.S.; Breese, R.; Walbridge, S.; Wang, H.; Cui, J.; Heiss, J.D.; Gilbert, M.R.; Kovach, J.S.; Lu, R.O.; et al. Inhibition of protein phosphatase-2A with LB-100 enhances antitumor immunity against glioblastoma. J. Neurooncol. 2020, 148, 231–244. [Google Scholar] [CrossRef]
- Chung, V.; Mansfield, A.S.; Braiteh, F.; Richards, D.; Durivage, H.; Ungerleider, R.S.; Johnson, F.; Kovach, J.S. Safety, Tolerability, and Preliminary Activity of LB-100, an Inhibitor of Protein Phosphatase 2A, in Patients with Relapsed Solid Tumors: An Open-Label, Dose Escalation, First-in-Human, Phase I Trial. Clin. Cancer Res. 2017, 23, 3277–3284. [Google Scholar] [CrossRef]
- Sacharidou, A.; Chambliss, K.L.; Ulrich, V.; Salmon, J.E.; Shen, Y.M.; Herz, J.; Hui, D.Y.; Terada, L.S.; Shaul, P.W.; Mineo, C. Antiphospholipid antibodies induce thrombosis by PP2A activation via apoER2-Dab2-SHC1 complex formation in endothelium. Blood 2018, 131, 2097–2110. [Google Scholar] [CrossRef] [PubMed]
- Gergs, U.; Boknik, P.; Buchwalow, I.; Fabritz, L.; Matus, M.; Justus, I.; Hanske, G.; Schmitz, W.; Neumann, J. Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J. Biol. Chem. 2004, 279, 40827–40834. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Li, L.; Gu, P.; Jin, T.; Wen, M.; Yuan, C.; Gao, X.; Liu, C.; Zhang, Z. Profiling metabolic remodeling in PP2Acα deficiency and chronic pressure overload mouse hearts. FEBS Lett. 2015, 589, 3631–3639. [Google Scholar] [CrossRef]
- Hoehn, M.; Zhang, Y.; Xu, J.; Gergs, U.; Boknik, P.; Werdan, K.; Neumann, J.; Ebelt, H. Overexpression of protein phosphatase 2A in a murine model of chronic myocardial infarction leads to increased adverse remodeling but restores the regulation of β-catenin by glycogen synthase kinase 3β. Int. J. Cardiol. 2015, 183, 39–46. [Google Scholar] [CrossRef]
- Neumann, J.; Eschenhagen, T.; Jones, L.R.; Linck, B.; Schmitz, W.; Scholz, H.; Zimmermann, N. Increased expression of cardiac phosphatases in patients with end-stage heart failure. J. Mol. Cell Cardiol. 1997, 29, 265–272. [Google Scholar] [CrossRef]
- Brewis, N.; Ohst, K.; Fields, K.; Rapacciuolo, A.; Chou, D.; Bloor, C.; Dillmann, W.; Rockman, H.; Walter, G. Dilated cardiomyopathy in transgenic mice expressing a mutant A subunit of protein phosphatase 2A. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H1307–H1318. [Google Scholar] [CrossRef] [PubMed]
- Reynhout, S.; Janssens, V. Physiologic functions of PP2A: Lessons from genetically modified mice. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 31–50. [Google Scholar] [CrossRef]
- Zaveri, S.; Srivastava, U.; Qu, Y.S.; Chahine, M.; Boutjdir, M. Pathophysiology of Cav1.3 L-type calcium channels in the heart. Front. Physiol. 2023, 14, 440. [Google Scholar] [CrossRef]
- Galbo, T.; Olsen, G.S.; Quistorff, B.; Nishimura, E. Free fatty acid-induced PP2A hyperactivity selectively impairs hepatic insulin action on glucose metabolism. PLoS ONE 2011, 6, e27424. [Google Scholar] [CrossRef]
- Matsumoto, M.; Pocai, A.; Rossetti, L.; Depinho, R.A.; Accili, D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 2007, 6, 208–216. [Google Scholar] [CrossRef]
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Ogawa, W.; Akimoto, K.; Inoue, H.; Miyake, K.; Furukawa, K.; Hayashi, Y.; Iguchi, H.; Matsuki, Y.; Hiramatsu, R.; et al. PKClambda in liver mediates insulin-induced SREBP-1c expression and determines both hepatic lipid content and overall insulin sensitivity. J. Clin. Investig. 2003, 112, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.A.; Lee, D.P. Enzymes of triacylglycerol synthesis and their regulation. Prog. Lipid Res. 2004, 43, 134–176. [Google Scholar] [CrossRef]
- Standaert, M.L.; Sajan, M.P.; Miura, A.; Kanoh, Y.; Chen, H.C.; Farese, R.V., Jr.; Farese, R.V. Insulin-induced activation of atypical protein kinase C, but not protein kinase B, is maintained in diabetic (ob/ob and Goto-Kakazaki) liver. Contrasting insulin signaling patterns in liver versus muscle define phenotypes of type 2 diabetic and high fat-induced insulin-resistant states. J. Biol. Chem. 2004, 279, 24929–24934. [Google Scholar] [CrossRef] [PubMed]
- Cazzolli, R.; Carpenter, L.; Biden, T.J.; Schmitz-Peiffer, C. A role for protein phosphatase 2A-like activity, but not atypical protein kinase Czeta, in the inhibition of protein kinase B/Akt and glycogen synthesis by palmitate. Diabetes 2001, 50, 2210–2218. [Google Scholar] [CrossRef]


{kind=link}
| Disease | PP2A Status | Mechanisms for Altered PP2A | Downstream of PP2A Signaling | Reference |
|---|---|---|---|---|
| COPD | Reduced PP2A responses | Increased CIP2A and SET signaling, reduced antioxidant responses, viral induced suppression of PP2A, inhibition of PTP1B responses, phosphorylation of the C subunit of PP2A | Increased: Immune cell infiltration, MAPK signaling, Cathepsin S expression, phosphorylation of TTP, innate inflammation, NFκB signaling | [43,44,52,53,54,55] |
| AAT deficiency | Reduced PP2A responses | Loss of AAT, inhibition of PTP1B responses, reduced PKA responses | Increased: Innate immune responses, proteolytic responses | [45] |
| Lung cancer | Reduced PP2A responses | Loss of PP2A subunits via gene mutations, increased CIP2A expression, and EGFR mutations | Dysfunction of downstream GTPase RalA, dysfunctional cell growth, migration, and apoptosis. Increased ERK, MKK4, ATF2, and c-Jun Akt, and c-Myc signaling | [46,56,57,58,59,60,61,62,63,64] |
| Asthma | Reduced PP2A responses | Eosinophil peroxidase enhances PP2A phosphorylation but other mechanisms may exist for suppression of PP2A | Increased: Phosphorylation of p38, JNK-1 and GR at site Ser226, CCL4, IL-13, and iNOS expression, serum IgE levels | [65,66,67,68,69] |
| IPF | Reduced PP2A responses | Low α2β1 integrin receptor concentrations lead to decreased PP2A levels; elevated MID1 and TRAIL expression | Increased: Inflammation, profibrotic genes, pulmonary fibrosis, and matrix collagen deposition in mouse models | [50,51,70,71] |
| Compound/Drug | Mode of Action | Impact on PP2A | FDA-Approval Status | Downstream Effects | Reference |
|---|---|---|---|---|---|
| FTY720 | FTY720 binds I2PP2A/SET at the K209/Y122 residue, inactivating SET | Indirectly increases PP2A activity | Fingolimod/Gilenya®, FDA-approved drug by Novartis to treat multiple sclerosis | Suppression of c-myc and upregulation of NDRG1, Reduces EMT | [127,128,129] |
| FTY720 derivative: AAL(s), OSU-2S | Disruption of the SET-PP2A complex | Indirectly increases PP2A activity | Experimental use only | AAL(s) enhances TTP responses | [79,130] |
| FTY720 derivatives: MP07-66 | Disruption of the SET-PP2A complex | Indirectly increases PP2A activity | Experimental use only | Antiproliferative activity in human hepatocellular carcinoma without immunosuppressive effects | [131] |
| ApoE-derived peptides: COG1410, COG112 | Binding to the C-terminal end of SET | Indirectly increases PP2A activity | Experimental use only | Inhibition of Akt signaling, cellular proliferation, cellular migration, and invasion | [132,133,134] |
| TGI1002 | Disrupts SET-PP2A interaction | Indirectly increases PP2A activity | Experimental use only | Increases dephosphorylation of BCR-ABL, inhibits tumor growth | [135] |
| EGFR kinase inhibitors: erlotinib | Erlotinib inhibits CIP2A responses | Indirectly increases PP2A activity | Erlotinib (Tarceva®) is approved for treating EGFR-mutant NSLC | Induces apoptosis in hepatocellular carcinomas, reduces smoke induced innate immune and protease responses | [44,136,137] |
| Erlotinib derivative: TD52 | Inhibits CIP2A independently of EGFR signaling | Indirectly increases PP2A activity | Experimental use only | Reduces CIP2A signaling, tumor burden, and increased apoptosis | [138,139,140] |
| Proteasome inhibitor: bortezomib | Suppresses CIP2A by undefined mechanism | Indirectly increases PP2A activity | Bortezomib (Velcade®), approved for treating multiple myeloma | Tumor growth inhibition | [141,142,143] |
| Metformin | CIP2A inhibition | Indirectly increases PP2A activity | Approved as an antidiabetic agent used in type 2 diabetes mellitus | Inhibition of GSK3β, represses tumor growth, indirectly leads to dephosphorylation of many proteins | [144,145] |
| Celastrol (tripterine) | CIP2A inhibition through the ubiquitin-proteasome pathway | Indirectly increases PP2A activity | Experimental use only | Inhibited cell proliferation and induced apoptosis in NSCL | [102] |
| Ethoxysanguinarine | CIP2A inhibition | Indirectly increases PP2A activity | Experimental use only | Downregulates c-Myc and pAkt, inhibits proliferation and induces apoptosis of lung cancer cells | [101] |
| PME-1 inhibitors: AMZ30 and ML174 | Inhibit PME-1 signaling | Reduces demethylation of PP2A, increases PP2A activity | Experimental use only | Decreases cell proliferation and invasive growth in vitro | [146] |
| SMAPs: DBK-1154, DT-382, DT-794, DT-061, and ATUX-792 | SMAP binding stabilizes and promotes PP2A heterotrimeric holoenzyme assembly | Directly activate PP2A | Experimental use only | Increased ADP-ribose cleavage, increased tumor cell death, increase tumor necrosis, reduce cathepsin S expression, MAP kinases responses | [43,46,147,148] |
| Xylulose-5-phosphate | Increases free phosphate | Indirectly increases PP2A activity | Experimental use only | Possible regulation of glucose metabolism and fat synthesis | [149] |
| α-Tocopheryl succinate | Unknown mechanism | Unknown if direct or indirect PP2A activation | Experimental use only | Inhibition of JNK, Akt, MAPK, NFκB, Sp1 and the androgen receptor | [150,151] |
| Forskolin | Unknown mechanism | Unknown if direct or indirect PP2A activation | Experimental use only | Dephosphorylation of PP2A substrates such as EF-2 and RB | [152] |
| Chlorpromazine (Thorazine) | Same mechanism as SMAPs | Direct activation of PP2A | Phenothiazine neuroleptic, FDA approved for short-term management of severe anxiety and psychotic aggression | Dephosphorylation of multiple PP2A substrates and subsequently induces apoptosis | [153] |
| Salmeterol | Unknown mechanism | Unknown if direct or indirect PP2A activation | FDA approved in the management and treatment of asthma and COPD | Reduced immune cell infiltration and innate immune responses in HDM mouse model | [84] |
| Theophylline | Unknown mechanism | Unknown mechanism to activate PP2A but is independent of its inhibition of PDE | FDA approved for the treatment of asthma and COPD | Inhibits type III and type IV phosphodiesterase (PDE). It also binds to the adenosine A2B receptor | [82,154,155,156] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Zaveri, S.; Sattar, Z.; Schaible, M.; Perez Gandara, B.; Uddin, A.; McGarvey, L.R.; Ohlmeyer, M.; Geraghty, P. Protein Phosphatase 2A as a Therapeutic Target in Pulmonary Diseases. Medicina 2023, 59, 1552. https://doi.org/10.3390/medicina59091552
Yu H, Zaveri S, Sattar Z, Schaible M, Perez Gandara B, Uddin A, McGarvey LR, Ohlmeyer M, Geraghty P. Protein Phosphatase 2A as a Therapeutic Target in Pulmonary Diseases. Medicina. 2023; 59(9):1552. https://doi.org/10.3390/medicina59091552
Chicago/Turabian StyleYu, Howard, Sahil Zaveri, Zeeshan Sattar, Michael Schaible, Brais Perez Gandara, Anwar Uddin, Lucas R. McGarvey, Michael Ohlmeyer, and Patrick Geraghty. 2023. "Protein Phosphatase 2A as a Therapeutic Target in Pulmonary Diseases" Medicina 59, no. 9: 1552. https://doi.org/10.3390/medicina59091552